
Abstract
Septoria tritici blotch (STB) caused by Zymoseptoria tritici is one of the most important foliar diseases of wheat causing significant yield losses worldwide. In this study, a panel of bread wheat genotypes comprised 185 globally diverse genotypes were tested against 10 Z. tritici isolates at the seedling stage. Genome-wide association study (GWAS) using high-throughput DArTseq markers was performed and further gene expression analysis of significant markers trait association (MTAs) associated with resistance to STB was analyzed. Disease severity level showed significant differences among wheat genotypes for resistance to different Z. tritici isolates. We found novel landrace genotypes that showed highly resistance spectra to all tested isolates. GWAS analysis resulted in 19 quantitative trait loci (QTLs) for resistance to STB that were located on 14 chromosomes. Overall, 14 QTLs were overlapped with previously known QTLs or resistance genes, as well as five potentially novel QTLs on chromosomes 1A, 4A, 5B, 5D, and 6D. Identified novel resistance sources and also novel QTLs for resistance to different Z. tritici isolates can be used for gene pyramiding and development of durable resistance cultivars in future wheat breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Common wheat (Triticum aestivum L.) is one of the most important cereal crop in the world and plays important role in the diets of humans and livestock. The global wheat production was reported to be 759.9 million tons in 2019/20 (https://www.fao.org/worldfoodsituation/csdb/en/). This makes wheat the third important crop in terms of production after maize and rice. Global wheat production can be negatively influenced by abiotic and biotic stresses. Septoria tritici blotch (STB) caused by Zymoseptoria tritici is one of the most destructive fungal diseases of wheat worldwide (Kema et al. 1996; Hardwick et al. 2001). The fungus causes expanded necrotic lesions from early-emerging leaves to flag leaves, and the overall damage caused by STB can result in grain yield losses up to 50% under optimal environmental conditions (Mehrabi et al. 2006; Goodwin, 2007; Kema and van Silfhout, 1997; Suffert et al. 2011). Under epidemic conditions, fungicide application is required to control STB, but this strategy is not adequately effective due to quick adaptation of the pathogen to fungicides by sexual recombination and mutation (Torriani et al. 2009; Mohammadi et al. 2017; Kema et al. 2018). Today, there is evidence that the frequently use of fungicides causes long-term severe negative effects on human health and the environment (Gikas et al. 2022). Therefore, the characterization of new resistance sources and the development of resistant wheat cultivars is the most economical and environmentally friendly approach and fundamental strategy in breeding programs for sustainable agriculture and food security (Talebi et al. 2010; Mehrabi et al. 2015). Up to now, 22 major genes and 89 QTLs for resistance to Z. tritici have been reported and mapped on the wheat genome along with their closely linked markers (reviewed by Brown et al. 2015; Yang et al. 2018).
Both qualitative and quantitative resistance has been reported for resistance to STB in wheat. Qualitative resistance is usually controlled by major genes that confer complete resistance and follows by the gene for-gene model (Brown et al. 2015). This model has been reported for the first time on Z. tritici isolate IPO323 and cv. Flame (Kema et al. 2000; Brading et al. 2002). Quantitative resistance is controlled by many minor genes and widely reported in wheat cultivars at both seedling and adult growth stages (Arraiano and Brown 2006; Chartrain et al. 2004; Goodwin 2007). The qualitative resistance has been shown to be effective strategy for controlling different fungal diseases in wheat, but on the other hand, the rapid evolution and adaptation of Z.tritici populations result in overcoming resistance in most improved cultivars (Cowger et al. 2000; Stukenbrock et al. 2007; Makhdoomi et al. 2015; Muqaddasi et al. 2019). Therefore, in order to increase the durability of wheat resistance to STB, combining qualitative and quantitative resistance genes is required to preserve resistance effectiveness over time (Brown et al. 2015; Vagndorf et al. 2017). Availability of high-definition genotyping using genotype-by-sequence (GBS) technologies enables breeders to identify resistance genes in diverse germplasm that can be employed for the development of new resistant cultivars using the gene pyramiding approach (Kidane et al. 2017; Vagndorf et al. 2017; Arraiano and Brown, 2017; Muqaddasi et al. 2019).
The genetic architecture of resistance to STB has been mostly evaluated in different bi-parental populations for detection of large-effect genes or quantitative trait loci (QTLs) (Chartrain et al. 2004, 2005a,b,c; Simón et al. 2005; Tabib Ghaffary et al. 2011, 2012). Genome-wide association mapping (GWAS) using a large number of markers with high genome coverage is a powerful tool for detecting the resistance loci associated with diverse germplasm possessing natural variation of resistance genes (Bartoli and Roux 2017). Various GWAS studies have been performed on diverse wheat germplasm (Mirdita et al. 2015; Vagndorf et al. 2017; Kidane et al. 2017; Muqaddasi et al. 2019; Arraiano and Brown 2017). Most previous GWAS studies for revealing QTLs against STB have been done for detecting resistance loci against a mixture of isolates under natural field conditions (Goudemand et al. 2013; Kidane et al. 2017; Arraiano and Brown, 2017; Muqaddasi et al. 2019). Therefore, GWAS analysis for specific isolate resistance using globally diverse pathotypes of Z. tritici is of great interest for breeders to identify new genes/loci that can be used in wheat breeding programs.
The present study relies on genotyping and phenotyping of a globally diverse panel of 185 wheat genotypes that were evaluated at the seedling stage against ten Z. tritici isolates (collected from Iran, Algeria, Turkey, France, and The Netherlands). DNA fingerprinting of the wheat genotypes was performed using high-throughput DArTseq technology (SilicoDArT and SNP markers) aiming to identify and localize possible novel QTLs associated with isolate-specific resistance against Z. tritici in wheat.
Materials and methods
Plant material and evaluation of STB infection
A total of 185 wheat genotypes comprised commercial cultivars, breeding lines, and landraces with a wide range of genetic background which was used in this study (Supplementary Table S1). The germplasm set included (1) 63 Iranian cultivars and breeding line that mainly used in wheat growing zones in Iran and also in national wheat breeding programs, (2) 103 landraces genotypes from globally diverse origins (20 countries), and (3) 18 wheat differential lines with previously known STB genes and Bezostaya which is originated from Russia and still grown in large wheat cultivation zones in northeastern Iran. To ensure the purity of the seeds, all landraces were grown in Iranian Seed and Plant Improvement Institute (SPII) field station and each genotype was selected from a single-spike.
Phenotypic evaluation of wheat genotypes against ten Z. tritici isolates (Table 1) was described in detail by Mahboubi et al. (2020). Briefly, STB isolates were pre-cultured in yeast-extract glucose (YG) liquid medium, and then these pre-cultures were used to inoculate 100 ml YG media per isolate. YG media were placed in an orbital shaker (set at 125 rpm) and incubated at 18 °C for 5–6 days. The propagated yeast-like spores were collected and their concentrations were adjusted to 107 spores/ml (Abrinbana et al. 2012). Five seeds of each genotype were sown in plastic pots and first leaf of the10-day-old seedlings was spray-inoculated using hand sprayers. Inoculated plants were kept in dark plastic bags for 48 h at 20–25 °C and then transferred to a greenhouse with the environmental condition of 18–22 °C and > 85% humidity (Kema et al. 1996; Makhdoomi et al. 2015). After 21 days post-inoculation, percentage necrotic leaf area covered with pycnidia of the first leaf for each genotypes (at least three plants per pot) was scored visually as described before (Kema et al. 1996; Ghaneie et al. 2012). Experiments were set up with a randomized complete block design with three replications.
Phenotypic data analysis
Data were normalized using arcsin square root transformation (Sokal and Rohlf 1995) . Normalized data were for ANOVA analysis using a linear mixed model (LMM), in which the isolates and wheat genotypes were the main effects and their two-way interaction as fixed effects.
The BLUE value for disease severity (% of leaf area covered by pycnidia) for all isolates was calculated using the PROC MIXED procedure in SAS v9.3 (SAS Institute Inc., Cary, NC, USA) considering the genotype as a fixed effect and replication (block) considered as random effect. These BLUE values were then used for broad sense heritability estimates and correlations between isolates, cluster analysis of wheat genotypes, and also to perform GWAS (Godoy et al. 2018).
Cluster analysis of wheat genotypes according to pycnidia coverage data by each isolate was performed using the un-weighted pair-group method (UPGMA) and the dissimilarity matrix was measured using the Ward’s method implemented in PAST software (Hammer et al. 2001). Pearson correlation coefficients among 10 STB isolates were calculated for the percentage of leaf covered by pycnidia in 185 wheat genotypes.
Genotypic data
DNA was extracted from seedling plants of each genotype using the CTAB protocol (Lassner et al. 1989) . The quality and quantity of DNA were checked by a spectrophotometer, and DNA concentration was adjusted to 100 ng/µl. DNA samples were plated in 96-plex and shipped to DArT Pty Ltd, Canberra, Australia, and genotyped using the DArTseq technology (Sansaloni et al. 2011; Alam et al. 2018). The detailed methodology for the generation of DArTseq markers (SNP and SilicoDArT) was described in Egea et al. (2017). We received a total of 94,535 (54,309 SilicoDArT and 40,225 SNP) markers, which were polymorphic across 185 wheat genotypes (Supplementary Table S2). The physical position of markers obtained by aligning to the reference genome of to the reference genome of Chinese Spring (CS) IWGSC Ref Seq v1.0 (IWGSC et al. 2018).
Marker loci with unknown chromosome positions (based on genome assembly) were removed from the analysis, and the remaining markers were filtered using a minimum minor allele frequency (MAF) of 0.05 in TASSEL v.5.2.37 software (Bradbury et al. 2007). Wheat is a self-pollinated crop and we assumed that all genotypes are homozygous. Therefore, markers showing heterozygous were indicated as missing and markers with > 20% missing were excluded. In total, 21,773 (15,856 SilicoDArT and 5917 SNP) distributed across the 21 chromosomes were maintained for analysis.
Linkage disequilibrium and population structure analysis
Linkage disequilibrium (LD) for DArTseq markers was implemented in TASSEL v.5.2.37. The critical r2-value was determined by root transforming the unlinked r2-values and taking the 95th percentile of the distribution as the threshold beyond which LD is likely caused by genetic linkage (Nielsen et al. 2014; Monostori et al. 2017). The graphical LD decay was imputed by the GAPIT R package (VanRaden, 2008; Lipka et al. 2012). Population structure was performed in STRUCTURE 2.1 based on an admixture model (Evanno et al. 2005). The optimal value of K ranges from 1 to 10, with three independent runs. For each run, the analysis was performed with 10,000 burn-in replicates and 10,000 Markov chain Monte Carlo (MCMC) iterations. Principal coordinate analysis (PCoA) and cluster analysis among the wheat genotypes conducted by DARwin ver. 5.0 software using the Unweighted Neighbor-Joining (UNJ) algorithm.
Genome-wide association analysis
Genome-wide association mapping (GWAS) was conducted using 21,773 DArTseq markers and the best linear unbiased estimates (BLUE) for phenotypic scoring data of 10 Z. tritici isolates. The R package Genome Association and Prediction Integrated Tool (GAPIT) (Lipka et al. 2012) was used for GWAS by fitting four different models such as mixed linear model (MLM), multiple loci mixed model (MLMM), compressed mixed linear model (CMLM), and fixed and random model Circulating Probability Unification (FarmCPU) methods to select the best fitting model and reduced false discovery rate (FDR). FarmCPU method uses both the fixed-effect model (FEM) and random effect model (REM) iteratively to eliminate confounding factors, which prevents model overfitting and control false positives simultaneously (Liu et al. 2016). This model was highly efficient in computation for complex traits by eliminates confounding issues arising due to population structure, controlling false positive and reduce their effect in GWAS analysis (Gahlaut et al. 2019; Kaler et al. 2020). Therefore, GWAS was performed on the BLUE for each trait (diseases severity data for each isolate) to identify DArTseq markers associated with both P levels and efficiency traits using FarmCPU model. In order to detect the marker-trait association (MTA) in GWAS, if the significant markers cross the false discovery rate threshold (P = 0.05), a uniform suggestive genome-wide significance threshold level of P-value ≥ 0.0001 (− log10 P = 4.00) was selected for MTA considering the deviation of the observed test statistics values from the expected test statistics values in the Q–Q plots.
Map positions of significant MTAs were determined according to their genetic positions in a high-resolution DArT-seq consensus map version 4, including 105,122 markers distributed across the 21 hexaploid wheat chromosomes (https://www.diversityarrays.com/technology-and-resources/geneticmaps/) and bread wheat IWGSC RefSeq v1.0 with BLAST + v2.7.1 (Camacho et al. 2009). Overlapping significant markers on the same chromosome for resistance to different STB isolates were considered to tag a single QTL if their positions were closer than 10 cm. Then, for comparison of the QTLs identified in this study, the map position of significant markers in each QTL was projected onto the two different wheat SSR consensus maps (Somers et al. 2004; Maccaferri et al. 2015) for cross-reference with previous SSR maps. Each QTL considered new if its position was ≥ 10 cm from previously reported STB resistance genes or QTLs.
Candidate genes identification and expression analysis
Promising candidate genes for individual MTA were identified by aligning the physical position of markers to the sequence of the wheat genome assembly IWGSC v.1.0 (https://plants.ensembl.org/Triticum_aestivum/Info/Index). High-confidence annotated genes were retrieved from a ± 3-Mb window of left and right of each identified MTA. For the expression analysis of annotated candidate genes (CG), the transcripts per kilobase million (TPM) values for every CG were retrieved from the public wheat expression database at http://wheatexpression.com (Ramírez-González et al. 2018) and the Log transformed (Log2X) value was used to generate a heatmap using online tool ClustVis (Metsalu and Vilo 2015).
Results
Response of wheat genotypes to Zymoseptoria tritici isolate at seedling stage
Analysis of disease severity data showed significant differences (P˂0.001) among wheat genotypes for resistance to Z. tritici (Table 2). Genotype × isolate interaction was highly significant (P˂0.001) and indicated the differences in wheat genotypes for their responses to Z. tritici isolates. Heritability values for disease severity were high for all isolates. The high heritability values indicated that there was a limited replication variation relative to genotypic variation for all isolates. This is supported by high Pearson correlation coefficients for disease severity between all isolates (Table 3). The Pearson correlation coefficient between STB isolates ranged from 0.26 (IPO02166 and IPO92034) to 0.90 (IPO323 and IPO86013) with an average value of 0.61 (Table 3). All isolates showed high virulence, which among them IPO02166 was the most aggressive isolates with the highest mean disease severity (63%) across all wheat genotypes and only 15 wheat genotypes (8%) were resistant to this isolate, while IPO323 was the least aggressive isolate (mean disease severity = 40%) and 35 wheat genotypes (20%) were resistant to this isolate (Supplementary Table S1). In total, 239 interactions among all (n = 1850) were found for resistance to one isolate, of which 183 showed disease severity P ≤ 5% (highly resistance), and 56 were disease severity 5 < P ≤ 10% (resistance) (Supplementary Table S1).
The mean disease severity (DS) among wheat genotypes ranged from 0 (M3 synthetic) to 68% (KC4821 from Iran). Among 63 Iranian commercial cultivar and lines, 51 cultivars (80%) were susceptible to all isolates, while ER-M-92–20 was resistant to all isolates. In addition, Sayson and Hirmand showed resistance responses to all isolates, except for IPO02166 and IPO99031. Although “Zare” was resistance to all isolates, expect for low disease susceptibility rate for two Iranian isolates RM22 and RM230 (Supplementary Table S1). In total, 39 isolate-specific resistances were found among all interactions (n = 640). The overall mean disease severity among Iranian commercial cultivar and lines was 45% ranging from 2 (ER-M-92–20) to 62% (Parsi). Among 103 landrace genotypes, 80 genotypes were susceptible to all isolates. The landrace IPK40740 (France) showed immune responses to all isolates, and in addition, five landraces (IPK45227, IPK46116, IPK40793, IPK41079, and IPK16452) were resistance to nine isolates. Overall, the mean disease severity of wheat landraces ranged from 0.9 to 68% with an average value of 44%, and in total, 116 isolate-specific resistances were found among landrace-isolate interactions (n = 1030). Cluster analysis and principal component analysis using omitted data from the percentage of leaf area covered by pycnidia grouped wheat genotypes in three distinct clusters (Fig. 1). The first cluster comprised 63 wheat genotypes, of which 19 genotypes were Iranian cultivar and breeding lines, 7 genotypes with previously known Stb genes (Bulgaria, Israel 493, Cs-Synthetic, Shafir, Es-Federal, M6, and Balance), and the remaining genotypes were from different sources. Resistance spectra of genotypes for this cluster ranged from 30.98 (IPO323) to 56.61% (IPO02166) (Table 4). Cluster-II contained 85 genotypes, with 32 Iranian cultivars, one differential cultivar (Curtot), and 52 genotypes from diverse origins. All genotypes in this cluster were highly susceptible to most of the isolates and the mean disease severity ranged from 45.14 (IPO323) to 66.57% (IPO02166) (Table 4). Cluster III comprised 37 genotypes, most of the differentials with known Stb genes (Veranopolis, Tadinia, Kavkaz-K4500, TE9111, Salamoni, Arina, Riband, and M3) grouped in this cluster. In general, genotypes from this cluster showed low mean disease severity that ranged from 7.76 (IPO323) to 43.59% (IPO02166) (Table 4). Cluster-III comprised of 37 genotypes, including 13 Iranian cultivar and breeding lines, eight differentials (Veranopolis, Tadinia, Kavkaz-K4500, TE9111, Salamoni, Arina, Riband, and M3), and 16 landraces from different origins. In general, genotypes grouped in this cluster had a low mean disease severity ranging from 7.76 (IPO323) to 43.59% (IPO02166) (Table 4).

Cluster analysis (a) and principal component analysis (PCA) of 185 wheat genotypes based on mean disease severity data against ten Zymoseptoria tritici isolates
Population structure and linkage disequilibrium
Extensive genotyping on 185 wheat genotypes resulted in 21,773 (15,856 SilicoDArT and 5917 SNP) markers. The unweighted Neighbor-Joining cluster analysis (Fig. 2a) and Bayesian model-based structure analysis (Fig. 2b) grouped wheat genotypes into four distinct subpopulations. Sub-population 1 (50 genotypes) consisted of 17 Iranian landraces and genotypes from Western Asia and Eastern Europe (Turkey, Tajikistan, Romania, Hungary). Sub-population 2 (29 genotypes) contained genotypes from diverse origins and two Iranian cultivars. Sub-population 3 (86 genotypes) comprised most of the Iranian improved genotypes (45 genotypes) and eight wheat differentials for resistance to STB. Sub-population 4 (20 genotypes) consisted of landraces accessions from different origins. In general, there was no significant relationship between cluster grouping and origin of wheat genotypes, most probably due to the ancient and recent international exchange of germplasm, while the Iranian improved cultivars mainly grouped in the same cluster. Several of the genotypes used in this study have been utilized as parental lines or have the same background pedigree. Therefore, a mixture of origins was observed in all clusters. Nevertheless, a clear distinction on the abovementioned four main subpopulations was clearly observed based on molecular data analysis. Principal component analysis (PCA) was used to confirm the results of population structure and this also showed a distinct pattern of subpopulations. The first two PCs represented 66.72% of the total variation (Fig. 2c). A comparable result similar to cluster analysis and PCA was also observed by the heatmap plot of kinship matrix where four distinct clusters were identified (Fig. 3). Different subpopulations showed different resistance levels for most of the isolates. Subpopulation 1 (mostly of Iranian landraces and landraces from West Asia) has the highest susceptibility (mean DS = 48.2), followed by the subpopulation 2 (mean DS = 43.4) and subpopulation 3 and 4 (mean DS are 43.3 and 40.4, respectively) (Fig. 4). These associations between population structure and STB resistance indicated that corrections for population structure were required for association mapping analysis. In general, the PCA analysis was consistent with the results of STRUCTURE analysis, while the cluster analysis showed more consistence with subpopulations identified by STRUCTURE analysis (Fig. 2).

The neighbor-joining cluster analysis based on genetic dissimilarity in 185 wheat genotypes (a). The color of branches indicates accessions corresponding to the subpopulations in population structute analysis. Determination of the optimal value of K and population structure analysis (b) and principal coordinate analysis (c) of 185 wheat genotypes based on DArTseq markers

Heatmap plot of kinship matrix displaying relationships of 185 wheat genotypes based on DArTseq markers

Different sub-population of 185 wheat genotypes panel that showed a different level of septoria tritici blotch disease severity (measured using the percentage of leaf area covered by pycnidia)
In LD analysis, the square of the correlation coefficient of alleles between loci (r2) was not significant for most of the pairwise comparisons, whereas out of 104,8575 marker pairs, only 297,546 (28%) intra-chromosomal pairs showed a significant level (P < 0.001) of LD. Marker pairs on the genome-B showed a higher number of significant pairs in comparison to the genome-A and genome-D. Mean and critical r2 values were 0.09 and 0.16, respectively. LD declined with increasing physical genetic distances between markers and r2 value falling below the critical value over distances of 1.6 Mb (Fig. 5).

Linkage disequilibrium (LD) measured r2 plotted vs. the physical map (bp) between pairs of DArTseq markers in a panel of 185 wheat genotypes
Genome-wide association analysis
Among different GWAS models, FarmCPU model shows reliable results and presented low spurious associations. For the analysis, BLUE value of phenotypic data (percentage of necrotic leaf area covered by pycnidia) for each isolate and 21,773 mapped DArTseq markers on 185 wheat genotypes were used. The highest number of markers was mapped on A genome (8031) and B genome (9537) compared to those mapped on D genome (4205). Association analysis was performed separately for each isolate. A total of 27 significant MTA were identified for STB resistance on 14 chromosomes (Table 5). Manhattan plots for the association between markers and STB responses to ten Z. tritici isolates are displayed in Fig. 6 (Supplementary Fig. 1).

Circular Manhattan plot for association analysis between DArTseq markers and ten different Z. tritici isolates in 185 hexaploid wheat genotypes. (1) IPO323, (2) IPO02166, (3) IPO86013, (4) IPO98022, (5) IPO92034, (6) IPO99031, (7) RM6, (8) RM22, (9) RM183, and (10) RM230
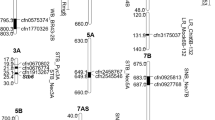
The QTLs identified for different isolates but located at overlapping genomic region on a chromosome were considered a single QTLs and assigned the same name using the nomenclature Qstb.iau- followed by the number of QTLs in chromosome order (Table 5; Supplementary Figure S2), and finally, 19 significant genomic regions associated with resistance to Z. tritici were detected on 14 chromosomes (Table 5). Most of the significant MTAs showed small effects, and regarding significant associations (P < 0.0001), only the MTA that explaining > 5% of the phenotypic variations are presented in Table 5.
The position of these QTLs was compared to the position of mapped QTLs and known genes reported in previous studies (summarized in Supplementary Fig. 2). Overall, 13 QTLs were associated with resistance to one isolates, although 6 QTLs were associated with resistance to multiple isolates that were on chromosome 2B (Qstb.iau-3), 2D (Qstb.iau-5), 3B (Qstb.iau-6), 4A (Qstb.iau-7 and Qstb.iau-8), and 6B (Qstb.iau-15) (Table 5).
The corresponding locations of the identified QTLs were checked on a consensus map and their overlaps compared with previously known STB resistance genes and QTLs. Out of 19 QTLs, 15 QTLs were identified in overlap regions or very close (≤ 10 cM) to genomic regions of the previously reported QTLs/genes. Five genomic regions were identified as putatively new QTLs for resistance to STB on chromosomes 1A, 4A, 5B, 5D, and 6D (Supplementary Fig. 2). Among these putatively novel genomic regions, Qstb.iau1, Qstb.iau-11, Qstb.iau-13, and Qstb.iau-16 were specific-isolate resistance QTLs for isolate RM6 (Iran),
IPO02166 (Iran), IPO86013 (Turkey), and IPO98022 (France), respectively, and interestingly the Qstb.iau7 were associated with resistance to four isolates (IPO98022, IPO99031, RM6, RM183) (Table 5).
Putative candidate gene identification and expression analysis
All the MTA associated with resistance to STB isolates identified by FarmCPU method were mapped to the wheat physical genome. The physical reference genome of Chinese Spring cv. was used to survey the genes in the flanking regions on each MTA (IWGSC RefSeq v1.0 with BLAST + v2.7.1). For each MTA, ± 3 Mb regions toward the left and right side was used to identify the putative candidate genes (CGs) using Ensembl Plant database. A search for candidate genes resulted in identification of multiple genes (70 CGs, ranges from 1 to 12 CGs per MTA); by annotation, these CGs based on gene ontology (GO) using the IWGSC RefSeq v1.0, 24 genes were found to have putative role in diseases stress on the basis of literature search (Table 6). These genes are involved in different biological activities like protein kinase-like domain, Cytochrome P450, leucine-riche repeat domain superfamily, Fbox domain, and Homeobox-like domain superfamily (Table 6). Gene expression analysis for 24 CGs was conducted using RNA-seq expression data retrieved from Wheat Expression Browser (http://www.wheat-expression.com/). The results are indicated for expression of 10 of the 24 genes that expressed at different developmental stages under disease stress like septoria, fusarium, and leaf rust (Supplementary Fig. 3). Five genes (TraesCS2A02G582500, TraesCS3B02G348200, TraesCS5A02G367700, TraesCS5B02G380400, and TraesCS6D02G327400) had high expression (up to 3 Transcripts Per Million; TPM) at different developmental stages of wheat under septoria tritici blotch, leaf rust, and fusarium head blight diseases, and interestingly, all these genes belonged to protein kinase-like domain superfamily (Supplementary Fig. 3). Some candidate genes like TraesCS1A02G004400, TraesCS2B02G001600, and TraesCS2D02G029700 are expressed uniquely only in reproductive stage (Supplementary Fig. 3).
Discussion
Novel sources for resistance to Z. tritici
Z. tritici is one of the most important foliar diseases in many wheat-growing areas, including Europe, Northern America, and Asia (Hardwick et al. 2001; Mehrabi et al. 2015). The use of genetic resistance is the most appropriate strategy to control the disease. However, the rapid adaptation of Z. tritici populations leads to a quick breakdown of resistance. Thus, continuous characterization and utilization of new sources of resistance in breeding programs are prerequisites (Abrinbana et al. 2010; Ghaneie et al. 2012). Iran is one of the primary centers of origin of wheat, and it is proposed that the co-evolution of wheat and Z. tritici occurred in this region. Therefore, the characterization of wheat genotypes using isolates from this region is likely required to add new resources to gene pool, which can be used for pyramiding resistance genes (Ghaneie et al. 2012; Makhdoomi et al. 2015; Aghamiri et al. 2015) . We have recently studied the interactions of 185 wheat genotypes against ten Z. tritici isolates from different sources (Mahboubi et al. 2020). Most wheat genotypes showed were susceptible to all isolates. High broad-sense heritability suggested that the resistance variation is heritable, which are in agreement with previous studies on septoria resistance in different wheat germplasm both at seedling and adult plant stages (Dreisigacker et al. 2015; Muqaddasi et al. 2019; Riaz et al. 2020).
Among the isolates, IPO323 showed the highest number of incompatible interactions (n = 35), while IPO02166 (originated from Iran) showed a high level of aggressiveness on wheat genotypes. All the differential wheat genotypes (except M3, Riband, Arina, and Kavkaz-K4500) possessing known Stb genes were susceptible to most of Z. tritici isolates, which are in agreement with previous reports for the ineffectiveness of known Stb genes against Z. tritici populations (Abrinbana et al. 2012; Makhdoomi et al. 2015; Mahboubi et al. 2020). Interestingly, two genotypes (ER-M-92–20 and IPK40740) and four differentials (Kavkaz-K4500, Arina, Riband, and M3) were resistant to all isolates. These genotypes could be of interest as resistance sources that contain resistance genes or a combination of diverse yet unknown Stb genes. Therefore, it can be concluded that the high resistance pattern of these genotypes should be due to the presence of Stb6, Stb15, or new unknown resistance genes. Besides these highly resistant genotypes, five genotypes (IPK45227, IPK26116, IPK41079, IPK16452, and IPK40793) showed resistance to nine isolates and can be used as valuable resistance sources in wheat breeding programs. Among wheat genotypes, M3 showed highly resistance (immune) responses to all isolates. This genotype contains Stb16 and Stb17 (Tabib Ghaffary et al. 2012; Mahboubi et al. 2020). Therefore, it can be concluded that this gene still is effective against STB, which is consistent with previous reports (Hosseinnezhad et al. 2014; Makhdoomi et al. 2015). The resistance sources used in this study that we previously reported as likely novel sources of resistance (Mahboubi et al. 2020) can be used in breeding programs for the development of modern wheat cultivars.
QTL validation and alignment to previously reported STB genes and QTLs
In line with our previous study, we used GWAS analysis to identify novel QTLs against Z. tritici isolates. This approach enables breeders to enhance crop genetic improvement by incorporating suitable QTLs into wheat breeding programs (Ibrahim et al. 2020). To this aim, DArTseq markers were successfully used to genotype a globally diverse wheat germplasm. The use of high-density markers with broad genome coverage in GWAS improved the accuracy of identified QTLs for resistance to STB, which is a highly quantitative disease trait with a minor contribution of each QTL (Mirdita et al. 2015; Muqaddasi et al. 2019). Overall, we found 19 QTLs for resistance to STB that were located on 14 chromosomes (Table 5). The phenotypic variation explained by each QTLs mapped on different chromosomes was relatively low (R2 ≤ 0.17), suggesting that the resistance to this pathogen follows a highly quantitative nature, which is consistent with previous reports (Kidane et al. 2017; Arraiano and Brown 2017; Yates et al. 2019).
The precise comparisons of these QTLs with known QTLs were difficult due to different populations, isolates, and markers used elsewhere. However, using consensus wheat maps, it was possible to compare QTLs with the mapped chromosomal location of previously known genes/QTLs. Most of the QTLs identified in this study were localized in adjacent regions with known QTLs that have previously been identified (Goudemand et al. 2013; Brown et al. 2015).
A QTLs on chromosome 1A (Qstb.iau-1) was isolate-specific for resistance to RM6 isolate from Iran. This QTLs did not align with any previously reported QTLs or STB resistance genes; therefore, we assume it as a potentially novel QTL. One isolate-specific QTLs was identified on chromosome 2A, co-located with previously known MQTL5 reported by Goudemand et al. (2013) for resistance under natural infection as also for two different isolates (IPO323 and IPO98099), but interestingly, this QTLs was not in association with IPO323 used in our study. This can be concluded by the different nature of wheat germplasm and also the molecular markers used in our study.
Two QTLs mapped on chromosomes 2B, Qstb.iau-3, was associated with resistance to two isolates (IPO323 and IPO86013) co-located with previously known QTLs reported by Goudemand et al. (2013) with the same isolate, IPO323. Another QTL was isolate-specific for IPO98022, co-localized with previously known resistance gene (Stb9) and QTLs under natural field condition (Eriksen et al. 2003; Risser et al. 2011). Another QTLs mapped on chromosomes 2D was in association with resistance to two Iranian Z. tritici isolates (RM6 and RM230) colocalized with previously reported meta-QTLs for resistance to septoria as seedling stage (Goudemand et al. 2013). As well as, identified QTLs on chromosome 3B in this study for resistance to isolates IPO323 and IPO98022 was overlapped with previously reported meta-QTLs on this chromosome with the same isolates (Goudemand et al. 2013).
Three genomic regions were identified on chromosome 4A, of which two QTLs (Qstb.iau-8 and Qstb.iau-9) were in association with resistance to Iranian Z. tritici isolates (RM22, Rm183, and RM230) overlapped with previously reported QTLs on this region at adult stage (Goudemand et al. 2013), while another QTLs, Qstb.iau-7, was in association with multiple isolates that did not align with any previously reported QTLs or STB resistance genes; therefore, we consume it as a potentially novel QTLs.
Two QTLs on chromosomes 5A (Qstb.iau-10) and 5B (Qstb.iau-12) overlapped with known STB resistance genes, Stb17 and Stb1, respectively. Two QTLs on chromosomes 5B (Qstb.iau-11) and 5D (Qstb.iau-13) were isolate-specific resistance and did not align with any previously reported QTLs or STB resistance genes; therefore, we consume that these are potentially novel QTLs. Recently reported QTLs of chromosome 5D (Langlands-Perry et al. 2022) did not align with the physical position of our finding QTL on this chromosome, which may be related to differences in wheat GWAS panel, isolates, and molecular markers that used in both studies. Therefore, we could not confirm that these are the same QTLs or different, because the physical position of our detected QTL on chromosome 5D is different from the previous report. This region on chromosome 5D is also known as the introgressed region from Aegilops umbellulata and Ae. tauschii into wheat and is an important chromosomal location having resistance genes against leaf rust, stripe rust, soil-borne mosaic virus, and powdery mildew (Bansal et al. 2020; Mohler et al. 2020; Liu et al. 2020; Fu et al. 2014), although the mapped QTLs on chromosomes 5A, 5B, and 6A were overlapped with Stb17, Stb1, and Stb15 genes, respectively. This can confirm the results of the phenotyping experiment using the differential cultivars that the high resistance pattern of genotypes that showed broad resistance to all isolates should be due to the presence of Stb15.
A QTLs identified on chromosome 6B (Qstb.iau-15) overlapped with previously reported QTLs and meta-QTLs for resistance to septoria under natural field infection at both seedling and adult stages (Eriksen et al. 2003; Goudemand et al. 2013). Novel genomic region (Qstb.iau-16) was identified on chromosome 6D, which was not aligned with any previously reported resistance genes or QTLs; therefore, we assume it as a potentially novel isolate-specific resistance QTLs. Three genomics regions associated with isolate-specific resistance were co-localized with previously reported QTLs at adult stages (Goudemand et al. 2013).
To better understand the functional roles of QTL regions, significant MTA were annotated and reviewed for putative candidate genes associated with diseases resistance in plant, and the expression analysis of these candidate genes also was examined. The results suggested that among 24 identified candidate genes, 10 genes showed expression under diseases stresses in different developmental stages of wheat. Overall, five genes belonged to protein kinase-like domain superfamily and a few genes belonged to leucine-rich repeat domain proteins exhibited a significantly higher expression under different diseases like septoria, leaf rust, and fusarium head blight (Supplementary Fig. 3).
These genes are well known as typical broad-spectrum genes that provide resistance to biotic stresses in different crop species (Yan et al. 2016; Andersen et al. 2018; Han 2019; Pandian et al. 2020) as well as for p-loop ATPase domain which has been associated with STB resistance in the previous studies (Louriki et al. 2021). Furthermore, we found several QTLs in close position of previously reported QTLs for resistance to Z. tritici isolates at both seedling and adult stages, which can be used as valuable sources for introgression of these QTLs into advanced wheat lines (Odilbekov et al. 2019). In addition to previously known QTLs, we found several potentially novel QTLs on chromosomes 1A, 4A, 5B, 5D, and 6D. Detection of these putative new QTLs provided useful information that could be used to track favorable alleles for developing wheat cultivars resistant to STB. This knowledge can be used for generation of new allelic combination through cross between novel sources for resistance to STB (Riaz et al. 2020). As we used different isolates with diverse origins and identified potentially novel QTLs that were associated with multiple isolates, this suggests that these QTLs remain effective as putative broad spectrum sources for resistance to STB, which can be used for future wheat breeding programs globally.
Conclusion
In this study, we investigated the resistance spectra of a diverse wheat global panel against ten Z. tritici isolates. Many of the Iranian cultivars and landraces were susceptible to most of the isolates. GWAS results revealed that 27 significant MTA within 19 QTLs, of which 6 QTLs (Qstb.iau-3, Qstb.iau-5, Qstb.iau-6, Qstb.iau-7, Qstb.iau-8, and Qstb.iau-15), are more interesting as they are associated with resistance to two or more Z. tritici isolates. Although most of the QTLs identified in this study co-localized with previously known STB QTLs/genes, five novel genomic regions associated with resistance to multiple Z. tritici isolates were identified. The QTLs localized on chromosome 5D confers that resistance to IPO86013 isolate was not aligned with recently reported QTLs for STB on this chromosome. Given that the physical position of our identified QTLs is different from those reported previously, therefore, it can be considered as a potentially novel QTLs on this chromosome. By aligning the significant MTA against available wheat reference genome sequence and gene expression analysis, we characterized several candidate genes involved in plant defense mechanisms against pathogens. These genes are of interest and their exact roles in STB resistance remain to be functionally analyzed in the future. Molecular and functional characterization of these QTLs/genes eventually will enhance our understanding of how resistance is achieved and sheds light on biochemical mechanisms underlying resistance against STB. As well as, the significant QTLs and MTA identified in this study will be further validated and can be used in marker-assisted selection for resistance to STB in wheat breeding programs.
Data availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
References
Abrinbana M, Mozafari J, Shams-bakhsh M, Mehrabi R (2010) Genetic structure of Mycosphaerella graminicola populations in Iran. Plant Pathol 59:829–838
Abrinbana M, Mozafari J, Shams-Bakhsh M, Mehrabi R (2012) Resistance spectra of wheat genotypes and virulence patterns of Mycosphaerella graminicola isolates in Iran. Euphytica 186:75–90
Aghamiri A, Mehrabi R, Talebi R (2015) Genetic diversity of Pyrenophera tritici-repentis isolates, the causal agent of wheat tan spot disease from Northern Iran. Iran J Biotech 13(2):e1118
Alam M, Neal J, O’Connor K, Kilian A, Topp B (2018) Ultra-high-throughput DArTseq-based silicoDArT and SNP markers for genomic studies in macadamia. PLoS ONE 13(8):e0203465
Andersen EJ, Ali S, Byamukama E, Yen Y, Nepal MP (2018) Disease resistance mechanisms in plants. Genes 9:39
Arraiano L, Brown J (2006) Identification of isolate-specific and partial resistance to septoria tritici blotch in 238 European wheat cultivars and breeding lines. Plant Pathol 55(6):726–738
Arraiano LS, Brown JKM (2017) Sources of resistance and susceptibility to Septoria tritici blotch of wheat. Mol Plant Pathol 18(2):276–292
Bansal M, Adamski NM, Toor PI, Kaur S, Molnar I, Holušova K, Vrana J, Doležel D, Valarik M, Uauy C, Chhuneja P (2020) Aegilops umbellulata introgression carrying leaf rust and stripe rust resistance genes Lr76 and Yr70 located to 9.47-Mb region on 5DS telomeric end through a combination of chromosome sorting and sequencing. Theor Appl Genet 133:903–915
Bartoli C, Roux F (2017) Genome-wide association studies in plant pathosystems: toward an ecological genomics approach. Front Plant Sci 8:763
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping ofcomplex traits in diverse samples. Bioinformatics 23:2633–2635
Brading PA, Verstappen ECP, Kema GH, Brown JKM (2002) A gene-for-gene relationship between wheat and Mycosphaerella graminicola, the Septoria tritici blotch pathogen. Phytopathology 92:439–445
Brown JK, Chartrain L, Lasserre-Zuber P, Saintenac C (2015) Genetics of resistance to Zymoseptoria tritici and applications to wheat breeding. Fungal Genet Biol 79:33–41
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopouloset J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinformatics 10:421
Chartrain L, Brading PA, Widdowson JP, Brown JKM (2004) Partial resistance to Septoria tritici blotch (Mycosphaerella graminicola) in the wheat cultivars Arina and Riband. Phytopathol 94:497–504
Chartrain L, Berry ST, Brown JKM (2005a) Resistance of wheat line Kavkaz-K4500 L.6.A.4 to Septoria tritici blotch controlled by isolate-specific resistance genes. Phytopathol 95:664–671
Chartrain L, Brading PA, Brown JKM (2005b) Presence of the Stb6 gene for resistance to Septoria tritici blotch (Mycosphaerella graminicola) in cultivars used in wheat-breeding programs worldwide. Plant Pathol 54:134–143
Chartrain L, Joaquim P, Berry ST, Arraiano LS, Azanza F, Brown JKM (2005c) Genetics of resistance to septoria tritici blotch in the Portuguese wheat breeding line TE9111. Theor Appl Genet 110:1138–1144
Cowger C, Hoffer ME, Mundt CC (2000) Specific adaptation by Mycosphaerella graminicola to a resistant wheat cultivar. Plant Pathol 49:445–451
Dreisigacker S, Wang X, Cisneros BAM, Jing R, Singh PK (2015) Adult-plant resistance to septoria tritici blotch in hexaploid spring wheat. Theor Appl Genet 128:2317–2329
Egea LA, Mérida-García R, Kilian A, Hernandez P, Dorado G (2017) Assessment of genetic diversityand structure of large garlic (Allium sativum) germplasm bank, by Diversity Arrays Technology “Genotyping-by-Sequencing” Platform (DArTseq). Front Genet 8:98
Eriksen L, Borum F, Jahoor A (2003) Inheritance and localization of resistance to Mycosphaerella graminicola causing septoria tritici blotch and plant height in the wheat (Triticum aestivum L.) genome with DNA markers. Theor Appl Genet 107:515–527
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fu S, Ren Z, Chen X, Yan B, Tan F, Fu T, Tang Z (2014) New wheat-rye 5DS-4RS_4RL and 4RS-5DS-5DL translocation lines with powdery mildew resistance. J Plant Res 127:743–753
Gahlaut V, Jaiswal V, Singh S, Baylan H, Gupta PK (2019) Multi-locus genome wide association mapping for yield and its contributing traits in hexaploid wheat under different water regimes. Sci Rep 9:19486
Ghaneie A, Mehrabi R, Safaie N, Abrinbana M, Saidi A, Aghaee M (2012) Genetic variation for resistance to septoria tritici blotch in Iranian tetraploid wheat landraces. Eur J Plant Pathol 132(2):191–202
Gikas GD, Parlakidis P, Mavropoulos T, Vryzas Z (2022) Particularities of fungicides and factors affecting their fate and removal efficacy: a review. Sustainability 14:4056
Godoy JG, Rynearson S, Chen X, Pumphrey M (2018) Genome-wide association mapping of loci for resistance to stripe rust in north American elite spring wheat germplasm. Phytopathology 108(2):234–245
Goodwin SB (2007) Back to basics and beyond: increasing the level of resistance to septoria tritici blotch in wheat. Aust Plant Pathol 36:532–538
Goudemand E, Laurent V, Duchalais L, Ghaffary SMT, Kema GHJ, Lonnet P, Margalé E, Robert O (2013) Association mapping and meta-analysis: two complementary approaches for the detection of reliable septoria tritici blotch quantitative resistance in bread wheat (Triticum aestivum L.). Mol Breed 32:563–584
Hammer Ø, Harper DAT, Ryan PD (2001) Paleontological statistics software package for education and data analysis. Palaeontol Electron 4:9–18
Han GZ (2019) Origin and evolution of the plant immune System. New Phytol 222:70–83
Hardwick NV, Jones DR, Slough JE (2001) Factors affecting diseases in winter wheat in England and Wales, 1989–98. Plant Pathol 50:453–462
Hosseinnezhad A, Khodarahmi M, Rezaee S, Mehrabi R, Roohparvar R (2014) Effectiveness determination of wheat genotypes and Stb resistance genes against Iranian Mycosphaerella graminicola isolates. Arch Phytopathol Plant Protec 47(17):2051–2069
Ibrahim AK, Zhang L, Niyitanga S, Afzal MZ, Xu Y, Zhang L, Zhang L, Qi J (2020) Principles and approaches of association mapping in plant breeding. Trop Plant Biol 13:212–224
Kaler AS, Gillman JD, Beissinger T, Purcell LC (2020) Comparing different statistical models and multiple testing corrections for association mapping in soybean and maize. Front Plant Sci 10:1794
Kema GHJ, van Silfhout CH (1997) Genetic variation for virulence and resistance in the wheat Mycosphaerella graminicola pathosystem. III Comparative seedling and adult plant experiments. Phytopathology 87:266–272
Kema GHJ, Sayoud R, Annone JG, van Silfhout CH (1996) Genetic variation for virulence and resistance in the wheat-Mycosphaerella graminicola pathosystem. 2. Analysis of interactions between pathogen isolates and host cultivars. Phytopathol 86:213–220
Kema GHJ, Verstappen ECP, Waalwijk C (2000) Avirulence in the wheat septoria tritici leaf blotch fungus Mycosphaerella graminicola is controlled by a single locus. Mol Plant Microbe Interact 13:1375–1379
Kema GHJ, Gohari AM, Aouini L, Gibriel HA, Ware SB, van Den Bosch F, Manning-Smith R, Alonso-Chavez V, Helps J, M’Barek SB (2018) Stress and sexual reproduction affect the dynamics of the wheat pathogen effector AvrStb6 and strobilurin resistance. Nature Genet 50(3):375
Kidane YG, Hailemariam BN, Mengistu DK, Fadda C, Pè ME, Dell’Acqua M (2017) Genome wide association study of septoria tritici blotch resistance in Ethiopian durum wheat landraces. Front Plant Sci 8:1586
Langlands-Perry C, Cuenin M, Bergez C, Krima SB, Gélisse S, Sourdille P, Valade R, Marcel TC (2022) Resistance of the wheat cultivar ‘Renan’ to septoria leaf blotch explained by a combination of strain specific and strain non-specific QTL mapped on an ultra-dense genetic map. Genes 13:100
Lassner MW, Peterson P, Yoder JI (1989) Simultaneous amplification of multiple DNA fragments by polymerase chain reaction in the analysis of transgenic plants and their progeny. Plant Mol Biol Report 7:116–128
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang Z (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28(18):2397–2399
Liu X, Huang M, Fan B, Buckler ES, Zhang Z (2016) Iterative usage of fxed and random efect models for powerful and efcient genome-wide association studies. PLoS Genet 12:e1005767
Liu S, Bai G, Lin M, Luo M, Dadong D, Jin F, Tian B, Trick HN, Yan L (2020) Identification of candidate chromosome region of Sbwm1 for Soil-borne wheat mosaic virus resistance in wheat. Sci Rep 10:8119
Louriki S, Rehman S, El Hanafi S, Bouhouch Y, Al-Jaboobi M, Amri A, Douira A, Tadesse W (2021) Identification of resistance sources and genome-wide association mapping of septoria tritici blotch resistance in spring bread wheat germplasm of ICARDA. Front Plant Sci 12:600176
Maccaferri M, Zhang J, Bulli P, Abate Z, Chao SH, Cantu D, Bossolini E, Chen X, Pumphery M, Dubcovsky J (2015) A genome-wide association study of resistance to stripe rust (Puccinia striiformis f. sp. tritici) in a worldwide collection of hexaploid spring wheat (Triticum aestivum L.). G3 5:449–465
Mahboubi M, Talebi R, Aghaee Sarbarzeh M, Naji AM, Mehrabi R (2020) Resistance and virulence variability in wheat–Zymoseptoria tritici interactions. Crop Pasture Sci 71(7):645–652
Makhdoomi A, Mehrabi R, Khodarahmi M, Abrinbana M (2015) Efficacy of wheat genotypes and Stb resistance genes against Iranian isolates of Zymoseptoria tritici. J Gen Plant Pathol 81(1):514
Mehrabi R, van der Lee T, Waalwijk C, Kema GHJ (2006) MgSlt2, a cellular integrity MAP kinase gene of the fungal wheat pathogen Mycosphaerella graminicola, is dispensable for penetration but essential for invasive growth. Mol Plant Microbe Interact 19:389–398
Mehrabi R, Makhdoomi A, Jafar-Aghaie M (2015) Identification of new sources of resistance to septoria tritici blotch caused by Zymoseptoria tritici. J Phytopathol 163(2):84–90
Metsalu T, Vilo J (2015) ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res 43:W566–W570
Mirdita V, Liu G, Zhao Y, Miedaner T, Longin CFH, Gowda M, Mette MF, Reif JC (2015) Genetic architecture is more complex for resistance to septoria tritici blotch than to fusarium head blight in central european winter wheat. BMC Genom 16:430
Mohammadi N, Mehrabi R, Gohari AM, Mohammadi GE, Safaie N, Kema GHJ (2017) The ZtVf1 transcription factor regulates development and virulence in the foliar wheat pathogen Zymoseptoria tritici. Fungal Genet Biol 109:26–35
Mohler V, Schmolke M, Zeller FJ, Hsam SLK (2020) Genetic analysis of Aegilops tauschii-derived seedling resistance to leaf rust in synthetic hexaploid wheat. J Appl Genet 61:163–168
Monostori I, Szira F, Tondelli A, Arendas T, Gierczik K, Cattivelli L, Galiba G, Vagujfalvi A (2017) Genome-wide association study and genetic diversity analysis on nitrogen use efficiency in a Central European winter wheat (Triticum aestivum L.) collection. PLoS ONE 12(12):e0189265
Muqaddasi QH, Zhao Y, Rodemann B, Plieske J, Ganal MW, Röder MS (2019) Genome-wide association mapping and prediction of adult stage blotch infection in European winter wheat via high-density marker arrays. Plant Genome 12:180029
Nielsen NH, Backes G, Stougaard J, Andersen SU, Jahoor A (2014) Genetic diversity and population structure analysis of European hexaploid bread wheat (Triticum aestivum L.) varieties. PLoS ONE 9:e94000
Odilbekov F, Armoniené R, Koc A, Svensson J, Chawade A (2019) GWAS-assisted genomic prediction to predict resistance to septoria tritici blotch in nordic winter wheat at seedling stage. Front Genet 10:1224
Pandian BA, Sathishraj R, Djanaguiraman M, Prasad PVV, Jugulam M (2020) Role of Cytochrome P450 enzymes in plant stress response. Antioxidants 9:454
Ramírez-González RH, Borrill P, Lang D, Harrington SA, Brinton J, Venturini L, Davey M, Jacobs J, van Ex F, Pasha A, Khedikar Y, Robinson SJ, Cory AT, Florio T, Concia L, Juery C, Schoonbeek H, Steuernagel B, Xiang D, Ridout CJ, Chalhoub B, Mayer KFX, Benhamed M, Latrasse D, Bendahmane A, International Wheat Genome Sequencing Consortium., Wulff BBH, Appels R, Tiwari V, Datla R, Choulet F, Pozniak CJ, Provart NJ, Sharpe AG, Paux E, Spannagl M, Bräutigam A, Uauy C (2018) The transcriptional landscape of polyploid wheat. Science 361(6403):eaar6089
Riaz A, KockAppelgren P, Hehir JG, Kang J, Meade F, Cockram J, Milbourne D, Spink J, Mullins E, Byrne S (2020) Genetic analysis using a multi-parent wheat population identifies novel sources of septoria tritici blotch resistance. Genes 11:887
Risser P, Ebmeyer E, Korzun V, Hartl L, Miedaner T (2011) Quantitative trait loci for adult-plant resistance to Mycosphaerella graminicola in two winter wheat populations. Phytopathology 101:1209–1216
Sansaloni CP, Petroli CD, Carling J, Hudson CJ, Steane DA, Myburg AA, Grattapaglia D, Vaillancourt RE, Kilian A (2011) A high-density Diversity Arrays Technology (DArT) microarray for genome-wide genotyping in Eucalyptus. Plant Methods 6:16
Simón MR, Worland AJ, Struik PC (2005) Chromosomal location of genes encoding for resistance to septoria tritici blotch (Mycosphaerella graminicola) in substitution lines of wheat. NJAS Wagening J Life Sci 53:113–129
Sokal RR, Rohlf FJ (1995) Biometry: the principles and practice of statistics in biological research, 3rd edn. W. H. Freeman and Company, New York
Somers DJ, Isaac P, Edwards K (2004) A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor Appl Genet 109:1105–1114
Stukenbrock EH, Banke S, Javan-Nikkhah M, McDonald BA (2007) Origin and domestication of the fungal wheat pathogen Mycosphaerella graminicola via sympatric speciation. Mol Biol Evol 24:398–411
Suffert F, Sache I, Lannou C (2011) Early stages of septoria tritici blotch epidemics of winter wheat: build-up, overseasoning, and release of primary inoculum. Plant Pathol 60:166–177
Tabib Ghaffary SM, Robert O, Laurent V, Lonnet P, Margale E, van der Lee TAJ, Visser RGF, Kema GHJ (2011) Genetic analysis of resistance to Septoria tritici blotch in the French winter wheat cultivars Balance and Apache. Theor Appl Genet 123:741–754
Tabib Ghaffary SM, Faris JD, Friesen TL, Visser RGF, van der Lee TAJ, Robert O, Kema GHJ (2012) New broad-spectrum resistance to Septoria tritici blotch derived from synthetic hexaploid wheat. Theor Appl Genet 124:125–142
Talebi R, Mardi M, Jelodar NB, Razavi M, Pirseyedi SY, Kema G, Mehrabi R, Marcel T (2010) Specific resistance genes in wheat chinese landrace ‘Wangshuibai’ against two Iranian Mycosphaerella graminicola isolates. Int J Biol 2:181–188
The International Wheat Genome Sequencing Consortium (IWGSC), Appels R, Eversole K, Feuillet C, Keller B, Rogers J, Stein N, Pozniak CJ, Stein N, Choulet F, Distelfeld A, Eversole K, Poland J, Rogers J, Ronen G, Sharpe AG, Pozniak C, Ronen G, Stein N, Barad O, Baruch K et al (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:eaar7191
Torriani SF, Brunner PC, McDonald BA, Sierotzki H (2009) QoI resistance emerged independently at least 4 times in European populations of Mycosphaerella graminicola. Pest Manag Sci 65(2):155–162
Vagndorf N, Nielsen NH, Edriss V, Andersen JR, Orabi J, Jørgensen LN, Jahoor A (2017) Genome-wide association study reveals novel quantitativetrait loci associated with resistance towards septoria tritici blotch in North European winter wheat. Plant Breed 136(4):474–482
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
Yan Q, Cui X, Lin S, Gan S, Xing H, Dou D (2016) GmCYP82A3, a soybean cytochrome P450 family gene involved in the jasmonic acid and ethylene signaling pathway, enhances plant resistance to biotic and abiotic stresses. PLoS ONE 11(9):e0162253
Yang N, McDonald MC, Solomon PS, Milgate AW (2018) Genetic mapping of Stb19, a new resistance gene to Zymoseptoria tritici in wheat. Theor Appl Genet 131(12):2765–2773
Yates S, Mikaberidze A, Krattinger SG, Abrouk M, Hund A, Yu K, Studer B, Fouche S, Meile L, Pereira D, Karisto P, McDonald BA (2019) Precision phenotyping reveals novel loci for quantitative resistance to septoria tritici blotch. Plant Phenomics 2019:3285904
Acknowledgements
The authors acknowledge the Iranian Seed and Plant Improvement Institute (SPII) for their kind support in germplasm preparation and disease phenotyping experiments.
Funding
Financial support for genotypic analysis was received from Islamic Azad University, Sanandaj Branch, Iran.
Author information
Authors and Affiliations
Contributions
MM disease phenotyping, writing, data analysis; RT and RM conceptualization, methodology, resources, writing—review and editing, supervision, funding acquisition; AMJ methodology, software, data analysis; MM and GK review and editing.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by Izabela Pawłowicz
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mahboubi, M., Talebi, R., Mehrabi, R. et al. Genetic analysis of novel resistance sources and genome-wide association mapping identified novel QTLs for resistance to Zymoseptoria tritici, the causal agent of septoria tritici blotch in wheat. J Appl Genetics 63, 429–445 (2022). https://doi.org/10.1007/s13353-022-00696-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-022-00696-x