
Abstract
Residue analysis in the field of Palaeolithic studies represents a powerful source of information, as well as a challenging operation of deduction, based on the interpretation of what remains of organic and inorganic compounds that testify to the ancient use of lithic artefacts. The disposal of reliable methods for residue characterisation is thus essential in the approach to this discipline, along with the collection of comprehensive, high-quality data. In this paper, the development of a new analytical protocol is presented through the chemical characterisation of red residues coming from three lithic artefacts retrieved from the Upper Palaeolithic stratigraphic sequence of Grotta Paglicci (Apulia, Italy). The morphological description of each residue was followed by the application of an advanced set of complementary techniques (namely Synchrotron Radiation Fourier-Transform InfraRed microscopy, Synchrotron Radiation Low-Energy X-ray Fluorescence and Particle Induced X-ray Emission) to obtain the molecular and elemental composition. The results provided by this pilot study allowed the identification of various organic and inorganic fractions within the samples, proving the reliability and high sensitivity of our newly designed protocol in dealing with ancient residues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Residue analysis in the study of Palaeolithic represents a vast field of research, involving many different approaches, disciplines, and expertise [1]. One of the most significant issues in this domain is certainly the heterogeneity of methods, resulting in a very stimulating research environment, but also in data outputs not always easy to compare. Besides the variety in the used techniques, one must point out the differences in the adopted approaches, which often probe the residue on different levels (morphological, chemical, structural, elemental level, etc.). Amongst these, the morphological approach constitutes the first step of investigation and a method endorsed by several research groups, both through optical light microscopy [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21] and scanning electron microscopy (SEM) [21,22,23,24,25]. However, several authors have reported issues in the morphological identification of residues, due to misleading structures [26, 27], the absence of diagnostic structures in composite or smeared residues [1, 25, 26], the intrinsic subjectivity of attribution, and the effective disposal of complete reference collections [1]. A further step in morphological identification is provided by the implementation of SEM with Energy Dispersive X-ray Spectroscopy systems (SEM–EDS) [28,29,30,31,32,33,34,35], which is generally employed to support the identification of morphological features with the elemental composition of the sample. This technique is one of the most widespread in the field of residue analysis, since it integrates different information into a single analysis and does not require any special sample preparation in low vacuum mode [32]. Nevertheless, although this approach can be informative about the inorganic fraction of the sample, it does not provide a conclusive knowledge on organic matter, for which molecular information is crucial. Indeed, it represents a valuable technique when combined with other complementary methods (see for instance [36,37,38,39,40]). The need of molecular characterisation has been in part addressed by the emerging use of Fourier Transform InfraRed (FTIR) technique [36, 39,40,41,42,43,44] and especially FTIR microscopy in reflection mode [37, 45,46,47,48,49]. This non-destructive approach allows the residue to be studied in its original context and several collections of reference spectra on various experimental materials have been produced to date [45, 47, 48]. However, FTIR microscopy in reflection mode has shown important limitations in collecting clear and intense signals from the residues. The main issues are due to the contribution of the underlying lithic surface, as well as artefact surface roughness and the refractive index of residue components [50]. All these factors can contribute to the distortion of infrared signals, making the interpretation of results challenging and, moreover, not allowing the detection of trace components. Hence, whilst some of the approaches discussed so far may provide clues for residue interpretation, the use of most of them alone does not allow for a complete chemical characterisation and thus a definitive identification of residues. Given the complexity and challenging nature of the discipline, a methodological procedure as comprehensive and exhaustive as possible represents the only chance for a reliable result.
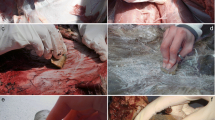
In this paper, we report the analysis of residues coming from three implements of the Upper Palaeolithic sequence of Grotta Paglicci (Apulia, southern Italy) as a case study to prove the effectiveness of a multi-technique approach in the field of residue analysis. As widely known, Grotta Paglicci is a reference site for the Palaeolithic in Italy, as well as for most of the Upper Palaeolithic (40,000–13,000 years ago) of Mediterranean Europe [51]. The site is a karstic system of multiple chambers located on the south-western side of the Gargano promontory, at 143 m above sea level (Fig. 1a). Its protected position on the northern flank and the opening onto the plain of Foggia below probably favoured human occupation through time, as well demonstrated by the huge anthropic deposit recovered in the cave’s atrium and in the so-called Rock shelter, once part of the cave system [52] (Fig. 1b). The site has yielded materials from the Lower and the Middle Palaeolithic, as well as an Upper Palaeolithic sequence that extends continuously through the Aurignacian, the Early, Middle and Final Gravettian and the whole Epigravettian [53, 54]. Amongst other findings, the site has yielded the only Palaeolithic wall paintings known to date in Italy, numerous scattered human remains, several burials and one of the oldest evidence of dog domestication [55,56,57,58,59].

a Location of the Archaeological site of Grotta Paglicci on the south-western slope of the Gargano promontory, southern Italy (Google Maps ©2022). b Cave plan of Grotta Paglicci (survey from Angelo Pasa, 1962) and the adjacent Grotta Dei Pilastri (survey from Franco Orofino) (source: http://www.catasto.fspuglia.it/). c Upper Palaeolithic stratigraphic sequence of the main trench, with the indication of the layers from which the artefacts under analysis were retrieved (original drawing by Giovanni Fabbri & Stefano Ricci). d, e, f Dorsal and ventral view of artefacts 580 (from layer 9c2), 59 and 67 (from layer 24a1), respectively, with the indication of the sampling sites
Of the three lithic artefacts discussed in the present work, two come from layer 24a1 (Aurignacian) and the third comes from layer 9c2 (evolved phase of the Epigravettian) of the main trench (Fig. 1b, c). The Aurignacian is a techno-complex related to a wave of colonisation of Europe by Homo sapiens populations from the Near East [60], and its occurrence at Grotta Paglicci is dated to between about 40,000 and 33,000 years ago. The Epigravettian represents the material culture related to the populations who survived the Last Glacial Maximum in southern Europe and is attested at Grotta Paglicci between approximately 20,000 and 13,500 years ago [51, 53].
The chromatic and morphological examination of the residues was conducted using 3D digital microscopy, a reliable method for the preliminary documentation of residues and their location, thanks to its capability to capture visual information on multiple focal planes [1, 32]. Bright, red residues were identified on all the artefacts, so abundant on two of them to be visible also to the naked eye (see Sect. 3.1 for residue optical description). This finding led to the application of a set of advanced complementary techniques, to ascertain the chemical nature of three examples of Upper Palaeolithic red residues. A specific micro-invasive protocol was devised to minimise intervention on the implement and the residue itself. Thanks to our workflow, each sample was analysed multiple times throughout the protocol, starting with the characterisation of its possible organic content and then, moving on to the study of the inorganic fraction and finally allowing its preservation and storage for eventual further studies. As a first step in the analytical process, FTIR microscopy was performed exploiting the high brilliance of Synchrotron Radiation (SR) for the identification of functional groups of organic and inorganic IR-active compounds [61]. The use of InfraRed Synchrotron Radiation (IRSR) has been proven to be a powerful resource for residue analysis [38, 50] thanks to its high brightness, which allows us to reach a spatial resolution nearing the diffraction limit and thus, select areas of a few micrometres within a single sample [50]. As demonstrated by previous studies [38, 50], the specific use of SR-FTIR microscopy in transmission mode allows the collection of highly intense signals, compared to the performance of conventional sources and the capabilities of the reflection approach [50] (for comparison see for instance [46,47,48]). To support SR-FTIR microscopy results and implement them with more detailed information on the inorganic fraction, two elemental techniques were furtherly applied: SR-Low Energy X-ray Fluorescence (LEXRF) microscopy was used to identify the distribution of light elements, whilst microbeam Particle Induced X-ray Emission (PIXE) was applied to extend the analysis to a wider range of chemical elements, providing further insights and a more complete overview of the elemental constituents of each sample.
2 Materials and methods
2.1 Materials
All the three unretouched artefacts analysed in the present work are made in fine grain chert, without patina and with fresh edges. According to technological analyses [62], the Epigravettian artefact 580 (16.5 mm × 34.3 mm × 5.6 mm) from layer 9c2 is a flake with a portion of lateral cortex that can be reconducted to a by-product of the laminar or lamellar débitage [63, 64] (Fig. 1d). On this artefact, the residue was mainly localised on the distal area of the ventral face, forming a nearly continuous cluster of around 11.0 mm alongside the margin. Smaller quantities of the same residue were observed on the butt and on one of the lateral margins in the proximal area of the ventral face. Artefacts 59 (36.2 mm × 38.8 mm × 5.3 mm) and 67 (16.3 × 8.4 × 2.3 mm) from layer 24a1 have been encompassed by the technological study of Borgia et al. [65]: artefact 59 is a mesial blade fragment, whilst artefact 67 is a proximal bladelet fragment. On artefact 59, two different clusters, one next to the other, were observed in the proximal area of the dorsal face, along one margin of the central blow (Fig. 1e). Finally, artefact 67 showed a residue in a single oval-shaped aggregate approximately 5.0 mm in diameter, located on a detachment alongside the lateral area of the dorsal face (Fig. 1f). A total of four samples (labelled as 580.1, 59.1, 59.2 and 67.1) were collected from the three artefacts, as described in Sect. 3.1.
2.2 Methods
2.2.1 3D digital microscopy (Hirox KH-7700)
The surface of the three artefacts was microscopically analysed by means of a Hirox KH-7700 3D digital microscope using the MXG-5040RZ (SZ) (Middle Range High Resolution Zoom Lens with Optical 3D Rotation) lens, equipped with the Normal (50 ×—400 ×) optical adapter. For the purpose of the present work, magnifications ranging from 50 × to 250 × were used, taking advantage of both ring vertical and coaxial lightning modes. Multifocal tiled images of the residues were acquired, together with the relative 3D models.
2.2.2 Synchrotron Radiation-Fourier Transform InfraRed (SR-FTIR) microscopy
FTIR measurements were performed at the Chemical and Life Sciences branch of SISSI beamline of Elettra Sincrotrone Trieste (Italy) [66]. Samples of red residues were collected from each artefact by the tip of a needle under stereo microscopic observation, using a Stand K LAB ZEISS Stemi 305 equipped with reflected light LED electronics and tiltable mirror-based transmitted light unit, with manual zoom magnifications ranging from 0.8 × to 4.0 × . The samples were then transferred onto a diamond compression cell (Diamond EX press by S.T. Japan, clear aperture 2.0 mm) to be flatten to a thickness suitable for FTIR measurements in transmission mode. The self-carrying double arm goosenecks of the ZEISS Stemi 305 stereo microscope were combined with brightfield, darkfield and oblique light illumination to observe the samples on the lower window of the diamond compression cell, which was opened after the compression. The samples were analysed with a VIS-IR Hyperion 3000 microscope equipped with an MCT-A detector operating in the MidIR range (4000—650 cm−1) and coupled with a BRUKER Vertex 70v interferometer. SR-FTIR measurements were collected in transmission mode setting the lateral resolution at 15 × 15 μm and collecting each spectrum at 4 cm−1 spectral resolution, averaging 512 scans with a scan velocity of 120 kHz. Approximately 20 spectra were collected for each sample.
To collect more detailed information on the presence of oxides and/or metal organic spectral features, the samples were also analysed in the sample compartment of the Bruker Vertex 70v interferometer, mounting the closed diamond compression cell in a 5-multiplication focusing unit (A524/Q, Bruker Optics) and using the Bruker wide range components (FIR-MIR beamsplitter and DTGS detector) for collecting the 4000—50 cm−1 in one single scan. Each spectrum was collected averaging 256 scans at 4 cm−1. A sample of soil from layer 9c2 has been also analysed in this modality and used as reference to discriminate possible inorganic contaminants deriving from the excavation environment.
All the infrared data, processed with Bruker OPUS 8.5 software, were baselined (16 pt. rubber band) and vector normalised. Average spectra were extracted from each sample, and second derivative spectra were calculated to better highlight the presence of in-traces organic compounds. With the same intent, single point spectra were also considered when necessary. The infrared spectra were compared with spectral libraries (Kimmel Centre for Archaeological Science Infrared Standards Library, IRUG Spectral Database and RRUFF database).
2.2.3 Synchrotron Radiation-Low Energy X-ray Fluorescence (SR-LEXRF)
For the distribution of light elements at micrometric spatial resolution, LEXRF microscopy was performed at the TwinMic beamline of the same facility [67]. Samples previously analysed by SR-FTIR microscopy were recovered by pressing adhesive kapton tape onto the diamond culet in correspondence of the sample. Kapton tape strips were then mounted on a specific sample holder suitable for TwinMic soft X-rays measurements. The microscope was operated in Scanning Transmission mode, with the specimens being scanned perpendicularly to the microprobe delivered by a zone plate diffractive optics. The transmitted X-rays were collected by a CCD camera, producing absorption and differential phase contrast images [68], whilst the emitted X-rays were collected simultaneously by 8 Silicon Drift Detectors, generating elemental distribution maps [69]. The samples were mapped at 2 keV with a spatial resolution of 2 μm to excite Si, Al, Mg, Na, Fe and O. The acquired LEXRF spectra were processed with the PyMca software package [70].
2.2.4 Particle Induced X-ray Emission (PIXE)
To extend elemental imaging to elements not accessible by LEXRF microscopy, the same sample holder designed for TwinMic measurements was mounted on the setup of the dual ion microprobe (DuMi) of the Laboratory for Ion Beam Interactions of the Ruđer Bošković Institute, Zagreb (Croatia), to perform microbeam PIXE [71]. The measurements were carried out by scanning a 2 MeV proton beam, focused to ~ 1 × 1 μm2 beam spot, over the same areas analysed with SR-LEXRF microscopy using a SDD detector of 30 mm2. The proton beam current did not exceed 60 pA. The samples were tilted by 30 degrees to the incident beam to reduce the self-absorption effects, whilst SDD detector was positioned at an angle of 60 degrees, respect to the horizontal plane. A 260 μm thick Al funny filter (a filter with a small hole, ~ 2 mm ⌀, in the centre) was placed in front of the detector. In this way, most of the low-energy X-rays were absorbed by the filter, except for the small portion passing through the hole, whilst high-energy X-rays were able to pass through. This setup made it possible to acquire the PIXE imaging of both light and heavy elements simultaneously, with a count rate of 1000 count/s. The corresponding data were then processed through PyMca [70].
3 Results
3.1 Optical and morphological description
On artefact 580, the residue concentrated on the distal portion of the ventral face was so abundant to be clearly visible to the naked eye (Fig. 1d). The microscopic analysis showed the association of its intense red colour with an incoherent powdery texture, so light as to exhibit voids in the most superficial portion of the residue, whilst below this limit it appeared to be well adhered to the lithic surface (Fig. 2a, e). The same superficial interval was characterised in some areas by the presence of duller-coloured granules and some transparent ones, both interspersed with the soft red matrix. Sample 580.1 was collected from the largest cluster and observed under the stereo microscope before and after compression. On the culet of the diamond compression cell, it appeared to be deep red in colour, quite homogeneous and of very loose and light consistency (Fig. 2i). After compression, some dark and whitish spots became visible in the central area (Fig. 2m).

a, b, c, d 3D models of the residues of interest collected with the Hirox KH-7700 microscope: a main cluster on artefact 580, b ruby red cluster on artefact 59, c brownish cluster on artefact 59, d central region of the oval-shaped aggregate on artefact 67. e, f, g, h Optical images collected with ZEISS Stemi 305 microscope of the same residues described above, respectively: sample 580.1 was collected from (e), sample 59.1 was collected from (f), sample 59.2 was collected from (g), and sample 67.1 was collected from (h). i, j, k, l Optical images of samples 580.1 i, 59.1 j, 59.2 k and 67.1 l before the compression. m, n, o, p Optical images of samples 580.1 m, 59.1 n, 59.2 o and 67.1 p after the compression in the diamond compression cell
On artefact 59, the two clusters observed (Fig. 1e) were found to differ one from the other in terms of colour and abundance. At microscopic level, the ruby red tone of cluster 1 was associated with a homogeneous and glowing texture (Fig. 2b, f), shared by the more dispersed cluster 2, which appeared less intense in colour and characterised by brownish tones (Fig. 2c, g). In both cases, the level of adhesion of the residues to the lithic surface was higher with respect to the general condition of the main residue on artefact 580, and especially, in the case of cluster 1, it was possible to recognise sediment particles covering the residue, without any mixing of the two phases (Fig. 2f). The smeared, bright appearance of the two clusters did not allow the recognition of any identifiable morphological feature. Samples 59.1 and 59.2 were collected from cluster 1 and 2, respectively, and deposited on the diamond culet to be observed again prior to compression. After the collection, sample 59.1 appeared to be strongly cohesive, darker than expected and predominantly blackish-red (Fig. 2j), whilst after compression some brighter areas became visible, alternating with a dark red tone probably corresponding to the thicker regions of the sample (Fig. 2n). At this stage, it is striking to note how the appearance of sample 59.1 converged with that of sample 580.1 (Fig. 2m), even though their chromatic and morphological characteristics on both the artefact and the diamond compression cell before compression were significantly dissimilar. Collected sample 59.2 (Fig. 2k), on the other hand, displayed an amber colour and a slightly different micro-arrangement compared to the previous one, with multiple smaller cohesive units than sample 59.1 and significantly less amounts of disperse particles with respect to sample 580.1. After compression, it turned into a very light brown and semi-transparent tone (Fig. 2o), reasonably suggesting a different composition from sample 59.1.
Artefact 67, as artefact 580, was characterised by the presence of a huge bright red deposit appreciable to the naked eye and very intense in colour, with brighter and darker areas adjacent to each other in its thicker region (Fig. 2d, h). Its powdery, but to some extent, coherent texture differed from the one observed in the main cluster on artefact 580, for its aggregation in laminar patches of different shape, size and shade of red (Fig. 2h). Sample 67.1 was collected from the central region of the residue and observed before compression on the diamond culet (Fig. 2l), onto which it exhibited a plain orange tone, much lighter compared to the other red samples at the same stage. When compressed, a bright orange colour emerged almost evenly, with shading areas near the borders and amber tones in the lower region (Fig. 2p).
For all the residues described, colour considerations relating to their appearance in the original micro-context were made based on the stereo microscopic images (Fig. 2e–h), for the subsequent comparison with the collected (Fig. 2i–l) and compressed samples (Fig. 2m–p).
3.2 Chemical characterisation
3.2.1 Sample 580.1
The average infrared spectrum of sample 580.1 (Fig. 3a) shows the absorption peaks at 1037 and 914 cm−1 due to Si–O stretching vibrations usually attributed to silicates. Despite the different colours of some regions, the sample appeared to be mainly composed of clay minerals. Amongst them, kaolinite (Al2Si2O5(OH)4), identified by the sharp absorption peaks at 3695 and 3624 cm−1, represents the main component of the sample and was detected in correspondence of the brighter red tones. Possible traces of montmorillonite ((Na,Ca)0.33(Al,Mg)2(Si4O10)(OH)2 · nH2O) and feldspars were also detected, with the former being mainly interspersed with kaolinite and the latter corresponding to the duller whitish areas within the sample. In addition, the averaged spectrum in Fig. 3a also shows absorption peaks at 2956, 2925 and 2854 cm−1 attributed to the CH2–CH3 stretching vibrations and indicative of the presence of organic matter. The second derivative spectrum shows peaks at 1738 and 1706 cm−1 due to C=O stretching and at 1460 cm−1 (Fig. 3a, lower panel, red frame) due to CH2 bending vibrations that could suggest the presence of a lipid substance. An intense peak at around 1645 cm−1, possibly due to the Amide I band, was also observed. However, the lack of a clear signal in the range of the Amide II band makes the presence of proteinaceous compounds uncertain. The signal at 1645 cm−1 and the broad absorption band in the range 3600–3000 cm−1 (in the absorbance spectrum) are more likely due to –OH bending and stretching, respectively. To deeply investigate the presence of oxides possibly conferring the bright red colour to the residue from artefact 580, its wide range spectrum has been compared with the spectrum of a soil sample from the same layer (9c2) (Fig. 3b) that showed a greyish-light brown colour. Despite both the spectra show very intense signals in the range 680–200 cm−1, a slight difference can be seen in the spectrum of the sample 580.1 showing an additional broad peak at around 326 cm−1 that could suggest the presence of iron oxides, such as hematite (Fe2O3) [72, 73]. Interestingly, the soil sample is characterised by a higher amount of carbonates (detected by a weak peak at 1799 cm−1, a broad band at around 1440 cm−1 and a shoulder at around 879 cm−1) and did not show any traces of organic matter.

a Average FTIR spectrum from sample 580.1 (upper panel) and its second derivative spectrum (lower panel), inset: enlargement of the spectral region 1780—1350 cm−1). b Wide range (WR) spectra of sample 1 (black trace) compared with a soil sample form layer 9c2 (red trace) from which artefact 580 was retrieved
LEXRF measurements provided the spatial distribution of Al, Mg, Na, Fe and O (Fig. 4a). The intense Fe signal appears to be overlapping with O, even if O is almost uniformly distributed, in good agreement with the presence of iron oxides. Na and Mg seem to be localised in distinct regions of the sample, with Na complementary to Fe. Al can be detected in a large area of the sample: its concentration appears higher in regions where Fe is lower and vice versa.

Elemental distribution in sample 580.1. a XRF maps of Al, Mg, Na, Fe and O acquired at 2 keV with a step size of 2 μm over an area of 80 μm × 80 μm (scale bar is 10 μm); b PIXE elemental maps of Ca, Al and Fe collected over an area of 258 μm × 258 μm (scale bar is 20 μm)
In PIXE elemental maps, the most abundant elements are Al and Fe, which overlap within the entire sample but have higher concentrations alternately (Fig. 4b). Ca is present with some sort of structure, associated with Al, which is more abundant and strongly alternating with Fe. This aspect, together with the previous data, could support the co-presence of iron oxides and kaolinite with traces of montmorillonite, which could explain the colocalisation of Al and Ca, as well as the presence of Na and Mg without spatial relation to Fe distribution.
3.2.2 Samples 59.1 and 59.2
The average IR spectra of the two samples collected from artefact 59 are shown in Fig. 5 (upper panel). The two samples turned out to be quite homogeneous showing, within their whole extension, the recurrent infrared features typical of carbonate hydroxyapatite (Ca5(PO4,CO3)3(OH)). This mineral is characterised by an intense band in the range 1200–900 cm−1, due to the ʋ3PO4, and a diagnostic weak peak at 963 cm−1 due to the ʋ1 PO4 stretching mode. –CO3 stretching modes have been detected by the broad band in the range 1500–1350 cm−1 and the weak peak at 872 cm−1, whilst the broad band peaking around 1585 cm−1 could be attributed to organic acids deriving from the degradation of an organic fraction. In one single spot of sample 59.2, corresponding to the absorbance spectrum No. 8, possible traces of proteinaceous materials were detected. Their presence was confirmed by the observation in the second derivative spectrum of the Amide I and II bands, peaking at 1653 and 1551 cm−1, respectively (Fig. 5, lower panel).

Average spectra of samples 59.1 and 59.2 (black and red traces, upper panel), spectrum No. 8 extracted from sample 59.2 and its second derivative spectrum (blue trace, upper and lower panels), inset: enlargement of the spectral region 1800—1350 cm−1, the Amide I e II bands are highlighted
LEXRF maps of Al, Mg, Na, Fe and O for sample 59.1 are shown in Fig. 6a. O signal is uniformly distributed and thus overlaps also with Fe, as in the case of the previous sample (580.1), indicating a possible presence of iron oxides. Mg distribution appears complementary to Fe one and co-localises in several areas of the sample with that of Na. Except for Fe and O, and the other elements seem to show smaller fluorescence counts compared to sample 580.1. Although only a qualitative XRF analysis was performed in the present study, a semi-quantitative evaluation can be done, by observing variation in counts amongst the elements detected in each sample.

Elemental distribution in sample 59.1. a XRF maps of Al, Mg, Na, Fe and O acquired at 2 keV with a step size of 2 µm over an area of 80 μm × 80 μm (scale bar is 10 μm); b PIXE elemental maps of Ca, P and Fe collected over an area of 165 μm × 290 μm (scale bar is 10 μm)
PIXE results for sample 59.1 show a very high concentration of Ca, followed by Fe and P (Fig. 6b). Notably, Ca and P co-localise throughout the whole area of the sample, whilst the Fe signal appears to be stronger in a specific region, within which Ca appears to be slightly less intense. This seems to strongly support the co-presence, within the sample, of iron oxides and carbonate hydroxyapatite as indicated by IR and LEXRF measurements.
3.2.3 Sample 67.1
Sample 67.1 showed the most uniform and homogeneous composition of the whole infrared dataset, with its average spectrum (Fig. 7) showing three very intense peaks at 1182, 1088 and 1006 cm−1 accompanied by a characteristic sharp shape of the –OH stretching band (spectral range 3600–3000 cm−1). The comparison with the RRUFF database suggested the sample could be constituted by the sodium iron sulphate natrojarosite (NaFe3(SO4)2(OH)6), which actually exhibits infrared spectral features similar to those of the more common jarosite (KFe3(SO4)2(OH)6) [74].

FTIR average spectrum of sample 67.1 (red trace) compared with natrojarosite reference spectrum from RRUFF database (black trace)
This result could be confirmed by a good colocalisation between Na, Fe and O in the LEXRF maps (Fig. 8a), which, notably, also show similar distributions for Mg and Al, probably deriving from the silicate component of the sediment. As in the case of sample 59.1, also in sample 67.1 Al, Mg and Na show smaller fluorescence counts compared to sample 580.1, whereas Fe and O show an increase in counts.

Elemental distribution in sample 67.1. a XRF maps of Al, Mg, Na, Fe and O acquired at 2 keV with a step size of 2 μm over an area of 80 μm × 80 μm (scale bar is 10 μm); b PIXE elemental maps of K, S and Fe collected over an area of 213 μm × 210 μm (scale bar is 10 μm)
The most represented elements in PIXE maps are Fe, K and S, with an overall good colocalisation amongst the three (Fig. 8b). This result could therefore suggest a major presence of jarosite within sample 67.1 and the derivation of the Fe–O association from this sulphate.
4 Discussion
The obtained results revealed some interesting aspects for residue analysis, both from a methodological and interpretative standpoint. For what concerns data interpretation, samples 580.1 and 59.1 revealed a clear association between Fe and O, suggesting the presence of iron oxides in some quantities. For sample 67.1, which together with sample 59.1 shows the highest rates of Fe and O, the presence of iron oxides is doubtful, given the high representation of potassium sulphate detected by PIXE. Sample 580.1 turned out to mainly consist of clay minerals such as kaolinite and montmorillonite, which are widely represented amongst the side constituents of ochres [75,76,77]. According to the literature, the term 'ochre' is basically used to designate earth pigments primarily composed of iron oxides and hydroxides, exploited by human groups mainly in relation to their colour and/or functional use, like hematite (red ochre) and goethite (yellow ochre) [75]. In addition to this, a broad meaning of the term is sometimes used to include all generic, red-coloured ferruginous materials retrieved from archaeological contexts [76, 78], with special reference to European Upper Palaeolithic. In the present case, the high representation of the association Fe–O on artefacts 580 and 59 allows us to consider the related samples as mainly composed of ochres sensu stricto, ranging in colour from deep to bright red. According to Croft et al. [27], the formation of iron oxides on lithic artefacts can be due to natural deposition of Fe from various sources, including pyrite (FeS2) oxidation in case of high sulphur concentrations in sediments within areas of human occupation. However, it is striking to note that in these samples (580.1 and 59.2) the presence of organic matter, of lipidic and proteinaceous nature, was also identified. Sample 59.2, in particular, was found to consist mainly of carbonate hydroxyapatite, which, given the presence of low but still detectable proteinaceous signals, could indicate its organic derivation from a bone fraction in contact with the artefact. The presence of strong organic signals within the sample 580.1, in turn, was underlined by the complete lack of organic traces in the soil sample of layer 9c2. Sample 67.1 produced the most uniform IR signals, indicating a homogeneous composition that could correspond to natrojarosite and/or jarosite, later confirmed to be mainly jarosite by PIXE measurements. Jarosite, as well as natrojarosite, forms in natural environments as a secondary mineral from the chemical or biological oxidation of pyrite and other sulphide minerals; the same process can underlie the formation of iron hydroxides, such as goethite [79]. Due to their yellow to dark brown colour, jarosite and natrojarosite are classified amongst the main constituents of yellow ochres [75, 79] and have historically been widely used as colouring agents [75]. Indeed, the minerals of the jarosite group can sometimes exhibit intense red hues, and some varieties (including natrojarosite) are still used as precursors in pigment production processes [80]. Given the strong uniformity of sample 67.1 and the absence of relevant traces of organic matter, any further discussion on the composition of this sample is difficult, apart from the probable contribution of silicates from the sediment detected by LEXRF (Mg and Al contributions).
To provide a wider picture on at least one of the analysed samples, we can mention that the association of red ochre and bone is reported in the literature for the Middle Stone Age African site of Blombos Cave, South Africa, where a 100,000 years old mixture including trabecular bone fragments, compact bone, red ochre and charcoal has been retrieved and interpreted as a painting substance [81]. Likewise, Cavallo et al. [82] indicate in the Protoaurignacian layers of Grotta di Fumane, Italy, the presence of hematite and goethite samples mixed with Ca phosphate, interpreted as bone. However, it is difficult to determine how the deposition of carbonate hydroxyapatite and iron oxides on artefact 59 occurred, or the timing with which the process took place. This represents one of the main challenges in residue analysis, which needs to be combined with evidence from other disciplines to support complex interpretations, as well as to rely on informative, specific and extensive analysis approaches.
As far as methodology is concerned, the multi-technique protocol presented here demonstrated to be able to provide a comprehensive knowledge of the composition of the analysed samples. Its main advantage is represented by its multilevel approach and by the complementarity of the included advanced spectromicroscopy techniques. SR-FTIR, SR-LEXRF and PIXE microscopies proved to be highly appropriate for their capabilities in dealing with heterogeneous samples, yielding point-related and highly sensitive information on specific areas within each sample. In addition, the development of an efficient transfer protocol made it possible to retrieve all samples and reuse them through the following step of the analytical process, remapping the same areas and preserving the spatial information. As evidenced by all samples analysed so far, the results of the techniques were consistent amongst each other, providing complementary insights without redundancy and sometimes clarifying open questions. This is particularly evident in sample 67.1, for which, as mentioned above, the dubious presence of natrojarosite and/or jarosite through SR-FTIR and SR-LEXRF measurements was disentangled by PIXE results, whilst in sample 59.1, the contribution of each technique on various fractions of the sample provides a remarkable example of a good match between the three. The spatial resolution of the SR-LEXRF elemental maps and the wide reading range of the imaging by PIXE thus combined well with the advantages of SR-FTIR microscopy for residue analysis already described in previous works [38, 50]. The overall outcome of the present work, which can be seen as a pilot study of a large-scale project exploring new approaches to the chemical characterisation of ancient residues, is an incentive for conceiving and designing equivalent analysis protocols in terms of comprehensiveness, applicability, micro-invasiveness and residue reuse.
5 Conclusions
In this manuscript, we propose a new protocol for the investigation of ancient residues, based on a multi-technique approach combining FTIR, LEXRF and PIXE analyses to complement conventional chromatic and morphological evaluations. To this end, a specific flow within these techniques was designed and applied to chemically characterise representative residues from Upper Palaeolithic artefacts, with the aim of providing guidance for its future applications. As our results have shown, residue appearance can be misleading and may lead to oversimplified conclusions on their attribution, if these are based solely on chromatic and morphological evaluation. In the present case, the generic attribution of the analysed residues to the category of 'ochres' based on their colour would not have allowed us to discriminate between the presence of iron oxides and iron sulphates. In addition, the advantage of exploiting a technique able to identify the presence and nature of organic matter provided further unexpected information: three similar-appearing residues showed widely differing IR results, whilst two residues with different chromatic appearance turned out to be chemically identical. As already pointed out by other authors, all these aspects suggest the adoption of a cautious approach towards the chromatic-morphological attribution of residues, even under the best conditions of preservation of their morphological features. This consideration is not intended to undermine preliminary colour-morphological assignments, which allow for a first necessary classification of residues, but rather as a call to further complement and deepen them with advanced analytical approaches. As demonstrated by the presented results, the choice of multiple analytical methods capable of supporting each other is imperative to this end, as one technique is not always sufficient to chemically characterise complex, ancient and degraded substances in a comprehensive way.
Data Availability Statement
This manuscript has associated data in a data repository. [Authors’ comment: Data will be made available on request].
References
S. Croft, Lithic residue analysis: a review and guide to techniques (BAR Publishing, Oxford, United Kingdom, 2021)
B.L. Hardy, M. Kay, A.E. Marks, K. Monigal, Proc Natl Acad Sci U S A 98, 10972 (2001)
B.L. Hardy, M.H. Moncel, PLoS ONE 6, e23768 (2011)
V. Rots, P. Van Peer, P.M. Vermeersch, J Hum Evol 60, 637 (2011)
V. Rots, B.L. Hardy, J. Serangeli, N.J. Conard, J Hum Evol 89, 298 (2015)
V. Rots, E. Hayes, D. Cnuts, C. Lepers, R. Fullagar, PLoS ONE 11, e0150437 (2016)
M. Lombard, L. Wadley, J Archaeol Sci 34, 155 (2007)
M. Lombard, J Hum Evol 48, 279 (2005)
M. Lombard, South. Afr. Humanit. 18, 57 (2006)
M. Lombard, J Hum Evol 53, 406 (2007)
M. Lombard, J Archaeol Sci 35, 26 (2008)
M. Lombard, L. Phillipson, Antiquity 84, 635 (2010)
G.H.J. Langejans, J Archaeol Sci 37, 971 (2010)
G.H.J. Langejans, J Archaeol Sci 39, 1694 (2012)
G.H.J. Langejans, South African Arch Bull 67, 200 (2012)
G.H.J. Langejans, M. Lombard, in Use-Wear and Residue Analysis, in Archaeology. ed. by J.M. Marreiros, J.F. Gibaja Bao, N. Ferreira Bicho (Springer International Publishing, Cham, 2015), pp.199–219
R. Fuentes, R. Ono, J. Carlos, C. Kerfant, Sriwigati, T. Miranda, N. Aziz, H. O. Sofian, and A. Pawlik, J Archaeol Sci Rep 30, 102207 (2020).
E. Assaf, I. Caricola, A. Gopher, J. Rosell, R. Blasco, O. Bar, E. Zilberman, C. Lemorini, J. Baena, R. Barkai, E. Cristiani, PLoS ONE 15, e0230972 (2020)
D. Cnuts, V. Rots, Archaeol Anthropol Sci 10, 1717 (2018)
D. Cnuts, S. Tomasso, V. Rots, J Archaeol Method Theory 25, 839 (2018)
A. Borel, A. Ollé, J.M. Vergès, R. Sala, J Archaeol Sci 48, 46 (2014)
B. Hardy and J. Svoboda, Archaeol Sci Microscope (2009).
A. Pedergnana, L. Asryan, J.L. Fernández-Marchena, A. Ollé, Micron 86, 1 (2016)
A. Pedergnana, A. Ollé, A.A. Evans, J Archaeol Sci Rep 30, 102237 (2020)
G.F. Monnier, J.L. Ladwig, S.T. Porter, J Archaeol Sci 39, 3284 (2012)
S. Croft, G. Monnier, A. Radini, A. Little, and N. Milner, Int Archaeol. (2016).
S. Croft, K. Chatzipanagis, R. Kröger, N. Milner, J Archaeol Sci Rep 19, 430 (2018)
V. Rots, C. Lentfer, V.C. Schmid, G. Porraz, N.J. Conard, PLoS ONE 12, e0175151 (2017)
A. Pedergnana, J Paleo Arch. 2019 3:3 3, 225 (2019).
A. Pedergnana, R. Blasco, Quatern. Int. 421, 255 (2016)
A. Pedergnana, A. Ollé, J Archaeol Method Theory 25, 117 (2018)
J.I. Martín-Viveros, A. Ollé, Quatern. Int. 569–570, 228 (2020)
R. Fuentes, R. Ono, N. Nakajima, H. Nishizawa, J. Siswanto, N. Aziz, Sriwigati, H. O. Sofian, T. Miranda, and A. Pawlik, J Archaeol Sci 112, 105031 (2019).
E. Hayes, D. Cnuts, V. Rots, J Archaeol Sci Rep 23, 116 (2019)
A.F. Pawlik, J.P. Thissen, J Archaeol Sci 38, 1699 (2011)
G. F. Monnier, T. C. Hauck, J. M. Feinberg, B. Luo, J. M. Le Tensorer, and H. al Sakhel, J Archaeol Sci 40, 3722 (2013).
A. Zupancich, S. Nunziante-Cesaro, R. Blasco, J. Rosell, E. Cristiani, F. Venditti, C. Lemorini, R. Barkai, and A. Gopher, Scientific Reports 2016 6:1 6, 1 (2016).
K. Sano, S. Arrighi, C. Stani, D. Aureli, F. Boschin, I. Fiore, V. Spagnolo, S. Ricci, J. Crezzini, P. Boscato, M. Gala, A. Tagliacozzo, G. Birarda, L. Vaccari, A. Ronchitelli, A. Moroni, and S. Benazzi, Nature Ecology & Evolution 2019 3:10 3, 1409 (2019).
F. Venditti, S. Nunziante-Cesaro, Y. Parush, A. Gopher, R. Barkai, J Hum Evol 131, 109 (2019)
F. Venditti, E. Cristiani, S. Nunziante-Cesaro, A. Agam, C. Lemorini, and R. Barkai, Scientific Reports 2019 9:1 9, 1 (2019).
E. Cristiani, A. Pedrotti, S. Gialanella, Documenta Praehistorica 36, 191 (2009)
L.C. Prinsloo, L. Wadley, M. Lombard, J Archaeol Sci 41, 732 (2014)
F. Venditti, A. Agam, J. Tirillò, S. Nunziante-Cesaro, R. Barkai, PLoS ONE 16, e0245595 (2021)
A. Nucara, S. Nunziante-Cesaro, F. Venditti, C. Lemorini, J Archaeol Sci Rep 33, 102526 (2020)
S. Nunziante Cesaro and C. Lemorini, Spectrochim Acta A Mol Biomol Spectrosc 86, 299 (2012).
N. Solodenko, A. Zupancich, S.N. Cesaro, O. Marder, C. Lemorini, R. Barkai, PLoS ONE 10, e0118572 (2015)
G. Monnier, E. Frahm, B. Luo, K. Missal, J Archaeol Sci 78, 158 (2017)
G. Monnier, E. Frahm, B. Luo, K. Missal, J Archaeol Method Theory 25, 1 (2018)
E. Frahm, D.S. Adler, B. Gasparyan, B. Luo, C. Mallol, G. Pajović, G.B. Tostevin, B. Yeritsyan, G. Monnier, PLoS ONE 17, e0266362 (2022)
C. Dominici, C. Stani, M. Rossini, L. Vaccari, J Phys Conf Ser 2204, 012050 (2022)
A. Palma di Cesnola, Le Paléolithique Supérieur En Italie (Éditions Jérôme Millon, Grenoble, France, 2002).
J. Crezzini, P. Boscato, S. Ricci, A. Ronchitelli, V. Spagnolo, F. Boschin, Archaeol Anthropol Sci 8, 227 (2016)
C. Berto, P. Boscato, F. Boschin, E. Luzi, A. Ronchitelli, Quat Sci Rev 168, 30 (2017)
F. Boschin, P. Boscato, C. Berto, J. Crezzini, A. Ronchitelli, Quat Res (United States) 90, 470 (2018)
S. Arrighi, V. Borgia, G. Guasparri, S. Ricci, A. Scala, A. Ronchitelli, Preistoria Alpina 46, 91 (2012)
F. Boschin, F. Bernardini, E. Pilli, S. Vai, C. Zanolli, A. Tagliacozzo, R. Fico, M. Fedi, J. Corny, D. Dreossi, M. Lari, A. Modi, C. Vergata, C. Tuniz, A. Moroni, P. Boscato, D. Caramelli, and A. Ronchitelli, Scientific Reports 2020 10:1 10, 1 (2020).
S. Ricci, G. Capecchi, F. Boschin, S. Arrighi, A. Ronchitelli, S. Condemi, Int J Osteoarchaeol 26, 281 (2016)
A. Ronchitelli, S. Mugnaini, S. Arrighi, A. Atrei, G. Capecchi, M. Giamello, L. Longo, N. Marchettini, C. Viti, A. Moroni, Quatern. Int. 359–360, 423 (2015)
F. Zorzi, Antiquity XXXVIII, 38 (1964).
A. Picin, A. Moroni, and S. Benazzi, Updating Neanderthals: Understanding Behavioural Complexity in the Late Middle Palaeolithic 321 (2022).
S. A. Khan, S. B. Khan, L. U. Khan, A. Farooq, K. Akhtar, and A. M. Asiri, Handbook of Materials Characterization 317 (2018).
M.-L. Inizan, M. Reduron-Ballinger, H. Roche, and J. Tixier, Technology and Terminology of Knapped Stone: Followed by a Multilingual Vocabulary Arabic, English, French, German, Greek, Italian, Portuguese, Spanish (Cercle de Recherches et d’Etudes Préhistoriques - CREP, Nanterre, 1999).
M. Rossini, A. Falcucci, C. Dominici, A. Ronchitelli, A. Tomasso, and F. Boschin, in 2022 IMEKO TC-4 International Conference on Metrology for Archaeology and Cultural Heritage, MetroArchaeo 2022 (2022), pp. 52–56.
M. Rossini, A. Falcucci, C. Dominici, A. Ronchitelli, A. Tomasso, and F. Boschin, Acta IMEKO in press, (n.d.).
V. Borgia, F. Ranaldo, A. Ronchitelli, and U. Wierer, Mémoire LII de La Société Préhistorique Française 161 (2011).
G. Birarda, D. Bedolla, F. Piccirilli, C. Stani, H. Vondracek, and L. Vaccari, Biomedical Vibrational Spectroscopy 2022: Advances in Research and Industry 11957, 27 (2022).
A. Gianoncelli, G. Kourousias, L. Merolle, M. Altissimo, A. Bianco, J. Synchrotron Radiat. 23, 1526 (2016)
A. Gianoncelli, G.R. Morrison, B. Kaulich, D. Bacescu, J. Kovac, Appl. Phys. Lett. 89, 251117 (2006)
A. Gianoncelli, G. Kourousias, A. Stolfa, B. Kaulich, J Phys Conf Ser 425, 182001 (2013)
V.A. Solé, E. Papillon, M. Cotte, P. Walter, J. Susini, Spectrochim Acta Part B At Spectrosc 62, 63 (2007)
M. Jakšić, G. Provatas, I.B. Mihalić, A. Crnjac, D. Cosic, T. Dunatov, O. Romanenko, Z. Siketić, Nucl. Instrum. Methods Phys. Res. B 539, 120 (2023)
G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd edn. (Wiley, Chichester, United Kingdom, 2001)
Sh. Yariv, E. Mendelovici, Appl Spectrosc 33, 410 (1979)
J. Liu, X. Xiu, P. Cai, Procedia Earth Planet Sci 1, 706 (2009)
N. Eastaugh, V. Walsh, T. Chaplin, and R. Siddall, Pigment Compendium: A Dictionary and Optical Microscopy of Historical Pigments (Elsevier Butterworth-Heinemann, Oxford, United Kingdom, 2004).
L. Dayet, P.J. Texier, F. Daniel, G. Porraz, J Archaeol Sci 40, 3492 (2013)
S. Wolf, R. Dapschauskas, E. Velliky, H. Floss, A.W. Kandel, N.J. Conard, Open Arch 4, 185 (2018)
G. Cavallo, F. Fontana, F. Gonzato, A. Guerreschi, M.P. Riccardi, G. Sardelli, R. Zorzin, Archaeol Anthropol Sci 9, 763 (2017)
P. Cogram, Reference Module in Earth Systems and Environmental Sciences (2018).
H. Vu, J. Jandová, T. Hron, Hydrometallurgy 101, 1 (2010)
C. S. Henshilwood, F. D’Errico, K. L. Van Niekerk, Y. Coquinot, Z. Jacobs, S. E. Lauritzen, M. Menu, and R. García-Moreno, Science (1979) 334, 219 (2011).
G. Cavallo, F. Fontana, F. Gonzato, M. Peresani, M.P. Riccardi, R. Zorzin, Geoarchaeology 32, 437 (2017)
Acknowledgements
We are grateful to Prof. Arturo Palma di Cesnola, from whose important research at Grotta Paglicci the studied material was derived and to Soprintendenza Archeologia Belle Arti e Paesaggio per le Province di Foggia, Barletta e Trani and Prof. Annamaria Ronchitelli for granting permission to study the material. We deeply thank Adriana Moroni for her helpfulness and knowledge and Stefano Ricci for the photos. This work is part of the PhD "Hunting technologies between the Middle and Upper Palaeolithic in Central-Southern Italy—Chemical and Morphological analyses on lithic and bone implements" supported by Central European Research Infrastructure Consortium, CERIC-ERIC. The authors acknowledge the CERIC-ERIC Consortium for the access to experimental facilities (projects number 20212179 and 20217185) and for the financial support.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Formal analysis and investigation were performed by CD (SR-FTIR, SR-LEXRF and PIXE measurements), CS (SR-FTIR measurements), AG and VB (SR-LEXRF measurements), IBM, GP and SF (PIXE measurements), MR (lithic technological study). The supervision of the project was performed by FB, LV and AG. The first draft of the manuscript was written by CD and CS, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dominici, C., Stani, C., Bonanni, V. et al. Combining SR-FTIR, SR-LEXRF and PIXE microscopies for residue analysis on Palaeolithic stone artefacts. Eur. Phys. J. Plus 138, 742 (2023). https://doi.org/10.1140/epjp/s13360-023-04320-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1140/epjp/s13360-023-04320-7