
Abstract
Nodal peripheral T-cell lymphomas are not well understood, and most of them are classified in the “not otherwise specified group” (PTCL-NOS). Data on their normal cellular derivation are ambiguous. Most peripheral T-cell lymphomas are composed of tumor cells and a (sometimes dominant) reactive background, which also includes resting and activated T-lymphocytes. We defined the phenotype of the tumor cells in 101 PTCL-NOS based on their cytological atypia and using immunohistochemical double stains on paraffin sections with CD4/Ki67 and CD8/Ki67. The results were correlated to clinical presentation and outcome. Lineage could be defined in 98 cases (97%). Tumor cells were CD4+ in 43 cases and CD8+ in 38. These presented at a younger age but a higher clinical stage compared with the CD4+ lymphomas. In 15 cases, the atypical cells were CD4−CD8−; two cases were CD4+CD8+. Of 17 lymphoepithelioid (Lennert’s) lymphomas, 15 expressed CD8, one each was CD4+ and CD4−CD8−.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Peripheral T-cell lymphomas (PTCL) comprise a heterogeneous group, and their classification is still debated. While the working formulation [1]—classifying malignant lymphomas on the basis of hematoxylin and eosin (H&E) sections alone—did not recognize PTCL as a separate group, the Kiel classification [44]—listing entities corresponding to a proposed normal counterpart [46]—distinguished PTCL from the more common B-cell lymphomas and defined various morphological subtypes. Despite their sound conceptual background, these categories were poorly reproducible [13, 18] and only vaguely correlated with clinical presentation and outcome [9, 38, 43]. While the Kiel classification comprises nodal lymphomas only, the extranodal PTCL could not be satisfactorily diagnosed in this scheme.
In the Revised European–American Classification of Lymphoid Neoplasms (REAL) [16] and World Health Organization (WHO) [21] classifications of malignant lymphoma, clinicopathological entities are defined by their morphological, immunophenotypical, genetic, and clinical characteristics. Applying these principles, the clinical presentation is an important feature and defines most extranodal PTCL [24, 38]. Among nodal PTCL, anaplastic large cell lymphoma and angioimmunoblastic T-cell lymphoma represent disease entities that are fairly frequent in the Western population. Other PTCL categories show considerable overlap in both their pathological and clinical characteristics [38]. These form the—admittedly heterogeneous—category of “not otherwise specified” PTCL (PTCL-NOS) [16], a category that comprises several morphological variants that correspond to entities defined in the Kiel classification: a medium-sized cell, a mixed medium and large cell, and a large cell variant as well as T-zone and lymphoepithelioid (Lennert’s) lymphoma (LeL). The international non-Hodgkin’s lymphoma classification project [2, 38] proved that PTCL can be reliably diagnosed according to the REAL classification. Variants of PTCL-NOS were not clinically distinguishable and are all equally characterized by a poor prognosis when using currently available therapy.
Lacking reproducible criteria to subclassify PTCL-NOS morphologically, tumor cell immunophenotype has been suggested to be a good basis for their classification. This approach seems particularly tempting because peripheral post-thymic T-cells, with few exceptions [14], exclusively express the lineage-specific molecules CD4 or CD8. Functional subsets comprise helper and cytotoxic T-cells, as well as T/NK-cells. While subsets of helper T-cell lymphomas (TH1 and TH2) cannot be defined in paraffin sections, cytotoxic granules and CD56 are easily detectable. However, phenotypical data on nodal PTCL are heterogeneous, regarding both tumor cell lineage and expression of cytotoxic granules [5, 7, 9, 30, 32, 33, 35, 39].
This heterogeneity might result from technical problems because, in addition to the neoplastic cells, PTCL also comprises varying numbers of reactive T-lymphocytes [16, 22, 27, 42]. Since most of these studies were performed, methods have become available to assess the morphology in immunostained slides and to double stain cells on paraffin sections. Applying these, we tried to define the phenotype of tumor cells in 101 PTCL-NOS and to correlate it to clinical presentation and follow-up.
Materials and methods
Altogether, we analyzed 101 cases of PTCL-NOS, including 18 LeLs. All cases were stained with H&E, Giemsa, periodic acid-Schiff, and silver impregnation (Gömöri) to assess morphology. Diagnoses were made according to the WHO classification [21]. Cases were included in this study as variants of PTCL-NOS after better-defined entities of PTCL had been ruled out. Cases with prominent CD30 expression and sinus infiltration in particular were diagnosed as anaplastic large cell lymphoma and excluded from the study. Angioimmunoblastic T-cell lymphomas were identified by their prominent proliferation of follicular dendritic cell networks and published separately [27].
Routine immunohistochemical stains were performed using mouse monoclonal antibodies against CD3, CD5, CD4, CD8, TIA-1, granzyme B, CD56, CD57, CD20, CD21, Ki-67, CD30, and latent membrane protein (LMP). T-cell receptor γ gene rearrangement [48] was studied if necessary to confirm the diagnosis.
Immunohistochemical double stains were performed for CD4/Ki67 and CD8/Ki67 to assess the proliferation of these subpopulations individually. For double stains, antibodies against CD4 or CD8 and Ki-67 were applied in consecutive steps and visualized with AEC (Zymed, Berlin, Germany) and Fast Blue (Zymed, Berlin, Germany), respectively. Prior to the application of the second antibody, free binding sites were blocked with Fab fragment goat anti-mouse IgG.
Tumor cell lineage was defined by assessing the staining of atypical T-cells in conventional stains and the proliferation rate in the double stains. Proliferation was assessed independently for the CD4+ and CD8+ population, counting Ki67 staining in 200 cells that stained positively for the respective antibody. In histiocyte-rich cases, morphology and staining intensity for CD4 were used as criteria to distinguish T-cells from histiocytes. Nuclear atypia was evaluated in additional single stains for CD4 and CD8. Tumors were regarded as positive for a particular antibody if more than 30% of morphologically identifiable tumor cells were clearly stained, regardless of their proliferation rate.
Clinical data could be obtained retrospectively for 66 patients from patient records. Estimates of overall survival distribution and failure-free survival were calculated using the method of Kaplan and Meier [23]; time-to-event distributions were compared using the log-rank test. The individual items for histological features, immunological, and clinical parameters, including international prognostic index (IPI) [40], were estimated in correlation with the other items and clinical outcome using χ2 tests or t-tests as appropriate. Statistical significance was assumed if P<0.05.
Results
Our study included 101 patients with PTCL-NOS diagnosed from lymph-node biopsies. The majority of these lymphomas consisted of a mixed medium-sized and large cell population (56 cases), while predominantly medium-sized or large tumor cells occurred in 10 and 17 cases, respectively. LeL was diagnosed in 18 cases according to the WHO classification when clusters of epithelioid histiocytes were prominent in the infiltrate.
While most of the tumors infiltrated into the perinodal fat (43%) or infiltrated the lymph-node capsule (19%), 38% were confined to the lymph-node. In 32% of our cases, the lymph-node tissue was only partially effaced, and areas of pre-existing lymphatic tissue were still present. Within the infiltrated areas, remnant regressive networks of follicular dendritic cells, partially infiltrated by the tumor, were identified in 32% of the cases. The lymph-node sinuses were partially or completely obliterated by tumor cells in 52% of cases. Cytologically, the tumor cell nuclei were mildly (54%), moderately (32%), or markedly (14%) atypical. Clear cells were prominent in 43% of all cases and detectable in another 27% (Fig. 1).

Morphological variability of peripheral T-cell lymphomas “not otherwise specified.” A Spread: lymphoma confined to the lymph node (left), infiltrating the capsule (middle) or perinodal fat (right). B Remnant reactive uninvolved areas. C Remnant follicles in the infiltrated areas (CD3 and CD21). D Mild (left), moderate (middle), and severe (right) cellular atypia
Accompanying reactive changes comprised various degrees of eosinophilia, plasmacytosis, and blood vessel proliferation encountered in 44%, 51%, and 79% of our cases, respectively. Epithelioid cells formed granulomas that were infiltrated by tumor cells in the 18 cases of LeL. Histiocytes were prominent in 20 additional cases, but did not form granulomas and were not epithelioid. Tumors were partially necrotic in 14 of 101 cases, and necrosis was extensive in an additional 9 tumors. In 3 of these 23 cases, these necroses were accentuated in lymph-node sinuses.
Immunophenotypes
Our PTCL were positive for CD3 in almost all cases (93%), but only 78% and 13% expressed CD5 and CD7, respectively. Altogether, 29% of all cases exhibited an antigen loss, i.e., were negative for CD3, CD5, or both lineage markers (CD4 and CD8). Only three cases were negative for both CD3 and CD5, and two of these expressed CD56. Immunophenotypes are detailed in Table 1.
Based on nuclear atypia and proliferation of T-cell subsets, lineage could be defined in 98 cases (97%): tumor cells were CD4+ in 43 cases and CD8+ in 38 cases (Fig. 2). In 15 cases, the atypical cells were negative for both antigenes, and these were considered to be double negative (DN). Two cases expressing both CD4 and CD8 on the tumor cells (double positive, DP) showed the morphology of PTCL-NOS and were negative for both TdT and CD99.

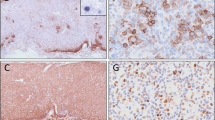
Definition of tumor cell lineage in a CD8+ peripheral T-cell lymphomas “not otherwise specified”: CD3− (upper left), CD5+ (upper right). Ki67 (red) labels CD8+ cells (blue, lower right), but not CD4+ cells (blue, lower left)
Correlating tumor cell lineage to the results of the double stains (that could both be evaluated in 69 cases), the proliferation index of the neoplastic subpopulation exceeded that of the reactive one (P<0.01, Fig. 3) in all but 14 cases (11 CD4+ and 3 CD8+). In these latter cases, the nuclear atypia was clear cut, and tumor cell lineage definition was based on this.

Mean proliferation and 95% confidence interval for tumors with lineage defined as CD4 or CD8
Concerning the background infiltrate, histiocytes and/or epithelioid cells occurred more frequently in CD8+ than in CD4+ lymphomas (P<0.001). This association remained significant (P=0.004) after excluding the LeL, which, by definition, contain epithelioid cell granulomas. Among the CD4+ lymphomas, epithelioid cell clusters were seen more often in those that coexpressed cytotoxic molecules than in others (P<0.001).
Cytotoxic molecules (either TIA1, granzyme B or both) were detected in 38 cases (38%) altogether. Their expression was strongly correlated with lineage (P<0.01): while 26 of 38 (68%) of the CD8+ tumors were cytotoxic, only 7 of 43 (16%) CD4+ lymphomas expressed cytotoxic granules. Additionally, 4 of 15 DN tumors and 1 of the 2 DP lymphomas expressed cytotoxic granules. As a group, cytotoxic lymphomas lacked CD5 more frequently (37%) than non-cytotoxic lymphomas (14%; P=0.03).
Compared with other patients, those with cytotoxic lymphoma presented at younger age (57 years versus 65 years; P=0.024) but more often at an advanced stage (stage 3/4: 85% versus 56%; P=0.034). However, their IPI scores and outcomes were similar to non-cytotoxic lymphoma.
CD30, an activation marker in T-cells, was detected in 18% of our cases in individual tumor cells. It was more frequently detected in tumors composed of large atypical cells; however, its expression was often weak and inconsistent among tumor cells in contrast to the strong membrane staining observed in anaplastic large cell lymphoma, LMP-positive B-cells were detected in the background of three cases.
Of the 101 nodal PTCL, 4 expressed CD56 (Table 2). None of them was associated with Epstein–Barr virus by LMP stain or by Epstein–Barr encoded RNA in situ hybridization. This group included two of the three CD3−CD5− cases. In two cases, T-cell receptor (TCR)γ was clonally rearranged. In the other cases, the DNA quality did not allow the respective investigation. Three of these patients presented in their 80s, but one patient was only 21 years old. Follow-up was available for three of these patients. They had clinical stage 3, and no sites of extranodal involvement were detected; however, there was marked blood leukocytosis in two patients. The young patient died from the disease within 8 days of diagnosis, but the two elderly patients were alive after 5 months and 23 months, respectively.
Variants
The subdivision of our cases, according to their cell size, did not reveal any significant differences between the respective groups with regard to immunophenotype. By definition, tumor cell atypia was more pronounced in larger cell tumors.
Among the morphological variants, LeL exhibited a distinct phenotype; 15 cases (88%) were derived from CD8+ cells. One case each was CD4+ and DN, and, in the remaining case, lineage could not be defined due to poor preservation of the material. In the CD8+ LeL, CD4+ cells often predominated in the infiltrate (Fig. 4), which was composed of epithelioid histiocytes, small and activated T-cells. Morphologically, these T-cells did not exhibit nuclear atypia or atypical mitoses, features that were confined to the CD8+ population. Therefore, the phenotype of LeL significantly differed from other PTCL-NOS (P<0.001) that were CD4+ in 51%, CD8+ in 28%, DN in 17%, and DP in 2% of the cases (Table 1). Cytotoxic molecules were detected in 11 of 18 LeL cases (61%), including the CD4+ LeL, compared with 43% in the other group (P=0.03). LeL also contained significantly fewer plasma cells than the other PTCL-NOS. The clinical presentation, however, was not significantly different for LeL versus other PTCL-NOS.

Lennert’s lymphoma. Hematoxylin and eosin (top), CD4− (bottom left), CD8+ (bottom right)
Clinicopathological correlations
Detailed clinical data were available for 66 patients (Table 3) who were mostly elderly and presented at a high clinical stage but ambulatory performance status. Most reached remission after therapy, but the long-term outcome was poor (Fig. 5).

Overall survival and freedom from treatment failure in 66 patients with available follow-up
Treatment was variable: while 51 patients received an intense treatment, 15 patients were treated with less intense regimens. Intensely treated patients achieved higher remission rates (P<0.01) than others (81% versus 41% complete or partial remission; P<0.01), resulting in a significantly better overall survival (OAS) (P=0.013), but not freedom from treatment failure (FFTF).
As detailed in Table 4, survival could be predicted by patient characteristics and response to therapy, but individual morphological or immunophenotypical parameters failed to predict survival. Compared with other CD8+ PTCL, patients with CD8+ LeL (CD8+ cases only) showed a significantly better FFTF (P=0.027) but not OAS.
Discussion
We studied 101 cases of nodal PTCL-NOS with special regard to the immunophenotype of the tumor cells. Nodal PTCL-NOS were derived from CD8+ or cytotoxic cells in 38% of our cases. Although we used a meticulous approach to define tumor cell phenotype, it did not distinguish clinically different groups. The clinical presentation is, rather, correlated to the expression of cytotoxic granules than to derivation from CD4+ or CD8+ cells. Patients’ outcome can be predicted using a morphological grading of PTCL-NOS. LeL is mainly derived from CD8+ cytotoxic cells and has a better survival than other CD8+ lymphomas.
Lineage of PTCL
The definition of the functional phenotype of the tumor cells of the 101 PTCL-NOS was a major goal of our study. Although some cases with large cell morphology are composed of an almost pure proliferation of tumor cells, most PTCL-NOS—in contrast to B-cell lymphomas—comprise a heterogeneous cellular population [16, 22, 42]. Apart from the tumor cells, there were abundant histiocytes, eosinophils, plasma cells, B-lymphocytes, and reactive T-lymphocytes. These reactive T-cells greatly complicate the study of PTCL. The mixed composition, comprising both neoplastic and reactive T-cells in varying proportions, may cause a lack of identifiable clonal TCR gene rearrangements in some cases of PTCL [50]. Reactive cells may exhibit a mild degree of nuclear atypia and, analogous to T-cell-rich B-cell lymphomas, may form the majority of the infiltrate in PTCL-NOS [37]. However, the neoplastic cells in PTCL cannot yet be specifically stained on histological sections and distinguished from infiltrating reactive T-cells. Therefore, studies attempting to define tumor cell lineage had to make various assumptions.
Some studies [12, 31, 47, 50] determined the proliferation fraction in immunohistochemical double stains and assumed the proliferating fraction to be neoplastic. However, proliferation alone is insufficient to define tumor cells, and the authors themselves found it difficult to associate their signals to morphology in frozen sections [50]. In such material, proliferating cells with their Ki67+ nuclei may be completely surrounded by CD4+ cells; this may easily give the impression that the Ki67+ nuclei are located within CD4+ cells. In LeL, atypical CD8+ cells were often situated within granulomas that were mainly composed of such CD4+ epithelioid cells.
In other studies, the proportion of T-cell subpopulations was compared, and the predominant population was regarded as neoplastic [9, 45]. However, the numbers of CD4+ and CD8+ T-lymphocytes in the infiltrate were often similar [26], and tumor lineage remained undetermined in 36% of 174 PTCL [32]. In a flow cytometry study, 6 of 31 PTCL did not show any subtype predominance [28]. Conceptually, a mere majority of a cell population in an infiltrate does not prove its clonality. For example, tumor cells are in the minority in Hodgkin’s lymphoma, and predominant subpopulations may occur in reactive lesions.
To better distinguish the neoplastic from reactive T-cells, we assumed that tumor cells could be characterized morphologically by atypical mitoses, nuclear atypia, and proliferation. These features can all be assessed in immunostained paraffin sections. To correlate the immunophenotype to morphology, we analyzed double stains for Ki67 and CD4 or CD8, respectively. Although it cannot be ruled out with certainty that two phenotypically different neoplastic populations existed in those 14 cases that showed a higher proliferation in the non-atypical versus atypical cells, proliferation may be considerable in reactive T-cell populations as in infectious mononucleosis. Therefore, we considered clear-cut atypia as definitive proof of malignancy regardless of the proliferation.
This approach allowed us to define tumor cell lineage in 98 of 101 cases (97%). The tumor cells were CD4+ in 43 cases, CD8+ in 38 cases, DN in 15 cases, and DP in 2 cases. None of these phenotypes are actually new to the literature, but the proportion of CD8+ tumors in our study is considerably higher than in previous ones [5, 7, 9, 30, 32, 33, 35, 39] that showed CD8+ tumors in 4–18% only. Apart from the far possibility of a selection bias that we cannot rule out with certainty, these discrepancies most likely result from different approaches to define the tumor cells, as discussed previously. These results suggested that a reliable identification of tumor cells in PTCL-NOS is a prerequisite to classify PTCL successfully.
Lennert’s lymphoma
LeL, with its typical prominent epithelioid cell reaction, was derived from CD8+ cytotoxic T-cells in 15 of 18 cases. In the two remaining evaluable cases, one was CD4+, and one was DN.
In contrast to our results of CD8+ tumor cells in LeL, in the original description [12], the tumor cells in four cases investigated with double stains on frozen sections were CD3+CD4+CD8−. In the early 1990s, CD4+ cells were regarded as the predominant cell type in most LeL [31, 32, 47], but intermingled CD8+ cells were reported. Proliferation seemed to be confined to the CD4+ population, but the authors suggested that CD4+ histiocytes may have distorted this result. While the percentage of CD4+Ki67+ cells was well correlated to clonal TCRβ gene rearrangements in both angioimmunoblastic lymphoma (AILT) and T-zone lymphoma, no such correlation could be established in LeL [47], probably because the clonal TCRβ rearrangement was correlated to the CD4+ population, which should be regarded as reactive.
In our LeL, we also found a quantitative predominance of CD4+ cells. However, using cytological criteria that were difficult to assess in frozen sections, the CD8+ cells, although comprising a minor population, were atypical (Fig. 4), and we, therefore, regarded them as neoplastic. Recently, Kagami et al. investigated paraffin sections and found six of eight nodal cytotoxic PTCL to be LeLs [22]. Subsequently, they could show that five of ten LeLs were CD8+, and TIA1 and/or granzyme B were expressed in seven of ten cases [51]. Adding our cases to these, evidence is accumulating that, based on the investigation of paraffin sections, LeL is possibly derived from a CD8+ cytotoxic T-cell.
CD8+ LeL also showed a significantly better OAS than other CD8+ PTCL-NOS in our study. Together with the established morphological criteria, CD8 expression and cytotoxic phenotype may well allow us to delineate LeL more precisely.
Other phenotypes
Having identified the tumor cells, it was possible to define the expression of other antigens on the tumor cells (Table 1). Cytotoxic granules (TIA1 or granzyme B) were expressed in 9 of 42 CD4+ tumors and in 32 of 38 CD8+ tumors. Excluding LeL, 31% of the remaining PTCL-NOS were still derived from CD8+ T-cells, and 30% of these expressed cytotoxic markers. This cytotoxic phenotype is well established for primary extranodal lymphomas; anaplastic large cell lymphomas also contain such granules, but they are derived from CD4+ cells [4, 6]. The coexpression of CD4 and cytotoxic granules has been described in nodal PTCL-NOS, but has been thought to be rare [6, 10, 11, 50].
In addition to these major observations, some of our cases exhibited phenotypes that presently cannot be included easily in our understanding of malignant lymphoma mirroring normal T-cell development.
Four cases strongly expressed CD56. While most published CD56+ lymphomas presented at extranodal sites, a few cases without extranodal involvement have been described [6, 7, 25, 34]. Extranodal sites were not involved in our cases; however, white blood cell counts in two patients suggested circulating tumor cells. Being confined to the lymph nodes, these tumors are distinct from CD4+CD56+ plasmacytoid dendritic cell precursor leukemia [8, 36].
Two other cases in our series of PTCL-NOS were positive for both CD4 and CD8. Six similar cases were described by Yamashita et al. [50]. Both morphologically and phenotypically, they represent a very rare and peculiar phenotype of PTCL-NOS.
Loss of any pan-T-cell antigen (CD3, CD5, CD4/8) has been proposed as a major criterion for the immunohistochemical diagnosis of PTCL [35], and it frequently concerns CD5 and CD7 [26]. In our series, CD7 was negative in 87% of cases, and at least one of the other pan-T-cell antigens was negative in 29% of our cases, similar to 31% in Chott’s series [9].
Clinicopathological correlations
The clinical presentation and outcome of our patients was similar to other published studies [9, 38, 49], after 3 years, approximately 30% of the patients were still alive (Fig. 5). In prospective studies, survival reached a plateau at 43% after 3 years [41], and 41% of the patients, treated with an intensive regimen, were alive after 5 years [15]. The poorer outcome in our cohort was probably caused by the heterogeneous treatment, because patients receiving either CHOP [cyclophosphamide, hydroxydaunomycin, vincristine (= Oncovin), predisone (regimen)] or a high-dose regimen with autologous stem cell transplant had a significantly better survival rate than those treated with a less intense regimen. In contrast, in our series of AILT, there was no correlation between primary therapy and survival [27]. These differences between PTCL-NOS and AILT are in concordance with the philosophy of the WHO to define prognostic factors within entities [17, 19, 20].
Outcome in patients with aggressive lymphomas can be predicted by clinical characteristics combined with IPI score [40]. This is also applicable to PTCL as a group [3, 29, 38] and to our cohort. However, the IPI score does not include tumor biology and is probably not very specific for malignant lymphoma.
We, therefore, correlated the phenotypes of our tumors with clinical presentation and outcome. Interestingly, tumors derived from helper and suppressor cells showed virtually identical clinical characteristics, as did the DN cases. LeL, in contrast, had a survival superior to other CD8+ lymphomas. The expression of cytotoxic markers was correlated with clinical presentation but not with outcome. Patients with cytotoxic lymphomas presented at younger age but with more advanced stages.
PTCL-NOS form a heterogeneous group of diseases, exhibiting a wide range of morphology and differentiating along both helper and suppressor lines. LeL is almost homogeneously CD8+, and its survival is different from other CD8+ PTCL-NOS. Immunophenotypical characteristics of the tumor cells cannot distinguish clinically relevant groups with available therapy, but for future studies, it will be crucial to reliably distinguish the tumor cells in PTCL-NOS from reactive T-lymphocytes.
References
(1982) National Cancer Institute sponsored study of classifications of non-Hodgkin’s lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin’s Lymphoma Pathologic Classification Project. Cancer 49:2112–2135
(1997) A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 89:3909–3918
Ansell SM, Habermann TM, Kurtin PJ, Witzig TE, Chen MG, Li CY, Inwards DJ, Colgan JP (1997) Predictive capacity of the International Prognostic Factor Index in patients with peripheral T-cell lymphoma. J Clin Oncol 15:2296–2301
Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugières L, Terrier-Lacombe M-J, Haralambieva E, Pulford K, Pileri S, Morris SW, Mason DY, Delsol G (1998) ALK-positive lymphoma. A single disease with a broad spectrum of morphology. Blood 91:2076–2084
Boulland ML, Kanavaros P, Wechsler J, Casiraghi O, Gaulard P (1997) Cytotoxic protein expression in natural killer cell lymphomas and in alpha beta and gamma delta peripheral T-cell lymphomas. J Pathol 183:432–439
Chan AC, Ho JW, Chiang AK, Srivastava G (1999) Phenotypic and cytotoxic characteristics of peripheral T-cell and NK-cell lymphomas in relation to Epstein–Barr virus association. Histopathology 34:16–24
Chan JK, Sin VC, Wong KF, Ng CS, Tsang WY, Chan CH, Cheung MM, Lau WH (1997) Nonnasal lymphoma expressing the natural killer cell marker CD56: a clinicopathologic study of 49 cases of an uncommon aggressive neoplasm. Blood 89:4501–4513
Chaperot L, Bendriss N, Manches O, Gressin R, Maynadie M, Trimoreau F, Orfeuvre H, Corront B, Feuillard J, Sotto JJ, Bensa JC, Briere F, Plumas J, Jacob MC (2001) Identification of a leukemic counterpart of the plasmacytoid dendritic cells. Blood 97:3210–3217
Chott A, Augustin I, Wrba F, Hanak H, Ohlinger W, Radaszkiewicz T (1990) Peripheral T-cell lymphomas: a clinicopathologic study of 75 cases. Hum Pathol 21:1117–1125
de Bruin PC, Kummer JA, van der Valk P, van Heerde P, Kluin PM, Willemze R, Ossenkoppele GJ, Radaszkiewicz T, Meijer CJ (1994) Granzyme B-expressing peripheral T-cell lymphomas: neoplastic equivalents of activated cytotoxic T cells with preference for mucosa- associated lymphoid tissue localization. Blood 84:3785–3791
Felgar RE, Macon WR, Kinney MC, Roberts S, Pasha T, Salhany KE (1997) TIA-1 expression in lymphoid neoplasms. Identification of subsets with cytotoxic T lymphocyte or natural killer cell differentiation. Am J Pathol 150:1893–1900
Feller AC, Griesser GH, Mak TW, Lennert K (1986) Lymphoepithelioid lymphoma (Lennert’s lymphoma) is a monoclonal proliferation of helper/inducer T cells. Blood 68:663–667
Feller AC, Lennert K, Zwingers T (1991) Peripheral T-cell lymphomas. Histopathology 19:481–484
Flamand L, Crowley RW, Lusso P, Colombini-Hatch S, Margolis DM, Gallo RC (1998) Activation of CD8+ T lymphocytes through the T cell receptor turns on CD4 gene expression: implications for HIV pathogenesis. Proc Natl Acad Sci U S A 95:3111–3116
Gisselbrecht C, Gaulard P, Lepage E, Coiffier B, Briere J, Haioun C, Cazals-Hatem D, Bosly A, Xerri L, Tilly H, Berger F, Bouhabdallah R, Diebold J (1998) Prognostic significance of T-cell phenotype in aggressive non-Hodgkin’s lymphomas. Groupe d’Etudes des Lymphomes de l’Adulte (GELA). Blood 92:76–82
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JKC, Cleary ML, Delsol G, De Wolf Peeters C, Falini B, Gatter KC, Grogan TM, Isaacson PG, Knowles DM, Mason DY, Müller-Hermelink HK, Pileri SA, Piris MA, Ralfkiaer E, Warnke RA (1994) A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 84:1361–1392
Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, Lister TA, Bloomfield CD (1999) The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. Report of the Clinical Advisory Committee meeting, Airlie House, Virginia, November, 1997. Ann Oncol 10:1419–1432
Hastrup N, Hamilton Dutoit S, Ralfkiaer E, Pallesen G (1991) Peripheral T-cell lymphomas: an evaluation of reproducibility of the updated Kiel classification. Histopathology 18:99–105
Jaffe ES (1999) Hematopathology: integration of morphologic features and biologic markers for diagnosis. Mod Pathol 12:109–115
Jaffe ES, Harris NL, Diebold J, Müller-Hermelink HK (1999) World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. A progress report. Am J Clin Pathol 111:8–12
Jaffe ES, Harris NL, Stein H, Vardiman JW (2001) World Health Organization classification of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. IARC Press, Lyon
Kagami Y, Suzuki R, Taji H, Yatabe Y, Takeuchi T, Maeda S, Kondo E, Kojima M, Motoori T, Mizoguchi Y, Okamoto M, Ohnishi K, Yamabe H, Seto M, Ogura M, Koshikawa T, Takahashi T, Kurita S, Morishima Y, Suchi T, Nakamura S (1999) Nodal cytotoxic lymphoma spectrum: a clinicopathologic study of 66 patients. Am J Surg Pathol 23:1184–1200
Kaplan EL, Meier P (1958) Nonparametric estimation from incomplete observations. J Am Stat Assoc 53:457
Kluin PM, Feller A, Gaulard P, Jaffe ES, Meijer CJ, Muller-Hermelink HK, Pileri S (2001) Peripheral T/NK-cell lymphoma: a report of the IXth Workshop of the European Association for Haematopathology. Histopathology 38:250–270
Kobashi Y, Nakamura S, Sasajima Y, Koshikawa T, Yatabe Y, Kitoh K, Mori S, Ueda R, Yamabe H, Suchi T (1996) Inconsistent association of Epstein–Barr virus with CD56 (NCAM)-positive angiocentric lymphoma occurring in sites other than the upper and lower respiratory tract. Histopathology 28:111–120
Krajewski AS, Myskow MW, Cachia PG, Salter DM, Sheehan T, Dewar AE (1988) T-cell lymphoma: morphology, immunophenotype and clinical features. Histopathology 13:19–41
Lee SS, Rudiger T, Odenwald T, Roth S, Starostik P, Muller-Hermelink HK (2003) Angioimmunoblastic T cell lymphoma is derived from mature T-helper cells with varying expression and loss of detectable CD4. Int J Cancer 103:12–20
Macon WR, Salhany KE (1998) T-cell subset analysis of peripheral T-cell lymphomas by paraffin section immunohistology and correlation of CD4/CD8 results with flow cytometry. Am J Clin Pathol 109:610–617
Melnyk A, Rodriguez A, Pugh WC, Cabannillas F (1997) Evaluation of the Revised European–American Lymphoma classification confirms the clinical relevance of immunophenotype in 560 cases of aggressive non-Hodgkin’s lymphoma. Blood 89:4514–4520
Montalban C, Obeso G, Gallego A, Castrillo JM, Bellas C, Rivas C (1993) Peripheral T-cell lymphoma: a clinicopathological study of 41 cases and evaluation of the prognostic significance of the updated Kiel classification. Histopathology 22:303–310
Nakamura S, Suchi T (1991) A clinicopathologic study of node-based, low-grade, peripheral T-cell lymphoma. Angioimmunoblastic lymphoma, T-zone lymphoma, and lymphoepithelioid lymphoma. Cancer 67:2566–2578
Nakamura S, Koshikawa T, Koike K, Kitoh K, Suzuki H, Oyama A, Motoori T, Kojima M, Ogura M, Kurita S et al (1993) Phenotypic analysis of peripheral T cell lymphoma among the Japanese. Acta Pathol Jpn 43:396–412
Ng CS, Lo ST, Chan JK (1999) Peripheral T and putative natural killer cell lymphomas commonly coexpress CD95 and CD95 ligand. Hum Pathol 30:48–53
Ohshima K, Suzumiya J, Sugihara M, Kanda M, Shimazaki K, Kawasaki C, Haraoka S, Kikuchi M (1999) Clinical, immunohistochemical and phenotypic features of aggressive nodal cytotoxic lymphomas, including alpha/beta, gamma/delta T-cell and natural killer cell types. Virchows Arch 435:92–100
Picker LJ, Weiss LM, Medeiros LJ, Wood GS, Warnke RA (1987) Immunophenotypic criteria for the diagnosis of non-Hodgkin’s lymphoma. Am J Pathol 128:181–201
Reimer P, Rudiger T, Kraemer D, Kunzmann V, Weissinger F, Zettl A, Konrad Muller-Hermelink H, Wilhelm M (2003) What is CD4+CD56+ malignancy and how should it be treated? Bone Marrow Transplant 32:637–646
Rüdiger T, Jaffe ES, Delsol G, deWolf Peeters C, Gascoyne RD, Georgii A, Harris NL, Kadin ME, MacLennan KA, Poppema S, Stein H, Weiss LE, Müller-Hermelink HK (1998) Workshop report on Hodgkin’s disease and related diseases (“grey zone” lymphoma). Ann Oncol 9[Suppl 5]:S31–S38
Rüdiger T, Weisenburger DD, Anderson JR, Armitage JO, Diebold J, MacLennan KA, Nathwani BN, Ullrich F, Müller-Hermelink HK (2002) Peripheral T-cell lymphoma. Results from the Non-Hodgkin’s Lymphoma Classification Project. Ann Oncol 13:140–149
Shih LY, Kuo TT, Dunn P, Liaw SJ (1992) HTLV-I-positive and HTLV-I-negative peripheral T-cell lymphomas in Taiwan Chinese. Int J Cancer 50:186–191
Shipp MA (1994) Prognostic factors in aggressive non-Hodgkin’s lymphoma: who has “high-risk” disease? Blood 83:1165–1173
Siegert W, Nerl C, Engelhard M, Brittinger G, Tiemann M, Parwaresch R, Heinz R, Huhn D (1994) Peripheral T-cell non-Hodgkin’s lymphomas of low malignancy: prospective study of 25 patients with pleomorphic small cell lymphoma, lymphoepitheloid cell (Lennert’s) lymphoma and T-zone lymphoma. The Kiel Lymphoma Study Group. Br J Haematol 87:529–534
Smith JL, Haegert DG, Hodges E, Stacey GN, Howell WM, Wright DH, Jones DB (1988) Phenotypic and genotypic heterogeneity of peripheral T-cell lymphoma. Br J Cancer 58:723–729
Sng I, Levin A, Jaffe ES, Ng HW, Sim CS, Blattner WB (1992) T-cell lymphoma in Singapore: pathology, clinical findings and association with HTLV-1 antibodies. Histopathology 21:101–113
Stansfeld AG, Diebold J, Noel H, Kapanci Y, Rilke F, Kelenyi G, Sundstrom C, Lennert K, van Unnik JAM, Mioduszewska O, Wright DH (1988) Updated Kiel classification for lymphomas. Lancet 1:292–293
Su IJ, Wang CH, Cheng AL, Chen YC, Hsieh HC, Chen CJ, Tien HF, Tsay W, Huang SS, Hu CY et al (1988) Characterization of the spectrum of postthymic T-cell malignancies in Taiwan. A clinicopathologic study of HTLV-1-positive and HTLV-1-negative cases. Cancer 61:2060–2070
Suchi T, Lennert K, Tu LY, Kikuchi M, Sato E, Stansfeld AG, Feller AC (1987) Histopathology and immunohistochemistry of peripheral T cell lymphomas: a proposal for their classification. J Clin Pathol 40:995–1015
Takagi N, Nakamura S, Ueda R, Osada H, Obata Y, Kitoh K, Suchi T, Takahashi T (1992) A phenotypic and genotypic study of three node-based, low-grade peripheral T-cell lymphomas: angioimmunoblastic lymphoma, T-zone lymphoma, and lymphoepithelioid lymphoma. Cancer 69:2571–2582
Trainor KJ, Brisco MJ, Wan JH, Neoh S, Grist S, Morley AA (1991) Gene rearrangement in B- and T-lymphoproliferative disease detected by the polymerase chain reaction Blood 78:192–196
Weisenburger DD, Linder J, Armitage JO (1987) Peripheral T-cell lymphoma: a clinicopathologic study of 42 cases. Hematol Oncol 5:175–187
Yamashita Y, Yatabe Y, Tsuzuki T, Nakayama A, Hasegawa Y, Kojima H, Nagasawa T, Mori N (1998) Perforin and granzyme expression in cytotoxic T-cell lymphomas. Mod Pathol 11:313–323
Yamashita Y, Nakamura S, Kagami Y, Hasegawa Y, Kojima H, Nagasawa T, Mori N (2000) Lennert’s lymphoma: a variant of cytotoxic T-cell lymphoma? Am J Surg Pathol 24:1627–1633
Acknowledgement
This work was partly supported by a grant from the Korea Science and Engineering Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Geissinger, E., Odenwald, T., Lee, SS. et al. Nodal peripheral T-cell lymphomas and, in particular, their lymphoepithelioid (Lennert’s) variant are often derived from CD8+ cytotoxic T-cells. Virchows Arch 445, 334–343 (2004). https://doi.org/10.1007/s00428-004-1077-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-004-1077-2