
Abstract
Hypoxia is a common feature in tumors and induces signaling that promotes tumor cell survival, invasion, and metastasis, but the impact of hypoxia inducible factor (HIF) signaling in the primary tumor on dissemination to bone in particular remains unclear. To better understand the contributions of hypoxia inducible factor 1 alpha (HIF1α), HIF2α, and general HIF pathway activation in metastasis, we employ a PyMT-driven spontaneous murine mammary carcinoma model with mammary specific deletion of Hif1α, Hif2α, or von Hippel-Lindau factor (Vhl) using the Cre-lox system. Here we show that Hif1α or Hif2α deletion in the primary tumor decreases metastatic tumor burden in the bone marrow, while Vhl deletion increases bone tumor burden, as hypothesized. Unexpectedly, Hif1α deletion increases metastatic tumor burden in the lung, while deletion of Hif2α or Vhl does not affect pulmonary metastasis. Mice with Hif1α deleted tumors also exhibit reduced bone volume as measured by micro computed tomography, suggesting that disruption of the osteogenic niche may be involved in the preference for lung dissemination observed in this group. Thus, we reveal that HIF signaling in breast tumors controls tumor dissemination in a site-specific manner.
Similar content being viewed by others


Introduction
Tumor cells frequently experience hypoxic conditions as the metabolic demands of the proliferating cells exceed the supply of oxygen and nutrients from the existing blood vessels. Cells must adapt to these stressful conditions by altering their metabolism and recruiting new blood vessels to increase oxygen supply1. The activation of hypoxia-inducible factor (HIF) signaling triggers these transcriptomic adaptations in response to low oxygen levels. Under normoxic conditions, HIF 1 alpha (HIF1α) and HIF2α are hydroxylated by prolyl hydroxylation domain (PHD)-containing enzymes2,3,4,5, which allows von Hippel-Lindau factor (VHL)6, an E3 ubiquitin ligase, to bind to and ubiquitinate HIF1α and HIF2α, marking them for proteasomal degradation7,8,9. In hypoxic conditions, PHD enzymes are inactive, leading to the stabilization of the alpha subunits, which can then dimerize with HIF1β and translocate to the nucleus10,11. Once in the nucleus, the HIF1 and HIF2 complexes bind to hypoxia response elements in the promoters of hypoxia-responsive genes and act as transcription factors12,13,14. HIF1α drives the expression of genes such as vascular endothelial growth factor (VEGF) to stimulate angiogenesis15,16, as well as glucose transporters, glycolytic enzymes, and lactose dehydrogenase A to shift the main energy source of the cell to glycolysis17,18,19,20,21. HIF signaling also promotes tumor cell metastasis by driving the expression of genes that control epithelial-to-mesenchymal transition (EMT), invasion, and extracellular matrix composition22,23,24. Furthermore, clinical evidence from breast cancer patients shows that high HIF1α levels in primary tumors correlate with poor patient outcomes25,26,27,28, and a hypoxic transcriptomic signature in tumor cells is associated with bone metastasis29,30. As such, HIF inhibitors are an attractive therapeutic avenue and are currently in development and undergoing clinical trials31.
High breast cancer patient morbidity and mortality arises from metastatic dissemination of tumor cells from the primary site, which occurs early in tumor progression32,33. Approximately 70% of breast cancer patients present with lung or bone metastases upon autopsy34,35. Thus, there is an urgent need to identify the molecular factors that drive tumor dissemination to distant metastatic sites. Previous studies suggest that HIF1α expression in breast cancer cells promotes lung dissemination in genetic models36 and bone colonization and osteolysis following intracardiac or orthotopic inoculation of MDA-MB-231 human breast cancer cells37,38,39. However, the comparative contributions of Hif1α and Hif2α on spontaneous bone dissemination are not well understood. Furthermore, dissemination patterns to bone and lung, the two leading sites of metastasis for breast cancer, have not been simultaneously evaluated in a spontaneous metastasis model to the best of our knowledge. We therefore generated three transgenic mouse models of spontaneous mouse mammary carcinoma with tumor-specific deletion of Hif1α, Hif2α, and Vhl and evaluated the effects of HIF modulation on tumor dissemination to multiple distant sites using highly sensitive detection techniques that have been optimized to detect low levels of disseminated tumor burden40. We found that deletion of Hif1α or Hif2α decreased dissemination to bone, while Vhl deletion increased bone dissemination. In contrast, Hif1α deletion increased lung metastatic tumor burden while Hif2α or Vhl deletion had no effect on lung dissemination. Thus, this study highlights the ability of HIF signaling to differentially modulate metastasis to certain sites.
Results
Deletion of Hif1α increases total tumor burden while slowing primary tumor growth
To assess the impact of primary tumor HIF1α expression on breast tumor cell dissemination to the bone, we generated an immune-competent spontaneous murine mammary carcinoma model with mammary-specific deletion of Hif1α using the Cre-lox system. In this model, mammary tumors grow spontaneously, driven by the polyoma middle T antigen (PyMT) expressed under the mouse mammary tumor virus long terminal repeats (MMTV-LTR), which restricts the oncogene expression to the mammary epithelium. In this study, we compared female Hif1α−/− PyMT+ mice (Hif1αf/f, Cre-positive, PyMT-positive) to control Hif1αf/f PyMT+ mice (Hif1αf/f, Cre-negative, PyMT-positive) (Fig. 1a). In this spontaneous model, mammary tumors were first palpable around 8 weeks of age and reached end point (any tumor reaching 1 cm in diameter) around 20 weeks of age for the Hif1αf/f PyMT+ mice, while the Hif1α−/− PyMT+ mice took significantly longer to reach end point, at an average of 23 weeks of age (Fig. 1b, c). This is consistent with previous reports using this model36. Interestingly, the total tumor weight was greater on average in Hif1α−/− PyMT+ mice upon sacrifice (Fig. 1d), while the number of tumors per mouse was not significantly increased (Fig. 1e).

a Example genotyping gel of Cre, floxed Hif1α, and PyMT from Hif1αf/f PyMT+, Hif1α−/− PyMT+, and Hif1αf/w PyMT− mice. “+ Ctrl” product indicates an internal positive control. b Survival analysis of Hif1αf/f PyMT+ and Hif1α−/− PyMT+ mice where end point represents sacrifice due to tumor size reaching collection threshold. Log-rank test. c–e Comparison of the age at sacrifice, total tumor burden at sacrifice, and number of tumors collected per mouse. Two-tailed Mann–Whitney test. f–h Quantitative PCR analysis of Hif1α, Hif2α, and Vegfa expression compared to B2m from whole-tumor homogenate RNA. Expression is normalized to the mean of the f/f control group. Two-tailed Mann–Whitney test. i The average pimonidazole staining intensity for each mouse across multiple images from a single tumor. Two-tailed Mann–Whitney test. j Representative images of pimonidazole staining, taken with ×10 and ×40 objectives. The ×40 field is denoted with a dashed box in the ×10 view. Scale bars represent 500 μm in ×10 fields, and 200 μm in ×40 fields. Negative controls lacked the primary antibody incubation step. Graphs represent mean per group and error bars represent s.e.m. n = 19 (or 16 in i) Hif1αf/f PyMT+ mice and n = 21 Hif1α−/− PyMT+ mice.
We confirmed through real-time quantitative polymerase chain reaction (PCR) analysis of tumor homogenate RNA that Hif1α−/− tumors had significant deletion of Hif1α (Fig. 1f), while transcript levels of Hif2α, the other major HIF pathway signaling factor, was unaffected (Fig. 1g). Recombination and deletion of Hif1α was further confirmed by PCR amplification of the Hif1α locus (Supplementary Fig. 1a, b). Hif1α and Hif2α transcript levels were unaffected in the lung, suggesting that we did not have off-target editing in other soft tissue sites (Supplementary Fig. 1c, d). Furthermore, the expression of Vegfa, a downstream target of Hif1α, was significantly lower in Hif1α−/− tumors (Fig. 1h), while the staining intensity of pimonidazole, a hypoxia marker, was comparable in Hif1αf/f and Hif1α−/− tumors (Fig. 1i, j). This confirms that Hif1α−/− tumors have a dampened hypoxia response despite experiencing similar levels of hypoxia as Hif1αf/f tumors.
Deletion of Hif1α in the mammary fat pad reduces trabecular bone volume
Since bone disseminated breast tumor cells can cause the formation of osteolytic lesions, we first assessed trabecular bone volume of tibiae from Hif1αf/f PyMT+ and Hif1α−/− PyMT+ mice by microcomputed tomography (microCT) as a readout of tumor-induced bone destruction. To ensure that Hif1α deletion in the mammary fat pad did not alter baseline bone microarchitecture, we also scanned tibiae from non-tumor bearing controls (PyMT−) of each genotype. Bone volume and trabecular number were significantly decreased in Hif1α−/− PyMT+ mice, with an accompanying increase in trabecular spacing (Fig. 2a–d), but bone volume was unaltered with Hif1α deletion in PyMT− mice, suggesting that the reduction in bone volume observed in Hif1α−/− PyMT+ mice was due to tumor-induced osteolysis. Hif1α−/− PyMT− mice did exhibit a significant increase in trabecular thickness (Fig. 2e), but this may not be biologically significant given the absence of changes in any other structural parameters for bone. Surprisingly, we found no discernible tumor burden present in the bone marrow of Hif1α−/− PyMT+ mice upon histological inspection by a certified veterinary pathologist, save in one mouse (Fig. 2f, g). Thus, Hif1a deletion in the primary tumor decreases trabecular bone volume but does not drive the development of macroscopic or osteolytic lesions in the bone.

a Representative 3D renderings of microCT scans of the proximal metaphysis of the right tibia. b–e Quantification of bone volume as a percentage of total volume, trabecular number, trabecular spacing, and trabecular thickness. Two-tailed Mann–Whitney test against corresponding f/f control. f Representative H&E-stained images of the proximal metaphysis of the tibia taken with ×4 and ×20 objectives. The ×20 field is denoted with a dashed box in the ×4 view. Scale bars represent 2.0 mm in ×4 view and 200 μm in the ×20 view. g H&E-stained images of the one histologically detectable bone metastatic lesion, identified in the distal region of the tibia from Mouse 27. Lower-magnification images taken with ×4 (i), ×20 (ii) objectives, while highest magnification image (iii) is taken at ×600 magnification. Scale bars represent 2.0 mm in ×4 view, 200 μm in the ×20 view, and 25 μm in ×600 view. Area of higher magnification is indicated in lower magnification images with a dashed box. Tumor area is labeled T and indicated by dashed circle. Atypical cells denoted with arrowheads. Graphs represent mean per group and error bars represent s.e.m. n = 19 Hif1αf/f PyMT+ mouse tibiae, n = 21 Hif1α−/− PyMT+ mouse tibiae, n = 16 Hif1αf/f PyMT− mouse tibiae, and n = 11 Hif1α−/− PyMT− mouse tibiae.
Deletion of Hif1α decreases bone dissemination while increasing lung metastasis
Given the lack of macroscopic tumor lesions in the bone and the fact that the main site of metastasis for the PyMT-driven mammary carcinoma model is the lung rather than the bone marrow41,42, we employed several sensitive strategies to determine whether there were differences in tumor dissemination to bone. First, tumor cells were detected by flow cytometry of bone marrow from the tibiae and femora from Hif1αf/f PyMT+ and Hif1α−/− PyMT+ mice based on epithelial cell adhesion molecule (EpCAM) positivity. While some background EpCAM staining was detected in non-tumor-bearing (PyMT−) bone marrow (Supplementary Fig. 2), a significant decrease in the number of EpCAM+ cells was detected in the bone marrow from Hif1α−/− PyMT+ mice compared to Hif1αf/f PyMT+ mice when normalized to total tumor weight at end point (Fig. 3a and Supplementary Fig. 3a). Second, the tumor-specific Pymt transcript was quantified from femur and spine homogenate RNA as a marker of tumor burden (Fig. 3b, c and Supplementary Fig. 3b, c). No significant difference in tumor burden was detected in the femur and spine of Hif1α−/− PyMT+ mice using this method. Taken together, these results suggest that Hif1α in the primary tumor promotes tumor cell dissemination to the bone.

a The percentage of EpCAM+ cells, out of the total number of live cells, detected by flow cytometric analysis of left hindlimb bone marrow. Numbers are normalized to the total tumor weight of each mouse. Two-tailed Mann–Whitney test. b, c Quantitative PCR analysis of Pymt transcript compared to Hmbs from right femur or spinal midsection, respectively. Pymt expression was then normalized to the total tumor weight of each mouse. Two-tailed Mann–Whitney test. d, e Metastatic lesion number or area detected by histological analysis of H&E-stained sections from the left lung. Numbers are normalized to the total tumor weight of each mouse. Two-tailed Mann–Whitney test. f Comparison of the proportion of mice from each group that had any detectable pulmonary lesions. Fisher’s exact test. g Representative images of H&E-stained lung sections. Scale bar in sub-gross photomicrographs is 500 μm. Scale bar in high-power micrographs is 100 μm. The high-power image field is denoted with a dashed box in the sub-gross view. Tumor area is denoted with a dashed oval in the high-power images (labeled T). Additional metastatic lesions in the sub-gross view are denoted with arrows. Graphs represent mean per group and error bars represent s.e.m. n = 19 Hif1αf/f PyMT+ mice, n = 21 Hif1α−/− PyMT+ mice.
In addition to quantifying tumor burden in bone sites, we quantified tumor burden in the lung by histological inspection in order to confirm that Hif1α−/− PyMT+ mice had decreased tumor burden, as reported previously36. Surprisingly, Hif1α−/− PyMT+ mice had a significant increase in the number of metastatic lesions in the lung (Fig. 3d and Supplementary Fig. 3d, e) and total metastatic lesion area (Fig. 3e and Supplementary Fig. 3f, g), regardless of whether the data were normalized to total tumor weight at end point, the mouse age at sacrifice, or left un-normalized. Individual lesion size was not significantly different between the two groups (Supplementary Fig. 3h), indicating that the increase in total tumor area is driven by the greater number of lesions. Hif1α−/− PyMT+ mice also had significantly greater incidence of pulmonary lesions compared to Hif1αf/f PyMT+ mice (Fig. 3f). A non-significant trend in increased tumor burden was observed in the contralateral lung of Hif1α−/− PyMT+ mice by Pymt transcript levels (Supplementary Fig. 3i, j). To determine whether the increased tumor burden in the lung was indicative of a global metastatic increase in soft tissue sites, tumor burden was also measured in the brain and liver. There was a non-significant trend toward a reduction in tumor burden in the brain by Pymt transcript levels in Hif1α−/− PyMT+ mice (Supplementary Fig. 3k, l), and no liver lesions were detected by histological analysis by a certified veterinary pathologist (Supplementary Fig. 3m). Thus, Hif1a deletion in the primary tumor reduces bone dissemination but also specifically promotes lung metastasis.
There was no difference in the expression of general breast cancer metastasis-associated genes (Fig. 4a), nor in EMT markers (Fig. 4b), between primary tumors from the Hif1αf/f PyMT+ and Hif1α−/− PyMT+ mice. This is consistent with site-specific differences in tumor burden, rather than a global increase or decrease in dissemination to all the distant sites measured. We also measured the expression of a panel of autophagy markers, since previous reports have shown that hypoxia-induced autophagy can affect pulmonary tumor burden. Inhibition of autophagy in hypoxic breast cancer cells has been shown to promote pulmonary metastasis43, while autophagy has also been shown to promote the survival of dormant breast cancer cells and promote metastatic tumor recurrence44. However, we saw no differences between Hif1αf/f and Hif1α−/− tumors in any of the autophagy markers tested (Fig. 4c).

a Heatmap depiction of the expression of a panel of metastasis-associated genes from n = 3 Hif1α−/− tumors compared to the average expression of n = 3 Hif1αf/f tumors. The difference in expression of each gene between the three Hif1αf/f and three Hif1α−/− tumors was compared using the Mann–Whitney test. b Expression of a panel of EMT-related genes (Cdh1, Cdh2, Egfr, Notch1, Snai1, Snai2, Snai3, Twist1, Zeb1, Zeb2) compared to B2m, measured by qPCR. Two-tailed Mann–Whitney test against corresponding f/f control. c Expression of a panel of autophagy-related genes (Atg7, Becn1, Map1lc3a, Map1lc3b, Sqstm1, Ulk1) compared to B2m, measured by qPCR. Two-tailed Mann–Whitney test against corresponding f/f control. d, e Correlation of Ye et al. hypoxia gene signature45 with mRNA levels of genes in the Kang et al. bone metastasis signature46 or the Minn et al. lung metastasis signature47, respectively, in TCGA Invasive Breast Carcinoma patient dataset. Pearson and Spearman correlation. n = 1100 patients. f Quantification of Ki-67+ area as a percentage of the lesion area. Two-tailed Mann–Whitney test. n = 17 individual Hif1αf/f metastatic lung lesions, n = 47 individual Hif1α−/− metastatic lung lesions. g Representative images of Ki-67-stained lung metastatic lesions, taken with ×20 objective. Scale bars represent 200 μm. h Quantification of the average number of CD4+ and CD8+ cells in the lung normalized to the total nuclear area for the image. Data points in red denote mice that had detectable lung tumor burden. Two-tailed Mann–Whitney test against corresponding f/f control. i Representative images of immunofluorescent staining of CD4 and CD8, taken with ×20 objective. Scale bars represent 200 μm. j Quantitative PCR analysis of Lox transcript compared to B2m from primary tumor homogenate RNA. Two-tailed Mann–Whitney test. Graph represents mean per group and error bars represent s.e.m. n = 19 Hif1αf/f PyMT+ mice, n = 21 Hif1α−/− PyMT+ mice.
We therefore examined gene expression data from The Cancer Genome Atlas (TCGA) Invasive Breast Carcinoma patient dataset to determine whether hypoxia differentially induced gene expression profiles associated with site-specific metastasis to the bone or lung. We found that a 42-gene hypoxia signature identified by Ye et al.45 significantly and positively correlated with the bone metastasis signature established by Kang et al.46 (Fig. 4d). Thus, hypoxia appears to drive a large transcriptomic program that promotes bone dissemination. However, a similar trend was observed for the lung metastasis signature from Minn et al.47 (Fig. 4e). Taken together, it appears that hypoxia upregulates tumor dissemination in general at the transcriptomic level, suggesting that the increase in lung tumor burden may be due to altered Hif1α−/− tumor cell response to signals from the lung microenvironment.
Proliferation and immune markers are unaltered in lung metastatic Hif1α−/− lesions
To reconcile the divergent role of Hif1α in tumor cell dissemination to bone and lung, we therefore sought to identify factors that may specifically be driving outgrowth of tumor cells in the lung. Since there is evidence that hypoxia promotes tumor dormancy specifically in the lung48, we investigated whether the increased lung tumor burden in Hif1α−/− PyMT+ mice was due to dormancy escape by the lung-disseminated tumor cells. First, we confirmed that there was no significant difference in pimonidazole-positive lung lesion area, indicating that Hif1αf/f and Hif1α−/− tumor cells experience similar levels of hypoxia in the lung (Supplementary Fig. 4a, b). There was, however, no difference in Ki-67+ lung lesion area between the groups, indicating that Hif1α expression does not alter the proliferative capacity of the disseminated cells once they exit dormancy (Fig. 4f, g). We next measured the abundance of CD4+ and CD8+ cells in the lungs, since altered immune cell numbers in the lung tissue may indicate differences in immune-mediated tumor cell clearance. However, there was no difference in CD4+ or CD8+ T cell infiltration in the lungs of Hif1αf/f PyMT+ and Hif1α−/− PyMT+ mice (Fig. 4h, i). Immune cell numbers were not significantly different between tumor-bearing lungs of Hif1αf/f PyMT+ and Hif1α−/− PyMT+ mice either (red points, Fig. 4h). To investigate whether pre-metastatic niche development could be driving the outgrowth of lung-disseminated tumor cells, we measured the expression of lysyl oxidase (LOX), a hypoxia-dependent tumor-secreted factor known to drive pre-metastatic niche development49. However, there was no change in Lox expression between Hif1αf/f and Hif1α−/− tumors (Fig. 4j).
Deletion of Hif2α decreases tumor dissemination to the bone but not to the lung
While Hif1α and Hif2α have some redundant functions, they have unique and sometimes opposing effects50. To determine whether the dissemination patterns observed in the Hif1α−/− PyMT+ mice were due to a general decrease in HIF signaling activity, or due to Hif1α-specific effects, we generated Hif2αf/f PyMT+ and Hif2α−/− PyMT+ mice using the same breeding strategy as the Hif1α−/− PyMT+ mice. We observed no difference between the Hif2αf/f PyMT+ and Hif2α−/− PyMT+ mice in the time it took tumors to reach collection size (Supplementary Fig. 5a, b), the total tumor weight upon sacrifice (Supplementary Fig. 5c), or the number of tumors (Supplementary Fig. 5d). We confirmed a significant reduction in Hif2α transcript levels from whole-tumor homogenate RNA of Hif2α−/− PyMT+ mice (Supplementary Fig. 5e) and no difference in Hif1α transcript levels (Supplementary Fig. 5f). Hif2α deletion did not significantly reduce Vegfa expression in the primary tumor (Supplementary Fig. 5g).
While microCT analysis of Hif2α−/− PyMT+ mice did not reveal any differences in trabecular bone parameters (Supplementary Fig. 5h–l), flow cytometric analysis of hindlimb bone marrow from Hif2α−/− PyMT+ mice revealed a significant reduction in the percentage of EpCAM+ tumor cells (Fig. 5a). However, there was no significant difference in the Pymt transcript abundance in the femur or spine (Fig. 5b, c). Unlike the Hif1α−/− PyMT+ mice, lung metastatic tumor burden in Hif2α−/− PyMT+ mice was not significantly different than Hif2αf/f PyMT+ mice, as measured by lesion number, total lesion area, individual lesion area, or incidence of lung metastases (Fig. 5d–g). Similarly, there was no significant difference in Pymt transcript abundance in the lungs or brains of Hif2α−/− PyMT+ mice (Fig. 5h, i). Metastatic tumor burden data for Hif2α−/− PyMT+ mice were un-normalized since there was no significant difference in tumor weight or age at sacrifice. Taken together, these results suggest that Hif2α in the primary tumor drives tumor cell dissemination to the bone but is dispensable for dissemination to the lung.

a The percentage of EpCAM+ cells, out of the total number of live cells, detected by flow cytometric analysis of left hindlimb bone marrow. Two-tailed Mann–Whitney test. n = 10 Hif2αf/f PyMT+ or Hif2α−/− PyMT+ mice. b, c Quantitative PCR analysis of Pymt transcript compared to Hmbs from right femur or spinal midsection, respectively. Two-tailed Mann–Whitney test. d, e Metastatic lesion number or area detected by histological analysis of H&E-stained sections from the left lung. Two-tailed Mann–Whitney test. f Comparison of individual lesion areas detected from histological inspection of left lung sections. Two-tailed Mann–Whitney test. n = 96 individual Hif2αf/f metastatic lung lesions, n = 35 individual Hif2α−/− metastatic lung lesions. g Comparison of the proportion of mice from each group that had any detectable pulmonary lesions. Fisher’s exact test. h, i Quantitative PCR analysis of Pymt transcript compared to B2m from the right lung or brain, respectively. Two-tailed Mann–Whitney test. Graphs represent mean per group and error bars represent s.e.m. For b–e, g–i, n = 19 Hif2αf/f PyMT+ mice, n = 18 Hif2α−/− PyMT+ mice.
Deletion of Vhl increases tumor dissemination to the bone but not to the lung
To determine the effect of Hif1α and Hif2α activation, rather than deletion, we generated a third transgenic mouse strain in which we could model constitutive HIF signaling activation through deletion of Vhl, a negative regulator of HIF signaling. Vhlf/f PyMT+ and Vhl−/− PyMT+ mice were generated using the same strategy as the other transgenic strains. Vhl−/− tumors took a significantly longer time to reach collection size compared to Vhlf/f controls (Fig. 6a, b) but had a significant reduction in total tumor weight upon sacrifice (Fig. 6c). The average number of tumors was not significantly different between Vhlf/f PyMT+ and Vhl−/− PyMT+ mice (Fig. 6d). While we were unable to detect a decrease in Vhl transcript in whole tumor RNA from Vhl−/− PyMT+ mice (Fig. 6e), we confirmed recombination of the Vhl locus at the genomic level (Fig. 6f) and observed a significant increase in the expression of Vegfa, phosphoglycerate kinase 1 (Pgk1), glucose transporter type 1 (Glut1), TEK receptor tyrosine kinase (Tek, also known as Tie2), and platelet-derived growth factor subunit B (Pdgfb) (Fig. 6g–k), indicating that HIF downstream signaling is elevated with Vhl deletion.

a Survival analysis of Vhlf/f PyMT+ and Vhl−/− PyMT+ mice where end point represents sacrifice due to tumor size reaching collection threshold. Log-rank test. b–d Comparison of the age at sacrifice, total burden at sacrifice, and number of tumors collected per mouse. Two-tailed Mann–Whitney test. e Quantitative PCR analysis of Vhl expression compared to B2m from whole-tumor homogenate RNA. Expression is normalized to the mean of the f/f control group. Two-tailed Mann–Whitney test. f Schematic of PCR-based validation of Vhl locus recombination using a three-primer PCR reaction. Genomic DNA was used as the input material. DNA electrophoresis gel represents the PCR products from the reactions depicted in the schematic diagram. The lower molecular weight of the band in the Vhl−/− lanes indicate successful recombination. The residual band in the Vhl−/− lanes are likely from non-recombined stromal cells present in the tumor. g–k Quantitative PCR analysis of HIF target genes (Vegfa, Pgk1, Glut1, Tek, Pdgfb) compared to B2m from whole-tumor homogenate RNA. Two-tailed Mann–Whitney test. Graphs represent mean per group and error bars represent s.e.m. n = 20 Vhlf/f PyMT+ mice, n = 19 (or 18 in e–k) Vhl−/− PyMT+ mice.
Despite unaltered trabecular bone parameters (Fig. 7a, b and Supplementary Fig. 7a–c), Vhl deletion increased the number of tumor cells detected in the bone marrow by flow cytometry when normalized to tumor weight (Fig. 7c and Supplementary Fig. 6d). No significant differences were observed in the Pymt transcript abundance in the femur or spine (Fig. 7d, e and Supplementary Fig. 6e, f). From histological analysis of the lung, no significant differences were observed in total metastatic lesion number or area between Vhlf/f PyMT+ and Vhl−/− PyMT+ mice, regardless of normalization to total primary tumor weight (Fig. 7f, g and Supplementary Fig. 6g, h). In addition, the incidence of lung metastases was no different between the two groups (Fig. 7h). There was a slight increase in the average individual lung lesion size in the Vhl−/− PyMT+ mice (Supplementary Fig. 6i), but this difference was not sufficient to drive an increase in the overall lung tumor burden. Additionally, there were no significant differences in Pymt transcript abundance in the lungs or brains of Vhl−/− PyMT+ mice (Fig. 7i, j and Supplementary Fig. 6j, k).

a Representative 3D renderings of microCT scans of the proximal metaphysis of the right tibia. b Quantification of bone volume from microCT analysis as a percentage of total volume. Two-tailed Mann–Whitney test against corresponding f/f control. n = 10 Vhlf/f PyMT+ mouse tibiae, n = 11 Vhl−/− PyMT+ mouse tibiae, n = 8 Vhlf/f PyMT− mouse tibiae, and n = 13 Vhl−/− PyMT− mouse tibiae. c The percentage of EpCAM+ cells, out of the total number of live cells, detected by flow cytometric analysis of left hindlimb bone marrow. Values have been normalized to the total tumor burden at end point of each mouse. Two-tailed Mann–Whitney test. n = 10 Vhlf/f PyMT+ mice, n = 10 Vhl−/− PyMT+ mice. d, e Quantitative PCR analysis of Pymt transcript compared to Hmbs from right femur or spinal midsection, respectively. Values have been normalized to the total tumor burden at end point of each mouse. Two-tailed Mann–Whitney test. f, g Metastatic lesion number or area detected by histological analysis of H&E-stained sections from the left lung. Values have been normalized to the total tumor burden at end point of each mouse. Two-tailed Mann–Whitney test. h Comparison of the proportion of mice from each group that had any detectable pulmonary lesions. Fisher’s exact test. i, j Quantitative PCR analysis of Pymt transcript compared to B2m from the right lung or brain, respectively. Values have been normalized to the total tumor burden at end point of each mouse. Two-tailed Mann–Whitney test. Graphs represent mean per group and error bars represent s.e.m. For d–j, n = 20 (or 19 in f–h) Vhlf/f PyMT+ mice, n = 19 Vhl−/− PyMT+ mice.
While Hif1α deletion was not found to alter the expression of genes involved in specific metastasis-related signaling pathways (Fig. 4), Vhl deletion was found to increase, although not significantly, the expression of Pthlh in the primary tumor (Fig. 8a). PTHLH and its gene product PTHrP are known to promote breast tumor progression and metastasis51 and are key drivers of tumor-induced bone disease52,53,54. Additionally, PTHLH is a direct target of HIF2α55. Accordingly, we observed that Hif2α, but not Hif1α, deletion drove a modest, non-significant decrease in Pthlh expression (Fig. 8b, c). Vhl deletion also modestly increased the expression of Cxcr4 (Fig. 8d), a gene known to drive breast cancer cell metastasis to bone56,57, though this increase did not reach the threshold of statistical significance. Hif1α deletion did not result in a reciprocal decrease in Cxcr4 expression, but Hif2α deletion yielded a non-significant decrease in Cxcr4 (Fig. 8e, f). Due to the subtlety of expression changes observed for Pthlh and Cxcr4, other HIF-driven pro-metastatic factors are most likely involved in the mechanism behind HIF-driven bone metastasis (Fig. 8g).

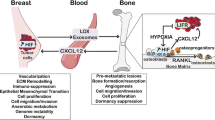
a–c Quantitative PCR analysis of Pthlh transcript compared to B2m in primary tumor with deletion of Vhl, Hif1α, or Hif2α, respectively. Two-tailed Mann–Whitney test. d–f Quantitative PCR analysis of Cxcr4 transcript compared to B2m in primary tumor with deletion of Vhl, Hif1α, or Hif2α, respectively. Two-tailed Mann–Whitney test. g Summary of findings. VHL and HIF1α expression in the primary tumor drive tumor growth, and VHL expressing tumors are larger, while HIF1α-expressing tumors are smaller on average. HIF1α expression in the primary tumor also disrupts bone homeostasis, leading to decreased bone volume. This disruption of the bone inhibits tumor cell dissemination to bone, while likely promoting tumor cell dissemination and outgrowth in the lung. HIF2α drives bone dissemination but does not influence lung metastasis. As the negative regulator of HIF signaling, VHL inhibits bone metastasis but VHL expression does not alter metastatic tumor burden in the lung. Image created with BioRender.com. Graphs represent mean per group and error bars represent s.e.m. n = 20 Vhlf/f PyMT+ mice, n = 18 Vhl−/− PyMT+ mice, n = 19 Hif1αf/f PyMT+ mice, n = 21 Hif1α−/− PyMT+ mice, n = 19 Hif2αf/f PyMT+ mice, n = 18 Hif2α−/− PyMT+ mice.
Discussion
While our main analysis was focused on dissemination patterns and quantifying tumor burden in various distant sites, we observed surprising patterns in primary tumor growth. The slower tumor progression we observed in the Hif1α−/− tumors has been reported previously in this model36 and has been attributed to delayed microvessel growth in the mammary fat pad due to insufficient HIF signaling. Interestingly, while tumor progression is delayed, total tumor weight was higher on average in Hif1α−/− PyMT+ mice than in Hif1αf/f PyMT+ mice. However, in the previous study using this model36, no significant difference in tumor weight at end point was noted. In contrast to Hif1α, our data indicate that Hif2α deletion does not significantly alter tumor progression, suggesting that HIF1α and HIF2α target distinct pathways that regulate tumor growth. Interestingly, while we expected Vhl deletion to yield the opposite tumor growth pattern from Hif1α deletion and cause tumors to grow more rapidly, Vhl-deleted tumors in fact progressed more slowly. VHL deletion has been reported to increase immune infiltration in mammary tumors58, which may contribute to increased tumor cell clearance and the slower tumor growth and decreased total tumor weight observed in the Vhl−/− PyMT+ mice.
Our finding that bone metastasis was significantly lower with Hif1α or Hif2α deletion are highly consistent with previous studies demonstrating that hypoxic signaling promotes tumor cell dissemination to the bone. HIF1α expression in breast cancer cells promotes bone colonization and osteolysis following intracardiac or orthotopic inoculation of MDA-MB-231 human breast cancer cells37,38,39, and hypoxic transcriptomic signatures in breast cancer cells has been associated with bone metastasis29,30.
It is important to note that our lung metastasis findings, which indicate that Hif1α inhibits lung metastasis while Hif2α has no effect, contradict a large body of work which demonstrates that both Hif1α and Hif2α promote lung metastasis. A prior study using the genetic MMTV-PyMT mouse model with mammary-specific deletion of Hif1α indicated that Hif1α expression promoted lung metastasis36. The reason for the differences in our findings from previous studies are likely multifaceted. We used similar methodology, and we had a board-certified, blinded veterinary pathologist perform the lung tumor analysis. It is possible that the differences may be due to differences in genetic strain, since the combination of these mice is on a mixed genetic background. Our colony is therefore genetically distinct from the previous study. Significant differences have also been noted in microbiome composition of genetically engineered mice with the same genotype housed at different facilities59. The commensal microbiota can significantly impact both local (gut) and systemic immunity60,61 and thus could also influence the clearance or growth of disseminated tumor cells. Minor variations in animal handling and housing conditions, such as cage density and diet, have also been shown to affect biochemical, hematological, metabolic, and endocrine parameters62.
Additional studies similarly indicate that increased HIF signaling increases metastasis, including a 45-gene hypoxia response gene signature that was predictive of breast cancer patients’ risk of developing lung metastases37. Several studies using MDA-MB-231 human breast cancer cells or EMT6 murine mammary carcinoma cells have also demonstrated that inhibition of HIF1α or HIF2α significantly diminishes metastasis to the lung37,63,64. HIF signaling has been shown to facilitate tumor cell extravasation by driving the expression of genes that increase breast cancer cell adhesion to endothelial cells, such as L1 cell adhesion molecule, as well as genes that decrease adhesion between endothelial cells, such as angiopoietin-like 463. Specific downstream effectors of HIF signaling that prepare the lung microenvironment for metastatic colonization, such as LOX and LOX-like proteins (LOXL2 and LOXL4), have also been identified23,65. A potential explanation for the difference in findings from these prior studies may be due to our use of a spontaneous tumor formation and dissemination model since the majority of the previous studies utilized xenograft or syngeneic injection models, or may be due to differences between PyMT tumor cells and the tumor lines utilized in the other studies. It is possible that, since the PyMT-driven spontaneous mammary cancer model has such a robust lung metastasis phenotype, contributions of HIF signaling may be negligible. This would explain why we do not see changes in lung tumor burden in the Hif2α- or Vhl-deletion models and could point to a yet unidentified mechanism at play in the Hif1α-deletion strain that exhibited increased lung metastasis.
Thus far, we have not been able to identify a molecular driver for the increased lung metastasis observed with Hif1α deletion and have eliminated a significant number of potential mechanisms. We demonstrated that Hif1α expression does not alter the proliferative capacity of macroscopic lung lesions, but due to the collection time point we were not able to detect single disseminated tumor cells in these mice. As we collected mice relatively late in disease progression in order to maximize the amount of disseminated cells, the lung lesions were already well developed. Thus, we could not determine whether the proliferative capacity of these disseminated tumor cells was altered by Hif1α expression prior to their development into histologically detectable lesions. We cannot rule out that disseminated tumor cells that had not yet grown into macroscopic lesions are detectable in mice collected earlier in disease progression and there may be differences in Ki-67 staining at that time point. Given the subtlety of our model and the large mouse numbers necessary for robust statistical analysis at earlier time points, it would be difficult to assess these differences earlier in the model. Importantly, normalizing lung tumor burden to age at sacrifice did not change our finding that Hif1α deletion increased lung metastasis, indicating that our data is likely not an artifact of the difference in collection age.
Additionally, this difference in outgrowth of disseminated tumor cells could be due to tumor cell-intrinsic properties driven by Hif1α expression or due to microenvironmental changes in the lung that result from Hif1α expression in the primary tumor. First, alterations in immune cell populations in the lung may alter the outgrowth of lung metastatic cells due to immune-mediated tumor cell clearance66. We measured the presence of CD4+ and CD8+ cells in the lungs of Hif1αf/f PyMT+ and Hif1α−/− PyMT+ mice but observed no differences. However, other immune cell types are also involved in tumor cell clearance. Modulating the activity or polarization of immune cells is also known to create microenvironments that oppose or support tumor growth. For example, T cell inactivation is known to create a permissive microenvironment for tumor growth67, and tumor-associated macrophages can promote tumor progression through many pathways, such as inducing tissue remodeling and fibrosis, taming of adaptive immunity, and providing protective niches for cancer stem cells68. Some immune cell types, especially CD11b+ cells, have been implicated in the establishment of the pre-metastatic niche49,69 that fosters tumor cell growth. Thus, a more extensive immune cell profiling of the lung may reveal differences that would explain the difference in tumor cell outgrowth. Second, primary tumor-secreted factors have been shown to affect the establishment of a pre-metastatic niche. Hypoxia-induced LOX secretion from breast cancer cells is a driver of pre-metastatic niche development in the lung49 as well as osteolytic lesion development in the bone29,70. While we did not observe a difference in Lox expression that would explain the increased lung tumor burden and decreased bone volume observed in Hif1α−/− PyMT+ mice, this is not the only factor that regulates pre-metastatic niche development. In prostate cancer, exosomes released from hypoxic cancer cells have been shown to promote matrix metalloproteinase activity at pre-metastatic sites71. The mechanisms that govern pre-metastatic niche development at various anatomical sites is not yet well characterized, and the impact of hypoxia on this process warrants further investigation.
Interestingly, a change in lung tumor burden was only observed in the Hif1α-deleted mice, and not in the Hif2α- or Vhl-deleted mice. Of note, a decrease in tibial bone volume was observed in the Hif1α−/− PyMT+ mice, but not in the Hif2α- or Vhl-deleted mice. The osteogenic niche is key to the establishment and growth of disseminated tumor cells in the bone marrow, as the interaction of tumor cells with osteoblast lineage cells through heterotypic adherens junctions activates pro-proliferation pathways72. However, osteoblast-secreted factors have also been shown to induce dormancy and quiescence in prostate cancer cells73,74, indicating that the osteogenic niche can promote quiescence of tumor cells that engraft in these regions of the bone. Since the osteogenic niche is reduced in the Hif1α−/− PyMT+ mice, as indicated by the reduction in bone volume, the shrinkage of suitable niches available for tumor cell engraftment in bone may cause a larger portion of the metastatic tumor cells to engraft in the lung instead. This may explain why we do not observe a difference in lung tumor burden in Hif2α- or Vhl-deleted mice. The mechanism behind the decreased bone volume in Hif1α−/− PyMT+ mice remains unclear; however, it is important to note that there was no difference in bone volume between Hif1αf/f and Hif1α−/− PyMT− mice, suggesting that the reduction in bone volume is not due to deletion of Hif1α in off-target cell lineages, but rather due to tumor-related factors.
While a loss of the osteogenic niche could explain the reduction in bone dissemination and increase in lung metastasis observed in the Hif1α−/− PyMT+ mice, other mechanisms must be involved in the bone dissemination phenotypes we observed in Hif2α−/− PyMT+ and Vhl−/− PyMT+ mice. Our data suggest that reduced expression of PTHrP in the primary tumor may, in part, mediate the decreased bone dissemination in Hif2α−/− PyMT+ mice, while increased C-X-C chemokine motif receptor 4 (CXCR4) expression may be involved in the regulation of bone dissemination in Vhl−/− PyMT+ mice, although it is important to note that neither gene was significantly changed. PTHrP (gene name PTHLH) is a key driver of osteolysis in bone-disseminated breast cancer, but its role in the primary tumor is more nuanced75. Previous work using the MMTV-PyMT model demonstrated that PTHrP deletion in the mammary epithelium delays primary tumor initiation, inhibits tumor progression, and reduces metastasis to distal sites51. Loss of primary tumor PTHrP expression in a MMTV-neu model of breast cancer, however, showed the opposite outcome, with PTHrP loss resulting in higher tumor incidence76. Clinical studies have also shown that patients with PTHrP-positive primary tumors have significantly improved survival and fewer metastases to distant sites, including the bone77,78. PTHrP is known to be expressed in a HIF2α, but not HIF1α, dependent manner55. In accordance with the modest decrease in Pthlh expression we observe in Hif2α−/− tumors, the trend toward increased Pthlh expression in Vhl−/− tumors may be due to Hif2α activation rather than Hif1α. CXCR4 is known to drive breast cancer metastasis to bone through engagement with its cognate ligand, CXCL12, which is highly expressed on mesenchymal stromal cells in the bone marrow56,57. Interestingly, although CXCR4 expression is known to be HIF1α dependent39, we did not observe a decrease in Cxcr4 transcript in response to Hif1α deletion. There is also evidence that HIF2α is required for CXCR4 expression in renal cancer cell lines79, but we did not observe a significant difference in Cxcr4 expression in response to Hif2α deletion. This suggests that, in our model, deletion of either of the HIFα factors on their own was not sufficient to decrease Cxcr4, but the combined stabilization of Hif1α and Hif2α may have been sufficient to drive Cxcr4 expression. Alternatively, the modest increase in Cxcr4 in response to Vhl deletion could be mediated by HIF-independent pathways. While VHL functions to regulate HIF signaling activity by modulating degradation of HIF1α and HIF2α, VHL also possesses HIF-independent functions80,81. For example, VHL has been shown to degrade or inhibit the catalytic activity of certain kinases, including some PKC isoforms82,83,84, AKT1, and AKT285. These kinases are involved in signaling pathways that regulate metabolism, angiogenesis, proliferation, differentiation, cell survival, tumor initiation, and metastasis86,87,88. Thus, it is possible that phenotypes observed in the Vhl−/− PyMT+ mice may be due, in part, to these HIF-independent pathways. To definitively address whether the phenotype and potential mechanisms observed in the Vhl−/− mice are specific for Hif1α or Hif2α, loss-of-function Vhl/Hif1α and Vhl/Hif2α double knockout mice will need to be investigated in future studies.
Our findings present important considerations for the development of clinical HIF inhibitors. While targeted inhibition of HIF1α or HIF2α could be beneficial in preventing bone metastasis, HIF1α inhibition may promote the outgrowth of lung-disseminated breast cancer cells. Thus, the site-specific effects of tumor cell HIF signaling must be considered. Furthermore, our results suggest that patients with high levels of HIF1α or HIF2α expression in their primary tumor may be at an increased risk of bone metastasis development. Thus, these markers may have prognostic value and patients with high expression may benefit from bone-focused clinical follow-up over time.
Methods
Animals
All experiments were performed following the relevant guidelines and regulations of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Vanderbilt University.
Mammary epithelial tumor cell-specific knock-out of Hif1α was achieved by breeding together three transgenic mouse strains. First, transgenic mice with loxP sites flanking both alleles of the Hif1α exon 2 (Jackson Laboratory Stock No. 007561, C57/B6 background)89 were bred with transgenic mice expressing Cre recombinase downstream of MMTV-LTR (Jackson Laboratory Stock No. 003553, C57/B6 background)90. The progeny from this cross were then bred with transgenic mice expressing the PyMT oncoprotein under the MMTV-LTR (Jackson Laboratory Stock No. 022974, C57/B6-FVB mixed background)91. Virgin female mice with Hif1α wild-type mammary fat pad tumors (Hif1αf/f PyMT+ = Hif1αf/f, MMTV-Cre negative, MMTV-PyMT positive) or Hif1α-null mammary fat pad tumors (Hif1α−/− PyMT+ = Hif1αf/f, MMTV-Cre positive, MMTV-PyMT positive) were used in these studies. Non-tumor bearing (PyMT−) controls of both Hif1αf/f and Hif1α−/− mice were also used.
Mammary epithelial tumor cell-specific knock-out of Hif2α was achieved using the same breeding strategy as above but using a transgenic mouse line with loxP sites flanking both alleles of the Hif2α exon 2 (Jackson Laboratory Stock No. 008407, C57/B6 background)92: Hif2αf/f PyMT+ = Hif2αf/f, MMTV-Cre negative, MMTV-PyMT positive; Hif2α−/− PyMT+ = Hif1αf/f, MMTV-Cre positive, MMTV-PyMT positive.
Similarly, mammary epithelial tumor cell-specific knock-out of Vhl was achieved by incorporating a transgenic mouse line with loxP sites flanking both alleles of the Vhl exon 1 (Jackson Laboratory Stock No. 012933, C57/B6 background)93: Vhlf/f PyMT+ = Vhlf/f, MMTV-Cre negative, MMTV-PyMT positive; Vhl−/− PyMT+ = Vhlf/f, MMTV-Cre positive, MMTV-PyMT positive.
At weaning, tail snips were collected from each mouse. Genomic DNA was extracted from the tail snip and genotyping was performed by PCR amplification of Hif1α (F: GGAGCTATCTCTCTAGACC, R: GCAGTTAAGAGCACTAGTTG)94, Hif2α (F: CTTCTTCCATCATCTGGGATCTGGGAC, R: CAGGCAGTATGCCTGGCTAATTCCAGTT)92, or Vhl (F: CTAGGCACCGAGCTTAGAGGTTGCG, R: CTGACTTCCACTGATGCTTGTCACAG)94, as well as PyMT (F: GGAAGCAAGTACTTCACAAGGG, R: GGAAAGTCACTAGGAGCAGGG), and Cre (F: GCGGTCTGGCAGTAAAAACTATC, R: GTGAAACAGCATTGCTGTCACTT). The PyMT genotyping reaction also included an internal positive control reaction (F: CAAATGTTGCTTGTCTGGTG, R: GTCAGTCGAGTGCACAGTTT). Primer sequences for PyMT and Cre genotyping were obtained from Jackson Laboratories. Once weaned, mice were palpated 2-3 times per week, and tumors were measured in three dimensions with digital calipers. Mice were collected when any tumor had grown to a size of 1 cm in diameter in any dimension, around 4–5 months of age. Littermate and cousin control mice were collected for each study. Forty-five minutes prior to collection, mice were inoculated with Hypoxyprobe (Hypoxyprobe-1 Omni Kit, Hypoxyprobe, Inc., catalog number HP3-1000Kit) via intraperitoneal injection at a dosage of 60 mg/kg body weight.
Real-time PCR
Tumor samples, right lung lobes, brains, and intact right femora were homogenized in 1 ml TRIzol (Life Technologies), spun down to clear the lysate, phenol–chloroform extracted, DNase digested (TURBO DNA-free Kit, Life Technologies), and cDNA synthesized (1 μg RNA, iScript cDNA Synthesis Kit, Bio-Rad) per the manufacturer’s instructions. Real-time PCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad) on a QuantStudio 5 (Thermo Fisher) with the following conditions: 2 min at 50 °C, 10 min at 95 °C, (15 s at 95 °C, 1 min at 60 °C) × 40 cycles followed by dissociation curve (15 s at 95 °C, 1 min at 60 °C, 15 s at 95 °C). For each biological replicate, three technical replicates were performed for each gene analyzed. Non-template controls were included as a negative control for each gene analyzed. Analysis was performed by normalizing the expression of the target gene to the average Hmbs or B2m expression within the same sample to determine ΔCt. The ΔCt was transformed (2−ΔCt) and the average of the three technical replicates was calculated. The average 2−ΔCt for each mouse is presented as target gene “(Pymt, Hif1a, Hif2a, etc.): (B2m or Hmbs) mRNA” or “Relative mRNA abundance” in the figures. Mouse primers for PyMT95 (Pymt—F: CTGCTACTGCACCCAGACAA, R: GCAGGTAAGAGGCATTCTGC) and hydroxymethylbilane synthase96 (Hmbs—F: TCATGTCCGGTAACGGCG, R: CACTCGAATCACCCTCATCTTTG) were previously published. Primer sequences for parathyroid hormone-related protein (Pthlh—F: ACATTGCTATGGGAGCCAC, R: TAGGAATCAGCGCCTCTAAC) were kindly provided by Dr. Natalie Sims and Dr. T. John Martin at St. Vincent’s Institute of Medical Research. Primers for beta-2-microglobulin, VEGFA, Hif1α (exon 2), Hif2α (exon 2), and Vhl (exon 1) were designed using PrimerBlast (NCBI) against the mouse genome (Mus musculus) and validated by dissociation: B2m (F: TTCACCCCCACTGAGACTGAT, R: GTCTTGGGCTCGGCCATA), Vegfa (F: GGAGATCCTTCGAGGAGCACTT, R: GGCGATTTAGCAGCAGATATAAGAA), Hif1α (F: TCGGCGAAGCAAAGAGTCTG, R: GCTCACATTGTGGGGAAGTG), Hif2α (F: TGAGGAAGGAGAAATCCCGTG, R: GGGCAACTCATGAGCCAACT), Vhl (F: GACCCGTTCCAATAATGCCC, R: CGTCGAAGTTGAGCCACAAA). Additional primer sequences were obtained from the Massachusetts General Hospital PrimerBank and specificity was confirmed by nucleotide BLAST (NCBI) and validated by dissociation: Cdh1 (F: CAGGTCTCCTCATGGCTTTGC, R: CTTCCGAAAAGAAGGCTGTCC), Cdh2 (F: AGCGCAGTCTTACCGAAGG, R: TCGCTGCTTTCATACTGAACTTT), Egfr (F: ATGAAAACACCTATGCCTTAGCC, R: TAAGTTCCGCATGGGCAGTTC), Notch1 (F: GATGGCCTCAATGGGTACAAG, R: TCGTTGTTGTTGATGTCACAGT), Snai1 (F: CACACGCTGCCTTGTGTCT, R: GGTCAGCAAAAGCACGGTT), Snai2 (F: CATCCTTGGGGCGTGTAAGTC, R: GCCCAGAGAACGTAGAATAGGTC), Snai3 (F: GGTCCCCAACTACGGGAAAC, R: CTGTAGGGGGTCACTGGGATT), Twist1 (F: GGACAAGCTGAGCAAGATTCA, R: CGGAGAAGGCGTAGCTGAG), Zeb1 (F: ACTGCAAGAAACGGTTTTCCC, R: GGCGAGGAACACTGAGATGT), Zeb2 (F: ATTGCACATCAGACTTTGAGGAA, R: ATAATGGCCGTGTCGCTTCG), Atg7 (F: TCTGGGAAGCCATAAAGTCAGG, R: GCGAAGGTCAGGAGCAGAA), Becn1 (F: ATGGAGGGGTCTAAGGCGTC, R: TCCTCTCCTGAGTTAGCCTCT), Map1lc3a (F: GACCGCTGTAAGGAGGTGC, R: CTTGACCAACTCGCTCATGTTA), Map1lc3b (F: TTATAGAGCGATACAAGGGGGAG, R: CGCCGTCTGATTATCTTGATGAG), Sqstm1 (F: GAACTCGCTATAAGTGCAGTGT, R: AGAGAAGCTATCAGAGAGGTGG), Ulk1 (F: AAGTTCGAGTTCTCTCGCAAG, R: CGATGTTTTCGTGCTTTAGTTCC), Pgk1 (F: TGGTGGGTGTGAATCTGCC, R: ACTTTAGCGCCTCCCAAGATA), Glut1 (F: CAGTTCGGCTATAACACTGGTG, R: GCCCCCGACAGAGAAGATG), Tek (F: CTGGAGGTTACTCAAGATGTGAC, R: TCCGTATCCTTATAGCCTGTCC), Pdgfb (F: CATCCGCTCCTTTGATGATCTT, R: GTGCTCGGGTCATGTTCAAGT), Cxcr4 (F: GAGGCCAAGGAAACTGCTG, R: GCGGTCACAGATGTACCTGTC). The expression of a panel of metastasis-associated genes was quantified using the tumor metastasis (SAB Target List, M384) qPCR array plate (Bio-Rad). Each gene in the array was run in duplicate. Three representative Hif1αf/f and Hif1α−/− tumor homogenate RNA samples were selected based on their Hif1α expression. Analysis was performed by normalizing the expression of the target gene to the average B2m expression within the same sample to determine ΔCt. The ΔCt was transformed (2−ΔCt) and the average of the two technical replicates was calculated. ΔCt values for each Hif1α−/− sample was then normalized against the average of the three Hif1αf/f samples to determine enrichment.
Polymerase chain reaction
Recombination and loss of the Hif1α exon 2 was confirmed by PCR amplification using a three-primer system split between two separate reactions. The forward primer (Fwd: CAGTGCACAGAGCCTCCTC) binds to Hif1α exon 1, the first reverse primer (Rev1: GCTCACATTGTGGGGAAGTG) binds to exon 2, and the second reverse primer (Rev2: ATGTAAACCATGTCGCCGTC) binds to exon 3. The first reaction (Fwd and Rev1 primers) will not amplify a band if recombination has occurred, while the second reaction (Fwd and Rev2) will amplify a lower molecular weight band in the case of recombination. PCR reactions were set up using the HotStarTaq system (Qiagen) using 200 ng of cDNA generated from RNA extracted from tumor homogenates as the template. PCR reactions were carried out in a T100 Thermal Cycler (Bio-Rad) with the following conditions: 15 min at 95 °C, (1 min at 94 °C, 1.5 min at 59 °C, 1 min at 72 °C) × 38 cycles, followed by 10 min at 72 °C. The B2m locus was also amplified as a loading control, using the same primers as used for real-time PCR above, using the following conditions: 15 min at 95 °C, (1 min at 94 °C, 1.5 min at 53 °C, 1 min at 72 °C) × 35 cycles, followed by 10 min at 72 °C.
Recombination of Vhl exon 1 was confirmed by PCR amplification using a one-reaction three-primer system97. The first forward primer (Fwd1: CTGGTACCCACGAAACTGTC) binds to the 3’ UTR upstream of Vhl exon 1, the second forward primer (Fwd2: CTAGGCACCGAGCTTAGAGGTTTGCG) binds to Vhl exon 1, and the reverse primer (Rev: CTGACTTCCACTGATGCTTGTCACAG) binds to a region in the first intron. If no recombination has occurred, only one fragment will amplify (Fwd2 and Rev primer pair) since the amplification fragment that would form from the Fwd1 and Rev primer pair is too large to amplify. If recombination has occurred, the binding site for the Fwd2 primer is no longer present, and a lower molecular weight band will form (Fwd1 and Rev primers). PCR reactions were set up using the HotStarTaq system (Qiagen) using 200 ng of cDNA generated from RNA extracted from tumor homogenates as the template. PCR reactions were carried out in a T100 Thermal Cycler (Bio-Rad) with the following conditions: 15 min at 95 °C (1 min at 94 °C, 1.5 min at 52 °C, 1 min at 72 °C) × 35 cycles, followed by 10 min at 72 °C.
Histology
All tissues collected were fixed in 10% formalin for 24 h. Hind limb bones were then decalcified in EDTA (20% pH 7.4) solution for 72 h prior to embedding. Tissues analyzed via histology and immunohistochemistry were embedded in paraffin and 5 μm-thick sections were prepared for staining. Hematoxylin and eosin (H&E) staining was performed98 and H&E-stained slides of left lung, liver, and tibia sections were analyzed for the presence of metastatic mammary tumors under blinded conditions by a board-certified veterinary anatomic pathologist. All tumors were imaged on an Olympus BX43 microscope equipped with a SPOT Flex camera (Diagnostic Instruments Inc., Sterling Heights, MI). Tumor areas were measured in the SPOT software (Diagnostic Instruments Inc.), and total tumor area per animal was calculated.
Pimonidazole immunohistochemistry
Sections were deparaffinized and rehydrated by soaking the slides in Xylene Substitute (Thermo Fisher), 100, 95, 90, and then 70% ethanol, and finally deionized water. Peroxidase activity was then quenched by incubating the slides in 3% hydrogen peroxide for 15 min. After slowly displacing the hydrogen peroxide solution with deionized water, slides were rinsed three times with phosphate-buffered saline (PBS). The deparaffinized sections were blocked in serum-free protein blocking agent (Dako) for 5 min and incubated with primary antibody (Hypoxyprobe-1 Omni Kit, Hypoxyprobe, Inc., catalog number HP3-1000Kit, 1:100) in Dako protein block overnight at 4 °C. Negative control sections were incubated with Dako alone. The sections were then washed three times with PBS and incubated with biotinylated secondary antibody (goat-anti-rabbit IgG, Vector, catalog number BA-1000, 1:200) in Dako protein block for 30 min at 37 °C, followed by streptavidin-conjugated horseradish peroxidase (HRP; Millipore, OR03L, 1:200) in PBS for 30 min at 37 °C, and developed using the DAB ImmPACT Kit (Vector, catalog number SK-4105) for approximately 1 min. Chromogen development was then quenched by rinsing twice in deionized water. Sections were counterstained with hematoxylin and dehydrated by soaking them in 70, 90, 95, and 100% ethanol, followed by Xylene Substitute. The coverslips were mounted using Permount (Fisher Scientific). All images were collected on an Olympus BX41 Microscope equipped with an Olympus DP71 camera using the ×4, ×20, and ×40 objectives. Pimonidazole-positive tumor area was quantified based on visual inspection of multiple micrographs with ×4 objective based on a scale of 0–3: 0 = no nuclear staining, 1 = <10% positive nuclear staining, 2 = 10–50% positive nuclear staining, 3 = ≥50% positive nuclear staining. Each field was assigned a score and an average score for the tumor was calculated based on all fields. The number of fields inspected varied from 1 to 4, depending on the size of the tumor. Pimonidazole-stained area of lung lesions was quantified using the ImageJ software with manual tumor area contouring and automatic color thresholding.
Ki-67 immunohistochemistry
Staining was performed by the Vanderbilt University Medical Center Translational Pathology Shared Resource (Nashville, TN) as follows: Slides were placed on the Leica Bond Max IHC stainer. All steps besides dehydration, clearing, and coverslipping are performed on the Bond Max. Slides are deparaffinized. Heat-induced antigen retrieval was performed on the Bond Max using their Epitope Retrieval 2 solution for 20 min. Slides were placed in a Protein Block (Ref# x0909, DAKO, Carpinteria, CA) for 10 min. The sections were incubated with anti-Ki-67 (Catalog #12202 S, Cell Signaling Technology, Danvers, MA) diluted 1:250 for 1 h. The Bond Refine Polymer detection system was used for visualization. Slides were the dehydrated, cleared, and coverslipped. All images were collected on an Olympus BX41 Microscope equipped with an Olympus DP71 camera using the ×4, ×20, and ×40 objectives. Ki-67-stained area of lung lesions was quantified using the ImageJ software with manual tumor area contouring and automatic color thresholding.
CD4/CD8 immunofluorescent imaging
Slides were stained using the Opal 7-Color Manual IHC Kit (PerkinElmer). In brief, slides were deparaffinized and rehydrated in a series of xylene and ethanol washes, microwaved in Rodent Decloaker (Biocare Medical), and then cooled. Slides were washed with water and then TBST (TBS + 0.05% Tween-20) followed by blocking with BLOXALL Blocking Solution (Vector Laboratories). The following antibodies were used: CD4 (Invitrogen, clone 4SM95; 1:100), CD8 (Invitrogen, clone 4SM16; 1:100), ImmPRESS HRP Goat anti-rat IgG (Vector Laboratories; 1:4). Staining with primary and secondary antibodies and OPAL fluorophores were performed per the manufacturer’s instructions. All images were collected on a Zeiss 880/Airyscan Microscope using the ×20 and ×40 objectives. For quantitation, the ×20 images were used to manually count the number of CD4+ and CD8+ cells and calculate nuclear area using color thresholding in ImageJ.
Microcomputed tomography
Ex vivo microCT was performed on the proximal tibia using the Scanco μCT 50. Scans were initiated from the proximal end of the metaphyseal growth plate and progressed 250 slices distal. Tibiae were scanned at 7 μm voxel resolution, 55-kV voltage, and 200 μA current. Scans were reconstructed and analyzed using the Scanco Medical Imaging Software to determine the bone volume/total volume, trabecular number, thickness, and separation. The most distal slice of the growth plate was used as a reference slice and analysis was set to begin 20 slices distal from this point. A 150 slice region of interest was selected for analysis. An automated contouring procedure was applied to separate the trabecular bone from the cortical bone and visually verified for each sample.
Flow cytometry
The epiphyses of the femur and tibia from one hindlimb were cut and the bones were flushed using centrifugation to obtain the bone marrow. The bone marrow was filtered through a 40 μm cell strainer to separate the cells from bone debris. Cells were suspended in red blood cell lysis buffer for 5 min on ice, spun down, and washed twice with PBS. Bone marrow (2 × 106 cells) was stained in 100 μl of 1% bovine serum albumin in PBS with 100 ng EpCAM antibody (BD Pharmingen, catalog number 563478) for 1 h at 4 °C in the dark. Cells were washed with PBS and resuspended in PBS and 0.5 ng 4,6-diamidino-2-phenylindole (DAPI) for 15 min on ice. Flow cytometric experiments were analyzed in the VUMC Flow Cytometry Shared Resource using the 3-laser or 5-laser BD LSRII. Datasets were analyzed using the FlowJo software (FlowJo, LLC). Cells were gated based on forward scatter and side scatter to identify single cells and then live cells (DAPI−) were gated based on EpCAM positivity using non-tumor-bearing bone marrow as a negative control. Mammary fat pad tumors were mechanically digested and filtered through a 40 μm cell strainer to obtain a single-cell suspension. These tumor cells were stained as described above and used as a positive control.
TCGA patient data analysis
The Cancer Genome Atlas cBioPortal99,100 was accessed on 16 November 2020, 17 November 2020, and 15 April 2021 to determine whether the hypoxia gene signature45 from n = 1100 patients with Breast Invasive Carcinoma (Firehose Legacy dataset; http://www.cbioportal.org/study/summary?id=brca_tcga) correlated with bone46 and lung metastasis signatures47. Genes from the hypoxia, bone metastasis, and lung metastasis gene signatures were entered into the query gene field for cBioPortal separately. The mRNA expression (RNA Seq V2 RSEM) for each gene across all 1100 patients was downloaded and saved in Excel format. For each gene in the three gene signatures, the gene expression provided by RNAseq was normalized to the median expression of that gene across all patients101. We next averaged the expression score for all of the genes in that signature for each individual patient. A log conversion was applied to the normalized, averaged gene signature expression scores. These signature scores were plotted against each other as a correlation graph in the Prism software (Graphpad) with a linear regression curve.
Statistics and reproducibility
To calculate sample size, we previously consulted with a biostatistician within the Department of Radiation Oncology at Stanford University who performed power calculations. The power calculations were based on previously published in vivo studies33. For the transgenic mouse lines, a sample size of n = 10 mice/group will provide 85% power in a two-sided t test with an alpha level 0.05 with a variation of 8.5 and an expected difference of 12. Thus, we sought to collect a minimum of n = 10 mice/group to ensure adequate statistical analysis at end point for these experiments. For all studies, n per group is as indicated in the figure or figure legend and the scatter dot plots indicate the mean of each group and the standard error of the mean. All graphs and statistical analyses were generated using the Prism software (Graphpad). All in vitro and in vivo assays were analyzed for statistical significance using two-tailed Mann–Whitney test, log-rank test, or Fisher’s exact test, as denoted in figure legends. Pearson and Spearman correlation coefficients were calculated for gene signature correlation analyses. P < 0.05 was statistically significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
References
Nakazawa, M. S., Keith, B. & Simon, M. C. Oxygen availability and metabolic adaptations. Nat. Rev. Cancer 16, 663–673 (2016).
Semenza, G. L. & Wang, G. L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 12, 5447–5454 (1992).
Wang, G. L., Jiang, B. H., Rue, E. A. & Semenza, G. L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl Acad. Sci. USA 92, 5510–5514 (1995).
Tian, H., McKnight, S. L. & Russell, D. W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 11, 72–82 (1997).
Gu, Y. Z., Moran, S. M., Hogenesch, J. B., Wartman, L. & Bradfield, C. A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 7, 205–213 (1998).
Gossage, L., Eisen, T. & Maher, E. R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 15, 55–64 (2015).
Huang, L. E., Gu, J., Schau, M. & Bunn, H. F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl Acad. Sci. USA 95, 7987–7992 (1998).
Ohh, M. et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2, 423–427 (2000).
Tanimoto, K., Makino, Y., Pereira, T. & Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 19, 4298–4309 (2000).
Ivan, M. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468 (2001).
Jaakkola, P. et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 (2001).
Schodel, J. et al. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117, e207–e217 (2011).
Mole, D. R. et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 284, 16767–16775 (2009).
Xia, X. & Kung, A. L. Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol. 10, R113 (2009).
Shweiki, D., Itin, A., Soffer, D. & Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 359, 843–845 (1992).
Tsuzuki, Y. et al. Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia-inducible factor-1alpha–> hypoxia response element–> VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer Res. 60, 6248–6252 (2000).
Firth, J. D., Ebert, B. L. & Ratcliffe, P. J. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. J. Biol. Chem. 270, 21021–21027 (1995).
Gleadle, J. M. & Ratcliffe, P. J. Induction of hypoxia-inducible factor-1, erythropoietin, vascular endothelial growth factor, and glucose transporter-1 by hypoxia: evidence against a regulatory role for Src kinase. Blood 89, 503–509 (1997).
Firth, J. D., Ebert, B. L., Pugh, C. W. & Ratcliffe, P. J. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoietin 3’ enhancer. Proc. Natl Acad. Sci. USA 91, 6496–6500 (1994).
Ebert, B. L., Firth, J. D. & Ratcliffe, P. J. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J. Biol. Chem. 270, 29083–29089 (1995).
Courtnay, R. et al. Cancer metabolism and the Warburg effect: the role of HIF-1 and PI3K. Mol. Biol. Rep. 42, 841–851 (2015).
Petrella, B. L., Lohi, J. & Brinckerhoff, C. E. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2 alpha in von Hippel-Lindau renal cell carcinoma. Oncogene 24, 1043–1052 (2005).
Erler, J. T. et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 440, 1222–1226 (2006).
Yang, M. H. et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 10, 295–305 (2008).
Schindl, M. et al. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin. Cancer Res. 8, 1831–1837 (2002).
Dales, J. P. et al. Overexpression of hypoxia-inducible factor HIF-1alpha predicts early relapse in breast cancer: retrospective study in a series of 745 patients. Int. J. Cancer 116, 734–739 (2005).
Bos, R. et al. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 97, 1573–1581 (2003).
Generali, D. et al. Hypoxia-inducible factor-1alpha expression predicts a poor response to primary chemoendocrine therapy and disease-free survival in primary human breast cancer. Clin. Cancer Res. 12, 4562–4568 (2006).
Cox, T. R. et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 522, 106–110 (2015).
Woelfle, U. et al. Molecular signature associated with bone marrow micrometastasis in human breast cancer. Cancer Res. 63, 5679–5684 (2003).
Fallah, J. & Rini, B. I. HIF inhibitors: status of current clinical development. Curr. Oncol. Rep. 21, 6 (2019).
Hosseini, H. et al. Early dissemination seeds metastasis in breast cancer. Nature 540, 552–558 (2016).
Husemann, Y. et al. Systemic spread is an early step in breast cancer. Cancer Cell 13, 58–68 (2008).
Jin, L. et al. Breast cancer lung metastasis: molecular biology and therapeutic implications. Cancer Biol. Ther. 19, 858–868 (2018).
Johnson, R. W., Schipani, E. & Giaccia, A. J. HIF targets in bone remodeling and metastatic disease. Pharmacol. Ther. 150, 169–177 (2015).
Liao, D., Corle, C., Seagroves, T. N. & Johnson, R. S. Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 67, 563–572 (2007).
Lu, X. et al. In vivo dynamics and distinct functions of hypoxia in primary tumor growth and organotropic metastasis of breast cancer. Cancer Res. 70, 3905–3914 (2010).
Hiraga, T., Kizaka-Kondoh, S., Hirota, K., Hiraoka, M. & Yoneda, T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 67, 4157–4163 (2007).
Dunn, L. K. et al. Hypoxia and TGF-beta drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS ONE 4, e6896 (2009).
Sowder, M. E. & Johnson, R. W. Enrichment and detection of bone disseminated tumor cells in models of low tumor burden. Sci. Rep. 8, 14299 (2018).
Guy, C. T., Cardiff, R. D. & Muller, W. J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell Biol. 12, 954–961 (1992).
Fantozzi, A. & Christofori, G. Mouse models of breast cancer metastasis. Breast Cancer Res. 8, 212 (2006).
Dower, C. M., Bhat, N., Wang, E. W. & Wang, H. G. Selective reversible inhibition of autophagy in hypoxic breast cancer cells promotes pulmonary metastasis. Cancer Res. 77, 646–657 (2017).
Vera-Ramirez, L., Vodnala, S. K., Nini, R., Hunter, K. W. & Green, J. E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 9, 1944 (2018).
Ye, I. C. et al. Molecular portrait of hypoxia in breast cancer: a prognostic signature and novel HIF-regulated genes. Mol. Cancer Res. 16, 1889–1901 (2018).
Kang, Y. et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549 (2003).
Minn, A. J. et al. Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 (2005).
Fluegen, G. et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 19, 120–132 (2017).
Erler, J. T. et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44 (2009).
Keith, B., Johnson, R. S. & Simon, M. C. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 12, 9–22 (2011).
Li, J. et al. PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J. Clin. Investig. 121, 4655–4669 (2011).
Kiriyama, T. et al. Transforming growth factor beta stimulation of parathyroid hormone-related protein (PTHrP): a paracrine regulator? Mol. Cell Endocrinol. 92, 55–62 (1993).
Yin, J. J. et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 103, 197–206 (1999).
Sterling, J. A., Edwards, J. R., Martin, T. J. & Mundy, G. R. Advances in the biology of bone metastasis: how the skeleton affects tumor behavior. Bone 48, 6–15 (2011).
Manisterski, M., Golan, M., Amir, S., Weisman, Y. & Mabjeesh, N. J. Hypoxia induces PTHrP gene transcription in human cancer cells through the HIF-2alpha. Cell Cycle 9, 3723–3729 (2010).
Guo, F. et al. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 35, 816–826 (2016).
Masuda, T. et al. ANGPTL2 increases bone metastasis of breast cancer cells through enhancing CXCR4 signaling. Sci. Rep. 5, 9170 (2015).
Seagroves, T. N. et al. VHL deletion impairs mammary alveologenesis but is not sufficient for mammary tumorigenesis. Am. J. Pathol. 176, 2269–2282 (2010).
Parker, K. D., Albeke, S. E., Gigley, J. P., Goldstein, A. M. & Ward, N. L. Microbiome composition in both wild-type and disease model mice is heavily influenced by mouse facility. Front. Microbiol. 9, 1598 (2018).
Fessler, J., Matson, V. & Gajewski, T. F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 7, 108 (2019).
Gopalakrishnan, V., Helmink, B. A., Spencer, C. N., Reuben, A. & Wargo, J. A. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 33, 570–580 (2018).
Champy, M. F. et al. Mouse functional genomics requires standardization of mouse handling and housing conditions. Mamm. Genome 15, 768–783 (2004).
Zhang, H. et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 31, 1757–1770 (2012).
Goto, Y. et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1alpha. Nat. Commun. 6, 6153 (2015).
Wong, C. C. et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl Acad. Sci. USA 108, 16369–16374 (2011).
Dunn, G. P., Old, L. J. & Schreiber, R. D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21, 137–148 (2004).
Andrews, L. P., Yano, H. & Vignali, D. A. A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat. Immunol. 20, 1425–1434 (2019).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Sceneay, J. et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 72, 3906–3911 (2012).
Reynaud, C. et al. Lysyl oxidase is a strong determinant of tumor cell colonization in bone. Cancer Res. 77, 268–278 (2017).
Deep, G. et al. Exosomes secreted by prostate cancer cells under hypoxia promote matrix metalloproteinases activity at pre-metastatic niches. Mol. Carcinog. 59, 323–332 (2020).
Wang, H. et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 27, 193–210 (2015).
Yu-Lee, L. Y. et al. Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFbetaRIII-p38MAPK-pS249/T252RB pathway. Cancer Res. 78, 2911–2924 (2018).
Yu-Lee, L. Y. et al. Bone secreted factors induce cellular quiescence in prostate cancer cells. Sci. Rep. 9, 18635 (2019).
Martin, T. J. & Johnson, R. W. Multiple actions of parathyroid hormone-related protein in breast cancer bone metastasis. Br. J. Pharmacol. https://doi.org/10.1111/bph.14709 (2019).
Fleming, N. I. et al. Parathyroid hormone-related protein protects against mammary tumor emergence and is associated with monocyte infiltration in ductal carcinoma in situ. Cancer Res. 69, 7473–7479 (2009).
Henderson, M. et al. Parathyroid hormone-related protein production by breast cancers, improved survival, and reduced bone metastases. J. Natl Cancer Inst. 93, 234–237 (2001).
Henderson, M. A. et al. Parathyroid hormone-related protein localization in breast cancers predict improved prognosis. Cancer Res. 66, 2250–2256 (2006).
Micucci, C., Matacchione, G., Valli, D., Orciari, S. & Catalano, A. HIF2alpha is involved in the expansion of CXCR4-positive cancer stem-like cells in renal cell carcinoma. Br. J. Cancer 113, 1178–1185 (2015).
Zhang, J. & Zhang, Q. VHL and hypoxia signaling: beyond HIF in cancer. Biomedicines. https://doi.org/10.3390/biomedicines6010035 (2018).
Minervini, G., Pennuto, M. & Tosatto, S. C. E. The pVHL neglected functions, a tale of hypoxia-dependent and -independent regulations in cancer. Open Biol. 10, 200109 (2020).
Okuda, H. et al. The von Hippel-Lindau tumor suppressor protein mediates ubiquitination of activated atypical protein kinase C. J. Biol. Chem. 276, 43611–43617 (2001).
Iturrioz, X. et al. The von Hippel-Lindau tumour-suppressor protein interaction with protein kinase Cdelta. Biochem. J. 397, 109–120 (2006).
Pal, S., Claffey, K. P., Dvorak, H. F. & Mukhopadhyay, D. The von Hippel-Lindau gene product inhibits vascular permeability factor/vascular endothelial growth factor expression in renal cell carcinoma by blocking protein kinase C pathways. J. Biol. Chem. 272, 27509–27512 (1997).
Guo, J. et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 353, 929–932 (2016).
Gould, C. M. & Newton, A. C. The life and death of protein kinase C. Curr. Drug Targets 9, 614–625 (2008).
Newton, A. C. Protein kinase C: poised to signal. Am. J. Physiol. Endocrinol. Metab. 298, E395–E402 (2010).
Hinz, N. & Jucker, M. Distinct functions of AKT isoforms in breast cancer: a comprehensive review. Cell Commun. Signal 17, 154 (2019).
Ryan, H. E. et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 60, 4010–4015 (2000).
Wagner, K. U. et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 25, 4323–4330 (1997).
Davie, S. A. et al. Effects of FVB/NJ and C57Bl/6J strain backgrounds on mammary tumor phenotype in inducible nitric oxide synthase deficient mice. Transgenic Res. 16, 193–201 (2007).
Gruber, M. et al. Acute postnatal ablation of Hif-2alpha results in anemia. Proc. Natl Acad. Sci. USA 104, 2301–2306 (2007).
Haase, V. H., Glickman, J. N., Socolovsky, M. & Jaenisch, R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc. Natl Acad. Sci. USA 98, 1583–1588 (2001).
Biju, M. P. et al. Vhlh gene deletion induces Hif-1-mediated cell death in thymocytes. Mol. Cell Biol. 24, 9038–9047 (2004).
Smith, B. A. et al. Targeting the PyMT oncogene to diverse mammary cell populations enhances tumor heterogeneity and generates rare breast cancer subtypes. Genes Cancer 3, 550–563 (2012).
Johnson, R. W. et al. The primary function of gp130 signaling in osteoblasts is to maintain bone formation and strength, rather than promote osteoclast formation. J. Bone Min. Res. 29, 1492–1505 (2014).
Rankin, E. B. et al. Inactivation of the arylhydrocarbon receptor nuclear translocator (Arnt) suppresses von Hippel-Lindau disease-associated vascular tumors in mice. Mol. Cell Biol. 25, 3163–3172 (2005).
Johnson, R. W. et al. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. 71, 822–831 (2011).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013).
Li, B. et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513, 251–255 (2014).
Acknowledgements
The authors wish to thank Mr. Joshua Johnson for bone histological processing, sectioning, and H&E staining. V.M.T. and R.W.J. are supported by NIH award R00CA194198 (to R.W.J.), and L.A.V., M.E.C., and R.W.J. are supported by DoD Breakthrough Award W81XWH-18-1-0029 (to R.W.J.). M.R. is supported by NIH award R00CA201304. This project was supported in part by scholarship funds from the NIH award P30CA068485 Vanderbilt-Ingram Cancer Center Support Grant, which also supports the Translational Pathology Shared Resource. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource, which is supported by P30CA68485 and the Vanderbilt Digestive Disease Research Center (DK058404). The results shown here are in part based on data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Author information
Authors and Affiliations
Contributions
R.W.J. and V.M.T conceived the original idea. M.R. provided support and critical feedback to the project and manuscript. R.W.J. supervised the project and provided funding. V.M.T. designed and performed the majority of experiments and data analysis and wrote the manuscript. L.A.V. maintained animal colonies and performed the majority of the animal collections. K.P.S. and C.D.O. performed experiments and assisted V.M.T. L.H. and C.P. performed the lung and liver histology analysis. M.E.C. performed the immunofluorescence experiments and analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary handling editors: Ruby Huang and Eve Rogers. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Todd, V.M., Vecchi, L.A., Clements, M.E. et al. Hypoxia inducible factor signaling in breast tumors controls spontaneous tumor dissemination in a site-specific manner. Commun Biol 4, 1122 (2021). https://doi.org/10.1038/s42003-021-02648-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02648-3
- Springer Nature Limited
This article is cited by
-
DMT1-dependent endosome-mitochondria interactions regulate mitochondrial iron translocation and metastatic outgrowth
Oncogene (2024)
-
Epigenetic regulation of breast cancer metastasis
Cancer and Metastasis Reviews (2024)
-
Amino Acid Metabolism in Bone Metastatic Disease
Current Osteoporosis Reports (2023)
-
SUBATOMIC: a SUbgraph BAsed mulTi-OMIcs clustering framework to analyze integrated multi-edge networks
BMC Bioinformatics (2022)