
Abstract
The interaction of palladium nanoparticles (average size, ∼3 nm) deposited on the surface of highly oriented pyrolytic graphite (HOPG) with nitrogen dioxide at room temperature and a pressure of 10–6 or 10–5 mbar was studied by X-ray photoelectron spectroscopy (XPS). It was shown that the structure of several surface graphene layers was destroyed under these conditions due to the oxidation of carbon at the interface between Pd and HOPG. The reaction proceeded with the participation of oxygen atoms, which were formed as a result of the dissociation of NO2 molecules on the palladium surface. Palladium particles retained their metallic nature, but they penetrated deep into graphite in this case.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
The activity of supported palladium-based catalysts in a particular reaction strongly depends on the oxidation state of the metal. In particular, in NOx absorption–reduction catalysts, the rate of oxidation of NO to NO2 and its further conversion into nitrates and nitrites under conditions of an oxidizing atmosphere is much higher on metallic palladium than on its oxides [1]. The reduction of NO2 by methane in the presence of oxygen [2] and the decomposition of nitrogen oxides into N2 and O2 [3, 4] also proceed more actively on metallic palladium, while palladium oxides are more active than the metal in the oxidation reactions of hydrocarbons with larger numbers of carbon atoms than that in methane [1, 4]. The reaction medium itself can affect the oxidation state of palladium and, thus, lead to a change in the activity of the catalyst [5].
Earlier, we performed a number of studies on the interaction of NO2, which has strong oxidizing properties, with platinum, gold, and silver nanoparticles deposited on highly oriented pyrolytic graphite (HOPG) at room temperature [6–10]. Prior to metal deposition, the HOPG surface was previously activated by etching with Ar+ ions under mild conditions [11, 12]. Interaction with NO2 led to the oxidation of metal particles with the formation of Pt(II), Pt(IV) [6, 8, 9], Au(III) [7], and Ag(I) oxides [10]. The smaller the particle size of a metal, the easier its oxidation occurred [6].
The formation of surface oxygen compounds on the support itself (HOPG) was also observed upon the interaction of catalytic systems with NO2 at room temperature; it is likely that these compounds were localized in the region of contact of graphite with metal particles [6, 7, 9, 10]. In this regard, a study of the interaction of NO2 with systems containing a metal on the surface of a carbon support (HOPG) can be of interest for the regeneration of carbonized catalysts and for the development of catalysts for the oxidation of soot formed in the operation of diesel engines. In the latter case, the use of nitrogen dioxide is justified by the fact that it can initially be a component of diesel exhaust gas (~5% of the total amount of nitrogen oxides [13]) or can be purposefully obtained in it [14]. In this work, using X-ray photoelectron spectroscopy (XPS), we studied the interaction of NO2 with palladium particles deposited on the activated surface of HOPG under conditions comparable to those used previously [6–10] (room temperature and a pressure of 10–6 or 10–5 mbar). The Pd/HOPG system is also of interest due to the widespread use of palladium supported on carbon for conducting selective hydrogenation [15, 16] and reductive dehalogenation reactions [17] and the Heck reaction [15, 18], as well as for electrochemical oxidation processes in fuel cells [19–22].
EXPERIMENTAL
A SPECS spectrometer (Germany) equipped with a 9-channel detector and a PHOIBOS-150 analyzer was used to measure the XPS spectra. Photoemission was excited by nonmonochromatized AlKα radiation with the quantum energy hν = 1486.6 eV. Before measurements, the spectrometer was calibrated using Au 4f7/2 and Cu 2p3/2 lines whose binding energies (Eb) for the foils of the corresponding metals are 84.0 and 932.7 eV, respectively. With consideration for the good conductivity of graphite, the measurements of Eb were carried out without correction for the possible charging of the samples. In the initial HOPG sample, the binding energy of the C 1s line was 284.4 eV, which is in accordance with published data [23–30]. The spectra were measured at two photoelectron collection angles of 0° and 45° relative to a normal to the surface. An analysis of spectral lines with decomposition into separate components was performed using the PeakXPS software [31]. The background line was specified by the Shirley function.
The Pd/HOPG samples were obtained in the preparation chamber of the XPS spectrometer by vacuum deposition of palladium metal on HOPG surface preliminarily activated by etching with argon ions (kinetic energy, 0.5 keV; ion current, 50 μA) for 10 s according to a procedure similar to that used previously for M/HOPG sample preparation (M = Pt, Au, and Ag) [6, 7, 9, 10, 12]. Figure 1a shows an image of a Pd/HOPG sample obtained using a GPI-300.02 vacuum scanning tunneling microscope (NPF Sigma Skan, Russia) using a platinum needle as a probe at a bias voltage of 1 V and a tunneling current of 0.3 nA. It can be seen that the surface of HOPG was uniformly covered with rounded palladium particles. From the histogram shown in Fig. 1b, it follows that the particle size varied in a range from 1 to 6 nm, and the average value was 3 nm. For comparison, in Pd/HOPG samples prepared by Kettner et al. [32] with the use of a similar technique of vacuum deposition on the activated surface of HOPG, the metal particles had a lateral size of 5.7–6.8 nm and a height of 1.6–1.8 nm. On the surface of HOPG activated by etching with nitrogen ions (N+ + \({\text{N}}_{2}^{ + }\)), palladium particles of size ~1.5–2 nm were obtained [33].

(a) STM image 200 × 200 nm and (b) size distribution of palladium nanoparticles on the surface of HOPG.
A source the operating principle of which is based on the thermal decomposition of lead nitrate in a vacuum [6] was used for the treatment of Pd/HOPG samples with nitrogen dioxide. The treatment was carried out in the preparation chamber of the XPS spectrometer at room temperature and a pressure of 10–6 or 10–5 mbar.
RESULTS
Figure 2 shows the Pd 3d and Pd MNN spectra of the Pd/HOPG sample before and after interaction with NO2 at room temperature and a pressure of (2) 10–6 or (3) 10–5 mbar for 30 min. The spectra were measured at a photoelectron collection angle of 0° relative to a normal to the surface. In the initial sample, the binding energy Eb(Pd 3d5/2) was (1) 335.6 eV, which corresponds to metallic palladium in the deposited Pd particles of small size [2, 33–35]. Spectra 2 and 3 indicate that Eb(Pd 3d5/2) and the shapes of the Pd 3d photoemission line and the Pd MNN Auger line remained unchanged after sample treatment in NO2. At the same time, a decrease in the intensity of both lines was observed, which was especially noticeable after interaction at 10–5 mbar; the effect manifested itself more significantly in the Auger line. The reason for the decrease in the absolute intensity values of the 3d and MNN lines of palladium can be (1) the agglomeration of Pd particles, (2) the formation of a film of foreign material on their surface, or (3) the penetration of particles deep into the support. In the first case, the decrease in intensity was due to a decrease in the number of palladium atoms in the zone of XPS analysis with increasing particle size [36, 37]. In the other two cases, the attenuation of a palladium signal was caused by the screening of photoemission by a layer of carbon material.

(a) The Pd 3d XPS spectra and (b) the Pd MNN Auger spectra of the Pd/HOPG sample (1) before and after interaction with NO2 at room temperature and a pressure of (2) 10–6 and (3) 10–5 mbar for 30 min.
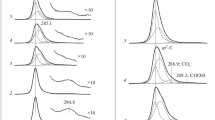
Figure 3 shows the C 1s spectra of (1) the initial HOPG, (2) HOPG after bombardment with argon ions, and (3) HOPG after evaporation of palladium onto it followed by heating at 300°C. Figure 3 also shows the spectra obtained after the interaction of the Pd/HOPG sample with NO2 at room temperature and a pressure of (4) 10–6 or (5) 10–5 mbar. The spectra were measured at a photoelectron collection angle of 0° relative to a normal to the surface. Figure 3a shows spectra recorded in a wider range of binding energies, which captures a surface plasmon characteristic of graphite at ~291 eV along with the C 1s line [24, 25, 27, 30, 33]. As can be seen in Fig. 3a, the plasmon intensity remained at the initial level after (2) the etching of the initial HOPG with Ar+ ions and (3) the subsequent deposition of palladium. This result contradicts the data of a study by Favaro et al. [33], where etching with nitrogen ions led to the complete disappearance of the plasmon from the C 1s spectrum, which was probably due to more stringent etching conditions. In our case, the plasmon peak intensity decreased (4) only after interaction with NO2. Its almost complete disappearance (5) after interaction with NO2 at 10–5 mbar indicated the amorphization of a surface layer of graphite caused by the violation of long-range order in the conjugated system of π‑bonded sp2-hybrid carbon atoms [38, 39].

The C 1s XPS spectra recorded for HOPG (1) in the initial state, after (2) bombardment with argon ions, (3) palladium deposition followed by heating at 300°C, and after the interaction of the resulting Pd/HOPG sample with NO2 at room temperature and a pressure of (4) 10–6 or (5) 10–5 mbar for 30 min: (a) spectra in a wide range of binding energies, including the region of surface plasmon detection (~291 eV); (b) spectra in a narrow range of binding energies with decomposition of the C 1s photoemission line into components.
The state of carbon with the sp2 hybridization manifested itself in the spectrum of the initial HOPG as a narrow asymmetric peak characteristic of graphite with a binding energy of 284.4 eV (Fig. 3a, curve 1), [23–25, 30, 40]. After ion etching and deposition of metallic palladium (Fig. 3a, curves 2 and 3), the position of the peak and its asymmetry were retained. The interaction of Pd/HOPG with NO2 (Fig. 3a, curves 4 and 5) led to a shift of the C 1s line toward higher binding energies and its broadening, which indicated the appearance of new states of carbon.
Figure 3b, where the spectra are presented in a narrower range of binding energies, shows in detail changes in the C 1s photoemission line during the interaction of the Pd/HOPG sample with NO2. Based on published data for graphite (HOPG), graphene, and its derivatives (mainly, for graphene oxide), spectra 4 and 5, which were obtained after the interaction of Pd/HOPG with NO2, were decomposed into components corresponding to sp2 carbon in the structure of graphite [23–30], sp3 carbon not bound to oxygen atoms (C–C, C–H bonds) [25, 27, 30, 41], and carbon atoms bound to oxygen by ordinary (C–OH, C–O–C) and double (C=O) bonds [26, 41–44]. In the decomposition of spectra with the use of the PeakXPS software, it was assumed that the width and asymmetry parameters of a component corresponding to the sp2 carbon are the same as those in the spectrum of the initial HOPG. The remaining components, including those of sp3 carbon, were characterized by symmetrical lines [45]. The spectra given in Fig. 3b indicate that the interaction with NO2 led to a significant decrease in the intensity of the sp2-carbon peak with a simultaneous increase in the intensity of peaks due to other components. After carrying out the reaction at a pressure of 10–6 mbar, spectrum 4 exhibited a peak with a binding energy of 285.1 eV belonging to the sp3 carbon not bound to oxygen and a peak at 286.2 eV from surface compounds with ordinary C–O bonds. The peak intensity at 286.2 eV increased after the reaction performed at 10–5 mbar (5) due to a further decrease in the peak intensity of the sp2 carbon, and an additional peak with a binding energy of 287.0 eV appeared, which can be attributed to carbonyl compounds with a double C=O bond.
To recognize surface components in the XPS spectrum, we compared the intensities of individual lines in the C 1s spectra obtained at different photoelectron exit angles. Figure 4 shows the C 1s spectra of a Pd/HOPG sample treated in NO2 at 10–5 mbar with photoelectron detection at angles of (1) 0° and (2) 45°. It can be seen that the contribution of a peak due to the sp2 carbon to the C 1s line intensity decreased (from 44 to 31%) with the angle of detection, whereas the contributions from the sp3 carbon and COx surface compounds simultaneously increased (from 44 to 53 and from 12 to 16%, respectively). Thus, the processes leading to the structure degradation of graphene layers and the formation of COx compounds mainly occurred on the surface. From a ratio between sp2-carbon signal intensities before and after interaction with NO2 and taking into account the mean free path of photoelectrons emitted from the C 1s level under the action of AlKα radiation (kinetic energy, ~1202 eV) in carbon materials λ = 3.31 nm [46], we estimated the fractured layer thickness at ~3–5 nm, which corresponds to ~10–15 graphene layers in the graphite structure.

The C 1s spectra of the Pd/HOPG sample after interaction with NO2 at room temperature and a pressure of 10–5 mbar with the detection of photoelectrons at angles of (1) 0° and (2) 45°.
Figure 5 shows the O 1s (Fig. 5a) and N 1s (Fig. 5b) spectra recorded for (1) HOPG and Pd/HOPG (2) before and after treatment in NO2 at (3) 10–6 and (4) 10–5 mbar. The lines of oxygen and nitrogen were absent from spectra 1 of the initial HOPG. In the region characteristic of O 1s, the Pd 3p3/2 line with a binding energy of 532.8–532.9 eV was detected after the deposition of palladium (2). After the interaction with NO2, the O 1s line with a binding energy of ~533.5 eV (Fig. 5a, curves 3 and 4) additionally appeared in this region, and a line with a binding energy of 400.4 eV appeared in the N 1s region (Fig. 5b, curves 3 and 4). The O 1s line with Еb = 533.5 eV corresponds to oxygen atoms bonded to carbon atoms by ordinary bonds in C–OH and C–O–C fragments [47–49]. The N 1s line with a binding energy of 400.4 eV corresponds to nitrogen atoms bonded to the sp2 carbon with the formation of fragments with a pyrrole structure [33, 40, 41, 50].

The (a) O 1s and (b) N 1s spectra of (1) HOPG and Pd/HOPG (2) before and after treatment in NO2 at room temperature and a pressure of (3) 10–6 or (4) 10–5 mbar.
DISCUSSION
Unlike platinum, silver, and gold nanoparticles deposited onto an HOPG surface activated by ion etching [6–10], the interaction of NO2 at room temperature with the Pd/HOPG system did not lead to the oxidation of palladium particles. At the same time, the intensity of lines belonging to palladium significantly decreased (Fig. 2), and the agglomeration of Pd particles can be a reason for this decrease. From the spectra shown in Fig. 2, it follows that the signal from the Pd MNN Auger electrons with kinetic energy Ekin ≈ 330 eV was attenuated to a much greater extent than the Pd 3d signal with Ekin ≈ 1150 eV. Thus, a ratio of the Auger line intensity to the photoemission line intensity in the initial Pd/HOPG sample was \({{{{I}_{{MNN}}}} \mathord{\left/ {\vphantom {{{{I}_{{MNN}}}} {{{I}_{{3d}}}}}} \right. \kern-0em} {{{I}_{{3d}}}}}\) = 1.71, and it decreased to 1.05 or 0.44 after treatment in NO2 at 10–6 or 10–5 mbar, respectively. Changes in the intensities of metal lines with very different Ekin with increasing the particle size of this metal on the support surface were analyzed using the Au/HOPG system as an example [51]. The intensity ratio between the Au 4f and Au 3d3/2 lines recorded using AgLα radiation and having Ekin of ~2900 and ~690 eV, respectively, was considered. It was shown that the intensity ratio \({{{{I}_{{{\text{Au}}3d}}}} \mathord{\left/ {\vphantom {{{{I}_{{{\text{Au}}3d}}}} {{{I}_{{{\text{Au}}4f}}}}}} \right. \kern-0em} {{{I}_{{{\text{Au}}4f}}}}}\) for nanometer-size particles was higher than that for bulk gold, but it asymptotically tended to that for bulk gold with increasing particle size.
We return to the Pd/HOPG system. The mean free path of electrons emitted from the Pd 3d level in metallic palladium is λ3d ≈ 1.55 nm, and that of Pd MNN Auger electrons is λMNN ≈ 0.66 nm [46]. Because λ3d > λMNN, the intensity ratio \({{{{I}_{{MNN}}}} \mathord{\left/ {\vphantom {{{{I}_{{MNN}}}} {{{I}_{{3d}}}}}} \right. \kern-0em} {{{I}_{{3d}}}}}\) for palladium particles several nanometers in size should be greater than the ratio \({{I_{{MNN}}^{0}} \mathord{\left/ {\vphantom {{I_{{MNN}}^{0}} {I_{{3d}}^{0}}}} \right. \kern-0em} {I_{{3d}}^{0}}}\) = 1.50 for palladium foil, which is consistent with the results of measurements performed in the Pd/HOPG sample before the interaction with NO2 (\({{{{I}_{{MNN}}}} \mathord{\left/ {\vphantom {{{{I}_{{MNN}}}} {{{I}_{{3d}}}}}} \right. \kern-0em} {{{I}_{{3d}}}}}\) = 1.71). If we assume that the size of Pd particles increases in the course of interaction with NO2, in accordance with an analysis performed previously [51], the ratio \({{{{I}_{{MNN}}}} \mathord{\left/ {\vphantom {{{{I}_{{MNN}}}} {{{I}_{{3d}}}}}} \right. \kern-0em} {{{I}_{{3d}}}}}\) should decrease with an asymptotic tendency to 1.50. Indeed, the ratio \({{{{I}_{{MNN}}}} \mathord{\left/ {\vphantom {{{{I}_{{MNN}}}} {{{I}_{{3d}}}}}} \right. \kern-0em} {{{I}_{{3d}}}}}\) decreased but to a substantially smaller value. Hence, it follows that the agglomeration of Pd particles, if it occurs, cannot be the only cause of the observed changes in the intensities of palladium lines in the XPS spectrum. The decrease in the intensities of the Pd 3d and Pd MNN lines with a simultaneous decrease in the intensity ratio \({{{{I}_{{MNN}}}} \mathord{\left/ {\vphantom {{{{I}_{{MNN}}}} {{{I}_{{3d}}}}}} \right. \kern-0em} {{{I}_{{3d}}}}}\) of these lines to a value lower than that of the foil can be related to the encapsulation of palladium particles due to the formation of a film from the fragments of support material on their surface or to the penetration of particles deeper into the support. In this case, screening by an outer layer of carbon material weakens both lines of palladium, but this concerns to a greater extent the Auger line due to a lower kinetic energy (and, therefore, λ) of the Auger electrons.
Structural changes in the samples of metal nanoparticles deposited onto the HOPG surface under the influence of an oxidizing atmosphere were studied [12, 52, 53]. The scanning tunneling microscopy (STM) method was used to study the morphology of the activated HOPG surface after treatment in air at a temperature of about 460°C in the presence of supported palladium particles [52]. It was shown that, in this case, graphite was oxidized with the participation of atmospheric oxygen as an oxidizing agent and palladium as a catalyst. As carbon burned out on an area of the surface, a palladium particle moved to an adjacent area; as a result of this, a track was formed in the surface layer of the support. Under these conditions, HOPG did not react with air in the absence of palladium. Similar results were obtained by Hugentobler et al. [53], who studied the oxidation of graphene layers in the HOPG structure catalyzed by supported gold particles. Demidov et al. [12] examined the oxidation of the HOPG surface in the course of its interaction with a mixture of ethylene and oxygen (1: 1) at a pressure of 0.25 mbar and a temperature of ≥250°C with the participation of particles of deposited silver using scanning electron microscopy (SEM). As in the case of palladium studied by Yuan et al. [52], the reaction was accompanied by the formation of tracks in burnt carbon places. It is likely that something similar was observed in this work, where the use of NO2 as a stronger oxidizing agent than oxygen allowed us to oxidize graphite even at room temperature. This assumption is based on the fact that the graphene structure of the surface HOPG layer was destroyed (Fig. 3), as a result of which metallic palladium particles were immersed in it. This is evident from a decrease in the ratio \({{{{I}_{{MNN}}}} \mathord{\left/ {\vphantom {{{{I}_{{MNN}}}} {{{I}_{{3d}}}}}} \right. \kern-0em} {{{I}_{{3d}}}}}\) between the intensities of palladium lines to an anomalously low value.
It was interesting to compare the behaviors of the Pd/HOPG system considered here and the Pt/HOPG system studied earlier [6, 8, 9]. Upon the treatment of the Pd/HOPG system in NO2 at room temperature, the palladium particles retained their metallic nature and graphite was intensely oxidized. On the contrary, the oxidation of platinum particles occurred in the Pt/HOPG system under similar conditions [6, 8, 9], whereas the structure of graphite remained unchanged. This was evidenced by the STM data [9] and the retention of the initial shape of the C 1s spectrum characteristic of graphite [6, 8, 9].
Apparently, the decisive role in the observed differences in the behaviors of the two systems was played by different interactions of oxygen atoms formed upon the dissociation of NO2 molecules with the surfaces of Pt and Pd particles. Massive palladium can absorb oxygen atoms in the course of treatment with oxygen or nitrogen oxides [54–59]. The interaction of Pd(111), (110), and (100) single crystals with O2 at temperatures of ≥150°C led to the appearance of a subsurface oxygen state [60–63]. It was shown [61, 62] that so-called subsurface oxygen atoms penetrate into Pd(110) at a depth of >1.5–2.0 nm. Higher temperatures (>300–350°C) are required for oxygen dissolution in the bulk of the metal [60, 64]. As the oxygen concentration in the bulk increased to a critical value, palladium oxide PdO was formed [64]. The necessity of using elevated temperatures is due to the fact that, on single crystals, the penetration of oxygen atoms into the subsurface region with the subsequent dissolution in the bulk at room temperature proceeds too slowly because of low concentrations of defects on the surface [60]. Both the formation of subsurface oxygen [63] and the dissolution of O atoms in the bulk [65] proceed more efficiently in polycrystalline palladium. It would be expected that oxygen absorption will occur even more easily on interaction with palladium nanoparticles due to a high-defect structure of their surface [5]. Not only the defect structure of the palladium surface, which is necessary for the transport of O atoms deep into the metal [63], but also the concentration of adsorbed oxygen play an important role in successful processes leading to the formation of subsurface oxygen and surface oxide and the dissolution of O atoms in the bulk with their further conversion into bulk oxide. In the case of molecular oxygen, the necessary concentration of adsorbed oxygen is provided by interactions at elevated temperatures and pressures; however, with the use of NO2 as a source of oxygen atoms, high O concentrations on the palladium surface can be achieved at room temperature and moderate pressures (such as those used in this work) [55, 57].
It is likely that the ability of palladium to readily dissolve oxygen is combined with the high mobility of the dissolved atoms in the bulk of a metal particle to provide an efficient approach to the carrier boundary, including that located under the metal particle. We can also assume that the dissolved oxygen atoms have a high oxidizing ability sufficient to oxidize graphite, at least at the sites where palladium particles are localized on defects created by ion etching. The efficient consumption of dissolved atoms for the oxidation of carbon limits the growth of their concentration inside palladium particles and, as a result, complicates the formation of an oxide phase.
Previously, we found that a state of atomic oxygen (according to XPS data, it differed from an oxide state) was formed at the initial stage of the interaction of platinum nanoparticles, which were obtained by vacuum evaporation onto the surface of oxides (Al2O3, SiO2, TiO2, and ZrO2), with NO2 [66, 67] or with a mixture of NO and O2 (1 : 1) at room temperature [68–71]. Oxygen atoms in this state have a higher oxidative activity than that of platinum oxide particles, and it manifested itself in the possibility of their reduction with hydrogen at room temperature [66, 69–71]. It was hypothesized that this state corresponds to oxygen atoms dissolved in platinum particles or, at least, embedded in the subsurface layer, and their appearance is preceded by the formation of platinum oxide particles. However, it was found that, in the case of platinum supported on HOPG, the state of dissolved (subsurface) oxygen did not form, and the surface carbon compounds with oxygen COx appeared instead; simultaneously, platinum metal particles were converted into the particles of Pt(II) and Pt(IV) oxides [6, 8, 9]. Based on the fact that, according to the XPS data [6, 9], the state of carbon after the treatment of Pt/HOPG in NO2 and, according to the STM data [9], the structure of graphite remained almost unchanged, it should be recognized that the reaction of active oxygen atoms with HOPG proceeded only to an insignificant degree and, apparently, only along the perimeter of supported platinum particles. It is likely that the oxygen atoms capable of reacting with carbon at room temperature in the Pt/HOPG system do not have an equally high mobility as that in palladium. They do not penetrate deep into the volume of platinum particles, but they diffuse in the subsurface region to reach a boundary with graphite, where they react to form surface COx compounds. In the case of the penetration of oxygen atoms into the volume of particles, low-activity platinum oxides are formed. We also did not detect the state of dissolved oxygen upon the interaction of NO2 with massive platinum (foil) [72].
CONCLUSIONS
The following results were obtained in an XPS study of the interaction of NO2 with palladium nanoparticles deposited onto the surface of highly oriented pyrolytic graphite (HOPG) at room temperature and a pressure of 10–6 or 10–5 mbar:
(1) Significant changes in the C 1s, O 1s, and N 1s spectra indicated the occurrence of graphite oxidation, which led to the destruction of the structure to 10–15 graphene layers and the appearance of surface oxygen compounds containing C–OH, C–O–C, and C=O groups and nitrogen compounds as fragments with a pyrrole structure.
(2) Under these conditions, palladium retained its metallic nature. Palladium particles were penetrated into the subsurface layer of graphite, as indicated by a decrease in the ratio of the intensity of the Pd MNN Auger line to the intensity of the Pd 3d photoemission line with a general weakening of the palladium lines in the XPS spectrum.
The behavior of palladium strongly distinguished this metal from platinum, gold, and silver, which were studied earlier in the form of particles supported onto HOPG. The deep oxidation of graphite was not observed upon the interaction of NO2 with M/HOPG (M = Pt, Au, or Ag) under similar conditions, and the metals were converted into an oxidized state. The Pd/HOPG system should be additionally studied in order to determine the detailed mechanism of carbon oxidation in the presence of palladium particles. Nevertheless, the data obtained in the study of the Pd/HOPG system allow us to propose now the use of palladium catalysts for the low-temperature oxidation of soot by NO2 molecules in the processes of neutralizing exhaust gases from diesel engines.
REFERENCES
Mirkelamoglu, B., Liu, M., and Ozkan, U.S., Catal. Today, 2010, vol. 151, p. 386.
Oktar, N., Mitome, J., Holmgreen, E.M., and Ozkan, U.S., J. Mol. Catal. A: Chem., 2006, vol. 259, p. 171.
Lin, S., Yang, L., Yang, X., and Zhou, R., Appl. Surf. Sci., 2014, vol. 305, p. 642.
Lin, S., Yang, X., Yang, L., and Zhou, R., RCS Adv., 2015, vol. 5, p. 37353.
Desikusumastuti, A., Happel, M., Qin, Z., Staudt, T., Lykhach, Y., Laurin, M., Shaikhutdinov, S., Rohr, F., and Libuda., J., J. Phys. Chem. C, 2009, vol. 113, p. 9755.
Kalinkin, A.V., Sorokin, A.M., Smirnov, M.Yu., and Bukhtiyarov, V.I., Kinet. Catal., 2014, vol. 55, p. 354.
Kalinkin, A.V., Smirnov, M.Yu., Bukhtiyarov, A.V., and Bukhtiyarov, V.I., Kinet. Catal., 2015, vol. 56, p. 796.
Smirnov, M.Yu., Kalinkin, A.V., Vovk, E.I., Simonov, P.A., Gerasimov, E.Yu., Sorokin, A.M., and Bukhtiyarov, V.I., Appl. Surf. Sci., 2018, vol. 428, p. 972.
Smirnov, M.Yu., Vovk, E.I., Nartova, A.V., Kalinkin, A.V., and Bukhtiyarov, V.I., Kinet. Catal., 2018, vol. 59, p. 653.
Kalinkin, A.V., Smirnov, M.Yu., Klembovskii, I.O., Sorokin, A.M., Gladky, A.Yu., and Bukhtiyarov, V.I., J. Struct. Chem., 2018, vol. 59, p. 1726.
Palmer, R.E., Pratontep, S., and Boyen, H.-G., Nat. Mater., 2003, vol. 2, p. 443.
Demidov, D.V., Prosvirin, I.P., Sorokin, A.M., and Bukhtiyarov, V.I., Catal. Sci. Technol., 2011, vol. 1, p. 1432.
Zheng, Y., Kovarik, L., Engelhard, M.H., Wang, Y., Wang, Y., Gao, F., and Szanyi, J., J. Phys. Chem. C., 2017, vol. 121, p. 15793.
Boutikos, P., Březina, J., Arvajova, A., and Koči, P., Chem. Eng. J., 2019, vol. 377, p. 119654.
Yoon, B. and Wai, C.M., J. Am. Chem. Soc., 2005, vol. 127, p. 17174.
Mori, A., Miyakawa, Y., Ohashi, E., Haga, T., Maegawa, T., and Sajiki, H., Org. Lett., 2006, vol. 8, p. 3279.
Chen, L., Yang, K., Liu, H., and Wang, X., Carbon, 2008, vol. 46, p. 2137.
Yuan, Z., Stephan, R., Hanf, M.C., Becht, J.M., Le Drian, C., Hugentobler, M., Harbich, W., and Wetzel, P., Eur. Phys. J. D, 2011, vol. 63, p. 401.
Li, F., Zhang, B., Wang, E., and Dong, S., J. Electroanal. Chem., 1997, vol. 422, p. 27.
Gao, G.-Y., Guo, D.-J., and Li, H.-L., J. Power Sources, 2006, vol. 162, p. 1094.
Mazumder, V. and Sun, S., J. Am. Chem. Soc., 2009, vol. 131, p. 4588.
Chen, L., Hu, G., Zou, G., Shao, S., and Wang, X., Electrochem. Commun., 2009, vol. 11, p. 504.
Schnyder, B., Alliata, D., Kotz, R., and Siegenthaler, H., Appl. Surf. Sci., 2001, vol. 173, p. 221.
Yang, D.-Q. and Sacher, E., Surf. Sci., 2002, vol. 504, p. 125.
Rousseau, B., Estrade-Szwarckopf, H., Thomann, A.-L., and Brault, P., Appl. Phys. A, 2003, vol. 77, p. 591.
Utsumi, S., Honda, H., Hattori, Y., Kanoh, H., Takahashi, K., Sakai, H., Abe, M., Yudasaka, M., Iijima, S., and Kaneko, K., J. Phys. Chem. C, 2007, vol. 111, p. 5572.
Paredes, J.I., Martınez-Alonso, A., and Tascon, J.M.D., Langmuir, 2007, vol. 23, p. 8932.
Demoisson, F., Raes, M., Terryn, H., Guillot, J., Migeon, H.-N., and Reniers, F., Surf. Interface Anal., 2008, vol. 40, p. 566.
Sandhu, J., Chauhan, A.K.S., and Govind, J. Nanopart. Res., 2011, vol. 13, p. 3503.
Blume, R., Rosenthal, D., Tessonnier, J.-P., Li, H., Knop-Gericke, A., and Schlogl, R., ChemCatChem, 2015, vol. 7, p. 2871.
http://xpspeak.software.informer.com/4.1/.
Kettner, M., Stumm, C., Schwarz, M., Schuschke, C., and Libuda, J., Surf. Sci., 2019, vol. 679, p. 64.
Favaro, M., Agnoli, S., Perini, L., Durante, C., Gennaro, A., and Granozzi, G., Phys. Chem. Chem. Phys., 2013, vol. 15, p. 2923.
Nosova, L.V., Stenin, M.V., Nogin, Yu.N., and Ryndin, Yu.A., Appl. Surf. Sci., 1992, vol. 55, p. 43.
Aiyer, H.N., Vijayakrishnan, V., Subbanna, G.N., and Rao, C.N.R., Surf. Sci., 1994, vol. 313, p. 392.
Pan, Y., Gao, Y., Kong, D., Wang, G., Hou, J., Hu, S., Pan, H., and Zhu, J., Langmuir, 2012, vol. 28, p. 6045.
Smirnov, M.Yu., Vovk, E.I., Kalinkin, A.V., and Bukhtiyarov, V.I., Kinet. Catal., 2016, vol. 57, p. 831.
Afanas’ev, V.P., Bocharov, G.S., Eletskii, A.V., Ridzel, O.Yu., Kaplya, P.S., and Koppen, M., J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas.,Phenom., 2017, vol. 35, p. 041804.
Zhu, C., Hao, X., Liu, Y., Wu, Y., and Wang, J., Appl. Surf. Sci., 2018, vol. 427, p. 1137.
Susi, T., Pichler, T., and Ayala, P., Beilstein J. Nanotechnol., 2015, vol. 6, p. 177.
Figueiredo, J.L. and Pereira, M.F.R., Catal. Today, 2010, vol. 150, p. 2.
Wang, Z.-M., Kanoh, H., Kaneko, K., Lu, G.Q., and Do, D., Carbon, 2002, vol. 40, p. 1231.
Fu, C., Zhao, G., Zhang, H., and Li, S., Int. J. Electrochem. Sci., 2013, vol. 8, p. 6269.
Saravanan, M., Girisun, T.C.S., and Rao, S.V., J. Mater. Chem. C, 2017, vol. 5, p. 9929.
Haerle, R., Riedo, E., Pasquarello, A., and Baldereschi, A., Phys. Rev. B, 2001, vol. 65, p. 045101.
Shinotsuka, H., Tanuma, S., Powell, C.J., and Penn, D.R., Surf. Interface Anal., 2015, vol. 47, p. 871.
Ganguly, A., Sharma, S., Papakonstantinou, P., and Hamilton, J., J. Phys. Chem. C., 2011, vol. 115, p. 17009.
Oh, Y.J., Yoo, J.J., Kim, Y.I., Yoon, J.K., Yoon, H.N., Kim, J.-H., and Park, S.B., Electrochim. Acta, 2014, vol. 116, p. 118.
Stobinski, L., Lesiak, B., Malolepszy, A., Mazurkiewicz, M., Mierzwa, B., Zemek, J., Jiricek, P., and Bieloshapka, I., J. Electron Spectrosc. Relat. Phenom., 2014, vol. 195, p. 145.
Biniak, S., Szymanski, G., Siedlewski, J., and Swiatkowski, A., Carbon, 1997, vol. 35, p. 1799.
Smirnov, M.Yu., Kalinkin, A.V., Bukhtiyarov, A.V., Prosvirin, I.P., and Bukhtiyarov, V.I., J. Phys. Chem. C, 2016, vol. 120, p. 10419.
Yuan, Z., Hanf, M.C., Stephan, R., Dulot, F., Denys, E., Florentin, A., Harbich, W., and Wetzel, P., Surf. Interface Anal., 2015, vol. 47, p. 82.
Hugentobler, M., Bonanni, S., Sautier, A., and Harbich, W., Eur. Phys. J. D, 2011, vol. 63, p. 215.
Conrad, H., Ertl, G., Kuppers, J., and Latta, E., Surf. Sci., 1977, vol. 65, p. 245.
Parker, D.H. and Koel, B.E., J. Vac. Sci. Technol., A, 1990, vol. 8, p. 2585.
Banse, B.A. and Koel, B.E., Surf. Sci., 1990, vol. 232, p. 275.
Zheng, G. and Altman, E.I., Surf. Sci., 2000, vol. 462, p. 151.
Zemlyanov, D., Azalos-Kiss, B., Kleimenov, E., Teschner, D., Zafeiratos, S., Havecker, M., Knop-Gericke, A., Schlogl, R., Gabasch, H., Unterberger, W., Hayek, K., and Klotzer, B., Surf. Sci., 2006, vol. 600, p. 983.
Gabasch, H., Unterberger, W., Hayek, K., Klotzer, B., Kleimenov, E., Teschner, D., Zafeiratos, S., Havecker, M., Knop-Gericke, A., Schlogl, R., Han, J., Ribeiro, F.H., Aszalos-Kiss, B., Curtin, T., and Zemlyanov, D., Surf. Sci., 2006, vol. 600, p. 2980.
Leisenberger, F.P., Koller, G., Sock, M., Surnev, S., Ramsey, M.G., Netzer, F.P., Klotzer, B., and Hayek, K., Surf. Sci., 2000, vol. 445, p. 380.
Titkov, A.I., Salanov, A.N., Koscheev, S.V., and Boronin, A.I., React. Kinet. Catal. Lett., 2005, vol. 86, p. 371.
Titkov, A.I., Salanov, A.N., Koscheev, S.V., and Boronin, A.I., Surf. Sci., 2006, vol. 600, p. 4119.
Nagarajan, S., Thirunavukkarasu, K., and Gopinath, C.S., J. Phys. Chem. C, 2009, vol. 113, p. 7385.
Han, J., Zemlyanov, D.Y., and Ribeiro, F.H., Surf. Sci., 2006, vol. 600, p. 2752.
Suprun, E.A. and Salanov, A.N., Kinet. Catal., 2017, vol. 58, p. 92.
Vovk, E.I., Kalinkin, A.V., Smirnov, M.Yu., Klembovskii, I.O., Bukhtiyarov, V.I., J. Phys. Chem. C., 2017, vol. 121, p. 17297.
Smirnov, M.Yu., Kalinkin, A.V., Nazimov, D.A., Toktarev, A.V., and Bukhtiyarov, V.I., Kinet. Catal., 2015, vol. 56, p. 540.
Smirnov, M.Yu., Kalinkin, A.V., and Bukhtiyarov, V.I., J. Struct. Chem., 2007, vol. 48, p. 1053.
Smirnov, M.Yu., Vovk, E.I., Kalinkin, A.V., Pashis, A.V., and Bukhtiyarov, V.I., Kinet. Catal., 2012, vol. 53, p. 117.
Smirnov, M.Yu., Kalinkin, A.V., Vovk, E.I., and Bukhtiyarov, V.I., Kinet. Catal., 2015, vol. 56, p. 801.
Smirnov, M.Yu., Kalinkin, A.V., Vovk, E.I., and Bukhtiyarov, V.I., J. Struct. Chem. 2016, vol. 57, p. 1127.
Kalinkin, A.V., Smirnov, M.Yu., and Bukhtiyarov, V.I., Kinet. Catal., 2016, vol. 57, p. 826.
ACKNOWLEDGMENTS
The studies are conducted using the equipment of the Center of collective use “National Center of Catalyst Research”.
Funding
This work was carried out at the Boreskov Institute of Catalysis, Siberian Branch, Russian Academy of Sciences within the framework of a state contract (project no. AAAA-A17-117041710078-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors have no conflicts of interest to disclose in this article.
Additional information
Translated by Valentin Makhlyarchuk
Abbreviations and designations: HOPG, highly oriented pyrolytic graphite; XPS, X-ray photoelectron spectroscopy; STM, scanning tunneling microscopy; SEM, scanning electron microscopy; Eb, binding energy; Ekin, kinetic energy of photoelectrons; I, line intensities in the XPS spectrum; λ, mean free path of photoelectrons.
Rights and permissions
About this article

Cite this article
Smirnov, M.Y., Kalinkin, A.V., Sorokin, A.M. et al. Room Temperature Oxidation of the Surface of Highly Oriented Pyrolytic Graphite (HOPG) with Nitrogen Dioxide in the Presence of Supported Palladium Particles. Kinet Catal 61, 637–645 (2020). https://doi.org/10.1134/S0023158420040126
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0023158420040126