
Abstract
Recent developments in the one-step synthesis of structurally controlled hyperbranched polymers by radical polymerization in terms of molecular weight, dispersity, number of branching points, branching density, and number of chain-end groups are reported. The structural character of HB-polyacrylates and HB-polystyrenes synthesized by organotellurium-mediated radical polymerization (TERP) resembles that of dendrimers and dendrons, which, although enabling complete control over branched structures, requires tedious stepwise synthesis. Successful control is realized by a new molecular design for the monomer inducing the branching structure, in which the reactivity of the dormant group changes from inactive to active upon the reaction and incorporation of the monomer into the polymer backbone. The principle of the monomer design and the scope and limitation of the polymerization method are described here.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
The primary structure of macromolecules plays a pivotal role in determining their higher-order structures in solution and their bulk; eventually, this structure affects many of the important physical properties of the resulting polymer materials [1,2,3]. Therefore, control of the primary macromolecular structure has been an important topic in polymer synthesis. Various “controls” in the primary structure, particularly molecular weight, dispersity, and tacticity, have been achieved through the invention of new polymerization techniques, such as living and stereoselective polymerization [4,5,6]. It is worth noting that many of these polymerization techniques are robust and have already been implemented in industry for fabricating high-value polymer materials and products. In contrast, topology control other than that for linear polymers, such as branched and cyclic structures, has been far less explored than that for linear polymers despite their characteristic physical properties [7].
Highly branched polymers have several unique physical properties, such as lower hydrodynamic volume, lower intrinsic viscosity, lower glass transition temperature, and a larger number of terminal groups than the corresponding linear polymers [8,9,10,11,12]. Therefore, they are promising for various applications, such as viscosity controllers and vehicles for active components, such as catalysts and drugs [13,14,15,16,17]. Control of the three-dimensional (3D) structure of highly branched polymers, i.e. molecular weight, dispersity, number of branching points, branching density (BD), and number of chain-end groups, can significantly modify polymer properties and contribute to the design and synthesis of new polymer materials. However, the development of materials science based on highly branched polymers lags far behind that for linear polymers due to the deficiencies of practical synthetic methods used to fabricate structurally controlled highly branched polymers.
Dendrimers and dendrons are the most 3D-structurally controlled highly branched polymers [18,19,20]. They are synthesized by a stepwise condensation reaction involving functional group transformation using either divergent or convergent methods (Scheme 1). For example, the first synthesis of dendrimers, polyamine dendrimers by Tomalia et al. [21], used divergent synthesis by the repetitive reaction of amine groups to methyl acrylate (MA) and subsequent transformation of the methyl ester to aminoethylacrylamide by treatment with ethylene diamine (Scheme 1a). In contrast, Hawker and Frechet [22] reported the convergent synthesis of a 3,5-diphenoxybenzyl dendrimer by stepwise condensation of 3,5-disubstituted benzyl bromide and 3,5-dihydroxybenzyl alcohol and subsequent transformation of the benzyl alcohol moiety of the product to the corresponding benzyl bromide (Scheme 2b). These stepwise syntheses enable (almost) perfect structural control of dendrimers and dendrons, which consist solely of branched constitutional repeating units with a degree of branching of 1.0 and a dispersity (Đ) of 1.00. However, despite perfect 3D structural control, their applications to materials science have been severely limited due to insufficient supply.


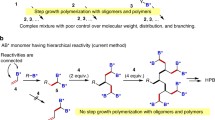
One-step synthetic route for HBPs utilizing a the AB2 monomer method, in which the reaction of the A and B groups forms an ab bond, b self-condensing vinyl polymerization using the AB* monomer, and c the current method
Hyperbranched polymers (HBPs) are another class of highly branching polymers that are usually synthesized in one step but with low 3D structural control. The synthetic method was proposed by Flory [23] using polycondensation of AB2 monomer (Scheme 2a, A, and B refer to two functional groups reacting with each other and 2 represents the number of B groups), but a synthetic breakthrough was recently realized by Kim and Webster [24]. Since then, various HBPs have been synthesized by polycondensation of ABx monomers and the use of A2 and Bx monomers [8]. Frechet et al. [25] also reported self-condensing vinyl (co)polymerization (SCV(C)P) using AB* monomer 2, in which A and B* refer to alkene and initiating groups, respectively (Scheme 2b). The method is a hybrid of step-growth and chain-growth polymerizations, and the homopolymerization of 2 or copolymerization of 2 and vinyl monomers under living cationic, radical, and group transfer polymerization conditions gives the corresponding HB-homo or copolymers, respectively [26,27,28,29,30]. The method was also extended to utilize ring-opening polymerization as chain-growth polymerization. Despite the simple synthetic operation, the control of the 3D structure of the resulting HBPs is very limited. For example, the branched constitutional repeating unit contains both branched and unbranched units, and the degree of branching is usually < 1. In addition, the dispersity of the resulting HBPs is usually very high (Đ > 4). Notably, SCV(C)P under cationic and radical polymerization conditions is a living polymerization condition that effectively controls the synthesis of linear polymers. However, these methods are inefficient for 3D structural control in HBP synthesis. Some control over the 3D structure was achieved by using special polymerization conditions, such as stopping the polymerization at low monomer conversion, slow monomer addition [31], or the use of monomer confinement under microemulsion polymerization conditions [32]. However, no general method has been realized until recently.
Recently, two research groups independently reported the controlled polymerization of HBPs via condensation polymerization using well-designed AB2 monomers. Ohta et al. [33, 34] reported aromatic AB2 monomer 5, in which the reactivity of the B group, an ester group, was enhanced after 5 was incorporated into a polymer chain (Scheme 3a). This is because the ester group of 5 is deactivated by the amide anion’s negative charge so that 5 does not react by itself. However, the reactivity of the ester group is recovered after the reaction and incorporation of 5 into a polymer backbone due to the disappearance of the negative charge. Shi et al. [35] and Cao et al. [36] reported the “click” polymerization of AB2 monomer 6 with alkyne and azide groups in the presence of a ligand-free Cu catalyst (Scheme 3b). While the origin of the control remains unclear, the proximity effect, in which the propagation reaction takes place at the proximity of the polymer-end group through the catalyst transfer reaction, has been proposed. Despite the high dispersity control (Đ < 2), however, the lack of structural versatility in monomers significantly limits their utilization in materials science.

Synthesis of 3D-structurally controlled HBPs by the AB2 monomer method reported by a Yokozawa and b Gao
We are interested in synthesizing structurally controlled HBPs by radical polymerization because this method is one of the most widely used polymerization techniques in the industry. The poor structural control observed in the SCV(C)P using an AB* monomer should be ascribed to the presence of a step-growth mechanism arising from the independent reactivity of A and B* groups (Scheme 2b). Therefore, we envisioned that the polymerization could be controlled if the reactivity of the A and B* groups is linked (Scheme 2c). For example, B* only participates in polymerization after A has reacted. Then, the repetition of this process under living polymerization conditions can give HBPs with controlled 3D structures since the step-growth mechanism is no longer involved. Monomer 5 developed by Yokozawa can be classified into this class of monomers despite the polymerization method being different.
An apparent challenge was how to realize the linking of reactivity between the A and B* groups. We hypothesized that monomer 3, in which the B* group is directly attached to the vinyl group, would be suitable for this purpose (Scheme 2c). This is because vinyl radicals are highly unstable species, so 3 does not initiate polymerization by vinyl radical generation. However, once 3 has reacted as a monomer and given 4, both B* groups in 4 can now initiate polymerization by generating a stable alkyl radical. In other words, the transformation of sp2-carbon-B* bonds to sp3-carbon-B* bonds activates radical formation. The repetition of the addition reaction leads to the formation of a living polymer, in which the polymer end is capped by the B* group.
The above concept should be applicable to all reversible deactivation radical polymerization methods [37, 38]. However, we used organotellurium-mediated radical polymerization (TERP) [39,40,41] because of its high synthetic versatility, especially its ability to synthesize random and block copolymers from monomers with different reactivities [42,43,44,45,46]. Our results for the synthesis of HB-polyacrylates [47], polymethacrylates [48], and polystyrenes (PSts) [49] are mainly discussed in this article. HB-polyacrylates and HBPSts were synthesized under significant 3D structural control as characterized by the control of molecular weight and low dispersity (Đ < 2), branching number, and branched constitutional repeat unit with a degree of branching of ~1.0. As the method relies on statistical copolymerization, the BD, which is the average number of monomer units inserted between the branched points, was also controlled. These structural properties resemble those of dendrons and dendrimers and are significantly different from those of HBPs synthesized by conventional methods. Therefore, the HB-polyacrylates and HBPSts can be defined as dendritic HBPs, which can define (average) generation and (average) branching numbers. In contrast, 3D structural control in HB-polymethacrylate synthesis is challenging and results in a rather low degree of branching. Therefore, HB-polymethacrylate can be classified into pseudodendritic HBPs with pseudodendritic generation. After we reported our initial study, Li et al. [50] reported the same strategy for synthesizing HB-polyacrylates and HBPSts using atom transfer radical polymerization (ATRP), and these results were also included.

Ideal schematic structure of the HBPs having a dendron structure with a generation number of N = 1, 3, 5, and 7. ♦, ●, B* refer to the α-polymer-end group derived from a chain transfer agent, branching point, and ω-polymer-end dormant group, respectively
According to a recent report on the nomenclature of dendrimers and HBPs by IUPAC, the structural characteristics of such compounds are mutually exclusive so that the use of dendritic HBPs is proscribed [51]. However, the report assumes that HBPs are synthesized by the conventional method involving step-growth polymerization, as discussed above. In contrast, the method described here is not classified the same as for traditional HBPs because of the lack of a step-growth pathway, and the HB-polyacrylates and HBPSts described here are a new class of HBPs that can define average (but not pseudo) generation as dendrons and dendrimers (Fig. 1). Therefore, generation N in this manuscript refers to the average generation of dendritic HBPs. Generation N leads to (2N – 1) being the average number of branching points and 2(N+1) being the average number of end groups for dendrons having a single focal point (Fig. 1). Furthermore, the BD of the dendrons can be estimated using the following equation, assuming ideal statistical copolymerization with a comonomer inducing a branching point (Conv1 and Conv2 refer to the conversion of monomer and comonomer, respectively):
Design of branching monomer for TERP
The feasibility of the concept shown in Scheme 2c was quantitatively analyzed based on the carbon–tellurium bond dissociation energy (BDE) of organotellurium compounds and their models involving HBP synthesis estimated from the density functional theory calculation (Fig. 2). The BDE of vinyl telluride 7a (R = Me) is 216 kJ mol−1, which is 56 and 62 kJ mol−1 higher than those of model compounds 8 and 9, respectively. These differences correspond to 4 and an intermediate after propagation of a single tellurium group of 4 (Fig. 2a) [47]. The significant change in the BDE from 7a to 8 and 9 strongly supports the validity of the hypothesis. A similar energy transition was also calculated for unsubstituted vinyl telluride 10, in which the BDE was >60 kJ mol−1 higher than that of intermediate models 11 and 12 (Fig. 2b) [48]. For the case of vinyl telluride 13, the BDE decreases by more than 100 kJ mol−1 by transforming to 14 and 15 (Fig. 2c) [49]. This is because 14 and 15 generate conjugated allyl radicals, while the vinyl radical generated from 13 cannot be stabilized by allylic conjugation. These results support the positive “linkage” of reactivity between the A and B* groups shown in Scheme 2c. In other words, the propagation reaction of vinyl tellurides serves as a stimulus to change the reactivity of carbon–tellurium bonds. Therefore, these monomers can be called stimuli-responsive monomers, in which polymerization serves as a stimulus.

Calculated carbon–tellurium BDE (kJ mol−1) for organotellurium compounds related to HBP synthesis by density functional theory at B3LYP/6–31G(d,p)(C,H) + LAN2DZ(Te). a Methyl-substituted vinyltelluride, b non-substituted vinyltelluride, c dienyl telluride monomers and the corresponding model dormant species after incorporation of the vinyltelluride monomers to polymer main chains and d organotellurium chain transfer agents
Polymethacrylate and PSt-end models 16a, 16b (R = Et), and 18 are popular chain transfer agents in TERP (Fig. 2d) [39, 42]. Their BDEs and those of polyacrylate end model 17a are higher than those of allyl telluride intermediates 14 and 15. These results indicate the efficient activation of dormant species having the same structure as 14 and 15 under copolymerization of methacrylate, acrylate, and styrene (St). In contrast, the BDEs of 8 and 9 are ~40 kJ mol−1 higher than those of 16a and 18, suggesting that the activation of the dormant species derived from 7a is less effective than the activation of the polymethacrylate and PSt ends. The BDEs of 11 and 12 are 15–20 kJ mol−1 higher than those of 8 and 9, indicating that the dormant species’ activation derived from 10 is more difficult than that from 7. The effect of BDEs on 3D structural control in dendritic HBPs is discussed below.
Synthesis of dendritic HB-polyacrylates by TERP [47]
The synthesis of HB-poly(methyl acrylate) (HBPMA) 19aA (R = Me, X = TeMe) was carried out by the copolymerization of vinyl telluride 7a and MA (E = CO2Me) in the presence of TERP chain transfer agent 16b (Fig. 3a). The polymerization behavior for synthesizing the sixth generation (N = 6) of 19 by [16b]0/[7a]0/[MA]0 = 1/63/500 in the presence of AIBN at 60 °C is summarized in Fig. 3b–d [52]. Since there is no direct analytical method to prove the 3D structure of HBPs, various indirect methods have to be combined for structural analyses. First, monomers’ consumption is important to predict the regularity of the incorporation of the branching point in a polymer backbone; 7a and MA consume nearly the same rate (Fig. 3b), indicating the statistical copolymerization and regulated incorporation of the branching point. When the activation of two carbon–tellurium bonds in 20a occurs simultaneously, as indicated by the theoretical calculation of the model intermediates 8 and 9 (Fig. 2a), the formation of a highly symmetrical branching structure is expected through 21a.

a Synthesis and b–g characterization of dendritic HBPMA 19. b Time dependence for the consumption of 7a and MA, c correlation between monomer conversion, number average molecular weight, and dispersity, and d time dependence for SEC traces for the synthesis of N = 6. e Mark–Houwink–Kuhn–Sakurada plot for linear PMA (N = 0) and HBPMAs with N = 2–6. f 2H NMR for 19aC with N = 4, and g height image obtained by AFM for 19B with N = 7
Next, the time dependence for the molecular weight was analyzed by size exclusion chromatography (SEC), indicating that the number average molecular weight (Mn[SEC]) increased with increasing monomer conversion. However, Mn[SEC] showed good agreement with theoretical values (Mn[theo]) only at low monomer conversion (<20%) and deviated significantly as the monomer conversion increased (Fig. 2d). This observation is consistent with forming a branched polymer, which has a smaller hydrodynamic volume than linear polymers. At 95% and 74% conversion of 7a and MA, respectively, Mn[SEC] (9800) was significantly smaller than Mn[theo] (34,600). In contrast, the Mn determined by multiangle laser light scattering (MALLS, Mn[MALLS] = 53,900) was close to Mn[theo] (note that Mn[MALLS] was determined from the weighted average molecular weight obtained from MALLS (Mw[MALLS]) by dividing by the dispersity (Mn/Mw) obtained by SEC). The dispersities were below 2.0 throughout the polymerization, and the values were significantly smaller than those observed by SCVP and SCVCP. In addition, the SEC traces were unimodal throughout the polymerization and shifted to the high molecular weight region over time (Fig. 2e). All results are consistent with the formation of HBPMA with a well-controlled 3D structure.
The generality of this method was demonstrated by the controlled synthesis of HBPMAs with different dendritic generations (N = 2–7) by changing the [16b]0/[7a]0 ratio (Table 1). For the same Mn[theo] of ~40,000, Mn[SEC] decreased as dendritic generation N increased, while Mn[MALLS] was in good agreement with Mn[theo] (runs 1–6). The SEC traces (not shown here) were unimodal, and the dispersity (Đ) was below 2 in all cases. Furthermore, the intrinsic viscosity ([η]) estimated from the SEC-MALLS measurement was lower than that of the linear polymer and decreased systematically as the generation and DB increased (Fig. 3e). The exponent a in the Mark–Houwink–Kuhn–Sakurada (MHKS) equation is 0.36–0.44, which is significantly smaller than that for linear PMA (0.82).
The degree of branching was determined by the labeling experiment to be >0.99 for 19aA (X = TeMe) with N = 4 by reducing to 19aC by MeTeD, which was generated in situ from MeTeSiMe3 and MeOD. The 2H NMR spectrum of the polymer showed a single peak at 2.3 ppm, corresponding to the acrylate chain ends, and no signal due to the alkyl ends, which can form from the ω-end structures of 20a and mid-chain structure of 21 (Fig. 3f). The results strongly support the formation of HBPMAs despite the higher BDEs of the carbon–tellurium bond of the branching point in 20 and 21 compared to those of the PMA ends in 19A and 21. The BD calculated from Eq. (1) varies from 2.9 (N = 6) to 68 (N = 2) for the polymer targeting Mn[theo] of ~40,000, emphasizing the flexibility for the control of the branched structure.
Control of the molecular weight and BD was also achieved. For example, HBPMAs with N = 4 and Mn[MALLS] ranging from 9100 to 162,200 with low dispersity (Đ < 2) were synthesized by changing the [16b]0/[MA]0 ratio from 100 to 2000 while keeping the same [16b]0/[7a]0 ratio of 15 (runs 4, 7–9). The BD was controlled from 3 to 60 under these conditions.
In conventional dendrimer synthesis, as discussed in Scheme 1, the highest N achievable is usually 4 because of the increase in steric congestion induced by the branched structure. However, N can be increased by increasing the targeted molecular weight in this method. For example, HBPMA with N = 7 with 127 average branching points was synthesized by targeting an Mn[theo] of ~160,000 (run 10). The SEC trace of the obtained polymer was unimodal, the dispersity was low (Đ ~ 2), and Mn[MALLS] was very similar to Mn[theo] but was ~3 times larger than Mn[SEC]. All of these results are consistent with the successful synthesis of 3D-structurally controlled HBPMA. Note that the BD of this polymer is 7.3, which is very similar to that of N = 5 and 4 having different molecular weights (runs 5 and 8), further illustrating the current method’s versatility.
The single polymer molecule of N = 7 was directly visualized by atomic force microscopy (AFM) as spherical dots (Fig. 3g). The globular shape and similar size of the dots further support the formation of structurally controlled HBPMAs. The estimated volume of the dot from the height and width is consistent with the observation of a single polymer.
The synthetic versatility of the current method enables the synthesis of HBPMAs with different structures (Scheme 4). For example, linear-block-HBPMA was synthesized starting from PMA macroinitiator 22d (E = CO2Me) prepared by TERP with 7a and MA (Scheme 4a). Furthermore, copolymerization utilizing bifunctional and trifunctional initiators 23 and 24 [53] resulted in dumbbell- and clover-shaped HBPMAs, respectively (Scheme 4b, c). All polymers exhibited unimodal SEC traces, narrow dispersities, and similar Mn(MALLS)s and Mn(theo)s, while the Mn(SEC)s were significantly smaller than the Mn(theo)s.

Synthesis of hyperbranched PMA with different molecular structures starting from a linear PMA macroinitiator, b bifunctionalized, and c trifunctionalized initiators
The method inherits the synthetic advantage of reversible deactivation radical polymerization. For example, the method is compatible with polar functional groups, and copolymerization of 2-(dimethylamino)ethyl acrylate (E = CO2C2H4NMe2) or N,N-dimethylacrylamide (DMAA, E = CONMe2) with 7a affords the corresponding dendritic HBPs with nitrogen functional groups (Table 1, runs 11 and 12). Furthermore, the polymer-end tellanyl group can not only be reduced to 19B (X = H) but can also be transformed into a variety of functional groups. For example, amine-functionalized HBPMA 19D was synthesized by treating 19A with amino-TEMPO 25E (Fig. 4) under photoirradiation [41, 54]. As many synthetic transformations of polymer-end groups synthesized by TERP have been developed [39, 55,56,57], such methods can also be applicable.

Structure of TEMPO derivatives used for polymer-end group transformation
The role of vinyl telluride was unambiguously determined by the use of 13C-labeled 7c* (R = Oct-n, * refers to the 13C-labeled compound), in which the quaternary vinyl carbon was selectively labeled by 13C. The 13C NMR spectrum of the resulting 13C-labeled HBPMA 19c*B (X = H) shows a quaternary carbon signal at 39.4 ppm as the major labeled signal and a tertiary carbon signal at 33.4 ppm as the minor labeled signal (Fig. 5a). The results clearly reveal that vinyl telluride indeed serves as a major branching point (Fig. 5b). The hydrogen atom attached to the minor tertiary carbon would derive from intramolecular hydrogen abstraction, i.e., the back-biting reaction [58]. Indeed, the 2H and 13C NMR of doubly labeled 19c*C (X = D) showed that the deuterium was located only at the acrylate ends, excluding the possibility that the minor structure was derived from intermediate 21. The results also suggest that the mid-chain radical formed by the back-biting reaction induces branching because the local structure of this radical is very similar to polymethacrylate chain-end radicals, which easily cross-propagate to acrylate monomers. All of these results further indicate that vinyl telluride induces branching points quantitatively.

a Structural analyses of branching by 13C NMR and DEPT 135° for 19cB and 19c*B. Newly observed signals are highlighted as red circles. b The proposed major and minor branched structures and their formation mechanism
Synthesis of HBPSt by TERP [49]
The above concept can be extended to the synthesis of dendritic HBPSts. While vinyl telluride 7a does not copolymerize with St, 13, having a conjugated diene group, participates in the copolymerization and gives dendritic HBPSts (Fig. 6a). Figure 6b–d illustrate the time dependence for the synthesis of HBPSt with N = 3 by mixing [16b]0/[13]0/[St]0 = 1/7/500 (Table 1, run 14). Characteristic features for successful 3D control are suggested, as is the case for HBPMA synthesis. For example, the nearly equal consumption rate of 13 and St (Fig. 6b), the deviation of Mn(SEC) from Mn(theo), and the similarity of Mn(MALLS) with Mn(theo) (Fig. 6c) suggest the formation of a regular branched structure. Furthermore, low dispersity (Đ < 2) and unimodal SEC traces and their shift with time (Fig. 6d) support the living character and good control throughout the polymerization period. A deuterium-labeling experiment revealed the selective formation of PSt-end dormant species 26C, and the branching efficiency was determined to be >95%, which further supports the dendritic structure.

a Synthesis and b–f characterization of dendritic HBPSt 26. b Time dependence for the consumption of 13 and St, c correlation between monomer conversion, Mn, and dispersity, and d time dependence for SEC traces for the synthesis for N = 3. e MHKS plot for linear PSt (N = 0) and HBPSts with N = 3 with different BDs. f Fluorescence spectra for 26E (N = 3) with different BDs at the same concentration of pyrene in DMF
This result also suggests that intermediate 27 selectively propagates at the terminal carbon to 28, which further gives desired 26. If 27 propagated at the internal carbon and gave 29, the resulting vinyl telluride group in 29 would not further propagate due to the difficulty of generating a vinyl radical. Therefore, selective propagation giving 28 is also the key for achieving control of the branching structure, although the origin of the selectivity giving 28 versus 29 needs further study.
The synthesis of HBPSts is fairly general. For example, control of molecular weight and BD was successfully achieved for the synthesis of N = 3 by changing [St]0/[16b]0 from 100 to 1000 (runs 14, 16–18). The copolymerization behavior, SEC-MALLS results, and low dispersity (Đ < 2.1) suggest the formation of dendritic HBPSts with BD values of 5.0–57. The effect of BD on intrinsic viscosity [η] was clearly observed by the SEC-MALLS measurement, in which [η] systematically decreased as BD decreased (Fig. 4e). The exponent a in the MHKS equation is 0.32–0.46, which is significantly smaller than that for linear PSt (0.72).
This method is also applicable to the synthesis of a tadpole block copolymer starting from linear-PSt macroinitiator 22e (E = Ph, Scheme 4a). Additionally, the method is also highly compatible with functional groups. For example, p-chlorostyrene and p-acetoxystyrene were also successfully copolymerized with 13, giving the corresponding dendritic HBPSts (runs 19 and 20). Although dispersities were relatively high, the copolymerization behavior and SEC traces suggest the formation of controlled branching structures in both cases.
In contrast, the maximum generation to be controlled was limited to 4 under [St]0/[16b]0 = 500 (runs 11–13), as indicated by the increase in dispersity with an increasing generation. For example, the dispersity was >2.5 for N = 4, while SEC-MALLS suggested the formation of HBPSt with a controlled branching structure. In the synthesis of N = 5, partial gelation was observed with increasing monomer conversion. This is probably due to the occurrence of a termination reaction for the propagating radical species by the combination [59, 60]. These results are in sharp contrast to those obtained from HBPMA synthesis, in which the generation can reach 6 under the same [MA]0/[16b]0 ratio, probably because the termination reaction of acrylate-end radicals is exclusively disproportionation [61].
The 3D structural control was further clarified by the excimer emission of pyrene-functionalized HBPSt 22E (X = TEMPO-pyrene), which was synthesized by treatment of 22A (X = TeMe) with 25F under UV–Vis irradiation. Three samples of 22E with N = 3 having different BD values of 15, 27, and 52 and linear PSt were synthesized and tested. The fluorescence spectrum of 22E showed a characteristic emission peak at λmax = 476 nm corresponding to the pyrene excimer [62], and the intensity of the excimer emission increased with decreasing BD (Fig. 6f). In contrast, no excimer emission was observed for the linear PSt. These results clearly demonstrate an increase in pyrene’s local concentration due to the decrease in BD and support the controlled branching of the synthesized HBPSts. The 1H NMR and absorption spectra for 22E also revealed the high end-group fidelity of HBPSts (86–90%), almost identical to that for linear PSt (92%).
Synthesis of HB-polymethacrylates by TERP
The synthesis of HB-poly(methyl methacrylate) (HBPMMA) was attempted using vinyl telluride 7a or 13 as a comonomer. However, while the conversion of vinyl telluride and MMA took place, no polymer growth was observed in either case. Analyses of the products by matrix-assisted laser desorption ionization-time of flight mass spectrometry revealed the occurrence of fragmentation reactions. For example, desired HBPMMA 31 was formed as a minor component in the experiment using 13 as a comonomer, and a mixture of linear polymers 32, 34, 35, and 37 was detected as the major products. These results can be explained by the β-carbon fragmentation of mid-chain radical intermediate 30, which forms from the expected intermediate 29, either path a or b giving radical 33 and 34 or 35 and radical 36 (Scheme 5). Radicals 33 and 36 are deactivated to the corresponding dormant species 32 and 37, respectively. The same fragmentation was also observed in the copolymerization using 7a as a comonomer. Among the radicals involved in fragmentation, i.e., 30, 33, and 36, allyl radical 30 is more thermodynamically stable than ester-substituted tertiary radicals 33 and 36. Therefore, thermodynamics cannot explain the observed fragmentation reaction.

Proposed mechanism for the β-carbon fragmentation of radical intermediate 24
In contrast, vinyl telluride 10′ copolymerized with MMA without β-carbon fragmentation at 30 °C, which afforded HBPMMA (Fig. 7a, implies TePh group instead of TeMe group). Our previous results revealed that the reactivity of methyltellanyl and phenyltellanyl groups in TERP is very similar [63, 64]. The formation of structurally controlled HBPMMA was suggested by monitoring the polymerization for the synthesis of N = 4 (Table 1, run 17); the statistical copolymerization of MMA and 13′ (Fig. 7b), Mn behavior analyzed by SEC and MALLS, and low dispersity (Fig. 7c), unimodal SEC traces and their shift (Fig. 7d) were observed, while the final conversion of 13′ and MMA was rather low (62% and 81%, respectively). When the temperature increased, such as to 60 °C, the dispersity increased, and the formation of β-carbon fragmentation started to occur.

a Synthesis and b–e characterization of dendritic HBPMMA 38. b Time dependence for the consumption of 10′ and MMA, c correlation between monomer conversion, number average molecular weight, and dispersity, and d time dependence for the SEC traces for the synthesis of N = 4. e MHKS plot for linear PMMA (N = 0) and HBPMMs for runs 21–25, Table 1
On the other hand, the end-group analyses carried out by deuterium-labeling experiments reveal that the dormant tellurium groups are present as the bis-telluride group, such as 39, and a mid-chain telluride foam, such as the unbranched structure 40 at a ratio of 40:60. No PMMA-end structures, such as 38 and the branched end of 40, were detected. These results are reasonable considering the low stability of the radicals generated from 39 and the unbranched tellurium group of 40 compared to the PMMA-end radical, as estimated from their BDEs (Fig. 2). The generation of radicals from these dormant species is probably the rate-determining step. BD was not determined due to the low degree of branching.
These results also indicate the necessity for careful evaluation of dispersity in HBP synthesis because dispersity in HBPMMA synthesis is very similar to or better than that in HBPMA and HBPSt syntheses, even though the branched structure in the former is less controlled than that in the latter. These results are not surprising because when the degree of branching decreases, the structure of the HBPs becomes similar to that of linear polymers. As the dispersity in the synthesis of linear polymers can usually be controlled below 1.5 and even below 1.1 by reversible deactivation radical polymerization, the apparent dispersity control observed in HBP synthesis improves as the degree of branching decreases. Further studies are needed to quantitatively clarify this point.
While this method has a limitation in the degree of branching, the 3D structure and properties of HBPMMAs can be systematically modulated. For example, the control of branching numbers was achieved by changing the [10′]0/[16b′]0 ratio to 30 and 60 while keeping the [MMA]0/[16b′]0 ratio at 500 (runs 22 and 23). The conversion of 10′ reached >60% in both cases under statistical copolymerization, and structurally controlled HBPMMAs over Mn with low dispersity (Ð < 1.7) were obtained. Additionally, the control of molecular weight was achieved by changing the [MMA]0/[16b′]0 ratio from 250 to 1000 while maintaining the [10′]0/[16b′]0 ratio as 15 with high conversion of MMA and 10′ (runs 24 and 25). HB-polymethacrylates consisting of t-butyl methacrylate and dimethylaminoethyl methacrylate were also synthesized by copolymerization using 10′ as a comonomer (runs 26 and 27). The MHKS plot for the synthesized HBPMMAs reveals the systematic change in intrinsic viscosity [η] as a function of the 10′/MMA ratio (Fig. 5e). The exponent coefficient a in the MHKS equation (a = 0.55–0.35) is also significantly lower than that for linear PMMA (a = 0.71).
The origin for the suppression of the β-carbon fragmentation using 10′ was estimated by calculating the model reactions from model radical 41 (a: R = Me, d: R = H) undergoing β-carbon fragmentation to yield alkene 42 or propagating to MMA to yield branched radical 43 (Fig. 8). The activation enthalpy for fragmentation from 41d to 42d is slightly higher than that from 41a to 42a (68.3 and 64.3 kJ mol−1, respectively), but the difference is relatively small considering the observed remarkable difference between 10′ and 7a. In contrast, the activation enthalpy for the addition reaction of 41d to MMA to give 43d is significantly lower than that for 41a to 43a (22.1 and 44.6 kJ mol−1, respectively). In addition, the formation of 43d is much more exothermic than that of 43a. These results suggest that the kinetic preference for the propagation reaction of 41d to yield 43d is responsible for the desired branch formation.

Energy diagram (kJ mol−1) of the propagation and β-fragmentation reactions from radical intermediate 35 obtained by DFT calculations at the (U)B3LYP/6–31G* level of theory. The numbers in parentheses are the Gibbs energies at 298.15 K
The effect of the branching structure on the molecular volume of 38B (X = H) in the gas phase was also clarified by ion mobility spectroscopy (IMS)-TOF MS measurements [65, 66], in which an ionized polymer molecule is propelled through an IMS tunnel by gas flow (Fig. 9). A 2D plot (heat map) of the m/z ratio versus the inverse of the ion mobility (1/K0 [V s cm−2]) for the 38B sample having 1–3 branching units (m) derived from 10′ and 19–22 MMA units (n) showed a series of intense bright spots corresponding to the doubly charged molecular ion mass of 38B with different m and n values. Each molecular ion having the same m and n is connected by lines to estimate the effect of the branching structure on the ion mobility, which reveals that the mobility of ionized 38B increases with increasing branching number m. These results also indicate that the collision cross section for 38B systematically increases with increasing m, which further supports the successful synthesis of structurally controlled HBPMMA under the current conditions.

Heat map of the selected mass region for HBPMMA 38B with branching units m = 1–3 and MMA units n = 19–22. The doubly charged molecular ion signals appearing as local maximum spots are highlighted by white circles. The signals having the same repeating units m and n are connected by straight lines. Reprinted with permission from ref. [43]. Copyright (2020) American Chemical Society [48]
Synthesis of HBPs by ATRP [50]
Zhong reported that butyl α-bromoacrylate (44) serves as a branching point for the synthesis of HB-polyacrylates and HBPSts (46) under Cu-catalyzed ATRP conditions (Fig. 10a). The BDE for 44 is ~80 kJ mol−1 higher than that for conventional ATRP initiators, such as ethyl α-bromoisobutylate (45), so that branching is induced only after 45 reacts as a monomer.

In the copolymerization of 44 and butyl acrylate (BA), the consumption of 44 proceeds approximately ten times faster than that of BA, giving polymers a high gradient composition under one-shot conditions because 44 is activated by an ester group. Therefore, 44 was slowly fed into the polymerization solution at various rates to regulate the cross-propagation reaction, leading to relatively uniform incorporation of 44. For example, eight equivalents of 44 with respect to 45 were fed into the ATRP of BA at a feed rate of 0.2 equivalent/h (Table 2, runs 1 and 2). The formation of structurally controlled HBPBAs with low dispersity (Đ < 1.3) was obtained. Similarities among Mn[theo], Mn[SEC], and Mn[MALLS] may be attributed to the low branching number as the conversion of 44 was limited (<50%). When the relative amount of 44 was increased to 12 and 20, the difference between Mn[theo] and Mn[SEC] became significant, while Mn[MALLS] showed good agreement with Mn[theo] (runs 3 and 4). HBPSts were also synthesized by copolymerization of 44 and St under slow feeding of 44 (run 5). The SEC-MALLS data and low dispersity also support the controlled synthesis of HBPSts. However, due to the low conversion of 44, BA, and St, the control of the 3D structure, i.e. molecular weight, dendritic generation, and BD, was limited. For example, the maximum generation synthesized was approximately three, and the BD range was 6.1–9.2.
The presence of the desired branched quaternary carbon in 46 and the absence of the quaternary carbon attached to bromine in 48 were observed by 13C NMR, suggesting that the degree of branching is very high. However, quantitative analysis is needed to quantify the branching efficiency considering the low sensitivity of quaternary carbons under routine 13C NMR measurements.
A star polymer consisting of 14 branched PBA arms was synthesized starting from a β-cyclodextrin-derived ATRP initiator with Đ = 1.05 (Fig. 10b). A bottlebrush polymer with a branch-on-brush architecture was also synthesized via a similar graft-from strategy using a linear backbone macroinitiator (Fig. 10c). The clear shift in the SEC traces for the macroinitiators and the absence of low molecular weight polymer indicates the absence of PBAs initiated from 44. Block copolymers consisting of linear polydimethylsiloxane (PDMS) and HBPBA or HBPSt were also synthesized starting from a PDMS-modified ATRP macroinitiator.
Summary
A method for the synthesis of controlled HBPs over a 3D structure by TERP and ATRP was developed using vinyl telluride and bromide, respectively, as key monomers to induce branching after incorporation into the polymer backbone. The synthesis of HB-polyacrylates by TERP is highly general in synthesizing various 3D structures in terms of molecular weight, number of branching points, BD, and number of chain-end groups. The synthesis of HBPSts by TERP also possesses good generality, while the synthesis of HB-polymethacrylate by TERP and HB-polyacrylates and PSts by ATRP still needs further development to achieve better 3D control. Despite its limitations, the proposed method clearly opens up new possibilities for designing new polymer architectures and functional materials based on HBPs. As TERP and ATRP are already implemented in industry, the large-scale production of HBPs and their application in materials science is within reach.
References
Elias H-G. Macromolecules. Vol. 3, Physical Structures and Properties. Weinheim: Wiley-VCH; 2008.
Matyjaszewski K, Gnanou Y, Leibler L, editors. Macromolecular engineering Vol. 3, structure-property correlation and characterization techniques. Weinheim: Wiley-VCH; 2007.
van Krevelen DW, te Nijenhuis K. Properties of polymers. 4th ed. Amsterdam; Elsevier; 2009.
Elias H-G. Macromolecules. Vol. 1, Chemical Structures and Syntheses. Weinheim: Wiley-VCH; 2005.
Matyjaszewski K, Gnanou Y, Leibler L, editors. Macromolecular engineering. Vol. 1, Synthetic techniques. Weinheim: Wiley-VCH; 2007.
Matyjaszewski K, Gnanou Y, Leibler L, editors. Macromolecular engineering. Vol. 2, Elements of macromolecular structural control. Weinheim: Wiley-VCH; 2007.
Hadjichristidis N, Hirao A, Tezuka Y, Du Prez F, editors. Complex macromolecular architectures. Synthesis, characterization, and self-assembly. Singapore: John Wiley & Sons (Asia) Pte Ltd; 2011.
Yan D, Gao C, Frey H, editors. Hyperbranched polymers. Synthesis, properties, and applications. New Jersey: John Wiley & Sons, Inc.; 2011.
Hadjichristidis N, Pitsikalis M, Pispas S, Iatrou H. Polymers with complex architecture by living anionic polymerization. Chem Rev. 2001;101:3747–92.
Jikei M, Kakimoto M. Hyperbranched polymers: a promising new class of materials. Prog Polym Sci. 2001;26:1233–85.
Gao C, Yan D. Hyperbranched polymers: from synthesis to applications. Prog Polym Sci. 2004;29:183–275.
Voit BI, Lederer A. Hyperbranched and highly branched polymer architectures-synthetic strategies and major characterization aspects. Chem Rev. 2009;109:5924–73.
Astruc D, Chardac F. Dendritic catalysts and dendrimers in catalysis. Chem Rev. 2001;101:2991–3023.
Boas U, Heegaard PMH. Dendrimers in drug research. Chem Soc Rev. 2004;33:43–63.
Lee CC, MacKay JA, Frechet JMJ, Szoka FC. Designing dendrimers for biological applications. Nat Biotechnol. 2005;23:1517–26.
Yamamoto K, Imaoka T, Chun W-J, Enoki O, Katoh H, Takenaga M, et al. Size-specific catalytic activity of platinum clusters enhances oxygen reduction reactions. Nat Chem. 2009;1:397–402.
Wang X, Gao H. Recent progress on hyperbranched polymers synthesized via radical-based self-condensing vinyl polymerization. Polymers. 2017;9:188.
Grayson SM, Frechet JMJ. Convergent dendrons and dendrimers: from synthesis to applications. Chem Rev. 2001;101:3819–67.
Tomalia DA, Christensen JB, Boas U. Dendrimers, dendrons, and dendritic polymers: discovery, applications, and the future. Cambridge: Cambridge University Press; 2012.
Bosman AW, Janssen HM, Meijer EW. About dendrimers: structure, physical properties, and applications. Chem Rev. 1999;99:1665–88.
Tomalia DA, Baker H, Dewald J, Hall M, Kallos G, Martin S, et al. A new class of polymers—starburst-dendritic macromolecules. Polym J. 1985;17:117–32.
Hawker CJ, Frechet JMJ. Preparation of polymers with controlled molecular architecture. A new convergent approach to dendritic macromolecules. J Am Chem Soc. 1990;112:7638–47.
Flory PJ. Molecular size distribution in three dimensional polymers. 6. Branched polymers containing A-R-Bf-1 type units. J Am Chem Soc. 1952;74:2718–23.
Kim YH, Webster OW. Water-soluble hyperbranched polyphynylene—a unimolecular micelle. J Am Chem Soc. 1990;112:4592–3.
Frechet JMJ, Henmi M, Gitsov I, Aoshima S, Leduc MR, Grubbs RB. Self-condensing vinyl polymerization—an approach to dendritic materials. Science. 1995;269:1080–3.
Hawker CJ, Frechet JMJ, Grubbs RB, Dao J. Preparation of hyperbranched and star polymers by a living, self-condensing free-radical polymerization. J Am Chem Soc. 1995;117:10763–4.
Gaynor SG, Edelman S, Matyjaszewski K. Synthesis of branched and hyperbranched polystyrenes. Macromolecules. 1996;29:1079–81.
Sakamoto K, Aimiya T, Kira M. Preparation of hyperbranched polymethacrylates by self-condensing group transfer polymerization. Chem Lett. 1997;26:1245–6.
Simon PFW, Radke W, Müller AHE. Hyperbranched methacrylates by self-condensing group transfer polymerization. Macromol Rapid Commun. 1997;18:865–73.
Alfurhood JA, Bachler PR, Sumerlin BS. Hyperbranched polymers via RAFT self-condensing vinyl polymerization. Polym Chem. 2016;7:3361–9.
Hanselmann R, Holter D, Frey H. Hyperbranched polymers prepared via the core-dilution/slow addition technique: computer simulation of molecular weight distribution and degree of branching. Macromolecules. 1998;31:3790–801.
M K, Gao H. New method to access hyperbranched polymers with uniform structure via one-pot polymerization of inimer in microemulsion. J Am Chem Soc. 2012;134:15680–3.
Ohta Y, Fujii S, Yokoyama A, Furuyama T, Uchiyama M, Yokozawa T. Synthesis of well-defined hyperbranched polyamides by condensation polymerization of AB(2) monomer through changed substituent effects. Angew Chem. 2009;48:5942–5.
Ohta Y, Sakurai K, Matsuda J, Yokozawa T. Chain-growth condensation polymerization of 5-aminoisophthalic acid triethylene glycol ester to afford well-defined, water-soluble, thermoresponsive hyperbranched polyamides. Polymer. 2016;101:305–10.
Shi Y, Graff RW, Cao X, Wang X, Gao H. Chain-growth click polymerization of AB(2) monomers for the formation of hyperbranched polymers with low polydispersities in a one-pot process. Angew Chem. 2015;54:7631–5.
Cao X, Shi Y, Gao H. A novel chain-growth CuAAC polymerization: one-pot synthesis of dendritic hyperbranched polymers with well-defined structures. Synlett. 2017;28:391–6.
Matyjaszewski K, Möller M, editors. Polymer science: a comprehensive reference. Vol. 3, Chain polymerization of vinyl monomers. Elsevier; Amsterdam; 2012.
Chatgilialoglu C, Studer A, editors. Encyclopedia of radicals in chemistry, biology and materials. Vol. 4, polymers & materials. John Wiley & Sons; 2012.
Yamago S, Iida K, Yoshida J. Organotellurium compounds as novel initiators for controlled/living radical polymerizations. Synthesis of functionalized polystyrenes and end-group modifications. J Am Chem Soc. 2002;124:2874–5.
Yamago S. Precision polymer synthesis by degenerative transfer controlled/living radical polymerization using organotellurium, organostibine, and organobismuthine chain-transfer agents. Chem Rev. 2009;109:5051–68.
Yamago S. Photoactivation of organotellurium compounds in precision polymer synthesis: controlled radical polymerization and radical coupling reactions. Bull Chem Soc Jpn. 2020;93:287–98.
Yamago S, Lida K, Yoshida J. Tailored synthesis of structurally defined polymers by organotellurium-mediated living radical polymerization (TERP): synthesis of poly(meth)acrylate derivatives and their Di- and triblock copolymers. J Am Chem Soc. 2002;124:13666–7.
Yusa S, Yamago S, Sugahara M, Morikawa S, Yamamoto T, Morishima Y. Thermo-responsive diblock copolymers of poly(N-isopropylacrylamide) and poly(N-vinyl-2-pyrroridone) synthesized via organotellurium-mediated controlled radical polymerization (TERP). Macromolecules. 2006;40:5907–15.
Mishima E, Yamago S. Controlled random and alternating copolymerization of (Meth)acrylates, acrylonitrile, and (Meth)acrylamides with vinyl ethers by organotellurium-, organostibine-, and organobismuthine-mediated living radical polymerization reactions. J Polym Sci A Polym Chem. 2012;50:2254–64.
Mishima E, Tamura T, Yamago S. Controlled copolymerization of acrylate and 6-methyleneundecane by organotellurium-mediated living radical polymerization (TERP). Macromolecules. 2012;45:2989–94.
Mishima E, Tamura T, Yamago S. Controlled copolymerization of 1-octene and (Meth)acrylates via organotellurium-mediated living radical polymerization (TERP). Macromolecules. 2012;45:8998–9003.
Lu Y, Nemoto T, Tosaka M, Yamago S. Synthesis of structurally controlled hyperbranched polymers using a monomer having hierarchical reactivity. Nat Commun. 2017;8:1863.
Lu Y, Yamago S. Synthesis of structurally controlled, highly branched polymethacrylates by radical polymerization through the design of a monomer having hierarchical reactivity. Macromolecules. 2020;53:3209–16.
Lu Y, Yamago S. One-step synthesis of dendritic highly branched polystyrenes by organotellurium-mediated copolymerization of styrene and a Dienyl Telluride Monomer. Angew Chem. 2019;58:3952–6.
Li F, Cao M, Feng Y, Liang R, Fu X, Zhong M. Site-specifically initiated controlled/living branching radical polymerization: a synthetic route toward hierarchically branched architectures. J Am Chem Soc. 2019;141:794–9.
Fradet A, Chen J, Hellwich K-H, Horie K, Kahovec J, Mormann W, et al. Nomenclature and terminology for dendrimers with regular dendrons and for hyperbranched polymers (IUPAC Recommendations 2017). Pure Appl Chem. 2019;91:523–61.
Goto A, Kwak Y, Fukuda T, Yamago S, Iida K, Nakajima M, et al. Mechanism-based invention of high-speed living radical polymerization using organotellurium compounds and azo-initiators. J Am Chem Soc. 2003;125:8720–1.
Fan W, Nakamura Y, Yamago S. Synthesis of multivalent organotellurium chain-transfer agents by post-modification and their applications in living radical polymerization. Chemistry. 2016;22:17004–8.
Yamago S, Ukai Y, Matsumoto A, Nakamura Y. Organotellurium-mediated controlled/living radical polymerization initiated by direct C−Te bond photolysis. J Am Chem Soc. 2009;131:2100–1.
Yamada T, Mishima E, Yamago S. Phenyltellanyl triflate (PhTeOTf) as a powerful tellurophilic activator in the Friedel–Crafts reaction. Chem Lett. 2008;37:650–1.
Kayahara E, Yamago S. In: Matyjaszewski K, Sumerlin BS, Tsarevsky NV, editors. Progress in controlled radical polymerization: materials and applications Vol. 1101 ACS symposium series. American Chemical Society; Washington, DC; 2012.
Nakamura Y, Arima T, Yamago S. Modular synthesis of mid-chain-functionalized polymers by photoinduced diene- and styrene-assisted radical coupling reaction of polymer-end radicals. Macromolecules. 2014;47:582–8.
Willemse RXE, van Herk AM, Panchenko E, Junkers T, Buback M. PLP-ESR monitoring of midehain radicals in n-butyl acrylate polymerization. Macromolecules. 2005;38:5098–103.
Nakamura Y, Yamago S. Termination mechanism in the radical polymerization of methyl methacrylate and styrene determined by the reaction of structurally well-defined polymer end radicals. Macromolecules. 2015;48:6450–6.
Nakamura Y, Ogihara T, Hatano S, Abe M, Yamago S. Control of the termination mechanism in radical polymerization by viscosity: selective disproportionation in viscous media. Chemistry. 2017;23:1299–305.
Nakamura Y, Lee R, Coote ML, Yamago S. Termination mechanism of the radical polymerization of acrylates. Macromol Rapid Commun. 2016;37:506–13.
Farhangi S, Casier R, Li L, Thoma JL, Duhamel J. Characterization of the long-range internal dynamics of pyrene-labeled macromolecules by pyrene excimer fluorescence. Macromolecules. 2016;49:9597–604.
Yamago S, Iida K, Nakajima M, Yoshida J. Practical protocols for organotellurium-mediated living radical polymerization by in situ generated initiators from AIBN and ditellurides. Macromolecules. 2003;36:3793–6.
Kayahara E, Yamago S, Kwak Y, Goto A, Fukuda T. Optimization of organotellurium transfer agents for highly controlled living radical polymerization. Macromolecules. 2008;41:527–9.
Ridgeway ME, Lubeck M, Jordens J, Mann M, Park MA. Trapped ion mobility spectrometry: a short reviewMark. Int J Mass Spectrom. 2018;425:22–35.
Kanu AB, Dwivedi P, Tam M, Matz L, Hill HH. Ion mobility-mass spectrometry. J Mass Spectrom. 2008;43:1–22.
Acknowledgements
The author deeply appreciates the intellectual and physical contributions of collaborators and coworkers, especially Drs. Atanu Kotal (now at JIS University, India), Shenyong Ren (now at the China University of Petroleum), and Yangtian Lu (Kyoto University). Drs. Atanu Kotal and Shenyong Ren pioneered this work, and Dr. Yangtian Lu realized the idea and helped prepare the paper. Financial support from the Japan Society for the Promotion of Science KAKENHI (16H06352) is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yamago, S. Practical synthesis of dendritic hyperbranched polymers by reversible deactivation radical polymerization. Polym J 53, 847–864 (2021). https://doi.org/10.1038/s41428-021-00487-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-021-00487-x
- Springer Nature Limited