
Abstract
A series of carbon-mineral composites with the carbon loading varying from 1.5 to 14.2 wt % has been synthesized by MgO carbonization in 1,3-butadiene at 600°C. The synthesized carbon-mineral composites have been studied by ESR, X-ray powder diffraction, and transmission electron microscopy. ESR has demonstrated that the MgO surface is completely covered with carbon after depositing 8–10 wt % C. MgO from the composite was dissolved by treatment with hydrochloric acid. The surface area of the carbon samples obtained after the acid treatment has been studied by thermal desorption of argon. It has been shown that the synthesized carbon material consists of a few graphene layers. The specific surface area of the synthesized graphene passes through a maximum of about 1800–1900 m2/g for samples obtained from C–MgO composites containing 8–10 wt % C.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Graphene is a two-dimensional hexagonal lattice composed of carbon atoms. Since this material is the basis for constructing a three-dimensional model of a graphite crystal, theoretical studies of graphene began long before its synthesis. In early works [1, 2], it has been argued that strictly two-dimensional (2D) carbon crystals are thermodynamically unstable and cannot exist. In 1962, a speculative 2D model of a carbon sheet was called graphene [3]. It has later been shown that the thermodynamic stability of real graphene is achieved due to thermal fluctuations that create a “ripple,” i.e., the waviness of a 2D surface [4].
In 2010, Andre Geim and Konstantin Novoselov were awarded the Nobel Prize in Physics for “groundbreaking experiments regarding the two-dimensional material graphene.” Despite the fact that the first experimental graphene samples were obtained relatively recently [5], there are already many studies on the use of graphene in various fields, and the number of publications devoted to graphene is growing exponentially. Graphene is a new carbon material with unique physical properties [4]. Various methods for producing graphene are known.
Exfoliation is the simplest way to produce graphene [6]. In this case, graphene is obtained by reducing the number of monolayers in graphite. The so-called Scotch tape method was the first method for producing graphene and was developed by K. Novoselov and A. Geim. In this method, Scotch tape is repeatedly used to separate graphite crystals into thinner layers [5]. However, graphene synthesized by the Scotch method cannot be scaled and limited by a size of a few micrometers (width of about 100 μm) [6]. Another method of exfoliation is the ultrasonic treatment of graphite. In particular, graphene was obtained by processing a dispersion of graphite powder in an ultrasonic bath [7]. After sonication, a gray liquid was obtained consisting of a homogeneous phase and a large number of macroscopic aggregates. These aggregates were then removed by centrifugation to obtain a homogeneous dark dispersion.
Reduction methods for producing graphene include pyrolysis of sodium ethylate and graphite oxide reduction [8]. Graphite oxide, when heated quickly, produces graphene flakes. The reduction of monolayer graphite oxide plates can be carried out chemically using hydrazine or thermally upon reduction in an argon–hydrogen medium [9]. However, graphene obtained by such methods is characterized by a lower quality than graphene produced by the Scotch method.
High conductivity, transparency, and layer control are key issues in the growth of graphene layers. The epitaxial growth of graphene has proven to be one of the promising approaches to achieve these goals. Graphene can be synthesized on 6H-SiC (0001) or 4H-SiC (0001) substrates [10]. This process consists of two stages. First: high temperature annealing decomposes SiC. This leads to the desorption of Si from the surface and the accumulation of carbon atoms to form a surface carbon layer [11]. Using this approach, one can obtain high-quality graphene sheets with a size of several hundred nanometers [12]. However, this method has drawbacks due to the lack of continuity and uniformity of the grown graphene film.
Chemical vapor deposition (CVD) is a promising method for production of graphene in high yield. In this method, graphene is a product of chemical decomposition of precursors, such as methane, acetylene, methanol, and ethanol, on catalytic surfaces of transition metals, such as nickel [13] or copper [14].
It should be noted that, despite the presence of a significant number of methods for producing graphene, the development of a method that would make it possible to obtain high-quality graphene with a high surface remains an urgent task.
EXPERIMENTAL
Graphene was synthesized by the template method. 1,3-Butadiene was selected as a source of catalytic carbon since it is very prone to condensation and polymerization reactions. Magnesium oxide (pure for analysis grade, State Standard GOST 4526-75, VEKTON Company) with a specific surface area of 80 m2/g was carbonized with 1,3-butadiene in a flow-type quartz reactor equipped with a McBain balance at 600°С. First, MgO was coated with a monolayer carbon film. Then, the initial MgO was removed from the carbon film by treatment with a hydrochloric acid solution. Hydrochloric acid of special purity grade 20-4, State Standard GOST 14261-77 was purchased from SoyuzKhimprom.
Catalyst samples were studied by transmission electron microscopy (TEM) on a JEOL JEM-2010CX electron microscope with a line resolution of 1.4 Å.
The phase composition of C/MgO samples was studied by X-ray powder diffraction on a Thermo Fisher Scientific ARL X’TRA diffractometer with CuKα radiation at a wavelength of 1.54184 Å and pointwise scanning. The scan range was ~15°–85°(2θ), scanning step 0.1°, and the counting time was 5 s per point.
ESR spectra were recorded on an ERS-221 spectrometer. The С/MgO samples were activated by calcination in air at 500°С for 1 h for desorption of chemisorbed water, then rapidly cooled to room temperature, and filled with a 4 × 10–2 M solution of 1,3,5-trinitrobenzene in toluene; after that, the ESR spectrum was recorded. The concentration of the forming trinitrobenzene radical anions was determined from the intensity of the low-field spectral line according to the previously suggested procedure [15].
The specific surface area of the С/MgO samples was determined by the BET method based on the thermal desorption of argon on a Sorbtometer (KATAKON company, Novosibirsk).
RESULTS AND DISCUSSION
Trends in the formation of С/MgO composites have been studied. The samples with carbon loadings of 1.5, 3.0, 5.0, 8.0, 10.0, and 12.2 wt % were prepared.
The X-ray powder diffraction patterns of all the samples show the presence of the MgO phase (Fig. 1) with a unit cell parameter of 4.218 Å and a coherent scattering length of 14 nm. Reflections of graphite are absent.

X-ray powder diffraction patterns of MgO samples and C/MgO composites produced at different carbonization times (from 15 to 120 min).
In the etched sample, the graphite-like phase is X-ray amorphous (Fig. 2) with a coherent scattering length of less than 2 nm and is observed as broad halos in the ranges 15°–30° and 35°–55°(2θ).

X-ray powder diffraction pattern of the C/MgO sample after etching in HCl compacted in an aluminum cuvette.
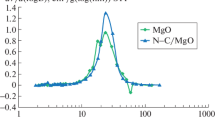
The ESR method has been used to estimate the concentrations of strong basic sites on the surface of carbon-mineral materials. Trinitrobenzene radical anions acted as spin probes. Previously [15], when studying trinitrobenzene adsorption onto magnesium and aluminum oxides, it has been found that strong basic sites exhibiting electron-donating properties react with the hydrocarbon to form adsorbed trinitrobenzene radical anions, which have a characteristic ESR spectrum with nitrogen hyperfine splitting (Fig. 3).

ESR spectra of initial MgO and 10 wt % С/MgO composite.
From the intensity of this ESR signal, one can judge the degree of MgO coverage with carbon [15]. Table 1 presents data on the change in the concentration of nitroxyl radicals depending on the carbon loading of the C/MgO composite.
It follows from Table 1 that with increasing the carbon content in the С/MgO composite, the concentration of trinitrobenzene radical ions sharply decreases. Since no nitroxyl radical form on the С/MgO sample with a carbon loading of 10.0 wt % or higher, we can draw the conclusion that the MgO surface is completely covered with a carbon layer. A single symmetric Lorentzian line with g = 2.0029, attributable to carbon deposits has been observed in the ESR spectrum of carbon-mineral materials. This line gradually narrows as the carbon loading of the samples increases from 1.5 to 12.2 wt %.
After treatment in hydrochloric acid, MgO converts to magnesium chloride and goes into solution, and the remaining carbon is a “crumpled” graphene sheet, which is confirmed by electron microscopy studies (Fig. 4).

TEM image of the carbon remaining after etching the 10 wt % С/MgO composite in hydrochloric acid.
In order to determine the effect of the carbon loading of the C/MgO composite on the properties of the resulting graphene, its specific surface was measured. The carbonization reaction temperature was 600°C. The results are presented in Table 2.
It can be seen that the measured specific surface area of the resulting graphene passes through a maximum depending on the carbon loading of the carbon-mineral composite. At small degrees of MgO coverage with carbon, graphene plates do not form a single framework and, after etching, are likely to stick together in packs. The maximum specific surface area of graphene is observed after etching the 8–10 wt % C/MgO composites and reaches 1800–1900 m2/g.
CONCLUSIONS
A series of carbon-mineral composites with the carbon loading varying from 1.5 to 12.2 wt % has been synthesized by MgO carbonization in 1,3-butadiene at 600°C. ESR has demonstrated that the MgO surface is completely blocked after depositing 8–10 wt % C. MgO from the composite was dissolved by treatment with hydrochloric acid. The surface area of the carbon samples obtained after the acid treatment has been studied by thermal desorption of argon. It has been shown that the synthesized carbon material consists of a few graphene layers. The specific surface area of the synthesized graphene passes through a maximum depending on the carbon concentration in the carbon-mineral composite. At small degrees of MgO coverage with carbon, graphene plates do not form a single framework and, after etching, are likely to stick together in packs. At higher loadings (8–10 wt %), carbon covers the outside surface of MgO particles with a monolayer film. After MgO etching from the C/MgO composite, the carbon structure is retained. The maximum specific surface area of graphene is observed after etching the 8–10 wt % C/MgO composites and reaches 1800–1900 m2/g. Thus, a method has been developed that allows one to obtain mono- and bilayer graphene.
REFERENCES
Peierls, R.E., Ann. I.H. Poincare, 1935, vol. 5, pp. 177–222.
Landau, L.D., Phys. Z. Sowjetunion, 1937, vol. 11, pp. 26–35.
Boehm, H.P., Clauss, A., Fischer, G.O., et al., Anorg.Allg. Chem., 1962, vol. 316, pp. 119–127.
Alekseenko, A.G., Grafen (Graphene), Moscow: BINOM, 2014.
Novoselov, K.S., Geim, A.K., Morozov, S.V., et al., Science, 2004, vol. 306, no. 5696, pp. 666–669.
Geim, A.K. and Macdonald, A.H., Phys. Today, 2007, vol. 60, no. 8, pp. 35–41.
Choi, K., Ali, A., and Jo, J., J. Mater. Sci. Mater. Electron., 2013, vol. 24, no. 12, pp. 4893–4900.
Choucair, M., Thordarson, P., and Stride, J., Nat. Nanotechnol., 2008, vol. 4, pp. 2–5.
Schniepp, H.C., Li, J.-L., McAllister, M.J., et al., J. Phys. Chem. B, 2006, vol. 110, no. 17, pp. 8535–8539.
Machac, P., Fidler, T., Cichon, S., et al., J. Mater. Sci. Mater. Electron., 2013, vol. 24, no. 10, pp. 3793–3799.
Huang, H., Chen, W., Chen, S., et al., ACS Nano, 2008, vol. 2, no. 12, pp. 2513–2518.
Emtsev, K.V., Bostwick, A., Horn, K., et al., Nature Mater., 2009, vol. 8, no. 3, pp. 203–207.
Reina, A., Thiele, S., Jia, X., et al., Nano Res., 2009, vol. 2, pp. 509–516.
Guermoune, A., Chari, T., Popescu, F., et al., Carbon, 2011, vol. 49, no. 13, pp. 4204–4210.
Heroux, D.S., Volodin, A.M., Zaikovskii, V.I., et al., J. Phys. Chem. B, 2004, vol. 108, no. 10, pp. 3140–3144.
Funding
The study was performed in the framework of State Assignment of the Institute of Catalysis, Siberian Branch, RAS (project no. AAAA-А17-117041710084-2).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Translated by G. Kirakosyan
Rights and permissions
About this article

Cite this article
Chesnokov, V.V., Chichkan, A.S., Bedilo, A.F. et al. Template Synthesis of Graphene. Dokl Phys Chem 488, 154–157 (2019). https://doi.org/10.1134/S0012501619100038
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0012501619100038