
Abstract
The geometrical structure of CuL2 (I), where L = R1N(O)–(O)CR2, R1 = Me, R2 = Ph, was studied by X-ray diffraction in the crystalline state and by stationary ESR spectroscopy in solution. In the crystalline state, I is a chain polynuclear complex. According to ESR data, in frozen solutions, complex I exists as three species, two being mononuclear and one being binuclear. The magnetic resonance parameters and concentrations of the species in frozen solutions were determined. The electronic structure of the complex with full geometry optimization of all systems was calculated in terms of the unrestricted density functional theory (DFT) method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Both hydroxamic acids and their metal complexes can exist as various isomers and polymolecular structures [1]. Most N-substituted copper(II) hydroxamates in the crystalline state are mononuclear square planar complexes with trans-coordinated ligands [2–5]. The binuclear compound [CuL2]2 (L = R1N(O)–(O)CR2, R1 = R2 = Me) was prepared and structurally characterized [6]. In organic solvents at room temperature, crystalline copper hydroxamates exist as two species, the ESR spectra of which are characteristic of mononuclear complexes. The difference between the ESR parameters of the monomers are attributed to the coexistence of cis- and trans-isomers of the complexes in solutions. According to ESR data, both isomers are present in solutions both at room temperature and in low-temperature glass. As solutions of copper(II) hydroxamate complexes are cooled below the solvent freezing point, [CuL2]2 dimers can be manifested in the ESR spectrum, apart from the CuL2 monomers [6, 7]. The stationary ESR spectra do not exhibit the hyperfine structure (HFS) caused by coupling of the unpaired electron with ligand atoms. However, there are published data on pulsed ESR, pointing to additional hyperfine coupling of the unpaired electron with the ligand nitrogen atoms in Cu(II) hydroxamates [8].
In this study, the structure of copper(II) hydroxamate CuL2 (L = R1N(O)–(O)CR2, R1 = Me, R2 = Ph) (I) was determined by X-ray diffraction analysis. The structure of the complex in a toluene solution was found by ESR spectroscopy. The electronic structure of copper complexes was calculated by density functional theory (DFT) calculations with full geometry optimization for all systems. These results were used to determine the magnetic parameters of compound I, which were compared with experimental ESR data.
EXPERIMENTAL
Complex I was synthesized by a procedure reported in [9]. For more accurate interpretation of the ESR spectra, the samples were synthesized using the single 65Cu isotope.
Single crystal X-ray diffraction study of compound I obtained by slow evaporation of a toluene solution was carried out at the Center for Collective Use of Physical Investigation Methods, Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, on a Bruker D8 Venture Photon diffractometer in the φ- and ω-scan modes at a temperature of 150 K (λ = 0.71073 Å, Incoatec IµS 3.0 microfocus X-ray source). Determination and refinement of unit cell parameters and integration of experimental set of reflection intensities for I were performed using the Bruker APEX3 program package [10]. The absorption corrections were applied using the SADABS program [10]. The structure was solved by direct methods [11] and refined by full-matrix least-squares method on F 2 [12] in the anisotropic approximation for all non-hydrogen atoms without geometric or thermal constraints for atoms in the model. The hydrogen atoms were placed in the geometrically calculated positions and refined in the riding model with Uiso(H) equal to 1.5Ueq(C) for methyl group hydrogens and 1.2Ueq(C) for phenyl group hydrogens. The calculations were carried out using the SHELXTL program package [12] in the OLEX2 data processing and visualization software [13].
The crystal data and structure refinement parameters for I are summarized in Table 1 and selected bond lengths and bond angles are given in Table 2.
The full set of X-ray crystallographic parameters of structure I was deposited with the Cambridge Crystallographic Data Centre (CCDC no. 2128056; https://www.ccdc.cam.ac.uk/structures/).
The ESR spectra were run on an Elexsys-E680X BRUKER radiospectrometer at the Center for Collective Use of Physical Investigation Methods, Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences (Moscow, Russian Federation). The spectrometer operation frequency was ~9.8 GHz (X-range). The solutions of the complexes were cooled down to T = 100 K with an Oxford Instruments block.
The spin Hamiltonian (SH) parameters of the complexes were determined by finding the best fit between the experimental and theoretical spectra via minimization of the error functional [14]:
where \(Y_{i}^{E}\) is the array of experimental ESR signal intensities with a constant step in the magnetic field H; \(Y_{i}^{T}\) are the theoretical values at the same field H; and N is the number of points. The sum of the Lorentz and Gauss functions was used as the line shape function [15]. According to the relaxation theory [16], the line width was specified by the expression:
where mI is the projection of the nuclear spin on the magnetic field direction, α, β, γ are parameters. Minimization was performed by varying the relative concentrations of complexes, g-factors, HFS constants, and line width and shape parameters for each complex. The other parameters that were varied for the dimer include the zero-field splitting parameter (D), orthorhombic distortion parameter (E), the distance between point dipoles (R), the angle between the radius-vector connecting the metal atoms and the z axis of the local environment of the metal (ξ), and the angle between the X axis and projection of the radius-vector connecting the metal atoms on the XY plane (η).
The electronic structure calculations with full geometry optimization of all systems were performed using unrestricted DFT method with the BP86 functional [17, 18] and split valence def2-TZVP basis set [18]; Grid4 was used for integration (in ORCA). The calculated vibrational frequencies characterize the optimized structures as energy minima. The magnetic parameters were calculated according to published recommendations [19] in the B3PW91/def2-TZVP approximation [20–22] in combination with the aug-cc-pVTZ-J auxiliary basis set [23] on the metal atom and Grid6. All calculations were carried out using the ORCA program, version 4.2.3 [24].
RESULTS AND DISCUSSION
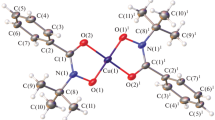
According to X-ray diffraction data, complex I crystallizes without solvent molecules in the orthorhombic space group Pbca. All atoms of the independent part of the unit cell are located in general positions (Fig. 1), and the local coordination environment of copper(II) atoms, which are central atoms of the complex, consists of the ligand oxygen atoms with the uniform distribution of Cu(1)–O interatomic distances in the 1.9314(12)–1.9525(12) Å range, which does not differ much (although is somewhat greater) from the corresponding distances in the previously described structures [5]. The OCu(1)O angles are in the range from 83.72(5)° to 97.19(5)°. The Cu(1) atom virtually does not deviate from the O(1)–O(2)–O(3)–O(4) plane; the deviation is only 0.0113(6) Å, while the main planes of the opposing ligands that form the Cu(1)–O(1)–N(1)–C(2)–O(2) and Cu(1)–O(3)–N(2)–C(9)–O(4) five-membered rings are rotated by an angle of 3.32(11)° relative to each other.

Structure of the molecular fragment of complex I and scheme of atom numbering; the ellipsoids of root-mean-square deviations of the atoms are given at 50% probability level.
It should be noted that these molecular groups form stacks extended along the crystallographic a axis; the copper coordination environment is thus completed to a somewhat distorted square bipyramidal (SBP), with the O(1)–O(4) oxygen atoms of one molecule forming the bipyramid base and the O(1) and O(3) atoms of the neighboring moieties being at the vertices (Fig. 2). The Cu(1)–O(1)#1 and Cu(1)–O(3)#2 distances (#1 x + 1/2, y, –z + 3/2, #2 x + 1/2, y, –z + 3/2) are fairly long, amounting to 2.5963(14) and 2.6106(13) Å, respectively, which markedly exceeds the sum of covalent radii for copper and oxygen (rcov(Cu) = 1.32 Å, rcov(O) = 0.66 Å, Σrcov(Cu, O) = 1.98 Å) [25], but is within the sum of their van der Waals radii (rvdW(Cu) = 2.38 Å, rvdW(O) = 1.50 Å, ΣrvdW(Cu, O) = 3.88 Å) [26]. In combination with the molecular geometry, this generally ensures proximate positions of the metal centers and formation of the chain structure in which the distance between the metals in the Cu(1)–Cu(1) chain (3.38302(16) Å) is also shortened compared to those in the complexes with N-tert-butylbenzoylhydroxamic acid [5].

Fragment of the stacked molecules of complex I. The phenyl groups are shown schematically for clarity.
At room temperature, the ESR spectrum of the complex in toluene is a superposition of the spectra of two mononuclear complexes, each of them being described by the isotropic SH of the form
where β is the Bohr magneton; i = A, B; S is the electron spin equal to 1/2; I is the 65Cu nuclear spin equal to 3/2. In the simulated spectra, the SH parameters of the two compounds at room temperature are substantially different. The compound whose ESR spectrum is described by SH with greater HFS constant, will be designated as form A. Form B will refer to the complex whose ESR spectrum is described by SH with smaller HFS constant. For compound A, gA = 2.119, aA = 88.4 × 10–4 cm–1; for compound B, gB = 2.118, aB = 76.4 × 10–4 cm–1; the concentration ratio of the complexes cA/cB = 1.5. According to the measurements of the molecular weight of copper hydroxamates in solution, the complexes are mononuclear [27]; therefore, the presence of two monomeric species can be attributed to the existence of cis- and trans-isomers. Previously, it was assumed that the ESR spectra of complexes with cis-coordinated ligands are described by SH with higher hyperfine structure (HFS) constants than the spectra of complexes with trans-coordination [4, 5]. On this basis, compound A was identified as the cis-isomer of the complex, and compound B was assigned to the trans-isomer.
The ESR spectrum of a frozen solution of I in toluene is a superposition of the spectra of two forms (A and B) of mononuclear copper(II) compound and the spectrum of the dimer (Fig. 3). The ESR spectrum of the dimer has the following parameters: gz = 2.233, gx = 2.029, gy = 2.060; A = 190.9 × 10–4 cm–1, B = 13.9 × 10–4 cm–1, C = 36.0 × 10–4 cm–1. The parameters corresponding to the interaction of the monomeric parts of the dimer: R = 3.5 Å, ξ = 29.75°, η = 2.8°, D = 122 × 10–4 cm–1, E = 19.3 × 10–4 cm–1. The concentration of the monomers that form the dimer is 69%. The geometric and electronic structure of the dimeric Cu(II) N-substituted hydroxamates in the crystalline state and in frozen solutions was described in detail previously [6, 7]. According to X-ray diffraction data, CuL2 (L = R1N(O)–(O)CR2, R1 = R2 = Me) is a dimer in the crystalline state [6]. According to ESR data, complex dimers with spectral parameters close to the parameters of compound I were detected in the frozen solution of this compound.

ESR spectra of the frozen solution of compound I: (1) experimental, (2) simulated (concentration ratio cIA/cIB = 2).
The spectra of mononuclear complexes are described by the rhombic SH of the following form:
where β is the Bohr magneton; i = A, B; giz, gix, giy are the z-, x-, and y-components of the g-tensor; Ai, Bi, Ci are the z-, x-, and y-components of the HFS tensor; Siz, Six, and Siy are the projections of the electron spin operator on the coordinate axes; S = 1/2; Iiz, Iix, and Iiy are the projections of the nuclear spin operator of the central atom of the complex on the coordinate axes (I = 3/2). Spin Hamiltonian parameters of the ESR spectra of copper compounds in frozen solutions obtained by best fitting of the experimental data are summarized in Table 3. The same table also gives the values giso = (giz + gix +g iy)/3 and aiso = ( Ai + Bi + Ci)/3 and SH parameters of the ESR spectra of mononuclear complexes that do not form dimers in the frozen solutions: CuL2 (L = R1N(O)–(O)CR2, where R1 = tert-Bu, R2 = Me (compound II) [4] and R1 = tert-Bu, R2 = Ph (compound III)) [5]. Presumably, compounds whose ESR spectra are described by SH with lower g-factors and higher HFS constants (A) are the cis-isomers, while the compounds the spectra of which are described by SH with higher g-factors and lower HFS constants (B) are the trans-isomers.
Quantum chemical calculations of the electronic and geometrical structures of the complexes proved to be useful for interpreting experimental ESR data [28]. Table 4 summarizes the SH parameters describing the ESR spectra of cis- and trans-isomers of compound I in toluene. The g-factors for the two isomers are similar and weakly reflect the differences between the structures. The HFS constants proved to be more structure-sensitive. The constants giso and Acis for the cis-isomer are greater than giso and Atrans for the trans-isomer. Note also that the differences in the planar parameters B and C are lower for the cis-isomer than for the trans-isomer. In the stationary ESR spectra of copper hydroxamates, it is impossible to detect HFS caused by the interaction of the unpaired electron with the ligand atoms. However, there are known pulsed ESR data indicating additional hyperfine coupling of the unpaired electron with the ligand nitrogen atom in Cu(II) hydroxamates. According to [8], the additional hyperfine coupling constant aiso = 0.43 × 10–4 cm–1. The results of our calculations are also given in Table 4.
Thus, according to X-ray diffraction data, compound I exists in the crystalline state as a chain polynuclear structure. According to ESR data, in solution at room temperature, the compound exists as two mononuclear species, presumably cis- and trans-isomers. The assignment of the isomers was substantiated by analysis of ESR data and results of quantum chemical calculations. It was shown that HFS constants are most structure-sensitive to the geometry of isomers. Apart from the mononuclear species, dimer of the complex is detected in the frozen solution.
REFERENCES
Codd, R., Coord. Chem. Rev., 2008, vol. 252, p. 1387.
Baugman, R.G., Brink, D.J., Butle, J.M., and New, P.R., Acta Crystallogr, Sect. C: Cryst. Struct. Commun., 2000, vol. 56, p. 528.
Dzyuba, V.I., Koval, L.I., Dudko, A.V., et al., J. Coord. Chem., 2014, vol. 67, no. 8, p. 1437.
Rotov, A.V., Ugolkova, E.A., Lermontova, E.Kh., et al., Russ. J. Inorg. Chem., 2015, vol. 60, no. 7, p. 866. https://doi.org/10.1134/S0036023615070128
Rotov, A.V., Yakushev, I.A., Ugolkova, E.A., et al., Russ. J. Coord. Chem., 2021, vol. 47, no. 6, p. 376. https://doi.org/10.1134/S1070328421060051
Rotov, A.V., Ugolkova, E.A., Efimov, N.N., et al., Russ. J. Inorg. Chem., 2014, vol. 59, no. 12, p. 1474. https://doi.org/10.1134/S0036023614120201
Rotov, A.V., Ugolkova, E.A., Efimov, N.N., et al., Russ. J. Inorg. Chem, 2013, vol. 58, no. 7, p. 186. https://doi.org/10.1134/S0036023613020216
Kofman, V., Shane, J.J., Dikanov, S.A., et al., J. Am. Chem. Soc., 1995, vol. 117, p. 12771.
Dzyba, V.I., Ternovaya, T.V., Kostromina, N.A., and Ksaverov, A.N., Ukr. Khim. Zh., 1986, vol. 52, no. 5, p. 453.
APEX3, SAINT and SADABS, Madison: Bruker AXS Inc., 2016.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Adv., 2015, vol. 71, p. 3.
Sheldrick, G.M., Acta Crystallogr., Sect. C: Struct. Chem., 2015 vol. 71, p. 3.
Donomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Rakitin, Yu.V., Larin, G.M., and Minin, V.V., Interpretatsiya spektrov EPR koordinatsionnykh soedinenii (Interpretation of ESR Spectra of Coordination Compounds), Moscow: Nauka, 1993.
Wilson, R. and Kivelson, D., J. Chem. Phys., 1966, vol. 44, no. 1, p. 154.
Lebedev, Ya.S. and Muromtsev, V.I., EPR i relaksatsiya stabilizirovannykh radikalov (ESR and Relaxation of Stabilized Radicals), Moscow: Khimiya, 1972, p. 25.
Becke, A.D., Phys. Rev. A: At., Mol., Opt. Phys., 1988, vol. 38, p. 3098.
Perdew, J.P., Phys. Rev. B: Condens. Matter Mater. Phys., 1986, vol. 33, p. 8822.
Weigend, F. and Ahlrichs, R., Phys. Chem. Chem. Phys., 2005, vol. 7, p. 3297.
Gómez-Piñeiro, R.J., Pantazis, D.A., and Orio, M., Chem. Phys. Chem., 2020, vol. 21, p. 2667.
Becke, A.D., J. Chem. Phys., 1993, vol. 98, p. 5648.
Perdew, J.P. and Wang, Y., Phys. Rev., 1992, vol. 45, p. 13244.
Weigend, F., Phys. Chem. Chem. Phys., 2006, vol. 8, p. 1057.
Neese, F., Wennmohs, F., Becker, U., and Riplinger, C., J. Chem. Phys., 2020, vol. 152, p. 224108.
Cordero, B., Gómez, V., Platero-Prats, A.E., et al., Dalton Trans., 2008, p. 2832.
Alvarez, S., Dalton Trans., 2013, vol. 42, p. 8617.
Broun, D.A., McKeith, D., and Glass, W.K., Inorg. Chim. Acta, 1979, vol. 35, p. 35.
Garipov, R.R., Shtyrlin, V.G., Safin, D.A., et al., Chem. Phys., 2006, vol. 320, p. 59.
Funding
This study was performed within the state assignment for the Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, in the field of fundamental research. The chemical synthesis was carried out within the state assignment for the Institute of Problems of Chemical Physics, Russian Academy of Sciences (no. AAAA-A19-119071190045-0).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by Z. Svitanko
ADDITIONAL INFORMATION
This article is prepared for the memorial issue in tribute to the Corresponding Member of the Russian Academy of Sciences K.Yu. Zhizhin on his 50th birthday.
Rights and permissions
About this article

Cite this article
Rotov, A.V., Yakushev, I.A., Zhilov, V.I. et al. Structure of Copper(II) N-Methylbenzoylhydroxamate in the Crystalline State and in Solution. Russ J Coord Chem 49, 350–356 (2023). https://doi.org/10.1134/S1070328423700562
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328423700562