
Abstract
The geometric structures of compounds CuL2 (L = R1N(O)–(O)CR2; R1 = tert-Bu, R2 = Ph (I) and R1 = Ph, R2 = tert-Bu (II)) are studied by X-ray diffraction analysis (XRD) in the crystalline state and by stationary EPR spectroscopy in a dichloromethane solution. According to the XRD data (CIF files CCDC nos. 1875367 and 2022310, respectively), in the crystalline state the copper atoms of both complexes exist in the planar-square environment of the oxygen atoms with the trans-coordination of the hydroxamate ligands. According to the EPR data for frozen solutions of the complexes in dichloromethane, the compounds coexist as two mononuclear forms A and B differed in magnetic resonance parameters. It is shown that compound IB forms aggregates when freezing the solution. The form B of compound II is an associate of the copper complex with the solvent molecules. The EPR data indicate that both forms of compounds I and IIA form no associates with the solvent molecules.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Hydroxamic acids found wide use in biology, medicine, analytical chemistry, and metallurgy. Both hydroxamic acids themselves and their metal complexes can exist as various isomers and polymolecular structures [1]. The majority of the known structurally characterized copper(II) hydroxamates in the crystalline state exists as planar-square complexes with the trans-coordination of the ligands [2–5]. It was convincingly shown by the EPR method that copper hydroxamates dissolved in organic solvents existed as two forms [6, 7], which behaved as mononuclear in the EPR spectra. The differences in the spectral parameters were attributed [8, 9] to the coexistence of cis- and trans-isomers of the complexes in the solution. The both indicated forms of the complexes are present in the solution and reliably detected by the EPR method both at room temperature and in low-temperature glass. When solutions of the copper(II) hydroxamates are cooled to the temperatures lower than the freezing point of the solvent, dimeric forms [CuL2]2 can be detected in the EPR spectra along with monomeric forms CuL2, depending on the structure of ligand L [10].
In this work, the structures of two copper(II) hydroxamates CuL2 (L = R1N(O)–(O)CR2; R1 = tert-Bu, R2 = Ph (I) and R1 = Ph, R2 = tert-Bu (II)) in the crystalline state were determined by the XRD method, and the structures of the compounds in frozen solutions of dichloromethane at 100 K were determined using EPR spectroscopy.
EXPERIMENTAL
The copper(II) hydroxamate complexes were synthesized according to a described procedure [11]. For a more precise interpretation of the EPR spectra, the samples were synthesized using only one copper isotope: 63Cu. Single crystals suitable for XRD were obtained by crystallization from petroleum ether.
XRD of complexes I and II was carried out on the Belok beamline at the Kurchatov Synchrotron Radiation Source of the National Research Center “Kurchatov Institute” (Moscow, Russia) in the φ scan mode using a Rayonix SX165 CCD detector at 100 K [12]. The primary indexing, parameter refinement, reflection integration, and a reflection intensity absorption correction were applied using the XDS program package [13]. The structures were solved by direct methods and refined by full-matrix least squares for F 2 with the anisotropic approximation parameters for all non-hydrogen atoms. Hydrogen atoms were placed in the calculated positions and refined by the riding model with Uiso(H) = 1.5 × Uequiv(C) for the methyl groups and 1.2 × Uequiv(C) for the hydrogen atoms of the phenyl group. The structure parameters were refined without additional restraints imposed on the model. Calculations were performed using the SHELXTL program package [14] in the OLEX2 medium of structural data visualization and processing [15]. The crystallographic data and structure refinement parameters for compounds I and II are presented in Table 1. Selected bond lengths and bond angles are given in Table 2.
The crystallographic data for complexes I and II were deposited with the Cambridge Crystallographic Data Centre (CIF files CCDC nos. 1875367 and 2022310, respectively; deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk/data_request/cif).
EPR spectra were recorded on an Elexsys-E680X radiospectrometer (BRUKER) at the Center for Collective Use of the Kurnakov Institute of General and Inorganic Chemistry of the Russian Academy of Sciences (Moscow, Russia). The working frequency of the spectrometer was ~9.8 GHz (X range). Solutions of the complexes were cooled to Т = 100 K using an Oxford Instruments thermal block.
Spin-Hamiltonian (SH) parameters of the complexes were determined using the best fit between the experimental and theoretical spectra by the minimization of the error function [16] as follows:
where \(Y_{i}^{E}\) is the set of experimental intensities of the EPR signal with a constant increment over the magnetic field strength Н, \(Y_{i}^{T}\) is the theoretical values at the same field strengths Н, and N is the number of points. The sum of the Lorentzian and Gaussian functions was used as a lineshape function [17]. According to the relaxation theory [18], the lineshape was specified by the equation
where mI is the nuclear spin projection on the magnetic field direction; and α, β, and γ are the parameters determined by various relaxation mechanisms. Relative concentrations of the complexes, g factors, hyperfine structure (HFS) constants, and linewidth and lineshape parameters were varied during minimization.
RESULTS AND DISCUSSION
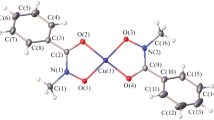
According to the XRD data, complexes I and II crystallize in the monoclinic space group P21/n without incorporation of the solvent into the crystal structure. In both compounds, the Cu(1) copper atom lies on the inversion center and exists in the planar-square environment of the oxygen atoms of the ligands with minor changes in the interatomic distances between the central metal atom and atoms of the coordination environment, Cu(1)–O(1) 1.888(3), Cu(1)–O(2) 1.909(3) Å (for I) and Cu(1)–O(1) 1.8764(18), Cu(1)–O(2) 1.9276(17) Å (for II), which are arranged in the trans-positions. The structures of compounds I and II are presented in Figs. 1 and 2, respectively. The five-membered metallocycle Cu(1)–O(1)–N(1)–C(1)–O(2) in compound I is close to an ideally planar one, whereas a slight deviation of the Cu(1) copper atom from the O(1)–N(1)–C(1)–O(2) (0.020(4) Å) plane is observed in compound II.

Molecular structure of complex I (thermal ellipsoids are given with 45% probability). Hereinafter superscript 1 designates atoms of the symmetrically independent part of the molecule.

Molecular structure of complex II (thermal ellipsoids are given with 45% probability).
The EPR spectra of frozen solutions of compounds I and II in dichloromethane represent a superposition of the spectra of two forms (A and B) of the mononuclear copper(II) compounds in a magnetically dilute solid matrix (Figs. 3, 4). Three HFS lines are observed in weak magnetic fields from the copper nucleus parallel to the g tensor component, and the fourth line is overlapped with an intense signal of the perpendicular component. An additional absorption peak is observed in the range of strong magnetic fields (~3300 G) [16].

Experimental and theoretical EPR spectra of the frozen solution of compound I (ratio of concentrations of the forms of the complex cIA/cIB = 1.50); theoretical spectra of compounds IA, IB, and IIIB.

Experimental and theoretical EPR spectra of the frozen solution of compound II (ratio of concentrations of the forms of the complex cIIA/cIIB = 1.86); theoretical spectra of compounds IIA and IIB.
The spectra of the complexes are described by the orthorhombic symmetry SH of the following form:
where β is Bohr’s magneton; i = 1, 2; giz, gix, and giy are the z, x, and y components of the g tensor; Ai, Bi, and Ci are the z, x, and y components of the HFS tensor; Siz, Six, and Siy are the projections of the electron spin operator on the coordinate axes; S = 1/2; and Iiz, Iix, and Iiy are the projections of the nuclear spin operator of the central atom of the complex on the coordinate axes (I = 3/2). The SH parameters of the EPR spectra of the copper compounds in the frozen solutions obtained by the best approximation of the experimental data are presented in Table 3.
The compounds, whose EPR spectra are described by the SH with higher HFS constants and lower g factors, hereinafter will be designated as compounds in form A. The complexes, whose EPR spectra are described by the SH with higher g factors and lower HFS constants, are named compounds in form B. The SH parameters for the spectra of compounds IA and IIA are close and characterized by a slight difference in the planar components gx, gy and B, C. In the bis(chelate) square cis-complex, the diagonals of the square formed by the oxygen atoms of the ligands are equivalent to Cu–O(N) + Cu–O(С), whereas they are not equivalent to Cu–O(N) + Cu–O(N) and Cu–O(С) + Cu–O(С) in the trans-complex. The equivalence of the planar components of the SH parameters indicates that the diagonals of the square are equivalent, which, in turn, corresponds to the cis-coordination of the ligands. The theoretical spectra of form A obtained when using the SH parameters in Table 3 are visually perceived as the spectra described by the axial symmetry SH (Figs. 3, 4). According to the published results [19], copper hydroxamates with the cis-coordination of the ligands are characterized by the following SH parameters: gz = 2.23, gx,y = 2.03, and А = 219 × 10–4 cm–1. Thus, forms A of complexes I and II can be ascribed to cis-isomers of the corresponding compounds on the basis of a comparison of the values obtained for the SH parameters.
The EPR spectrum of compound IB is described by the SH with a substantial difference in the planar parameters gx, gy, B, and C. Since the diagonals of the coordination environment of the central metal atom are nonequivalent, this difference corresponds to the spectrum of the complex with the trans-coordination of the ligands. The SH parameters of this compound are close to the EPR spectral parameters of complex CuL2 (L = R1N(O)–(O)CR2; R1 = tert-Bu, R2 = Ме (IIIB)) with the trans-coordination of the ligands [5]. The difference in the spectra of compounds IB and IIIB is a substantial broadening of the spectrum of complex IB in the range of low magnetic field strengths (Fig. 3). Similar broadening can be related only to the interaction of the mononuclear CuL2 complexes in a frozen solution, which indicates their aggregation.
The EPR spectra for complexes IB and IIB (Figs. 3, 4) differ substantially in the SH parameters, which can be due to different effects of the solvent molecules on the trans-isomers of compounds I and II. The solvent molecules can form stable solvates with the copper(II) bis(chelates). According to the NMR and EPR data, these solvates exist in a solution at room temperature and in low-temperature glass at 77 K [20]. Similar solvates of copper hydroxamates have previously been isolated in the crystalline state [2]. According to the XRD data, the solvent molecules form hydrogen bonds only with the О(N) atoms of the hydroxamate groups. This interaction leads to a decrease in the differences in the Cu–O(N) and Cu–O(C) metal–ligand distances. In compound IIB, a similar influence of the solvent molecules results in a decrease in the orthorhombic distortion of the coordination polyhedron, which, in turn, affects the EPR spectra for which the planar SH parameters (gx, gy and B, C) become close in value. Three-axial anisotropy of the SH parameters (see Table 3) of the EPR spectrum of compound IB indicates no such an interaction of the solvent and complex. In this case, the bulky substituent (R1 = tert-Bu) probably prevents the formation of a hydrogen bond of the dichloromethane molecule with the О(N) atom.
Thus, it was found from the XRD data that compounds I and II in the crystalline state were the mononuclear complexes with the trans-coordination of the ligands. At the same time, the EPR data indicate that in the frozen solutions the complexes coexist as two forms, presumably, as cis- and trans-isomers. The trans-form of copper(II) N-phenylpivaloyl hydroxamate forms a solvate with the solvent molecules.
REFERENCES
Codd, R., Coord. Chem. Rev., 2008, vol. 252, p. 1387.
Baugman, R.G., Brink, D.J., Butle, J.M., and New, P.R., Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, vol. 56, p. 528.
Rotov, A.V., Ugolkova, E.A., Efimov, N.N., et al., Russ. J. Inorg. Chem., 2014, vol. 59, no. 12, p. 1474. https://doi.org/10.1134/S0036023614120201
Dzyuba, V.I., Koval, L.I., Dudko, A.V., et al., J. Coord. Chem., 2014, vol. 67, no. 8, p. 1437.
Rotov, A.V., Ugolkova, E.A., Lermontova, E.Kh., et al., Russ. J. Inorg. Chem., 2015, vol. 60, no. 7, p. 866. https://doi.org/10.1134/S0036023615070128
Abragam, A. and Bleaney, B., Electron Paramagnetic Resonance of Paramagnetic Ions, Oxford: Clarendon, 1970.
Chizhik, V.I., Chernyshev, Y.S., Donets, A.V., et al., Magnetic Resonance and Its Applications, New York: Springer, 2014.
Larin, G.M., Rotov, A.V., and Dzyuba, V.I., Dokl. Akad. Nauk, 1995, vol. 334, no. 6, p. 779.
Larin, G.M., Rotov, A.V., and Dzyuba, V.I., Russ. Chem. Bull., 1999, vol. 48, no. 4, p. 722.
Rotov, A.V., Ugolkova, E.A., Efimov, N.N., and Minin, V.V., Russ. J. Inorg. Chem., 2013, vol. 58, no. 2, p. 186. https://doi.org/10.1134/S0036023613020216
Dzyuba, V.I., Ternovaya, T.V., Kostromina, N.A., and Ksaverov, A.N., Ukr. Khim. Zh., 1986, vol. 52, no. 5, p. 453.
Svetogorov, R.D., Dorovatovskii, P.V., and Lazarenko, V.A., Cryst. Res. Technol., 2020, vol. 55, no. 1900184.
Kabsch, W., Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, vol. 66, p. 125.
Sheldrick, G.M., Acta Crystallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 3.
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Rakitin, Yu.V., Larin, G.M., and Minin, V.V., Interpretatsiya spektrov EPR koordinatsionnykh soedinenii (Interpretation of the ESR Spectra of Coordination Compounds), Moscow: Nauka, 1993.
Wilson, R. and Kivelson, D., J. Chem. Phys., 1966, vol. 44, no. 1, p. 154.
Lebedev, Ya.S. and Muromtsev, V.I., EPR i relaksatsiya stabilizirovannykh radikalov (ESR and Relaxation of Stabilized Radicals), Moscow: Khimiya, 1972.
Barclay, S.J. and Raymond, K.N., Inorg. Chem., 1986, vol. 25, no. 20, p. 3561.
Talzi, E.P., Nekipelov, V.M., Kozyrev, N.V, and Zamoraev, K.I., Zh. Strukt. Khim., 1982, vol. 23, no. 6, p. 158.
Funding
This work was carried out in terms of state assignment of the Kurnakov Institute of General and Inorganic Chemistry (Russian Academy of Sciences) in the area of basic research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by E. Yablonskaya
Rights and permissions
About this article

Cite this article
Rotov, A.V., Yakushev, I.A., Ugolkova, E.A. et al. Structures of Copper(II) N-tert-Butylbenzoyl and N-Phenylpivaloyl Hydroxamates in the Crystalline State and in a Frozen Solution. Russ J Coord Chem 47, 376–381 (2021). https://doi.org/10.1134/S1070328421060051
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328421060051