
Abstract
Age-related cardiovascular disease is the leading cause of death in elderly populations. Coxibs, including celecoxib, valdecoxib, etoricoxib, parecoxib, lumiracoxib, and rofecoxib, are selective cyclooxygenase-2 (COX-2) inhibitors used to treat osteoarthritis and rheumatoid arthritis. However, many coxibs have been discontinued due to adverse cardiovascular events. COX-2 contains cyclooxygenase (COX) and peroxidase (POX) sites. COX-2 inhibitors block COX activity without affecting POX activity. Recently, quercetin-like flavonoid compounds with OH groups in their B-rings have been found to serve as activators of COX-2 by binding the POX site. Galangin-like flavonol compounds serve as inhibitors of COX-2. Interestingly, nabumetone, flurbiprofen axetil, piketoprofen-amide, and nepafenac are ester prodrugs that inhibit COX-2. The combination of galangin-like flavonol compounds with these prodrug metabolites may lead to the development of novel COX-2 inhibitors. This review focuses on the most compelling evidence regarding the role and mechanism of COX-2 in cardiovascular diseases and demonstrates that quercetin-like compounds exert potential cardioprotective effects by serving as cofactors of COX-2.
Similar content being viewed by others
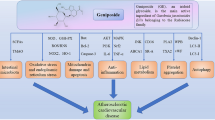

Introduction
Cardiovascular disease is the leading cause of death worldwide. Aging is a major risk factor for cardiovascular diseases (Lopez-Otin et al. 2013). By 2050, the worldwide population aged 60 years and older is expected to total 2 billion, increasing from 900 million in 2015, according to the World Health Organization (WHO). Today, 125 million people are aged 80 years or older, and by 2050, there will be almost as many people (120 million) in this age group living in China alone and 434 million people in this age group worldwide (Sendama 2020; Tyrrell and Goldstein 2020). Thus, the prevention and treatment of cardiovascular disease is a great challenge.
Cyclooxygenase-2 (COX-2) is the key rate-limiting enzyme required for the conversion of arachidonic acid (AA) to prostanoids (PGE2, PGD2, PGF2a, PGI2, and TAX2) Morre et al. (2020). The suppression of COX-2 is mediated by nonsteroidal anti-inflammatory drugs (NSAIDs), which are one of the most diverse classes of drugs clinically available to attenuate pain and inflammation. However, NSAIDs induce serious adverse events, including gastrointestinal (GI) and cardiovascular complications. Compared with nonselective NSAIDs, COX-2-selective drugs are known as coxibs, including rofecoxib, celecoxib, and lumiracoxib. Coxibs not only attenuate pain and inflammation but also reduce the incidence of serious GI adverse effects. However, coxibs also cause cardiovascular hazards, including atherosclerosis (coronary heart disease), hypertension, myocardial infarction, stroke, heart failure, arrhythmogenesis and sudden cardiac death (Bahmani et al. 2017; Mitchell et al. 2020). Celecoxib was removed from the market in 2004 by the Food and Drug Administration (FDA). The labels of COX-2 drugs must carry a “black box” warning to highlight the risks of serious cardiovascular events in many countries, including in the United States (US) and according to the Australian and European authorities related to the Therapeutic Goods Administration (TGA) (Arora et al. 2020; Zhu et al. 2020). Previous findings suggest that COX-2 may be a beneficial protein in the cardiovascular system. Interestingly, quercetin-like plant compounds can protect against cardiovascular diseases. Recently, quercetin-like plant compounds have been shown to act as natural cofactors of COX-2 by binding tightly to the peroxidase active site of COX-2 (Chen et al. 2020). These compounds could strongly stimulate the catalytic activity of COX-2 in vitro and in vivo at lower doses (Bai and Zhu 2008, 2010; Wang et al. 2010). We hypothesize that quercetin-like plant compounds decrease the risk of cardiovascular diseases by serving as cofactors of COX-2. In this article, we will review the most compelling evidence regarding the role of COX-2 in cardiovascular disease, and quercetin-like plant compounds exert potential cardioprotective effects by serving as cofactors of COX-2. These findings may be useful in understanding the molecular mechanism underlying the interaction between quercetin compounds and COX-2 in the cardiovascular system.
The protective role and mechanism of COX-2 in cardiovascular disease
COX-2 and atherosclerosis
Atherosclerosis is a major factor of coronary heart disease and is characterized by the formation of fat-laden plaques in large and medium vessels. Clinical data have shown that COX-2-selective inhibitors increase the atheroscerotic burden in patients (Bea et al. 2003; Belton et al. 2003; Burleigh et al. 2002; Burleigh et al. 2005). Global deletion of COX-2 in apoE-/- mice has been shown to accelerate atherogenesis (Yu et al. 2012). COX-2-/- mice have been shown to exhibit increased accumulation of proinflammatory factors and reduced abilities to prevent LDL oxidation and cholesterol efflux (Narasimha et al. 2007), suggesting that COX-2 protects against the development of atherosclerosis. In addition, pharmacological activation of COX-2 inhibitors also promotes the development of atherosclerosis. The COX-2 inhibitor MF-tricyclic increased the early atherosclerosis lesion area in apoE-/- mice (Rott et al. 2003). The inhibition of COX-2-derived PGE2 by celecoxib enhanced P. gingivalis LPS-induced atherosclerosis by increasing the macrophage production of TNFα (Gitlin and Loftin 2009). In another study, the COX-2-selective inhibitors celecoxib and rofecoxib also increased intermediate plaque formation in apoE-/- mice (Metzner et al. 2007). In addition, the effect of COX-2 on atherosclerosis depended on the cell type. The selective depletion of COX-2 in vascular smooth muscle cells (VSMCs) and endothelial cells (ECs) could accelerate atherosclerosis progression in low-density lipoprotein receptor (LDLR)-/-mice (Tang et al. 2014). The depletion of COX-2 in macrophages reduced atherosclerosis progression (Hui et al. 2010), suggesting that the role of COX-2 in atherosclerosis is most likely related to the cell type and atherosclerosis stage. Interestingly, COX-2 was most abundant in the thymus, brain, lung, kidney, stomach and gastrointestinal tract but not in blood vessels, as shown in COX-2fLuc/+ reporter mice (Kirkby 2013). COX-2 deletion accelerated atherosclerosis progression by increasing T lymphocytes in plaques (Kirkby 2014). However, COX-2 deletion did not alter vascular prostaglandin production in apoE-/- and healthy mice, suggesting that COX-2 protects against atherosclerosis independently of local vascular prostacyclin (Kirkby 2014; Kirkby et al. 2012). Taken together, these findings suggest that COX-2 can protect against atherosclerosis in vivo, but the mechanism should be further investigated.
COX-2 and hypertension
Hypertension is a risk factor for cardiovascular disease. Evidence indicates that COX-2 plays an important role in the regulation of blood pressure (Schjerning et al. 2020). The deletion of COX-2 in C57BL6/J mice increased the blood pressure in response to both low and high salt intakes, suggesting that COX-2 activity plays a key role in blood pressure homeostasis in response to salt loading (Ricciotti et al. 2018; Staehr et al. 2013; Zhang et al. 2018). The systolic blood pressure was elevated in response to the selective inhibition (celecoxib), knockout, or mutation of COX-2 in mice with a mixed C57BL/6 × 129/Sv genetic background fed a regular chow diet (Cheng et al. 2006). In addition, a specific COX-2 pharmacological inhibitor could increase blood pressure (Zhu et al. 2020; Yao et al. 2019). Celecoxib also significantly elevated blood pressure in both normal and hypertensive rats (Huang et al. 2019). Another COX-2 inhibitor, rofecoxib, caused an increase in blood pressure dependent on PGI2 synthesis in normotensive Wistar-Kyoto rats (WKYs) and young spontaneously hypertensive rats (SHRs) fed a normal-salt or high-salt diet (Hocherl et al. 2002). Rofecoxib also completely prevented the hypotensive effects of the ACEi inhibitor lisinopril in SHRs (Ricciotti et al. 2018; Dubey et al. 2005). Most importantly, clinical studies suggest that hypertension was more common in patients taking COX-2 inhibitors such as celecoxib and etoricoxib, and COX inhibition may attenuate the effects of some antihypertensive therapeutics (Mitchell et al. 2020; Chan et al. 2009). Therefore, COX-2 has the ability to decrease blood pressure.
COX-2 and myocardial ischemia–reperfusion injury
Ischemic heart disease, including acute myocardial infarction, is a major cause of death and disability worldwide. Early reperfusion is helpful for myocardial salvage but easily induces reperfusion injury, which then reduces the benefits of myocardial reperfusion. Epidemiological studies have clearly established that COX-2 alleviates myocardial ischemia–reperfusion (I/R) injury (Zhu et al. 2020; Bolli et al. 2002). Endothelial COX-2–derived PGI2 suppresses platelet aggregation. Coxibs promote thrombosis by depressing PGI2 synthesis without altering TxA2 synthesis. COX-2 promotes the recovery of left ventricular pressure after cardiac ischemia (Zhu et al. 2020). COX-2 also increases the protective effects of the late phase of ischemic preconditioning (PC) against both myocardial stunning and myocardial infarction by mediating the synthesis of PGE2 and/or PGI2. Inhibition of COX-2 activity augments myocardial cell death by obliterating the innate defensive response of the heart against I/R injury. COX-2 plays an indispensable role in protecting the heart against I/R injury (Bolli et al. 2002). COX-2 protects isolated myocytes from oxidative stress, and COX-2 inhibitors exacerbate doxorubicin-mediated myocardial injury (Adderley and Fitzgerald 1999; Dowd et al. 2001). Targeted disruption of the COX-2 gene in COX-2-knockout mice or selective deletion of COX-2 in cardiomyocytes has been shown to contribute to myocardial fibrosis and myocardial I/R injury (Dinchuk et al. 1995; Camitta et al. 2001; Papanicolaou et al. 2010). Transgene-mediated overexpression of human COX-2 protected against IR injury in mice (Inserte et al. 2009). In fact, apoptotic cell death promoted I/R injury. The inhibition of COX-2 enhanced I/R injury by promoting cell death (Dowd et al. 2001; Camitta et al. 2001). The protective role of COX-2 in myocardial I/R injury has also been identified with other molecules or drugs. Adiponectin induced COX-2 expression via a SphK-1–S1P receptor mechanism in the heart (Ikeda et al. 2008). Adiponectin protected against myocardial I/R injury by activating COX-2 and releasing PGE2 in cardiac cells (Li et al. 2003; Shibata et al. 2005; Minami et al. 2008). Adiponectin also promoted endothelial cell function and revascularization in ischemic muscle via a COX-2-dependent mechanism (Ohashi et al. 2009), suggesting that in the context of cardioprotection, adiponectin is closely associated with COX-2 activation. In addition, estrogen protected the heart from I/R injury via COX-2 activation and PGI2 synthesis (Booth et al. 2008; Xiao et al. 2001). High-density lipoprotein (HDL) has been reported to protect the heart against I/R injury by reducing cardiac TNFα levels and enhancing cardiac PGE2 and PGI2 release (Calabresi et al. 2003; Rossoni et al. 2004). HDL induced COX-2 expression and PGI2 release via a p38 MAPK/CRE-dependent pathway in endothelial cells (Norata et al. 2004), suggesting that HDL protected against myocardial injury through a COX-2-dependent mechanism. The beneficial effects of iNOS gene therapy on myocardial I/R injury are also associated with the upregulation of COX-2 activity (Li et al. 2003; Li et al. 2007). Peroxisome proliferator-activated receptor γ (PPARγ) agonists and recombinant human erythropoietin (rhEPO) were also effective in protecting against I/R injury in the heart by inducing COX-2 expression (Wang et al. 2012; Liu et al. 2006). Glucocorticoids protected against myocardial injury by activating COX-2 expression and lipocalin-type prostaglandin D synthase (L-PGDS)-derived PGD2 biosynthesis in cardiomyocytes (Tokudome et al. 2009). These findings suggest that COX-2 exerts beneficial effects on myocardial I/R injury. The beneficial effects of COX-2 on myocardial I/R injury are mainly mediated by PGI2, PGE2 and PGD2 through mechanisms including adenylyl cyclase antagonism, ATP-sensitive potassium channel activation, Ca2+ influx inhibition, and neutrophil infiltration attenuation (Bolli et al. 2002; Shinmura et al. 2002; Shinmura et al. 2000). PGE2 and PGI2 reduce myocardial I/R injury through the EP3, EP4 and IP receptors (Booth et al. 2008; Xiao et al. 2001; Xiao et al. 2004; Martin et al. 2005; Hohlfeld et al. 2000; Hishikari et al. 2009; Hirata et al. 2012). PGD2 and its dehydrated metabolite (15-deoxy-Δ12,14-PGJ(2)) protect the heart against I/R injury by activating Nrf2 predominantly via the FP receptor (Katsumata et al. 2014). Taken together, these findings suggest that cardiac COX-2 activity might be a promising tool for cardioprotection against myocardial I/R injury by producing PGE2, PGI2 and PGD2, which act through their own or other PG receptor signaling pathways.
The possible mechanism of COX-2 inhibitor-mediated cardiotoxicity
COX-2 includes cyclooxygenase (COX) and peroxidase (POX) active sites (Chan et al. 2019; Chandel et al. 2018). AA binds to the COX active site and is converted to PGG2. PGG2 has a high binding affinity for the POX site; thus, it tightly binds to this site and is converted to PGH2. Finally, cell synthases and isomerases convert PGH2 to prostaglandins. Interestingly, COX-2 inhibitors block COX activity without affecting POX activity (Radi and Khan 2019). The phenylalanine-385 mutant of COX-2 lacks COX activity but retains POX activity, suggesting that tyrosine 385 of COX-2 is a critical residue for the initiation of COX catalysis (Yu and Funk 2007). COX-2 Y385F mice have disrupted COX activity, while POX activity is fully intact. COX-2 knockout mice have disruptions in both COX and POX activity. Interestingly, both diastolic and systolic blood pressure were elevated in COX-2 Y385F mice, COX-2 knockout mice and COX-2 inhibitor celecoxib-treated mice. These three groups of mice exhibited increased platelet consumption and thrombogenesis. The mice exhibited decreased urinary PGI2 metabolites, but TxA2 metabolites did not show overt alterations (Yu and Funk 2007; Seta et al. 2009). These results suggest that COX-2 inhibitors cause cardiotoxicity by blocking COX activity but not the POX site of COX-2.
The possible cardioprotective effects of quercetin-like plant flavonoids as cofactors of COX-2
Quercetin-like plant flavonoids are natural cofactors of COX-2
Recently, quercetin-like plant compounds with OH groups in their B-rings have been shown to be strong activators of the catalytic activity of COX-2 as cofactors in vitro and in vivo (Bai and Zhu 2008, 2010; Wang et al. 2010, 2018, 2019). Quercetin-like plant compounds (including quercetin, myricetin, fisetin, morin, 5,4′-dihydroxyflavone, and 7,4′-dihydroxyflavone) at very low concentrations (< 1 μM) can stimulate the formation of prostaglandins in a concentration-dependent manner (Table 1). Quercetin-like plant compounds have a high potency for activating COX-2, with an apparent EC50 value of approximately 50 nM (Bai and Zhu 2008, 2010). Specifically, quercetin compounds have the ability to bind to the COX-2 POX active site and promote COX-2 reactivation by facilitating electron transfer from compounds to heme by directly interacting with heme during a catalytic cycle (Wang et al. 2010). Most importantly, the administration of quercetin compounds strongly increased the plasma and tissue levels of several PG products in normal Sprague–Dawley rats, suggesting that quercetin-like plant compounds are naturally occurring activators of COX-2 as cofactors in vivo (Bai and Zhu 2008, 2010; Wang et al. 2010, 2018, 2019). In addition, galangin, chrysin and flavone, which have no hydroxyl groups in their B-rings, suppressed COX-2 and its mediated formation of PGs by blocking the POX site of COX-2 (Bai and Zhu 2008; Wang et al. 2018, 2019; Hyoung -Woo Bai, BT. Z. 2009; Bai et al. 2021). Curcumin also increases COX-2 expression in a time- and concentration-dependent manner (Tan et al. 2011). However, the mechanism by which curcumin acts as a cofactor of COX-2 is unclear.
Notably, quercetin compounds at higher concentrations (> 10 μM) inhibited COX-2 activity, whereas at low concentrations (10 nM), they stimulated COX-2 activity (Bai and Zhu 2008, 2010; Paoletti et al. 2009). Surprisingly, high concentrations of quercetin compounds suppressed COX-2 expression in vitro, but these compounds did not affect COX-2 expression in vivo and could even stimulate COX-2 activity (Arias et al. 2014; Pascual-Teresa et al. 2004; Nieman et al. 2007; Choi et al. 2006). Taken together, these findings suggest that quercetin-like natural plant compounds stimulate COX-2 catalytic activity by acting as cofactors of COX-2, and this effect depends on the OH structural features of their B-rings.
Quercetin compounds exert cardioprotective effects by serving as cofactors of COX-2
Quercetin-like plant compounds have been shown to be beneficial to the cardiovascular system due to their antiatherogenic, anti-inflammatory, anticoagulative and antihypertensive effects (Deng et al. 2020; Pechanova et al. 2020; Sato and Mukai 2020). The AIN-93M diet is a flavonoid-deficient diet. The atherosclerotic plaque areas of apoE-/- mice fed the AIN-93M diet were increased by approximately 3–fourfold compared with those of C57BL/6J mice. Quercetin compounds almost completely abrogated AIN-93M-induced lesion formation in ApoE-/- mice (Loke et al. 2010). In fact, when the animals were fed a flavonoid-deficient diet, the catalytic activity of the COX-2 enzyme was very low, and the animals even died because the POX site lacked cofactors such as quercetin-like plant compounds. As mentioned above, COX-2 could reduce the atherosclerosis process. Thus, the plaque areas were increased in apoE-/-mice fed a flavonoid-deficient diet, despite the fat levels of these diets being very low (4%) (Loke et al. 2010), suggesting that quercetin-like plant flavonoids protect against the development of atherosclerosis as cofactors of COX-2.
Many studies suggest that quercetin compounds decrease blood pressure in hypertensive patients and animal models (Larson et al. 2012a). The NO and PGI2 pathways decrease blood pressure by relaxing blood vessels and inhibiting platelet activation. Quercetin compounds decreased the mean blood pressure by 5 mmHg in hypertensive men through a mechanism that was independent of changes in NO bioavailability (Larson et al. 2012b). Quercetin compounds also reduced the blood pressure, cardiac hypertrophy and vascular remodeling in NO-deficient rats (Duarte et al. 2002), suggesting that quercetin compounds mediate blood pressure through other mechanisms. Interestingly, quercetin compounds induced vasorelaxation through the COX-2/PGI2 pathway, which was not dependent on the NO pathway (Roghani et al. 2004). Consistent with these observations, we hypothesize that quercetin-like plant compounds act as cofactors of COX-2 to stimulate PGI2 release and then relax blood vessels to decrease blood pressure.
Quercetin compounds also play a protective role in alleviating myocardial injury (Lu 2020; Zhang et al. 2020). Quercetin compound postconditioning produced significant protective effects against myocardial I/R injury in rats by activating the PI3K/Akt signaling pathway. However, quercetin was used 5 min before reperfusion, and the heart was reperfused for 2 h (Wang et al. 2013). The PI3K/Akt signaling pathway was not activated quickly in vitro or in vivo (Liu et al. 2014). Interestingly, COX-2-mediated PGE2 formation reached a plateau 1 h after quercetin administration (Bai and Zhu 2010). COX-2 protects against myocardial injury by producing PGE2, PGI2 and PGD2, indicating that quercetin compounds induce cardioprotection via the COX-2/PG pathway in vivo. Therefore, we hypothesize that quercetin-like plant compounds may protect against myocardial I/R injury as cofactors of COX-2.
Conclusion
Accumulating evidence has indicated that COX-2 is a beneficial protein in cardiovascular disease. Clinical studies suggest that long-term exposure to COX-2 inhibitors known as coxibs may promote the initiation of cardiovascular disease (Jeong et al. 2020; Kang et al. 2020; Liao, et al. 2020). However, clinical and rodent-based studies using coxibs have shown differential toxicity levels in the cardiovascular system, and future work is required. Of interest, quercetin-like plant compounds that are beneficial to the cardiovascular system serve as activators and cofactors of COX-2 because of the OH structural features of their B-rings (Fig. 1). Based on these observations, we suggest a new hypothesis that quercetin-like plant compounds decrease the risk of cardiovascular diseases by serving as cofactors of COX-2. We also suggest that coxibs significantly increase the risk of cardiovascular diseases in animal models fed flavonoid-deficient diets. If these hypotheses are correct, it may explain the mechanism by which coxibs are associated with a high risk of cardiovascular events in response to diets lacking certain flavonoid compounds. In addition, quercetin-like natural plant compounds usually affect multiple targets to prevent cardiovascular events. Thus, the activity of quercetin-like plant compounds as cofactors of COX-2 is just one mechanism by which they decrease the risk of cardiovascular diseases, and more research is needed to confirm this hypothesis. Galangin is present at high levels in the Alpinia officinarum rhizome. It is of interest that the A. officinarum rhizome is an herb used for conditions such as the common cold, wound swelling and pain, stomachache and diarrhea. Given that all currently used NSAIDs target the COX active sites of COX-2, galangin-like compounds that lack B-ring OH groups may serve as good lead compounds for the rational design of novel COX-2 inhibitors for clinical use as anti-inflammatory drugs by targeting the POX active sites of COX-2. Nabumetone, flurbiprofen axetil, piketoprofen-amide, and nepafenac are prodrugs that inhibit COX-2 enzymes (Sehajpal et al. 2018). The effective metabolites of these prodrugs are 6-methoxy-2-naphthyl acetic acid, flurbiprofen, ketoprofen, and amfenac (Table 2). The combination of galangin with these metabolites may lead to the development of novel COX-2 inhibitors, as ester bonds are very easily broken in vivo. We hope that more scientists will focus on the potential roles and associations of COX-2 and quercetin-like natural plant compounds in cardiovascular diseases to identify new drugs for this disease.

COX and POX reactions are catalyzed by coxibs and quercetin-like natural plant compounds, respectively. COX-2 catalyzes arachidonic acid conversion to PGG2 by the COX activity site. The POX activity site of COX-2 reduces PGG2 to PGH2. Downstream prostaglandin products are formed from PGH2 via different synthases. The COX activity site of COX-2 but not the POXsite is inhibited by COX-2–selective inhibitors called coxibs. The labels of coxibs must carry a “black box” warning due to adverse cardiovascular events. Quercetin-like natural plant compounds decrease the risk of cardiovascular disease and serve as activators and cofactors of COX-2 to reduce the cardiotoxicity of coxibs by binding to the POX site
Availability of data and materials
Not applicable.
References
Adderley SR, Fitzgerald DJ. Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase-2. J Biol Chem. 1999;274:5038–46.
Arias N, Macarulla MT, Aguirre L, Martinez-Castano MG, Portillo MP. Quercetin can reduce insulin resistance without decreasing adipose tissue and skeletal muscle fat accumulation. Genes Nutr. 2014;9:361.
Arora M, Choudhary S, Singh PK, Sapra B, Silakari O. Structural investigation on the selective COX-2 inhibitors mediated cardiotoxicity: a review. Life Sci. 2020;251:117631.
Bahmani M, Sarrafchi A, Shirzad H, Asgari S, Rafieian-Kopaei M. Cardiovascular toxicity of cyclooxygenase inhibitors and promising natura l substitutes. Curr Pharm Des. 2017;23:952–60.
Bai HW, Zhu BT. Strong activation of cyclooxygenase I and II catalytic activity by dietary bioflavonoids. J Lipid Res. 2008;49:2557–70.
Bai HW, Zhu BT. Myricetin and quercetin are naturally occurring co-substrates of cyclooxygenases in vivo. Prostaglandins Leukot Essent Fatty Acids. 2010;82:45–50.
Bai HW, Yang C, Wang P, Rao S, Zhu BT. Inhibition of cyclooxygenase by blocking the reducing cosubstrate at the peroxidase site: discovery of galangin as a novel cyclooxygenase inhibitor. Eur J Pharmacol. 2021;899:174036.
Bea F, et al. Chronic inhibition of cyclooxygenase-2 does not alter plaque composition in a mouse model of advanced unstable atherosclerosis. Cardiovasc Res. 2003;60:198–204.
Belton OA, Duffy A, Toomey S, Fitzgerald DJ. Cyclooxygenase isoforms and platelet vessel wall interactions in the apolipoprotein E knockout mouse model of atherosclerosis. Circulation. 2003;108:3017–23.
Bolli R, et al. Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovasc Res. 2002;55:506–19.
Booth EA, Flint RR, Lucas KL, Knittel AK, Lucchesi BR. Estrogen protects the heart from ischemia-reperfusion injury via COX-2-derived PGI2. J Cardiovasc Pharmacol. 2008;52:228–35.
Burleigh ME, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation. 2002;105:1816–23.
Burleigh ME, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in ApoE-deficient and C57BL/6 mice. J Mol Cell Cardiol. 2005;39:443–52.
Calabresi L, et al. High-density lipoproteins protect isolated rat hearts from ischemia-reperfusion injury by reducing cardiac tumor necrosis factor-alpha content and enhancing prostaglandin release. Circ Res. 2003;92:330–7.
Camitta MG, et al. Cyclooxygenase-1 and -2 knockout mice demonstrate increased cardiac ischemia/reperfusion injury but are protected by acute preconditioning. Circulation. 2001;104:2453–8.
Chan CC, et al. Do COX-2 inhibitors raise blood pressure more than nonselective NSAIDs and placebo? An updated meta-analysis. J Hypertens. 2009;27:2332–41.
Chan PC, Liao MT, Hsieh PS. The dualistic effect of COX-2-mediated signaling in obesity and insulin resistance. Int J Mol Sci. 2019;20:3115.
Chandel P, Rawal RK, Kaur R. Natural products and their derivatives as cyclooxygenase-2 inhibitors. Future Med Chem. 2018;10:2471–92.
Chen W, et al. The physiologic activity and mechanism of quercetin-like natural plant flavonoids. Curr Pharm Biotechnol. 2020;21:654–8.
Cheng Y, et al. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116:1391–9.
Choi SY, Park JH, Kim JS, Kim MK, Aruoma OI, Sung MK. Effects of quercetin and beta-carotene supplementation on azoxymethane-induced colon carcinogenesis and inflammatory responses in rats fed with high-fat diet rich in omega-6 fatty acids. Biofactors. 2006;27(1–4):137–46.
de Pascual-Teresa S, et al. Quercetin metabolites downregulate cyclooxygenase-2 transcription in human lymphocytes ex vivo but not in vivo. J Nutr. 2004;134:552–7.
Deng Q, Li XX, Fang Y, Chen X, Xue J. Therapeutic potential of quercetin as an antiatherosclerotic agent in atherosclerotic cardiovascular disease: a review. Evid Based Complement Alternat Med. 2020;2020:5926381.
Dinchuk JE, et al. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–9.
Dowd NP, Scully M, Adderley SR, Cunningham AJ, Fitzgerald DJ. Inhibition of cyclooxygenase-2 aggravates doxorubicin-mediated cardiac injury in vivo. J Clin Investig. 2001;108:585–90.
Duarte J, et al. Protective effects of the flavonoid quercetin in chronic nitric oxide deficient rats. J Hypertens. 2002;20:1843–54.
Dubey K, et al. Adverse interactions of rofecoxib with lisinopril in spontaneously hypertensive rats. Clin Toxicol (phila). 2005;43:361–73.
Gitlin JM, Loftin CD. Cyclooxygenase-2 inhibition increases lipopolysaccharide-induced atherosclerosis in mice. Cardiovasc Res. 2009;81:400–7.
Hirata Y, et al. A synthetic prostacyclin agonist with thromboxane synthase inhibitory activity, ONO-1301, protects myocardium from ischemia/reperfusion injury. Eur J Pharmacol. 2012;674:352–8.
Hishikari K, et al. Pharmacological activation of the prostaglandin E2 receptor EP4 improves cardiac function after myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2009;81:123–32.
Hocherl K, Endemann D, Kammerl MC, Grobecker HF, Kurtz A. Cyclo-oxygenase-2 inhibition increases blood pressure in rats. Br J Pharmacol. 2002;136:1117–26.
Hohlfeld T, Meyer-Kirchrath J, Vogel YC, Schror K. Reduction of infarct size by selective stimulation of prostaglandin EP(3)receptors in the reperfused ischemic pig heart. J Mol Cell Cardiol. 2000;32:285–96.
Huang C, et al. MEG3, as a competing endogenous RNA, binds with miR-27a to promote PHLPP2 protein translation and impairs bladder cancer invasion. Mol Ther Nucleic Acids. 2019;16:51–62.
Hui Y, et al. Targeted deletions of cyclooxygenase-2 and atherogenesis in mice. Circulation. 2010;121:2654–60.
Hyoung WB, BTZ. Modulation of COX-1 and COX-2-mediated formation of various arachidonic acid metabolites in vitro and in vivo by dietary polyphenols. Doctoral thesis of the university of kansas; 2009.
Ikeda Y, et al. Cyclooxygenase-2 induction by adiponectin is regulated by a sphingosine kinase-1 dependent mechanism in cardiac myocytes. FEBS Lett. 2008;582:1147–50.
Inserte J, et al. Constitutive COX-2 activity in cardiomyocytes confers permanent cardioprotection constitutive COX-2 expression and cardioprotection. J Mol Cell Cardiol. 2009;46:160–8.
Kang DO, et al. Cardiovascular and bleeding risks associated with nonsteroidal anti-inflammatory drugs after myocardial infarction. J Am Coll Cardiol. 2020;76:518–29.
Katsumata Y, et al. Endogenous prostaglandin D2 and its metabolites protect the heart against ischemia-reperfusion injury by activating Nrf2. Hypertension. 2014;63:80–7.
Kirkby NS, et al. Cyclooxygenase-1, not cyclooxygenase-2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci U S A. 2012;109:17597–602.
Kirkby NS, et al. LC-MS/MS confirms that COX-1 drives vascular prostacyclin whilst gene expression pattern reveals non-vascular sites of COX-2 expression. PLoS One. 2013;8:e69524.
Kirkby NS, et al. COX-2 protects against atherosclerosis independently of local vascular prostacyclin: identification of COX-2 associated pathways implicate Rgl1 and lymphocyte networks. PLoS One. 2014;9:e98165.
Larson A, et al. Acute, quercetin-induced reductions in blood pressure in hypertensive individuals are not secondary to lower plasma angiotensin-converting enzyme activity or endothelin-1: nitric oxide. Nutr Res. 2012a;32:557–64.
Larson AJ, Symons JD, Jalili T. Therapeutic potential of quercetin to decrease blood pressure: review of efficacy and mechanisms. Adv Nutr. 2012b;3:39–46.
Li Q, et al. Gene therapy with inducible nitric oxide synthase protects against myocardial infarction via a cyclooxygenase-2-dependent mechanism. Circ Res. 2003;92:741–8.
Li Q, et al. Cardioprotection afforded by inducible nitric oxide synthase gene therapy is mediated by cyclooxygenase-2 via a nuclear factor-kappaB dependent pathway. Circulation. 2007;116:1577–84.
Liao YC, et al. Association between nonsteroidal anti-inflammatory drug use and major adverse cardiovascular events in patients with end-stage renal disease: a population-based cohort study. J Nephrol. 2020;34:441.
Liu X, et al. Cyclooxygenase-2 plays an essential part in cardioprotection of delayed phase of recombinant human erythropoietin preconditioning in rats. Postgrad Med J. 2006;82:588–93.
Liu H, Guo X, Chu Y, Lu S. Heart protective effects and mechanism of quercetin preconditioning on anti-myocardial ischemia reperfusion (IR) injuries in rats. Gene. 2014;545:149–55.
Loke WM, et al. Specific dietary polyphenols attenuate atherosclerosis in apolipoprotein E-knockout mice by alleviating inflammation and endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2010;30:749–57.
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217.
Lu L, et al. Quercetin for myocardial ischemia reperfusion injury: a protocol for systematic review and meta-analysis. Medicine (Baltimore). 2020;99:e20856.
Martin M, et al. Cardiospecific overexpression of the prostaglandin EP3 receptor attenuates ischemia-induced myocardial injury. Circulation. 2005;112:400–6.
Metzner J, et al. The effects of COX-2 selective and non-selective NSAIDs on the initiation and progression of atherosclerosis in ApoE-/- mice. J Mol Med (berl). 2007;85:623–33.
Minami M, et al. Prostaglandin E receptor type 4-associated protein interacts directly with NF-kappaB1 and attenuates macrophage activation. J Biol Chem. 2008;283:9692–703.
Mitchell JA, et al. Cyclooxygenases and the cardiovascular system. Pharmacol Ther. 2020;217:107624.
Moore N. Coronary risks associated with diclofenac and other NSAIDs: an update. Drug Saf. 2020;43:301–18.
Narasimha A, et al. A novel anti-atherogenic role for COX-2–potential mechanism for the cardiovascular side effects of COX-2 inhibitors. Prostaglandins Other Lipid Mediat. 2007;84:24–33.
Nieman DC, et al. Quercetin's influence on exercise-induced changes in plasma cytokines and muscle and leukocyte cytokine mRNA. J Appl Physiol (1985). 2007;103:1728–35.
Norata GD, Callegari E, Inoue H, Catapano AL. HDL3 induces cyclooxygenase-2 expression and prostacyclin release in human endothelial cells via a p38 MAPK/CRE-dependent pathway: effects on COX-2/PGI-synthase coupling. Arterioscler Thromb Vasc Biol. 2004;24:871–7.
Ohashi K, et al. Adiponectin promotes revascularization of ischemic muscle through a cyclooxygenase 2-dependent mechanism. Mol Cell Biol. 2009;29:3487–99.
Paoletti T, et al. Anti-inflammatory and vascularprotective properties of 8-prenylapigenin. Eur J Pharmacol. 2009;620:120–30.
Papanicolaou KN, et al. Preserved heart function and maintained response to cardiac stresses in a genetic model of cardiomyocyte-targeted deficiency of cyclooxygenase-2. J Mol Cell Cardiol. 2010;49:196–209.
Pechanova O, Dayar E, Cebova M. Therapeutic potential of polyphenols-loaded polymeric nanoparticles in cardiovascular system. Molecules. 2020;25:3322.
Radi ZA, Khan KN. Cardio-renal safety of non-steroidal anti-inflammatory drugs. J Toxicol Sci. 2019;44:373–91.
Jeong HE, Oh IS, Kim WJ, Shin JY. Risk of major adverse cardiovascular events associated with concomitant use of antidepressants and non-steroidal anti-inflammatory drugs: a retrospective cohort study. CNS Drugs; 2020.
Ricciotti E, et al. Cyclooxygenase-2, asymmetric dimethylarginine, and the cardiovascular hazard from nonsteroidal anti-inflammatory drugs. Circulation. 2018;138:2367–78.
Roghani M, Baluchnejadmojarad T, Vaez-Mahdavi MR, Roghani-Dehkordi F. Mechanisms underlying quercetin-induced vasorelaxation in aorta of subchronic diabetic rats: an in vitro study. Vascul Pharmacol. 2004;42:31–5.
Rossoni G, et al. Synthetic high-density lipoproteins exert cardioprotective effects in myocardial ischemia/reperfusion injury. J Pharmacol Exp Ther. 2004;308:79–84.
Rott D, et al. Effects of MF-tricyclic, a selective cyclooxygenase-2 inhibitor, on atherosclerosis progression and susceptibility to cytomegalovirus replication in apolipoprotein-E knockout mice. J Am Coll Cardiol. 2003;41:1812–9.
Sato S, Mukai Y. Modulation of chronic inflammation by quercetin: the beneficial effects on obesity. J Inflamm Res. 2020;13:421–31.
Schjerning AM, McGettigan P, Gislason G. Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat Rev Cardiol. 2020;17:574–84.
Sehajpal S, Prasad DN, Singh RK. Prodrugs of Non-steroidal Anti-inflammatory Drugs (NSAIDs): a long march towards synthesis of safer NSAIDs. Mini Rev Med Chem. 2018;18:1199–219.
Sendama W. The effect of ageing on the resolution of inflammation. Ageing Res Rev. 2020;57:101000.
Seta F, et al. Renal and cardiovascular characterization of COX-2 knockdown mice. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1751-1760.
Shibata R, et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–103.
Shinmura K, et al. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc Natl Acad Sci USA. 2000;97:10197–202.
Shinmura K, et al. Inducible nitric oxide synthase modulates cyclooxygenase-2 activity in the heart of conscious rabbits during the late phase of ischemic preconditioning. Circ Res. 2002;90:602–8.
Staehr M, et al. Deletion of cyclooxygenase-2 in the mouse increases arterial blood pressure with no impairment in renal NO production in response to chronic high salt intake. Am J Physiol Regul Integr Comp Physiol. 2013;304:R899-907.
Tan X, et al. Regulation of the expression of cyclooxygenases and production of prostaglandin I(2) and E(2) in human coronary artery endothelial cells by curcumin. J Physiol Pharmacol. 2011;62:21–8.
Tang SY, et al. Cyclooxygenase-2 in endothelial and vascular smooth muscle cells restrains atherogenesis in hyperlipidemic mice. Circulation. 2014;129:1761–9.
Tokudome S, et al. Glucocorticoid protects rodent hearts from ischemia/reperfusion injury by activating lipocalin-type prostaglandin D synthase-derived PGD2 biosynthesis. J Clin Investig. 2009;119:1477–88.
Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol. 2020;18:58.
Wang H, et al. Pioglitazone attenuates myocardial ischemia-reperfusion injury via up-regulation of ERK and COX-2. Biosci Trends. 2012;6:325–32.
Wang HR, Sui HC, Zhu BT. Ellagic acid, a plant phenolic compound, activates cyclooxygenase-mediated prostaglandin production. Exp Ther Med. 2019;18:987–96.
Wang P, Bai HW, Zhu BT. Structural basis for certain naturally occurring bioflavonoids to function as reducing co-substrates of cyclooxygenase I and II. PLoS One. 2010;5:e12316.
Wang HR, Sui HC, Ding YY, Zhu BT. Stimulation of the production of prostaglandin E(2) by ethyl gallate, a natural phenolic compound richly contained in longan. Biomolecules. 2018;8:91.
Wang Y, et al. Quercetin postconditioning attenuates myocardial ischemia/reperfusion injury in rats through the PI3K/Akt pathway. Brazil J Med Biol Res Revista Brasileira de Pesquisas Medicas e Biologicas/Sociedade Brasileira de Biofisica ... [et al.]. 2013; 46: 861–867.
Xiao CY, et al. Roles of prostaglandin I(2) and thromboxane A(2) in cardiac ischemia-reperfusion injury: a study using mice lacking their respective receptors. Circulation. 2001;104:2210–5.
Xiao CY, et al. Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4. Circulation. 2004;109:2462–8.
Yao L, et al. Regulation of YAP by mammalian target of rapamycin complex 1 in endothelial cells controls blood pressure through COX-2/mPGES-1/PGE2 cascade. Hypertension. 2019;74:936–46.
Yu Y, Funk CD. A novel genetic model of selective COX-2 inhibition: comparison with COX-2 null mice. Prostaglandins Other Lipid Mediat. 2007;82:77–84.
Yu Z, et al. Disruption of the 5-lipoxygenase pathway attenuates atherogenesis consequent to COX-2 deletion in mice. Proc Natl Acad Sci U S A. 2012;109:6727–32.
Zhang MZ, et al. Renal medullary interstitial COX-2 (Cyclooxygenase-2) is essential in preventing salt-sensitive hypertension and maintaining renal inner medulla/papilla structural integrity. Hypertension. 2018;72:1172–9.
Zhang YM, Zhang ZY, Wang RX. Protective mechanisms of quercetin against myocardial ischemia reperfusion injury. Front Physiol. 2020;11:956.
Zhu L, Zhang Y, Guo Z, Wang M. Cardiovascular biology of prostanoids and drug discovery. Arterioscler Thromb Vasc Biol. 2020;40:1454–63.
Acknowledgements
We thank our colleagues in Dr. Dongming Xing’s laboratory for the technical help provided and stimulating discussions during this investigation.
Funding
The authors are grateful for the financial support provided by the Qingdao Major Scientific and Technological Project for Distinguished Scholars (20170103), the Laoshan Major Scientific and Technological Project for Distinguished Scholars (20181030), the Natural Science Foundation of Shandong Province (ZR2020MH242, ZR2020MH369), the Youth Innovation Team Talent Introduction Program of Shandong Province (20190164).
Author information
Authors and Affiliations
Contributions
Conception and design: WC and DX. Manuscript writing: WC and YZ. Collection and assembly of data: SW, ZG and NF. Final discussions and approval of the manuscript: all authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
No individual personal data are involved in this review.
Competing interests
The authors declare no conflicts of interest associated with this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, W., Zhong, Y., Feng, N. et al. New horizons in the roles and associations of COX-2 and novel natural inhibitors in cardiovascular diseases. Mol Med 27, 123 (2021). https://doi.org/10.1186/s10020-021-00358-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10020-021-00358-4