
Abstract
Controlled atom-transfer radical polymerization and ring-opening polymerization methods are used for the first time to synthesize symmetric pentablock copolymers with a central block, which is a polymer brush with the polyimide backbone and poly(methyl methacrylate) side chains, and outer linear block copolymers of poly(ε-caprolactone) with poly(methyl methacrylate). The chemical structure of the polymers is studied by 1H NMR spectroscopy. The molecular weight characteristics of the synthesized pentablock copolymers are determined by multidetector size exclusion liquid chromatography. It is shown that these characteristics are in good agreement with the absolute molecular weights obtained by sedimentation-diffusion analysis.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
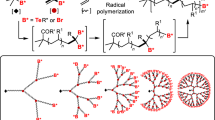
Linear three-component copolymers containing more than three thermodynamically incompatible blocks (i.e., ABCA and ABCBA [1]; A(BC)n or A(BC)nBA) [2] type block copolymers) attract a great deal of attention from researchers due to the possibility of forming hierarchically ordered microstructures in films and selective solvents [1–4]. Such hierarchically ordered structures include, in particular, multisection micelles formed in solutions of ABC ternary block copolymers [5–14]. Moreover, the topology of such multiblock copolymers has a significant effect on the morphology of the resulting hierarchically ordered structures [2]. For example, star-shaped triple block copolymers exhibit the ability to form nonconcentric multisection micelles, including hamburger-type micelles, segmented wormlike micelles, and nanostructured vesicles [15–21]. At the same time, realization of a particular type of the hierarchically ordered structure depends on the ratio of the lengths of solvophobic and solvophilic blocks and the way they are combined into a multiblock copolymer structure [15].
Thus, an important and urgent task in modern polymer chemistry is the synthesis of multiblock copolymers with controlled length and architecture of each block. Approaches to synthesizing such multiblock copolymers are very diverse [1]. Ternary block copolymers can be obtained by the sequential poly-merization of monomers without isolating the intermediate products (so-called one-pot processes) using all known methods for controlled anionic [22, 23], cationic [24], and radical polymerizations [25, 26].
Currently, approaches combining several controlled polymerization methods are increasingly used in the synthesis of multiblock copolymers. Although such approaches are more expensive than one-pot synthesis, since they require isolation of intermediate products and the use of a wide range of reagents, catalysts, and solvents, they offer greater possibilities for varying the chemical structure and architecture of multiblock copolymers. For example, the combination of ring-opening polymerization (ROP) and controlled reversible addition-fragmentation chain transfer (RAFT) polymerization have made it possible to synthesize the ternary copolymer of lactic acid, N,N-dimethylacrylamide, and styrene; and nanoporous hydrophobic polystyrene films with hydrophilic pores with walls covered by poly(N,N-dimethylacrylamide) have been obtained via etching of polylactide block [27]. The combination of controlled atom-transfer radical polymerization (ATRP) and ROP was used to synthesize molecular brushes with the poly(ε-caprolactone) (PCL) backbone and poly(methyl methacrylate) side chains [28]. It should be noted that the introduction of PCL blocks into multiblock copolymers is of considerable interest for further application of these copolymers, since PCL, like polylactides, is subjected to alkaline [29] and plasma [30] etching and biodegradation.
Also of undoubted interest is the combination of covalently linked blocks with a pronounced difference in nature in one macromolecule, since it enables variation of the properties of copolymers in a wide range. Such structures include copolymers with a combination of aromatic and aliphatic blocks. Previously, ROP catalyzed by difunctional polyimide macroinitiators was used to obtain two-component linear PCL–PI–PCL triblock copolymers with an inner polyimide block and outer PCL blocks [31]. In this study, we synthesized new three-component pentablock copolymers PMMA-block–PCL–block-(PI–graft–PMMA)–block–PCL–block–PMMA of the mixed linear-brush topology with the central block of the polyimide brush with PMMA side chains and outer blocks of PMMA–block–PLC block copolymers using ROP combined with ATRP, which had been used earlier to obtain a wide range of molecular polyimide brushes with side chains of vinyl polymers [32–49].
MATERIALS AND METHODS
Solvents
N-Methyl-2-pyrrolidone (MP) (98%, Aldrich), toluene (analytical grade, Vekton), and DMF (reagent grade, Vekton) were dried by heating with anhydrous calcium hydride (99.9%, Aldrich) and were distilled under vacuum. Dichloromethane (reagent grade, Vekton), THF (reagent grade, Vekton), and pyridine (≥99%, Aldrich) were dried over calcium hydride and distilled. Benzene (reagent grade, Vekton) was purified by a known method. Methanol (reagent grade, Vekton), petroleum ether (reagent grade, Vekton), and acetic acid (reagent grade, glacial, Vekton) were used without prior purification. The prepared eluents were additionally filtered with a mobile phase filtration apparatus (Supelco) equipped with a Teflon membrane filter.
Reagents
2-Bromoisobutyroyl bromide (98%, Aldrich), LiBr (≥99%, Aldrich), m-aminophenol (98%, Aldrich), and p-aminophenylethyl alcohol (98%, Aldrich) were used as received.
Monomers
4,4′-(1,3-Phenylene-dioxy)-bis(phthalic anhydride) (99%, Ambinter Stock Screening Collections) was dried at 140°C in vacuum. 2,4-Diaminophenol dihydrochloride (98%, Lancaster dried at 100°C in vacuum before synthesis). Methyl methacrylate (MMA) (99%, Aldrich) and ε-caprolactone (CL) (99%, Aldrich) was twice distilled in vacuum before use.
Catalytic Complexes
CuCl (≥99%, Aldrich) was purified from Cu(II) impurities by keeping in glacial acetic acid (two days, periodical stirring). The powder was filtered, washed with methanol (reagent grade, Vekton), and dried in vacuum at 35–40°C for 1 week. 2,2′-Bipyridyl (bpy) (≥99%, Aldrich) was used without further purification. Sn(II) 2-ethyl hexanoate (92.5–100.0%, Aldrich) was distilled in vacuum.
Synthesis of Multifunctional Polyimide Macroinitiators PIMI-1(OH) and PIMI-2(OH)
Multifunctional polyimide macroinitiators PIMI-1(OH) and PIMI-2(OH)

were synthesized as follows.
The synthesis of PIMI(OH) was carried out according to the standard two-step scheme by the initial production of a precursor poly(amic acid) (PAA) based on 4,4′-(1,3-phenylene-dioxy)-bis(phthalic anhydride) and 2,4-diaminophenol, followed by high-temperature (180–195°C) dehydrocyclization (imidization) of PAA in the native solution. For the introduction of hydroxyl groups into the polymer terminal units, part of the diamine was replaced with a hydroxyl-containing monoamine (m-aminophenol to obtain PIMI-1(OH) or p-aminophenyl ethyl alcohol to obtain PIMI-2(OH) while maintaining an equimolar ratio of anhydride and amine groups of the monomers. The general procedure for synthesizing PIMI(OH) included preparation of a diamine and monoamine solution in MP, the introduction of dianhydride as a fine powder in three portions at an interval of 15 min in an inert atmosphere (Ar) into the resulting solution, and stirring of the mixture for 40 h to obtain PAA (the solution concentration was 20%). To obtain PIMI(OH), high-temperature (180–195°C) dehydrocyclization (imidization) of PAA was performed over 4 h using the PAA solution preliminarily diluted to 5–7%. Water released during imidization was distilled off as an azeotropic mixture with toluene, which was preliminarily added to the PAA solution. After imidization was complete, the solution was cooled to room temperature and diluted with DMF, and the polymeric product was precipitated into methanol, filtered, and dried at 50°C under reduced pressure.
Synthesis of Graft Pentablock Copolymers of Mixed Topology PMMA–block–PCL–block– (PI–graft–PMMA)–block–PCL–block–PMMA
Graft pentablock copolymers of mixed topology were synthesized by the ATRP polymerization of MMA catalyzed by the triblock copolymer macroinitiator PCL(Br)-block–PI(Br)-block–PCL(Br) containing 2-bromoisobutyrate groups in each repeating unit of the central PI block and in the terminal units of the peripheral PCL blocks. To obtain such a macroinitiator, ROP polymerization of ε-caprolactone was carried out at the first stage in bulk or in MP solution using PIMI(OH) as a macroinitiator. To calculate the molecular weight of the macroinitiator units per initiating group, the MW of the used PIMI(OH) was divided by two. A typical experiment was conducted as follows. A 25-mL Schlenk flask equipped with a magnetic stirrer was loaded with 0.0745 g (1.08 × 10–5 mol) of PIMI(OH) and hermetically sealed with a rubber septum, and 1.2 mL (1.08 × 10–2 mol) of ε-caprolactone was added. The mixture was thermostatically controlled at 130°C with an oil bath. After complete dissolution of PI, 0.05 mL (1.52 × 10–4 mol) of Sn(Oct)2 was loaded in an argon flow using a mechanical microsampler. The molar ratio of PIMI (OH) : CL was 1 : (300–1000). Sn(Oct)2 was taken in an amount of 5 wt % with respect to the monomer weight. When the reaction was carried out in MP solution, a solvent was loaded in the flask simultaneously with the monomer; the monomer : solvent volume ratio was 1 : 1. Upon completion of polymerization, the reaction mixture was rapidly cooled to room temperature; after opening of the septum, the reaction mixture was diluted with methylene chloride. The resulting solution was passed through a silica gel column to purify the product from catalyst and monomer impurities. The solution was concentrated using a rotary evaporator, and the product precipitated into cooled petroleum ether. The polymer was dried in vacuum at 30°C.
In the second stage, esterification of hydroxyl groups was carried out in order to functionalize OH repeating and terminal units of the triblock copolymer with 2-bromoisobutyrate groups. The reaction conditions were as follows. Potassium iodide was added to a 5% PIMI(OH) solution in pyridine in an argon flow. Then a solution of 2-bromoisobutyroyl bromide in benzene was added dropwise under vigorous stirring and cooling to the resulting solution, which had been cooled to –2 to 0°C. The molar ratio of PI fragment : bromanhydride : potassium iodide per OH group in the terminal unit was 1 : 2 : 10. The reaction mixture was kept for 4 h with cooling and stirring, the temperature was raised to room temperature, and the reaction mixture was filtered. The reaction product was precipitated from the filtrate into methanol, and the precipitate was filtered and repeatedly washed with water and ethanol. The isolated polymer was dried in vacuum at 50°C.
The synthesized triblock copolymer was used as a macroinitiator for the polymerization of MMA. For polymerization mediated by the PIMI-1(OH) derivative, a weighed portion of the macroinitiator (0.050 g) and 2,2'-bipyridine (0.020 g) were placed in a 25-mL Schlenk flask equipped with the magnetic stirrer. The flask was sealed with a rubber septum, evacuated for 10 min, and filled with argon. Then, the MP (6.8 mL) and MMA (3.18 mL) were introduced into the flask using a syringe, and the mixture was stirred until the powder was completely dissolved. Then there were three freeze-pump-thaw cycles (evacuation for 15 min), after which the flask was filled with argon. The septum was opened, and CuCl (0.005 g) was added in an argon flow, after which the flask was closed again with the septum, another three freeze–pump–thaw cycles (evacuation for 15 min) of the reaction mixture were performed, and the flask was filled with argon and thermostatted (80°C) in an oil bath placed on a magnetic stirrer equipped with a temperature controller. After a given duration of polymerization, the reaction mixture was rapidly cooled to room temperature and, after opening of the septum, it was diluted twofold with THF. To remove copper salts from the mixture, it was passed through a column filled with Al2O3. The mixture was concentrated using a rotary evaporator, and the polymerization product was precipitated into a water–methanol mixture (volume ratio 1 : 6). The filtered powder was dried in vacuum at 50°C.
For polymerization mediated by the PIMI-2(OH) derivative, a weighed portion of the macroinitiator (0.070 g), triphenylphosphine (PPh3) (0.060 g), FeCl3 · 6H2O (0.020 g), and MMA (1.5 mL) were loaded in a 25-mL Schlenk flask equipped with a magnetic stirrer. The contents of the flask was dissolved in MP (3.8 mL), and the mixture was stirred until the powder was completely dissolved. The flask was hermetically sealed with the rubber septum and purged with argon for 1 h, the septum was opened, and ascorbic acid (0.01 g) was introduced into the reaction mixture in an argon flow. The flask was immediately hermetically sealed again with the septum, and the freeze–pump–thaw cycle was performed three times (evacuation for 15 min). Then the flask with the prepared reaction mixture was filled with argon and thermostatted at 80°C under stirring in an oil bath placed on the magnetic stirrer equipped with the temperature controller. The stirring rate for the reaction mixture was 800 rpm. After a predetermined duration of synthesis, the reaction mixture was rapidly cooled to room temperature and, after opening of the septum, was transferred into a beaker; the mixture was stirred for 0.5 h. The mixture was concentrated using the rotary evaporator, and the polymerization product was precipitated into a water–methanol mixture (volume ratio, 1 : 6). The filtered powder was dried in vacuum at 50°C.
1H NMR spectra were recorded on a Bruker AC-400 instrument (400.1 MHz) with respect to the signals of the solvent (DMSO-d6, CDCl3).
The IR spectra of polymers were recorded on a Vertex 70 FTIR spectrometer (Bruker) in the MATR (multiple attenuated total reflectance) mode with a resolution of 4 cm–1; the scan number was 30.
Polyimide macroinitiators and copolymers synthesized using these macroinitiators were analyzed by SEC using an Agilent-1260 Infinity chromatographic system equipped with two columns with a PLgel MIXED-C sorbent (column size 7.5 × 300 mm; particle size 5 μm). Measurements were conducted at 50°C in the isocratic elution mode using DMF containing 0.1 mol/L LiBr as an eluent (flow rate, 1 mL/min). In the course of chromatographic analysis, refractometric, viscometric, and light scattering detectors were used. MW values were calculated by three methods. In the first case, refractometric detection was used and MW was calculated using calibration according to PMMA standards. In the second case, the combination of refractometric and viscometric detectors was used and the MW values were determined using universal calibration. In the third case, a combination of refractometric, viscometric, and light scattering detectors (so-called triple detection) was used; MW values were evaluated without calibration.
Determination of the molecular weight of polymers by sedimentation-diffusion analysis. After preparation, the copolymer solutions in chloroform were kept at room temperature for 1 day. The copolymers were investigated by hydrodynamic methods (viscometry, translational diffusion, velocity sedimentation). The intrinsic viscosity [η] of samples was measured in chloroform at a temperature of 21°C by the standard method using an Ostwald capillary viscometer; the efflux time of the solvent was 63 s. Translational diffusion coefficients D in chloroform were determined by the solution/solvent boundary dispersion method on a Tsvetkov polarization diffusometer at 24°C. The solution/solvent boundary was created in a stratifying optical cell with a length of 3 cm along the light beam. Photographs of the interference fringes of the solution/solvent boundary were processed by the method of the maximum ordinate and the area under the interference fringe. Sedimentation coefficients s of macromolecules in the centrifugal field were measured at 24°C on a MOM 3180 ultracentrifuge (Hungary) equipped with a polarization-interferometric device at a rotor speed of 40 × 103 rpm. A two-sector cuvette was used, in which an artificial boundary was formed by the stratification method. The thickness of the cuvette along the light beam was 12 mm. The absolute molecular weights of the copolymer samples were determined from the sedimentation-diffusion data (Svedberg method MsD = (s/D)NAkT/(1 – \({v}\)ρ0), where k is the Boltzmann constant and T is absolute temperature). The partial specific volume of copolymers \({v}\) was calculated additively from the mass density (\({{{v}}^{{ - 1}}}\)) of the copolymer components taking into account its composition.
RESULTS AND DISCUSSION
Graft block copolyimides of the mixed linear-brush topology were synthesized in several stages. First, multicenter polyimide macroinitiators PIMI-1(OH) and PIMI-2(OH) containing phenol groups in each unit and either phenol (PIMI-1(OH)) or hydroxyethyl (PIMI-2(OH)) terminal groups were synthesized.
The presence of phenol groups in PIMI-1(OH) and PIMI-2(OH) was controlled by the presence of a signal at 10.3 ppm in the 1H NMR spectrum (Fig. 1). In the PIMI-2(OH) spectrum, the number of terminal groups was determined from the signals at 2.7 ppm due to methylene protons in the –CH2OH group.

1H NMR spectrum of multicenter polyimide macroinitiators PIMI-1(OH) and PIMI-2(OH).
The PIMI-1(OH) and PIMI-2(OH) samples according to SEC had Mn ∼ 32 × 103 and Ð ∼ 1.23 (PIMI-1(OH)) and Mn ∼ 31 × 103 and Ð ∼ 1.25 (PIMI-2(OH)).
The multicenter polyimide macroinitiators PIMI-1(OH) and PIMI-2(OH) were used to prepare linear triblock copolymers PCL-block-PI(OH)-block–PCL with the central polyimide block containing phenol groups in each repeating unit and peripheral poly(ε-caprolactone) blocks:

As shown in [50], the polymerization of ε-caprolactone catalyzed by multicenter polyimide macroinitiators of different structure containing phenol groups in each repeating unit and terminal hydroxyethyl groups occurs exclusively via these terminal groups without affecting the “middle” phenol groups [50]:

To test this result, we attempted to synthesize PI-graft-PLC molecular brushes with a polyimide backbone and PCL side chains using ring-opening polymerization of the ε-caprolactone catalyzed by the PIMI‑3(OH) multifunctional polyimide macroinitiator bearing phenol groups in each repeating unit.

Polymerization was carried out in the bulk of CL at 130°C or in DMF solution at 80°C.
On the chromatograms of the polymerization products obtained using refractometric, viscometric, and light scattering detectors (Fig. 2), two well-resolved peaks are seen. As a result of selective extraction (using THF), these polymerization products were divided into two polymer components. The chromatogram of the component isolated using THF showed one mode whose position coincided with that of one of the modes on the chromatogram of the polymerization product being analyzed (Fig. 2). Obviously, this mode refers to the linear homo-PCL formed due to the initiation of CL polymerization by water impurity.

SEC chromatograms obtained using light scattering detector for (1) the product of CL polymerization catalyzed by PIMI-3 (OH) and (2) its component extracted by THF.
According to the SEC data with refractometric detection (Fig. 3), the relative content of the graft copolymer and homopolymer was 5–12/95–88. When polymerization was carried out in the reaction system diluted with DMF, the proportion of the homopolymer increased to 95%.

SEC chromatogram obtained using the refractometric detector for the products of CL polymerization catalyzed by PIMI-3(OH). The graft copolymer to homopolymer ratio is 8 : 92.
Thus, it can be considered proven that the middle phenol groups of multicenter polyimide macroinitiators PIMI-1(OH), PIMI-2(OH), and PIMI-3(OH) hardly participate at all in the polymerization of CL in accordance with the previous data obtained for polyimide macroinitiators with different structures [50]. In other words, the polymerization of CL catalyzed by PIMI-1(OH) and PIMI-2(OH) results in the production of linear triblock copolymers PCL–block-PI(OH)-block–CPL. These triblock copolymers dissolve in a wide range of organic solvents, in contrast to the original polyimide macroinitiators. Table 1 presents the molecular weight characteristics of triblock copolymers PCL-block-PI(OH)-block–PCL determined by SEC using refractometric, viscometric, and light scattering detectors (so-called triple detection). According to Table 1, with an increase in the molar ratio of initiator : monomer from 1 : 1000 to 1 : 2000, the monomer conversion increases and the MWD of the polymerization product narrows.
The reliability of the MW values determined by the SEC method was confirmed by comparing them with the data of sedimentation-diffusion analysis of triblock copolymers PCL-block-PI(OH)-block–PLC obtained using PIMI-1(OH) and PIMI-2(OH) macroinitiators in chloroform solution. The absolute MsD values of triblock copolyimides agree well with the SEC data (Table 1).
To impart the brush structure to the central block of triblock copolymers PCL-block-PI(OH)-block–PCL, they were functionalized by esterifying hydroxyl groups located in each unit of the PI block and at the ends of the PCL chains using 2-bromoisobutyroyl bromide:

The presence of 2-bromoisobutyrate groups in triblock copolymers (Br)PCL-block–PI(Br)-block–PLC(Br) that initiate ATRP was detected according to 1H NMR data (Fig. 4). The degree of functionalization of (Br)PCL–block–PI(Br)-block–PLC(Br) by 2-bromoisobutyrate groups determined from the ratio of the signal intensities at 1.9 ppm due to methyl protons of 2-bromoisobutyrate groups and the signal intensities at 6.0–8.5 ppm due to aromatic protons was greater than 90%. Gravimetry data indicated an increase in the mass of functionalization products by ∼25%, which corresponded to the introduction of a specified number of 2-bromoisobutyrate groups in the copolyimide.

1H NMR spectrum of triblock copolymer macroinitiator (Br)PCL–block–PI(Br)-block–PCL(Br) with the central PI block based on the polyimide macroinitiator PIMI-1(OH).
Using the as-functionalized multicenter triblock copolymer macroinitiators (Br)PCL–block–PI(Br)-block–PLC(Br), ATRP polymerization of methyl methacrylate (MMA) was carried out in DMF solution:

The presence of PMMA blocks in the pentablock copolymers PMMA–block–PCL–block-(PI–graft–PMMA)-block–PCL-block–PMMA was confirmed by 1H NMR spectroscopy (Fig. 5) from the disappearance of the signals at 1.9 ppm due to methyl protons in 2-bromoisobutyrate groups and the appearance of signals due to protons of the PMMA block—the signals at 3.5–3.7 ppm due to OCH3 groups and at 0.7–1.1 ppm due to methyl groups—while maintaining the signals due to aromatic protons at 6.0–8.5 ppm, which correspond to the PI block in the main chain.

1H NMR spectrum of pentablock copolymer PMMA-block–PLC–block–(PI–graft–PMMA)-block–PCL–block–PMMA with the central PI block based on the PIMI-1(OH) polyimide macroinitiator.
Additionally, the structure of the obtained copolymers was studied by IR spectroscopy. In the spectrum of the polyimide macroinitiator PIMI(OH) (Fig. 6, spectrum 1), absorption bands were observed at 1720, 1780, 1380, and 720 cm–1, characteristic of the imide cycle. In the IR spectrum of the copolymers PCL-block–PI–block–PCL (Fig. 6, spectrum 2), along with the absorption bands of the imide cycle, there are absorption bands at 1190–1080 (C–O), 1700 (C = O), and 2800–3000 cm–1 (CH2), corresponding to aliphatic polyester. The grafting of PMMA blocks is characterized by a change in the type of bands in the region of 2800–3000 (due to the appearance of CH3 groups) and the appearance of bands at 1730 and 1140 cm–1.

IR spectra of (1) PIMI-1(OH) and (2) related triblock copolymer PCL-block–PI–block–PCL and (3) pentablock copolymer PMMA–block–PCL–block-(PI–graft–PMMA)-block–PCL–block-PMMA.
The molecular weight of the synthesized copolymer samples increased sequentially from PIMI(OH) to the triblock copolymer PCL-block-PI(OH)-block–PCL and pentablock copolymer PMMA-block–PCL–block-(PI–graft–PMMA)-block–PCL–block–PMMA (Table 2, Fig. 7). The MW values of the penta-block copolymer obtained using the initiator PIMI-1(OH) with the brush central block were also determined by sedimentation-diffusion analysis in chloroform. The obtained absolute MsD values agree well with the SEC data (Table 2).

MMD curves for (1) PIMI(OH) and (2) related linear triblock copolymers PCL–block-PI(OH)-block–PCL and (3) graft pentablock copolymers PMMA-block–PCL–block-(PI–graft–PMMA)-block–PCL–block–PMMA based on (a) PIMI-1(OH) and (b) PIMI-2(OH).
CONCLUSIONS
It was shown that the combination of ATRP and ring-opening polymerization catalyzed by multifunctional polyimide macroinitiators make it possible to synthesize copolymers of complex topology with the central brush-type block of graft copolyimide with PMMA side chains, which is terminated by the PCL–block–PMMA diblock copolymer. The thermodynamic incompatibility of blocks and related microphase separation in such block copolymers of complex architecture and topology can form domain structures with different degrees of crystallinity and gas permeability of domains in their films, which can be used in various, e.g., membrane technologies. It is also interesting to consider how the number of blocks, their topology, and the sequence of the connection of blocks influence the physicochemical properties of such multiblock copolymers. These studies will be the subject of a forthcoming publication.
REFERENCES
C. Tsitsilianis, in Controlled and Living Polymerizations, Ed. by A. H. E. Müller and K. Matyjaszewski (Wiley-VCH Verlag GmbH and Co. KGaA, Weinheim, 2009), Chap. 9, p. 445.
L. Wang, J. Lin, and X. Zhang, Polymer 54, 3427 (2013).
B. V. K. J. Schmidt and C. Barner-Kowollik, Nat. Chem. 5, 990 (2013).
C. Tschierske, Chem. Soc. Rev. 36, 1930 (2007).
T. Jiang, L. Wang, S. Lin, J. Lin, and Y. Li, Langmuir 27, 6440 (2011).
J. F. Gohy, N. Willet, S. Varshney, J. X. Zhang, and R. Jérôme, Angew. Chem., Int. Ed. 40, 3214 (2001).
Z. Ma, H. Yu, and W. Jiang, J. Phys. Chem. B 113, 3333.
W. Kong, W. Jiang, Y. Zhu, and B. Li, Langmuir 28, 11714 (2012).
Y. Zhu, X. Yang, W. Kong, Y. Sheng, N. Yan, Soft Matter 8, 11156 (2012).
F. Schacher, A. Walther, M. Ruppel, M. Drechsler, and A. H. E. Müller, Macromolecules 42, 3540 (2009).
A. Laschewsky, Curr. Opin. Colloid Interface Sci. 8, 274 (2003).
L. Wang and J. Lin, Soft Matter 7, 3383 (2011).
H. von Berlepsch, C. Böttcher, K. Skrabania, and A. Laschewsky, Chem. Commun. 2009, 2290 (2009).
K. Skrabania, H. von Berlepsch, C. Bottcher, and A. Laschewsky, Macromolecules 43, 271 (2010).
Z. Li, M. A. Hillmyer, and T. P. Lodge, Langmuir 22, 9409 (2006).
Z. Li, M. A. Hillmyer, and T. P. Lodge, Nano Lett. 6, 1245 (2006).
Z. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer, and T. P. Lodge, Science 306, 98 (2004).
N. Saito, C. Liu, T. P. Lodge, and M. A. Hillmyer, Macromolecules 41, 8815 (2008).
T. I. Löbling, O. Borisov, J. S. Haataja, O. Ikkala, A. H. Gröschel, and A. H. E. Müller, Nat. Commun. 7, article no. 12097 (2016).
A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther, and A. H. E. Müller, Nat. Commun. 3, article no. 710 (2012).
T. I. Lobling, O. Ikkala, A. H. Gröschel, and A. H. E. Müller, ACS Macro Lett. 5, 1044 (2016).
N. Hadjichristidis, H. Iatrou, M. Pitsikalis, S. Pispas, and A. Avgeropoulos, Prog. Polym. Sci. 30, 725 (2005).
F. Liu and A. Eisenberg, Angew. Chem., Int. Ed. 42, 1404 (2003).
S. Subihara, S. Kanaoka, and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem. 42, 2601 (2004).
K. A. Davis and K. Matyjaszewski, Macromolecules 34, 2101 (2001).
S. Kubowicz, J.-F. Baussard, J.-F. Lutz, A. F. Thune-mann, H. von Berlepsch, and A. Laschewsky, Angew. Chem., Int. Ed. 44, 5262 (2005).
J. Rzayev and M. A. Hillmyer, Macromolecules 38, 3 (2005).
D. Mecerreyes, B. Atthoff, K. A. Boduch, M. Trollsas, and J. L. Hedrick, Macromolecules 32, 5175 (1999).
D. Gupta, A. K. Singh, N. Kar, A. Dravid, and J. Bellare, Mater. Sci. Eng. 98, 602 (2019).
Y.-M. Ko, S.-W. Myung, and B.-H. Kim, J. Nanosci. Nanotechnol. 15, 6048 (2015).
T. K. Meleshko, A. V. Kashina, N. N. Saprykina, S. V. Kostyuk, I. V. Vasilenko, P. A. Nikishev, and A. V. Yakimanskii, Russ. J. Appl. Chem. 90, 602 (2017).
T. K. Meleshko, A. S. Ivanova, A. V. Kashina, I. V. Ivanov, T. N. Nekrasova, N. V. Zakharova, A. P. Filippov, and A. V. Yakimansky, Polym. Sci., Ser. B 59, 674 (2017).
V. D. Pautov, T. N. Nekrasova, T. D. Anan’eva, T. K. Meleshko, I. V. Ivanov, and A. V. Yakimansky, J. Polym. Res. 25, 1 (2018).
T. K. Meleshko, A. Yu. Pulyalina, N. S. Tyan, G. A. Polotskaya, I. V. Ivanov, N. V. Kukarkina, A. M. Toikka, and A. V. Yakimansky, Polym. Sci., Ser. B 59, 183 (2017).
A. P. Filippov, A. S. Krasova, E. B. Tarabukina, A. V. Kashina, T. K. Meleshko, and A. V. Yakimansky, J. Polym. Res. 23, 1 (2016).
A. P. Filippov, E. V. Belyaeva, N. V. Zakharova, A. S. Sasina, D. M. Ilgach, T. K. Meleshko, and A. V. Yakimansky, Colloid Polym. Sci. 293, 555 (2015).
A. P. Filippov, E. V. Belyaeva, T. K. Meleshko, and A. V. Yakimansky, J. Polym. Sci., Part B: Polym. Phys. 52, 1539 (2014).
T. K. Meleshko, D. M. Il’gach, N. N. Bogorad, N. V. Kukarkina, and A. V. Yakimansky, Polym. Sci., Ser. B 56, 118 (2014).
A. V. Yakimansky, T. K. Meleshko, D. M. Ilgach, M. A. Bauman, T. D. Anan’eva, L. G. Klapshina, S. A. Lermontova, I. V. Balalaeva, and W. E. Douglas, J. Polym. Sci., Part A: Polym. Chem. 51, 4267 (2013).
A. V. Yakimanskii, T. K. Meleshko, D. M. Il’gach, N. N. Bogorad, E. N. Vlasova, and T. D. Anan’eva, Russ. Chem. Bull. 61, 999 (2012).
A. Krasova, E. Belyaeva, E. Tarabukina, A. Filippov, T. Meleshko, D. Ilgach, N. Bogorad, and A. Yakimansky, Macromol. Symp. 316, 32 (2012).
D. M. Ilgach, T. K. Meleshko, and A. V. Yakimansky, Polym. Sci., Ser. C 57, 3 (2015).
S. A. Klimova, O. A. Inozemtseva, S. V. German, D. A. Gorin, D. M. Ilgach, T. K. Meleshko, and A. V. Yakimansky, Prot. Met. Phys. Chem. Surf. 51, 396 (2015).
A. S. Ivanova, N. V. Zakharova, A. P. Filippov, T. K. Meleshko, and A. V. Yakimansky, Polym. Sci., Ser. A 59, 281 (2017).
T. K. Meleshko, D. M. Il’gach, N. N. Bogorad, N. V. Kukarkina, E. N. Vlasova, A. V. Dobrodumov, I. I. Malakhova, N. I. Gorshkov, V. D. Krasikov, and A. V. Yakimanskii, Polym. Sci., Ser. B 52, 589 (2010).
A. P. Filippov, E. V. Belyaeva, A. S. Krasova, M. A. Simonova, E. B. Tarabukina, T. K. Meleshko, D. M. Ilgach, N. N. Bogorad, and A. V. Yakimansky, Polym. Sci., Ser. A 56, 1 (2014).
A. P. Filippov, E. V. Belyaeva, A. S. Krasova, M. A. Simonova, T. K. Meleshko, D. M. Ilgach, N. N. Bogorad, A. V. Yakimansky, S. V. Larin, and A. A. Darinskii, Polym. Sci., Ser. A 56, 393 (2014).
A. R. Ibragimova, A. B. Mirgorodskaya, E. A. Vasilieva, E. I. Khairutdinova, T. K. Meleshko, I. V. Ivanov, A. V. Yakimansky, I. R. Nizameev, M. K. Kadirov, and L. Y. Zakharova, Colloids Surf., A 526, 20 (2017).
N. Y. Shilyagina, N. N. Peskova, S. A. Lermontova, A. A. Brilkina, V. A. Vodeneev, A. V. Yakimansky, L. G. Klapshina, and I. V. Balalaeva, J. Biophotonics 10, 1189 (2017).
C. Liu, B. Liu, and M. B. Chan-Park, Polym. Chem. 8, 674 (2017).
Funding
This study was financed by a grant from the Government of the Russian Federation for State Support of Scientific Research Supervised by Leading Scientists (contract 14.W03.31.0022).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kashina, A.V., Meleshko, T.K., Bogorad, N.N. et al. Synthesis of Pentablock Copolymers of the Mixed Linear-Brush Topology by Controlled Radical Polymerization and Ring-Opening Polymerization Reactions. Polym. Sci. Ser. C 61, 174–185 (2019). https://doi.org/10.1134/S1811238219010090
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1811238219010090