
Abstract
Background
Septoria tritici blotch (STB) disease causes yield losses of up to 50 per cent in susceptible wheat cultivars and can reduce wheat production. In this study, genomic architecture for adult-plant STB resistance in a Septoria Association Mapping Panel (SAMP) having 181 accessions and genomic regions governing STB resistance in a South Asian wheat panel were looked for.
Results
Field experiments during the period from 2019 to 2021 revealed those certain accessions, namely BGD52 (CHIR7/ANB//CHIR1), BGD54 (CHIR7/ANB//CHIR1), IND92 (WH 1218), IND8 (DBW 168), and IND75 (PBW 800), exhibited a high level of resistance. Genetic analysis revealed the presence of 21 stable quantitative trait nucleotides (QTNs) associated with resistance to STB (Septoria tritici blotch) on all wheat chromosomes, except for 2D, 3A, 3D, 4A, 4D, 5D, 6B, 6D, and 7A. These QTNs were predominantly located in chromosome regions previously identified as associated with STB resistance. Three Quantitative Trait Loci (QTNs) were found to have significant phenotypic effects in field evaluations. These QTNs are Q.STB.5A.1, Q.STB.5B.1, and Q.STB.5B.3. Furthermore, it is possible that the QTNs located on chromosomes 1A (Q.STB.1A.1), 2A (Q.STB_DH.2A.1, Q.STB.2A.3), 2B (Q.STB.2B.4), 5A (Q.STB.5A.1, Q.STB.5A.2), and 7B (Q.STB.7B.2) could potentially be new genetic regions associated with resistance.
Conclusion
Our findings demonstrate the importance of Asian bread wheat as a source of STB resistance alleles and novel stable QTNs for wheat breeding programs aiming to develop long-lasting and wide-ranging resistance to Zymoseptoria tritici in wheat cultivars.
Similar content being viewed by others

Introduction
Wheat (Triticum aestivum L.), an important foodgrain with economic impact, reaped significant production gains during the Green Revolution. However, the spectacle in distended wheat production is drastically restrained and challenged by discrete biotic stresses, amongst which prominent fungal pathogens severely decrease the yield and quality of wheat crops [1, 2]. Among these biotic factors, wheat production is limited by a foliar disease caused by the haploid ascomycete pathogenic fungus Zymoseptoria tritici (syn. Mycosphaerella graminicola; anamorph: Septoria tritici), the causal agent of Septoria tritici blotch (STB) disease. The reduction in photosynthetic capacity due to chlorotic and necrotic blotches symptomized by STB causes yield losses of up to 50 per cent in susceptible wheat cultivars [1,2,3]. The disease causes persistent challenges to world wheat growers of temperate climate regimes like Europe, the Mediterranean, Africa, the Americas, and Australia [4, 5]. The changing climate favours moderate rainfall and temperature conditions (20–25 °C with germination at temperatures ranging from 2 °C to 37 °C), increasing STB incidences and yield losses [6,7,8]. Amongst multifaceted disease control strategies, resistance genes/loci discovery and their utilization in resistance breeding are an appealing perspective to achieve an economical, durable, and environmentally friendly control of STB in wheat.
Breeding for qualitative resistance is considerably easier but is subjected to breakdowns periled by the emergence of new virulence in pathogen populations. Such "boom-to-bust" resistances to STB have been observed and reported by different researchers [9,10,11,12,13,14]. A classic example was exhibited by the US cultivar "Gene" with the Stb4 gene, which displayed exceptional resistance to STB in the early 1990s [15]. Still, it became susceptible by the mid-1990s [16] due to the evolution of virulence amongst local Z. tritici populations. Similarly, the wheat variety "Tadinia", having a single dominant resistance gene, Stb4, was used in the California wheat breeding programme and remained effective for 15 years [17] but later became ineffective. Therefore, breeding for STB resistance encompassing quantitative resistance is the most effective, durable, and preferred method [3, 18].
Up to now, 23 resistance genes: Stb1 to Stb20, StbSm3, StbWW, and TmStb1 [19,20,21] have been designated, with two of these, Stb6 and Stb18q have been cloned, encoding a wall-associated receptor kinase-like protein and a plasma membrane cysteine-rich receptor-like kinase, respectively [11, 14]. Additionally, several quantitative trait loci (QTLs) and many marker-trait associations (MTAs) for resistance to Z. tritici have been identified and mapped on the different wheat chromosomes [20]. However, 3BL, 6BS and 7DL chromosome arms seem especially involved in quantitative resistance, highlighting the remarkable complexity and diversity of the genetic underpinnings of STB resistance [19].
So far, STB has not yet been reported in South Asia, and no reports on resistant sources in South Asian wheat germplasm and the underlying resistance mechanism are currently available. Therefore, characterization of the genomic regions governing STB resistance in a South Asian wheat panel is important to supplement the germplasm resources of wheat against STB. Here, we present a genome-wide association study for quantitative resistance to STB under field conditions in a diverse panel of 181 bread-wheat lines. We anticipate that this study provides breeders with a rich basis for improving durable STB resistances in future wheat cultivars.
Results
Resistance spectra of wheat accessions to Zymoseptoria tritici
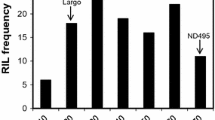
The 181 wheat genotypes tested for STB severity showed different AUDPC scores, with the mean AUDPC scores ranging from 356.58 (BGD 52) to 1384.84 (IND 51) (Table S1). The frequency distribution curve indicated a near-normal distribution for the STB severity (Fig. 1a). Among the 181 SAMP accessions, BGD52 (CHIR7/ANB//CHIR1), BGD54 (CHIR7/ANB//CHIR1), IND92 (WH 1218), IND8 (DBW 168) and IND75 (PBW 800) showed high levels of resistance to STB, with mean AUDPC score of 356.58, 369.86, 370.27, 372.43 and 484.16, respectively (Table S1, Fig. 1b), as compared to susceptible check "Huirivis #1" (1185.49), wheat lines from the cross CHIR7/ANB//CHIR1 showed increased resistance.

Septoria tritici blotch severity in individual years (2019–2021) and across the years (a) Weather data during the cropping season (standard metrological week 21 to 34) (b) area under the disease progress curve (AUDPC) of top ten wheat genotypes showing the least Septoria tritici blotch severity
Descriptive statistical analysis revealed that the mean STB severity showed seasonal fluctuation and was higher during the 2019 growing season (918.74) (Table S2). The genotypic effect was significant (p < 0.01) for STB resistance in individual years, and moderate to high heritability estimates were obtained (Table S3). The combined analysis of variance over years highlighted that the effect of genotype, year, and their two-way interaction (genotype × years) were all highly significant for STB severity (Table 1). The analysis of pooled data revealed that the genotypic variance (35,613.20) was higher than the environmental effect (1353.32) for STB. The broad sense heritability of STB severity was high (H= 88%).
Pearson's correlation analysis of the means over all environments revealed that STB severity was significantly negatively associated with days to heading (r = -0.61) and plant height (r = -0.46) traits (Table 2).
Population structure, diversity, and linkage disequilibrium (LD) analysis
Initially, 13,191 SNPs were scored, of which 9924 (75.23%) were mapped to known chromosomal positions and were well distributed along all the chromosomes of the reference wheat genome (Fig. 2a and b). Maintaining SNPs with higher call rate (> 70%) and MAF > 0.05 resulted in 8353 SNP markers, among which 78% (6524) had a known chromosome position, of which 2,501 were distributed on the A genome, 3,034 on the B genome, and 989 on the D genome. These 8,353 SNPs were used in downstream analyses. Among the 21 wheat chromosomes, the highest and the lowest numbers of SNPs were mapped to chromosomes 2B (594) and 4D (47), respectively (Fig. 2c).

Distribution of DArT Seq SNPs on 181 bread wheat chromosomes (a) SNPs density across the genome; The number of SNPs identified on each chromosome is described at the top of the chromosome. (b): Display of initial 13,191 SNPs used (c): Display of 8353 SNP markers maintained after higher call rate (> 70%) and MAF > 0.05
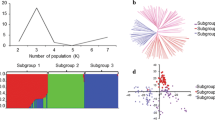
The SAMP was divided into two sub-populations, as confirmed by hierarchical clustering and constellation plots (Fig. S1a and S1b). The phenotyping division of the population based on STB AUDPC is shown in a hierarchical cluster. The wheat lines were randomly distributed across two groups, splitting into nine sub-clusters. Group I contained 51 wheat genotypes, while Group II comprised 130 accessions. All five resistant genotypes having low AUDPC scores for STB (BGD52, BGD54, IND92, IND8, IND75) were reported from Group I, and four of them fell under the same sub-cluster (BGD52, BGD54, IND92, IND8-light green). The Kinship and neighbour-joining cluster analysis also verified the presence of two clusters (Fig. 3). The kinship (Fig. 3C) showed a large proportion of yellow, representing greater diversity among genotypes.

Principal component and familial relatedness analyses of 181 bread wheat genotypes based on 8353 genome-wide scanned high-quality SNPs (a) A screen plot displaying the first 10 principal components with their corresponding fraction of variation explained, (b) 3D plot of the three principal components to depict the response of genotypes to STB (2019,2020 & 2021), days to heading and plant height, and (c) Heat map showing the kinship analysis. The kinship values showed a normal distribution (turquoise curve), and yellow and orange colours represent weak and high kinship relations in the panel, respectively. The resulting clustering tree is indicated outside of the matrix
Linkage disequilibrium varied considerably along individual chromosomes, among chromosomes and sub-genomes (Table S4). Genome A showed a higher proportion of significant marker pairs (38.25%) in comparison to genome B (37.15%) and genome D (23.86%). However, the SNPs on the A genome showed the strongest LD, with a mean value of r2 = 0.14. The marker pairs for chromosomes 4D (r2 = 0.04%) and 2D (r2 = 0.24%) showed the weakest and strongest LD values, respectively. Genome-wide LD decayed to its half at 4.91 Mb, and for A genome at 4.38 Mb, B genome at 5.08 Mb, and D genome at 7.13 Mb (Fig. S2).
Marker-trait association for STB, Pleiotropic regions, and stable QTNs
Across all experiments, a total of 99 quantitative traits SNP's (QTNs) were identified with significant -log10(p) value of ≥ 3 (Fig. 4, Table S5). Among them, 21 QTNs showing repeatability across years were located on all wheat chromosomes except 2D, 3A, 3D, 4A, 4D, 5D, 6B, 6D and 7A (Table 3). The most stable QTNs include Q.STB.5A.1 on 5AS associated with SNP 995502, Q.STB.5B.1 on 5BS associated with SNP 1122319, and Q.STB.5B.3 on 5BL associated with SNP 3222429, they were significant in all experiments as well as the overall mean (Table 3).

Manhattan plot for GWA scan for Septoria progress coefficient measured in different years. Each dot represents a single SNP. On the X-axis is the genomic position of the SNPs on the corresponding chromosomes indicated in different colours. On the Y-axis is the –log10 of the P-value, signifying the significance of the association test. The horizontal dotted line is the Bonferroni correction significance (α = 0.05) threshold used in the association study of STB. Figure 4a-d indicates the significant makers for the STB resistance during 2019, 2020, and 2021 and the mean AUDPC
In addition to QTNs for STB only, there were a few QTNs associated with both STB and DH, like Q.STB_DH.2A.1 and Q.STB_DH.3B.2 that were significant in two years and Q.STB_DH.5A.3 and Q.STB_DH.5D.4 that were significant in one year only (Table S6, Fig. S3). The same applies to Q.STB_DH.UN.5, but its chromosome location is unknown. No common QTN between STB and PH was observed.
In silico analysis
Most SNPs were close to transcripts or candidate genes (CGs) associated with disease resistance. For repetitive QTNs, 42 CGs (Table 3) were sorted, while for unique QTNs, 62 CGs (Table S7) were sorted in the IWGSC RefSeq v1.1 reference genome. The linked genes were associated with pathogen resistance in wheat and other crops, encoding ADP binding, protein-coding/binding, metal ion binding, chromosome binding or lipid binding activities. (Table 3).
Haplotype analysis and staking of R alleles
The 21 significant QTNs in Table 3 were used to understand the frequency of favourable alleles and their effects on STB resistance. The grand mean frequency of favourable alleles in the panel was 43.88%, with the highest percentage of favourable alleles identified in BGD52 (78.57%), followed by IND 75 (71.43) (Table S8). Haplotype analysis conducted on different allele combinations showed a significant reduction in STB severity with increased resistant alleles (Fig. 5). For example, the accession BGD 52 has 11 resistant alleles and exhibited the lowest mean AUDPC score of 356.58, and IND 75, BGD 53, BGD55, and IND 72, having 10 resistant alleles, had low values of 484.16, 579.01, 567.39, 495.88 respectively (Table S8). On the contrary, the most susceptible genotypes, like IND 51 with an AUDPC score of 1384.84, were associated with only one resistant allele.

The decreasing trend of Septoria tritici blotch infection with the stacking of favourable alleles of QTNs and allele frequency of resistant alleles. The Y-axis indicates the AUDPC of the Septoria tritici blotch, while the X-axis indicates the number of resistant alleles and their frequency in the population of the 181 genotypes
Seven significant QTNs i.e., Q.STB.2A.3, Q.STB.2B.4, Q.STB_DH.3B.2, Q.STB.5A.1, Q.STB.5B.1, Q.STB.5B.3, and Q.STB.6A.1, were used in haplotype analysis. As expected, accessions with the resistance alleles exhibited significant lower AUDPC than those with the susceptibility alleles (Fig. 6). The alleles C, G and T alleles of SNP markers 995,502, 1,122,319 and 3,222,429 associated with QTNs- Q.STB.5A.1, Q.STB.5B.1 and Q.STB.5B.3 displayed a significant reduction in STB severity (Fig. 6). The allele G associated with the SNP 1122319 on chromosome 5B (Q.STB.5B.1) had the highest allele frequency (94.77%), followed by allele A associated with SNP 1059080 (88.70%) on chromosome 3B (Q.STB_DH.3B.2) and contributed significantly for reduction in the pycnidia development (Table S8).

The haplotypes of highly significant QTNS linked to Septoria tritici blotch resistance, showing significant differences in AUDPC due to resistance and susceptible alleles. The X-axis indicates the response of the individual allele in QTN regarding the AUDPC. The QTN locus with the corresponding marker on the respective chromosome is named Q.STB., e.g. Q.STB.6A
Discussion
Finding resistance sources and genetic loci for marker-assisted breeding is crucial because most temperate wheat cultivars are susceptible to STB. Bread wheat accessions from India and Bangladesh have not been screened for STB, so this study identifies new sources of STB resistance. Our findings revealed complex genotypic and environmental interactions, quantitative STB resistance inheritance, and novel QTNs for STB breeding programs.
Genetic variation for STB resistance
This study found polygenic control of STB severity due to its near-normal distribution over three years. For future wheat breeding and improvement, phenotypic evaluation showed significant genetic variability among tested wheat genotypes for STB resistance. The broad sense heritability estimated across environments was high, as suggested by previous studies [18, 22,23,24,25]. However, there were significant year- and genotype-by-year effects, supporting the need for multiple-year germplasm evaluation to identify stable STB-resistant genotypes [25, 26].
Many lines from the cross CHIR7/ANB//CHIR1 showed increased resistance, highlighting the significance of the parents in this cross for the STB resistance breeding programme. STB resistance is negatively correlated with PH and DH, among the most confounding factors. Several studies reported increased disease severity in earlier heading and dwarf genotypes [22, 27]. The relationship suggests that genotypes with tall and late phenology may escape STB with reduced infection or that their genes may have pleiotropism or be tightly linked to STB genes. Many GWAS and QTL mapping studies have found genetic linkage to cause the relationship [28, 29]. We found genetic factors supporting the association between STB and DH in QTNs Q.STB_DH.2A.1, Q.STB_DH.3B2, Q.STB_DH.5A.3, and Q.STB_DH.5D.4.
Association analysis and candidate genes for STB resistance
In the current study, the ‘year’ effect significantly influenced disease expression, as observed in other studies on STB resistance, justifying the importance of multiple field trials. The temperature varied between 17 °C to 28 °C and rainfall between 36 to 94 cm during 3 different cropping seasons. The experiments were analyzed individually using pooled data to find the most stable QTNs. Of the 99 identified QTNs, only 21 were significant in more than one environment, and most were of minor effects. Three loci, Q.STB.5A.1, Q.STB.5B.1, and Q.STB.5B.3, were the most stable QTN consistent among all 3 years irrespective of the environmental variations. This agrees well with the idea that resistance against STB in field experiments is a quantitatively inherited trait [19, 30]. It is possible to develop germplasm resistant to STB by combining beneficial alleles from multiple loci. This can be achieved by adding the alleles from their respective donors to the current SAMP.
Out of the 21 repeatable STB QTNs that were identified, the presence of allele A on Q.STB.2A.3, Q.STB.2B.4, Q.STB_DH.3B.2, Q.STB.6A resulted in a decrease in STB severity. Additionally, the alleles C, G, and T on QTNs Q.STB.5A.1, Q.STB.5B.1, and Q.STB.5B.3 were also associated with significantly reduced STB severity. The seven QTNs mentioned above are located within the previously identified QTLs for STB resistance, specifically in the regions of MQTL14 on chromosome 3B (identified at both seedling and adult stages) and QTL10 on chromosome 5B (identified at the seedling stage) [19].
Some tagged QTNs concurred with the known mapping locations for STB resistance genes (Fig. 7). For example, the markers of Q.STB.1D.1 were positioned at similar chromosomal region as STB resistance genes QStb.ipk-1D and close to Stb10; Q.STB_DH.3B.2 and Q.STB.3B.1 was positioned at MQTL14; Q.STB.5B.1, Q.STB.5B.3 at QTL10; Q.STB.6A.1, Q.STB.6A.3 at MQTL20; and Q.STB.7D.5 at MQTL26 region of previously published QTLs [19]. The QTNs, Q.STB.1B.2 and Q.STB.1B3 on chromosome 1B were found close to SSR wmc206, which was linked to MQTL3. Similarly, QTNs, Q.STB.2A.6 and Q.STB.2A.7 were close (3-4 Mb) to marker gwm294, conferring MQTL5 for STB resistance. The Q.STB_DH2A.1 was located close to MQTL4 and is responsible for adult plant resistance [19]. On chromosome 5A, we detected QTNs, Q.STB.5A.5 positioned at 417.22 Mb and Q.STB_DH.5A.3 positioned at 588.45 Mb, which are in the region of a previously published MQTL19 and QTL9 [3] respectively, conferring resistance to STB. However, both QTL9 and MQTL19 provide STB resistance at the seedling stage, while in our study, the QTNs were associated with adult plant resistance. Moreover, the QTN, Q.STB_DH.5A.3 also coincided with the Stb17 gene having a quantitative effect on disease at the adult plants stage [31]. Stb10 in chromosome 1D is a qualitative resistance gene imparting durable resistance to STB [32]. The QTL10 and MQTL9 are responsible for seedling resistance, while MQTL14, MQTL20 and MQTL19 are effective at seedling and adult plant stages. Interestingly, four SNPs synchronizing with QTLs: MQTL3 at 1B chromosome, MQTL5 and MQTL4 on 2A chromosome and MQTL26 at 7D chromosome imparting adult plant resistance were also mapped. Brown et al. [19] also reported the involvement of QTLs on chromosomes 1B, 2A, and 7D, imparting quantitative resistance to STB, which is also true in our findings. Hence, strategic use of the identified major and minor genes/QTNs can be used to deploy STB-resistant varieties.

Location in the wheat genome of major genes, QTL, meta-QTL and QTNs involved in resistance to Septoria tritici blotch in 181 wheat genotypes
Q.STB.1A.1, Q.STB_DH 2A.1, Q.STB.2A.3, Q.STB.2B.4, Q.STB.5A.1, Q.STB.5A.2 and Q.STB.7B.2 didn't coincide the location on the previously reported STB resistance loci in respective chromosomes. At the 7B chromosome, the Q.STB.7B.2 didn't coincide with Stb13, Stb3 or Stb 8 genes and any of the previously reported QTLs (MQTL25, MQTL26). Similar was the case with other QTNs. We have found novel QTNs on chromosomes 1A, 2A, 2B,5A and 7B, which can be good candidates for MAS for the STB resistance wheat breeding programme.
Examining the putative CG associated with significant SNP markers and STB resistance is crucial. Q.STB_DH 2A.1, 2A.3, 2A.6, and 2A.7 were mapped on chromosome 2A. SNP 3955868 at 106.81 Mb is associated with the putative candidate gene linked to this marker, TraesCS2A02G159300 (glycoside hydrolase superfamily). The loci covered glycosyl group manipulation genes. Molecular modifications by glycosylation change protein properties, activity, and target location. Glycosylation of metabolites and hormones occurs during biotic and abiotic stress. He et al. [33] found that wheat Glycosyl transferases convert Fusarium graminearum toxin DON into non-toxic DON-3-glucoside. QTNs Q.STB.2A.3 encodes CG TraesCS2A02G307700, Q.STB.2A.6 encodes TraesCS2A02G561400 and TraesCS2A02G561300, and Q.STB.2A.7 encodes TraesCS2A02G563700 and TraesCS2A02G563900, which are associated with metal ion binding, zinc ion binding, serine/threonine kinase activity. These proteins aid plant-pathogen interactions [34]. Saintenac et al. [11] cloned a major STB resistance gene (Stb6) that encodes a conserved wall-associated receptor kinase-like protein. In wheat, the inactivating serine/threonine kinase gene TaPsIPK1 confers broad-spectrum resistance to Chinese Pst races endemic in 2020 and 2021 [35]. Additionally, we found that chromosome 5A harboured protein binding, protein-coding and protein kinase domain (Peak marker 995,502 of Q.STB.5A.1; 1,228,444 of Q.STB.5A.2; 1,023,146 of Q.STB.5A.5 and Q.STB_DH.5A.3 (5,411,867). In wheat, Wang et al. [35] reported that the inactivation of a wheat protein kinase gene, TaPsIPK1 confers broad-spectrum resistance to rust fungi.
Further, TraesCS3B02G440700 (Q.STB_DH.3B.2) encodes protein kinase activity, while TraesCS3B02G442100 encodes aldose 1-epimerase and TraesCS3B02G442400 encodes cysteine-kinase. The major STB resistance gene Stb16q encodes a cysteine-rich receptor-like kinase and resists Z. tritici broadly [14]. Most R genes already identified have the ATPase domain, which is involved in biotic stress response and found in our CGs. The present study outlined the CG-encoded proteins that confer plant disease resistance (Table 3).
Overlapping regions for STB resistance and disease-escape traits
One of the most perplexing factors affecting the selection for STB resistance could be the reported interaction between resistance and plant height (PH) or heading date (DH) [36, 37]. In the present study, a few loci were identified to be associated with both STB and DH on chromosomes 2A, 3B, 5A, 5D, and unknown location Q.STB_DH.UN.5. Louriki et al. [30] also reported QTLs for heading days in wheat on chromosome 2A. Previous studies confirmed chromosome 5 for flowering and the presence of the vernalization gene, VRN-1 having three homoeologous loci VRN-A1, VRN-B1, and VRN-D1, which are reported on the long arm of homologous chromosomes 5A, 5B, and 5D, respectively [38, 39]. The identified QTN in our study at 5A was close to VRN-A1. Also, for flower induction, the photoperiod response in wheat is mainly controlled by the PHOTOPERIOD1 (PPD1) loci located on the short arms of chromosomes 2A, 2B, and 2D. PPD1 genes identified in wheat are members of the pseudo-response regulator family [40]. The fact that in our panel, days to heading provide putative chromosome locations with QTNs for disease resistance at 2A, 5A and 5D chromosomes hypothesizes the presence of a common genetic base for plant structure, phenology, and disease susceptibility in the studied material. The present study also revealed that some of the significant QTNs for DH, co-mapped with previously identified STB-resistant regions like MQTL4 (Q.STB_DH.2A.1/3955868), MQTL14 (Q.STB_DH.3B.2/1059080), QTL9 and Stb17 gene (Q.STB_DH.5A.3/5411867) [19] thereby showed strong pleiotropism. QTN such as Q.STB_DH.3B.2/1059080 located near transcript TraesCS3B02G440800 related to magnesium ion transmembrane transporter activity has been reported to play a significant role in powdery mildew and stripe rust resistance in wheat [41]. The putative-associated regions identified in the present study need further study to confound indirect selection for STB resistance via late heading.
Conclusion
Septoria tritici blotch resistance breeding will improve wheat yield. Thus, genetic dissection of the STB-resistant genomic region is highly desired. The study found STB resistance sources, genomic regions, QTLs, haplotypes, pleiotropic SNPs, and candidate genes in Asian bread wheat genotypes. The SAMP had many STB resistance alleles for wheat improvement. BGD52, BGD54, IND92, IND8, and IND75, which are more resistant than "Huirivis#1", can be used to generate STB-resistant varieties and mapping populations to find STB resistance genes/QTLs. This study revealed 21 high-confidence STB resistance markers. Many MTAs were near the candidate gene and protein-coding transcript, which may affect desirable traits. Five of the discovered QTNs were potentially unique, one on each chromosome 1B (Q.STB.1A), 2A (Q.STB_DH2A.1, Q.STB.2A.3), 2B.4, 5A, and 7B. Our findings demonstrate the value of Asian bread wheat as a source of STB resistance alleles and novel stable QTNs for wheat breeding initiatives to generate durable and broad-spectrum Z. tritici-resistant wheat cultivars.
Materials and methods
Association mapping panel
The Septoria Association Mapping Panel (SAMP) was constituted of 181 bread wheat (T. aestivum L.) accessions (Table S1) from wheat breeding programs in India (88 genotypes) and Bangladesh (93 genotypes).
Adult plant screening for STB resistance
The field screening experiments were conducted at CIMMYT’s Sanjaya Rajaram Experimental Station, Toluca, Mexico. This location experiences warm and humid wheat seasons from May to September, with frequent rainfall, which is ideal for creating both artificial and natural epiphytotic conditions for STB infection. The SAMP panel was evaluated during the planting seasons of 2019, 2020, and 2021, and the corresponding experiments were designated STB19, STB20, and STB21, respectively. The accessions were planted by a randomized complete block design (RCBD), with two replications using double rows spaced 1 m apart and 25 cm apart within each row. Wheat genotypes "Murga" and "Huirivis#1" were sown as resistant and susceptible checks, respectively. A combination of six aggressive isolates of Z. tritici, namely St1 (B1), St2 (P8), St5 (OT), St6 (KK), 64 (St 81.1), and 86 (St 133.4), was sprayed at a concentration of 1 × 107 spores/ml for artificial inoculation in the field [42]. The first inoculation was done at the Zadoks growth stage (ZGS) 29, i.e. pseudo stem erect stage [43], using an ultra-low volume sprayer succeeded by two more applications at ZGS 33 (third node detectable) and ZGS 39 (flag leaf fully visible) to maintain sufficient inoculum load under the field conditions. STB severity was evaluated on leaves with necrotic lesions bearing pycnidia, using a double-digit scale (00 to 99) [44], wherein the first digit (D1) represents the relative height of the disease spread vertically, and the second digit (D2) represents the severity based on the diseased leaf area using the following formula:
Disease evaluations were conducted five times at weekly intervals, starting from approximately ZGS 60. The STB severity values were subsequently used to calculate the area under the disease progress curve (AUDPC) using the following equation [45]:
where STBi = STB severity on ith day, ti = time at ith observation, and n is the total number of observations.
Agronomic data scoring
Plant height (PH) and days to heading (DH) were scored in all the field experiments. PH was measured at physiological maturity, from the ground to the average spike tips, excluding awns. DH was scored as days from sowing to the date when approximately 75% of the spikes emerged.
DNA extraction and genotyping
Genomic DNA was extracted from two-week-old seedlings employing the CTAB method [46]. After that, extracted DNA quality and quantity were inspected on a Thermo Scientific™ NanoDrop™ 2000 Spectrophotometer (Thermo Scientific™, USA). Single nucleotide polymorphism (SNP) genotyping was carried out using DArTSeq genotyping-by-sequencing platform [47]. Markers were filtered for minor allele frequency (MAF) higher than 5%, missing data less than 30% and heterozygosity less than 10% for further analysis.
Population structure and linkage disequilibrium
Population structure was visualized by principal component analysis (PCA) using the KDCompute system version 1.0.1 (https://kdcompute.seqart.net/kdcompute/plugins), and kinship among the individuals was computed using the centred identity-by-state method. PCA over the genotypic data was conducted by plotting the PC1 over PC2 using the R software (v. R-4.4.1) [48]. The Linkage disequilibrium (LD) decay at the half-life (R = 0.5) analysis was performed separately for each sub-genome (A, B and D) and across the genome. The LD was calculated through TASSEL software version 5.2.53 [49] using SNP markers with known physical positions, and the LD decay plots were produced by plotting the R2 values against physical distance (bp).
Genome-wide association mapping
Association mapping was conducted with the mixed linear model (MLM) in the TASSEL software (v 5.2.53). The analysis for STB resistance was executed for individual years, and the BLUE means across years. In addition, GWAS on PH and DH was also conducted to investigate their potential association with STB resistance.
All marker-trait associations (MTAs) with the logarithm of the odds, LOD ≥ 3 (-log10 of P value), were declared to be significant for STB resistance. The Manhattan and Quantile–Quantile plots were recreated in R-studio using the CMplot-R tool. The physical position of the significant markers was further confirmed with the BLAST search tool of the Wheat@URGI portal (http://www.plants.ensembl.org/Triticum_aestivum/Tools/Blast) using marker sequences against the International Wheat Genome Sequencing Consortium (IWGSC) RefSeq v1.0 genome [50]. For comparison, QTNs identified in the present study and those already catalogued STB genes/QTLs [19] were projected onto a linkage map developed using MapChart software version 2.3 [51]. A QTN was considered potentially new if the physical distance was ≥ 50 Mb from the reported STB gene or QTL.
Putative candidate gene identification
SNP markers for the detected QTLs were used to identify candidate genes in the genomic regions encompassing the SNPs in the PGSB (Plant Genome and Systems Biology) database; the search was focused on genes or domains that are functionally related to disease resistance mechanisms. The SNP sequences were BLASTed against the wheat reference genome sequence IWGSC (RefSeq v1.0) in "Plant Ensembl" (https://plants.ensembl.org/Triticum_aestivum/) to retrieve the corresponding genes and their functional descriptions [52, 53]. For precise identification of candidate genes, the physical starting point of the marker and the chromosome name were entered into Ensembl Plants. An additional 300 bp was added before and after the SNP to increase accuracy. Some markers had SNPs within gene sequences, classified as direct gene hits. However, for markers without SNPs within a gene, potential candidate genes were selected 2 Mb upstream and downstream of the SNPs. These genes were related to the pathogenic process or known to regulate the induction of genes related to pathogenesis. In cases where annotations were unavailable in the Triticum aestivum genome, orthologous genes in related species with known predicted functions were screened using the comparative genomics tool in Ensembl.
Staking resistance alleles
The BLUP-corrected mean disease severity data was used to assess the effects of pyramiding different numbers of resistance alleles at the significant loci detected in the current study. Wilcoxon-test and t-test (p < 0.05) were used to examine the significance of phenotypic differences across groups in the R package 'multcom View' and ‘ggpubr’ [54, 55].
Haplotype analysis
Stable MTAs on chromosomes 2A, 2B, 3B, 5A, 5B, and 6A were chosen for haplotype analysis, for which seven markers that met the criterion of being significant in all environments were selected for analysis: 2A:1,091,069, 2B:3,026,452, 3B:1,059,080, 5A:995,502, 5B:1,122,319, 5B:3,222,429 and 6A:2,258,509. Using the R package, the Wilcoxon test was used to assess the corrected disease severities between haplotypes [55].
Statistical analysis
Data analysis was performed using R software. Analysis of variation (ANOVA) was employed to assess variability in disease severity, and the least significant difference (LSD) was used to compare the significant means. Phenotypic and genotypic coefficients of variation and broad-sense heritability were estimated using a formula described by Singh and Chaudhary [56]:
where σ2g stands for genetic variance, σ2gy for variance associated with genotype-by-year interaction, σ2e the experimental error, and y and r indicate the number of years and replication, respectively. Using SAS software version 9.2 [57], Pearson's correlation between STB and agronomic variables was determined.
Availability of data and materials
The original data presented in the study are included in the supplementary materials (phenotypic data) and are accessible via https://hdl.handle.net/11529/10548634 (genotypic data). Further inquiries can be directed to the corresponding author.
References
Goodwin SB. Back to basics and beyond Increasing the level of resistance to Septoria tritici blotch in wheat. Austral Plant Pathol. 2007;36:532–8.
Fones H, Gurr S. The impact of Septoria tritici blotch disease on wheat: An EU perspective. Fungal Genet Biol. 2015;79:3–7.
Goudemand E, Laurent V, Duchalais L, Ghaffary SMT, Kema GH, Lonnet P, Margale E, Robert O. Association mapping and meta-analysis: two complementary approaches for the detection of reliable Septoria tritici blotch quantitative resistance in bread wheat (Triticum aestivum L.). Mol Breed. 2013;32(3):563–84.
Kosina P, Reynolds M, Dixon J, Joshi A. Stakeholder perception of wheat production constraints, capacity building needs, and research partnerships in developing countries. Euphytica. 2007;57:475–83.
Dean R, Van Kan JAL, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J, Foster GD. The top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol. 2012;13:414–30.
Juroszek P, von Tiedemann A. Linking plant disease models to climate change scenarios to project future risks of crop diseases: A review. J Plant Dis Prot. 2015;122:3–15.
Birr T, Hasler M, Verreet JA, Klink H. Temporal changes in sensitivity of Zymoseptoria tritici field populations to different fungicidal modes of action. Agriculture. 2021;11:269.
Juroszek P, Laborde M, Kleinhenz B, Mellenthin M, Racca P, Sierotzki H. A review on the potential effects of temperature on fungicide effectiveness. Plant Pathol. 2022;71:775–84.
Zhong Z, Marcel TC, Hartmann FE, Ma X, Plissonneau C, Zala M, Ducasse A, Confais J, Compain J, Lapalu N, Amselem J, McDonald BA, Croll D, Guerrero JP. A small secreted protein in Zymoseptoria tritici is responsible for avirulence on wheat cultivars carrying the Stb6 resistance gene. New Phytol. 2017;214:619–31.
Brunner PC, McDonald BA. Evolutionary analyses of the avirulence effector AvrStb6 in global populations of Zymoseptoria tritici identify candidate amino acids involved in recognition. Mol Plant Pathol. 2018;19:1836–46.
Saintenac C, Lee WS, Cambon F, Rudd JJ, King RC, Marande W, Powers SJ, Berges H, Phillips AL, Uauy C, Hammond-Kosack KE. Wheat receptor-kinase-like protein Stb6 controls gene-for-gene resistance to fungal pathogen Zymoseptoria tritici. Nat Genet. 2018;50:368–74.
Kildea S, Byrne JJ, Cucak M, Hutton F. First report of virulence to the Septoria tritici blotch resistance gene Stb16q in the Irish Zymoseptoria tritici population. New Disease Reports. 2020;41:13.
Stephens C, Olmez F, Blyth H, McDonald M, Bansal A, Turgay EB, Hahn F, Saintenac C, Nekrasov V, Solomon P, Milgate A. Remarkable recent changes in genetic diversity of the avirulence gene AvrStb6 in global populations of the wheat pathogen Zymoseptoria tritici. Mol Plant Pathol. 2021;22:1121–33.
Saintenac C, Cambon F, Aouini L, Verstappen E, Ghaffary SMT, Poucet T, Marande W, Berges H, Xu S, Jaouannet M, Favery B, Alassimone J, Sanchez-Vallet A, Faris J, Kema G, Robert O, Langin T. A wheat cysteine-rich receptor-like kinase confers broad-spectrum resistance against Septoria tritici blotch. Nat Commun. 2021;12:433.
Mundt CC, Brophy LS, Schmitt MS. Choosing crop cultivars and cultivar mixtures under low versus high disease pressure: A case study with wheat. Crop Prot. 1995;14:509–15.
Cowger C, Hoffer ME, Mundt CC. Specific adaptation by Mycosphaerella graminicola to a resistant wheat cultivar. Plant Pathol. 2000;49:445–541.
Jackson LF, Dubcovsky J, Gallagher LW, Wennig RL, Heaton J, Vogt H, Gibbs LK, Kirby D, Canevari M, Carlson H, Kearney T, Marsh B, Munier D, Mutters C, Orloff S, Schmierer J, Vargas R, Williams J, Wright S. Regional barley and common and durum wheat performance tests in California. Agron Prog Rep. 2000;272:1–56.
Dreisigacker S, Wang X, Cisneros BAM, Jing R, Singh PK. Adult-plant resistance to Septoria tritici blotch in hexaploid spring wheat. Theor Appl Genet. 2015; 128:2317–29.
Brown JK, Chartrain L, Lasserre-Zuber P, Saintenac C. Genetics of resistance to Zymoseptoria tritici and applications to wheat breeding. Fungal Genet Biol. 2015;79:33–41.
Yang N, McDonald MC, Solomon PS, Milgate AW. Genetic mapping of Stb19, a new resistance gene to Zymoseptoria tritici in wheat. Theor Appl Genet. 2018;131:2765–73.
Langlands-Perry C, Cuenin M, Bergez C, Krima SB, Gelisse S, Sourdille P, Valade R, Marcel TC. Resistance of the Wheat Cultivar ‘Renan’ to Septoria Leaf Blotch Explained by a Combination of Strain Specific and Strain Non-Specific QTL Mapped on an Ultra-Dense Genetic Map. Genes. 2022;13:100.
Longin CFH, Wurschum T, Burstmayr H. Genetic variability, heritability, and correlation among agronomic and disease resistance traits in a diversity panel and elite breeding material of spelt wheat. Plant Breeding. 2014;133:459–64.
Juliana P, Singh RP, Singh PK, Crossa J, Rutkoski JE, Poland JA, Bergstrom GC, Sorrells ME. Comparison of models and whole-genome profiling approaches for genomic-enabled prediction of Septoria tritici blotch, Stagonospora nodorum blotch, and tan spot resistance in wheat. The Plant Genome. 2017;10:plantgenome2016.08.0082.
Muqaddasi QH, Reif JC, Roder MS, Basnet BR, Dreisigacker S. Genetic mapping reveals large-effect QTL for anther extrusion in CIMMYT spring wheat. Agronomy. 2019;9:407.
Yang N, Ovenden B, Baxter B, McDonald MC, Solomon PS, Milgate A. Multi-stage resistance to Zymoseptoria tritici revealed by GWAS in an Australian bread wheat diversity panel. Front Plant Sci. 2022;13:990915
He X, Azzimonti G, Sanchez-Vidana MDR, Pereyra SA, Sansaloni C, Hernandez-Anguiano AM, Chawade A, Singh PK. Mapping for adult-plant resistance against Septoria tritici blotch in a common wheat line Murga. Phytopathology. 2021;111:1001–7.
Miedaner T, Zhao Y, Gowda M, Longin CFH, Korzun V, Ebmeyer E, Kazman E, Reif JC. Genetic architecture of resistance to Septoria tritici blotch in European wheat. BMC Genomics. 2013;14:1–8.
Francki MG. Improving Stagonospora nodorum resistance in wheat: A review. Crop Sci. 2013;53:355–65.
Ruud AK, Dieseth JA, Ficke A, Furuki E, Phan HTT, Oliver RP, Tan KC, Lillemo M. Genome-wide association mapping of resistance to Septoria nodorum blotch in a Nordic spring wheat collection. The Plant Genome. 2019;12:180105.
Louriki S, Rehman S, El Hanafi S, Bouhouch Y, Al-Jaboobi M, Amri A, Douira A, Tadesse W. Identification of resistance sources and genome-wide association mapping of Septoria tritici blotch resistance in spring bread wheat germplasm of ICARDA. Front Plant Sci. 2021;12: 600176.
Ghaffary SMT, Faris JD, Friesen TL, Visser RGF, van der Lee TAJ, Robert O, Kema GHJ. New broad-spectrum resistance to Septoria tritici blotch derived from synthetic hexaploid wheat. Theor Appl Genet. 2012;124:125–42.
Tidd H, Rudd JJ, Ray RV, Bryant R, Kanyuka K. A large bioassay identifies Stb resistance genes that provide broad resistance against Septoria tritici blotch disease in the UK. Front Plant Sci. 2023;13:1070986.
He Y, Ahmad D, Zhang X, Wu L, Jiang P, Ma H. Genome-wide analysis of family-1 UDP glycosyltransferases (UGT) and identification of UGT genes for FHB resistance in wheat (Triticum aestivum L.). BMC Plant Biol. 2018;18:67.
Sharma A, Sharma D, Verma SK. Zinc binding proteome of a phytopathogen Xanthomonas translucens pv. undulosa. R Soc Open Sci. 2019;6(9):190369.
Wang N, Tang C, Fan X, He M, Gan P, Zhang S, Hu Z, Wang X, Yan T, Shu W, Yu L, Zhao J, He J, Li L, Wang J, Huang X, Huang L, Zhou JM, Kang Z, Wang X. Inactivation of a wheat protein kinase gene confers broad-spectrum resistance to rust fungi. Cell. 2022;185:2961–74.
Arraiano L, Balaam N, Fenwick PM, Chapman C, Feuerhelm D, Howell P, Smith SJ, Widdowson JP, Brown J. Contributions of disease resistance and escape to the control of Septoria tritici blotch of wheat. Plant Pathol. 2009;58:910–22.
Piaskowska D, Piechota U, Radecka-Janusik M, Czembor P. QTL Mapping of seedling and adult plant resistance to Septoria tritici blotch in winter wheat cv. Mandub (Triticum aestivum L.). Agronomy. 2021;11:1108.
Barrett B, Bayram M, Kidwell K. Identifying AFLP and microsatellite markers for vernalization response gene Vrn-B1 in hexaploid wheat (Triticum aestivum L.) using reciprocal mapping populations. Plant Breed. 2002;121:400–6.
Yan L, Loukolanov A, Tranquilli G, Helguera M, Fahima T, Dubcovsky J. Positional cloning of the wheat vernalization gene VRN1. Proc Natl Acad Sci USA. 2003;100:6263–8.
Beales J, Turner A, Griffiths S, Snape JW, Laurie DA. A pseudo-response regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theoret Appl Genet. 2007;115:721–33.
Tang Y, Yang X, Li H, Shuai Y, Chen W, Ma D, Lu Z. Uncovering the role of wheat magnesium transporter family genes in abiotic responses. Front Plant Sci. 2023;14:1078299.
Gilchrist L, Fuentes DG, Martinez-Cano C, Lopez-Atilano RM, Duveiller E, Singh RP, Henry M, Garcia AI. Practical guide to the identification of selected diseases of wheat and barley. Mexico, D.F.: CIMMYT, Second Edition. 2006 https://repository.cimmyt.org/handle/10883/1268.
Zadoks JC, Chang TT, Kozak CF. A decimal code for the growth stages of cereals. Weed Res. 1974;14:415–21.
Saari EE, Prescott JM. A scale for appraising the foliar intensity of wheat disease. Plant Dis Rep. 1975;59:377–80.
Jeger MJ, Viljanen-Rollinson SLH. The use of the area under the disease-progress curve (AUDPC) to assess quantitative disease resistance in crop cultivars. Theor Appl Genet. 2001;102:32–40.
Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical bulletin. 1987;19(1):11–5.
Li H, Vikram P, Singh RP, Kilian A, Carling J, Song J, Burgueno-Ferreira JA, Bhavani S, Huerta-Espino J, Payne T, Sehgal D. A high-density GBS map of bread wheat and its application for dissecting complex disease resistance traits. BMC Genom. 2015;16:1–15.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2021. https://www.R-project.org/.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5.
Alaux M, Rogers J, Letellier T, Flores R, Alfama F, Pommier C, Mohellibi N, Durand S, Kimmel E, Michotey C, Guerche C, Loaec M, Laine M, Steinbach D, Choulet F, Rimbert H, Leroy P, Guilhot N, Salse J, Feuillet C. International Wheat Genome Sequencing Consortium, Paux E, Eversole K, Adam-Blondon AF, Quesneville H. Linking the International Wheat Genome Sequencing Consortium bread wheat reference genome sequence to wheat genetic and phenomic data. Genome Biol. 2018;9:111.
Voorrips RE. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93(1):77–8.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345:1251788.
Graves S, Piepho H, Selzer L. 2015. Dorai-Raj, S. Multcomp View: visualizations of paired comparisons version 01–7 available online:https://cranr-project.org/web/packages/multcompView/indexhtml.
Kassambara A. ggpubr: 'ggplot2' Based Publication Ready Plots. R package version 0.6.0. 2023. https://rpkgs.datanovia.com/ggpubr/.
Singh RK, Chudhary BD. Biometrical methods in quantitative genetic analysis. New Delhi, India: Kalyani; 1985. p. 318.
SAS Institute Inc. SAS/STAT ® 9.2 User's Guide. Cary: SAS Institute Inc; 2008.
Acknowledgements
The authors thank Francisco Lopez and Nerida Lozano for their technical support in field trials and inoculum preparation, respectively.
Funding
Financial support from the Indian Council of Agriculture Research (ICAR), India, Accelerating Genetic Gain (AGG) in Maize and Wheat Project Grant INV-003439 funded by the Bill and Melinda Gates Foundation (BMGF), the Foreign and Commonwealth Development Office (FCDO) and Foundation for Food and Agriculture Research (FFAR), USAID, and One CGIAR Initiatives PHI and ABI for conducting this research is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
Madhu Patial and Sudhir Navathe contributed to manuscript writing and data analysis. Xinyao He contributed to conceptualization, disease scoring and data analysis, Umesh Kamble and Manjeet Kumar helped in data analysis and manuscript writing. Pawan Kumar Singh and Arun Kumar Joshi contributed to the conceptualization, project monitoring, fund acquisition and revising. All authors contributed to the article write-up and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate:
All the methods involving plants, and their materials complied with relevant institutional, national, and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Patial, M., Navathe, S., He, X. et al. Novel resistance loci for quantitative resistance to Septoria tritici blotch in Asian wheat (Triticum aestivum) via genome-wide association study. BMC Plant Biol 24, 846 (2024). https://doi.org/10.1186/s12870-024-05547-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-05547-x