
Abstract
Purpose of Review
This review provides an overview of the mechanisms induced by hypoxia that take place within primary tumors and within the bone microenvironment, and that promote bone metastasis.
Recent Findings
In primary tumors, hypoxia stimulates metastasis by promoting vascularization, epithelial-mesenchymal transition, immunosuppression, and by inducing pre-metastatic lesions in distant tissues. In bones, local hypoxia maintains bone homeostasis and has been recently shown to promote tumor colonization, to reactivate dormant tumor cells, and strikingly, to promote systemic cancer growth and dissemination beyond the skeleton.
Summary
Hypoxia is recognized as a critical component of the tumor microenvironment that strongly affects tumor growth and metastasis. However, our knowledge of the mechanisms induced locally by hypoxia in the skeleton that promotes tumor dissemination in bones and beyond is only emerging. Recent studies identified LIFR and CXCL12 as important factors downstream of hypoxia signaling in the bone, but other molecules and mechanisms will be likely discovered in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metastasis is the main cause of cancer-related deaths. Bone metastasis occurs frequently in several types of cancer. It is not curable, and causes intractable pain. Metastasis is a complex process (reviewed in [1•]) that starts with the acquisition of an invasive behavior by cancer cells in the primary tumors. Cancer cells that enter the blood or the lymph circulation are called circulating tumor cells (CTCs). Most CTCs do not survive but a small fraction succeeds to colonize organs distant from primary tumors. In bones, disseminated tumor cells (DTCs) can become quiescent and remain dormant for several years. Eventually, cancer cells start proliferating again to generate osteolytic (associated with bone destruction) or osteoblastic (associated with bone formation) bone metastases. At each step of the dissemination process, cancer cells interact closely with their microenvironment. The tumor stroma is composed of several cell types (immune cells, cancer-associated fibroblasts, endothelial and mesenchymal cells, adipocytes, etc.) but also the extra cellular matrix (ECM), and plays essential functions in tumor progression [2•]. Solid tumors inherently present regions of low oxygen tensions (hypoxia), and hypoxia is also an important component of the tumor microenvironment that influences primary tumor growth and metastasis. While the role of hypoxia in tumor growth and metastasis has been relatively well described at the primary tumor site, the role of tissue hypoxia present within the bone microenvironment that influences bone metastasis has been much less studied. This review highlights recent advances in our understanding of the mechanisms induced by hypoxia that promote bone metastasis, in primary tumors located outside the skeleton, and locally in the bone microenvironment.
Hypoxia Is an Important Component of Solid Tumors
The hypoxic tumor microenvironment is a common feature of solid tumors that results in part from increased oxygen consumption by a large number of highly proliferating tumor cells, and a compromised oxygen supply due to leaky and abnormal blood vessels commonly found in tumors [3, 4]. The cellular response to hypoxia is mediated by a family of three transcription factors called hypoxia-inducible factors (HIF1, 2, and 3), first identified as regulator of erythropoietin (Epo) gene expression, almost 30 years ago [5, 6].
HIFs are composed of a stable beta subunit (HIF1beta), associated with one of the three labile alpha subunits (HIF1alpha, HIF2alpha or HIF3alpha). The amounts of alpha subunits present in cells are regulated by the partial pressure of oxygen at a post-translational level [7, 8]. In presence of oxygen, prolyl-hydroxylase domain proteins (PHDs) hydroxylate HIF-alpha subunits. This leads to their recognition by von Hippel-Lindau (VHL), which forms with Elongin B, C and Cullin 2 a complex carrying an E3 ubiquitin ligase activity. Polyubiquitination of HIF-alpha results in its degradation by the proteasome. Notably, O2-independent regulations of HIF signaling also exist in cancer cells. Hydroxylation of HIF-alpha by PHDs requires alpha-ketoglutarate (a key intermediate in the Tricarboxylic Acid (TCA) or Krebs cycle), in addition to oxygen. In tumor cells, dysfunctions of the cell metabolism that lead to TCA cycle intermediates accumulation, or intracellular cysteine depletion, have been reported to inhibit PHD activity [9, 10]. HIF signaling can also be stimulated independently of O2 levels by several pathways activated in cancer cells. The transcription and translation of HIF-alpha subunits are increased by the mammalian target of rapamycin (mTOR) downstream of phosphoinositide 3-kinase (PI3K)–protein kinase B (PKB/AKT) signaling [11], which can occur independently of oxygen in cancers [12]. In triple negative breast cancer cells, the breast tumor kinase (BRK) has been reported to phosphorylate HIF1alpha, enhancing its stability in presence of oxygen [13]. Lastly, recent work has identified the ubiquitin C-terminal hydrolase-L1 (UCHL1) that is able to deubiquitinate HIF1alpha, thus preventing its degradation by the proteasome [14]. When stabilized, HIF transcription factors bind to a specific DNA sequence called hypoxia response element (HRE), located in regulatory regions of HIF target genes. This binding leads to the recruitment of the transcriptional co-activators p300/CBP (CREB binding protein), inducing gene transcription. The association of HIF with its cofactors is regulated by O2-dependent hydroxylation mediated by the factor inhibiting HIF (FIH) [7]. Although most bona fide target genes of HIFs are transactivated, HIFs can also inhibit gene expression, presumably through the induction of non-coding RNA or repressors.
HIF signaling is essential for the cellular adaptation to hypoxia, in tumors like in normal tissues. HIF is required in hypoxic cells to generate energy (adenosine triphosphate-ATP) and to limit the production of reactive oxygen species, and in hypoxic tissues to increase blood supply by promoting angiogenesis and erythropoiesis (reviewed in [15]). It is therefore not surprising that several studies using various experimental models demonstrated that HIF signaling promotes tumor development and progression (reviewed in [16]). In mouse models of hypopharyngeal cancer, lung cancer and breast cancer, tumor growth and metastasis are promoted by overexpression of HIF [17, 18], and reduced by the inhibition of HIF using dominant-negative HIF1alpha, or silencing RNA, or pharmacological inhibitors [17, 19,20,21].
Many genes regulated by hypoxia are directly involved in cancer progression, by promoting various mechanisms such as anaerobic glycolysis, angiogenesis, dormancy, cell survival, cell stemness, and metastasis [8]. The deleterious role of the HIF target genes encoding vascular endothelial growth factor (VEGF), angiopoietin, and platelet-derived growth factor (PDGF) that induce angiogenesis in tumors, and thus supply nutrients and survival cues to promote tumor progression has been particularly well described [22]. Another important adaptation of the tumor cells to hypoxia is the stimulation of the anaerobic metabolism to increase the production of ATP in absence of oxygen (reviewed in [23]). Interestingly, hypoxia also induces histone lysine demethylase 3A (KDM3A) expression, which could increase genome instability and favor the acquisition of metastatic properties by cancer cells [24].
Moreover, several studies indicate that hypoxia inhibits the immune response against tumors. For examples, reduced tumor hypoxia obtained by respiratory hyperoxia in preclinical models increased tumor infiltration by cytotoxic lymphocytes, and decreased infiltration by regulatory T cell, resulting in a better anti-tumoral immune response [25]. Other studies reported that activation of HIF signaling in tumor cells induces a resistance to immune surveillance by cytotoxic T lymphocyte and natural killer (NK) cells (reviewed in [26]). Immunosuppression is achieved through the recruitment of regulatory T cells, marrow-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) at the tumor site, a process that is promoted by hypoxia and HIF via the induction of transforming growth factor beta (TGFbeta), VEGF and several cytokines such as C-C motif chemokine 5 (CCL5) and C-X-C motif chemokine 12 (CXCL12, also called SDF1) [26,27,28,29,30,31,32]. MDSCs and TAMs promote immunosuppression but also produce inflammatory cytokines that are favorable to tumor progression [2, 33]. Activation of HIF signaling in tumor cells also induces the recruitment of mesenchymal stem cells (MSCs), which leads to a bidirectional signaling cascade between breast cancer cells and MSCs that promotes metastasis [34].
Consistent with these pre-clinical data, hypoxia and activation of HIF signaling in primary tumors have been clinically associated with a poor prognosis in patients with various types of cancers [35,36,37]. Several clinical studies also associate HIF1alpha expression with increased dissemination in lymph nodes [38, 39], and in visceral organs [18]. The expression of HIF2alpha in tumors has also been associated with metastasis in the lungs, in breast cancer patients [40, 41].
It has been proposed that hypoxia may induce dormancy in primary tumors by upregulating the expression of genes controlling cell dormancy, such as those coding for NR2F1, DEC2, and p27 (reviewed in [42]). Interestingly, hypoxia-induced dormancy is persistent, even after withdrawing tumor cells from hypoxia. This could explain the presence of “post-hypoxic” dormant DTCs in colonized tissues, which could be responsible for chemotherapy resistance and relapse [43••]. This, along with other mechanisms, could explain why HIF signaling has been reported to promote chemoresistance [44]).
Altogether these clinical and experimental results showed that activation of HIF signaling in the tumor of origin promotes tumor growth and metastasis. The next paragraph focuses on some of the mechanisms induced by HIF signaling in primary tumors that are responsible for cancer cell dissemination.
Hypoxia in Primary Tumors Promotes Metastasis, Including to Bones
At the earliest stage of metastasis, cancer cells acquire invasive and migratory properties. Several studies showed that these properties are associated with epithelial-mesenchymal transition (EMT), a change in cell morphology and junctions with the neighboring cells and with the ECM. Hypoxia and HIF signaling induce EMT and invasive behavior in several cancer models, via direct transcriptional activation of key EMT inducers like TWIST (twist basic helix-loop-helix transcription factor 1), ZEB1 (zinc finger E-box binding homeobox 1) or Snail (snail family transcriptional repressor 1) [18, 45]. In vitro studies showed that hypoxia exposure was sufficient to activate RhoA (Ras homolog gene family, member A)/FAK (Focal adhesion kinase) signaling to induce cancer cell motility [46]. Several genes involved in ECM synthesis and remodeling, which is essential for cancer cell invasion, are regulated by hypoxia: vimentin, fibronectin, matrix metalloproteinase 2 (MMP2), cathepsin D, urokinase plasminogen activator receptor (PLAUR), procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 (PLOD2) and lysyl oxidase (LOX) [47, 48]. As mentioned previously, hypoxia also promotes vascularization allowing tumor cells to disseminate, and favor immune system evasion that is crucial for tumor cell survival in the blood and in colonized tissues.
To enter and then exit the bloodstream, cancer cells must interact with endothelial cells. Interestingly, recent studies showed that extravasation of breast cancer cells is improved after their exposure to hypoxia, through induction of L1 cell adhesion molecule (L1CAM) expression that enhances their interaction with endothelial cells [49]. During this process, hypoxic tumor cells also express angiopoietin-like 4 (ANGPTL4) that inhibits cell-cell interactions between endothelial cells, allowing the tumor cells to migrate through the blood vessel wall [50].
Through systemic signals, primary tumors remotely instigate modifications in tissues targeted by CTCs prior to their arrival, establishing pre-metastatic niches. This process was first identified with the recruitment of bone marrow-derived hematopoietic progenitors expressing VEGF receptor 1 to the site of metastasis, before the arrival of cancer cells [51]. Interestingly, hypoxic breast cancer cells secrete LOX that is a critical mediator of the pre-metastatic niche [52•]. LOX produced by primary breast tumors accumulates in distant sites such as in the lungs, liver and bones. In the lungs, it has been shown that LOX and type IV collagens cross-linking occurs, allowing the recruitment of bone marrow-derived cells (BMDCs) that produce MMP2. In turn, MMP2 production favors the infiltration of more BMDCs, and the colonization of the tumor cells [53]. In the bones, LOX produced by hypoxic tumors induces pre-metastatic lesions that favor bone colonization by stimulating bone resorption mediated by osteoclasts [54••]. A recent study similarly highlights the role of LOX in colorectal cancer metastasis to bone. In this case, LOX produced by primary tumors induces bone lesions through interleukin 6 (IL6) induction, which promotes osteoclast-mediated bone resorption [55••].
A growing literature focuses on tumor cell-derived microvesicles or exosomes that are thought to precondition the metastatic niche by carrying lipids, proteins, mRNA and miRNA [56, 57]. These microvesicles are also involved in the tropism of certain breast cancer cell lines toward the lungs or the bones [58•]. Specific patterns of integrins expression in exosomes determine organ specificity, but other mechanisms may exist. Interestingly, hypoxia promotes the formation of microvesicles in a HIF dependent manner [59], and exposure of non-hypoxic breast cancer cells with microvesicles derived from hypoxic cells promotes their invasive and metastatic behavior [60]. This could result from microvesicles carrying microRNA that enhance HIF signaling activity [61], or carrying HIF1alpha itself [62]. Thus, microvesicles could be critical mediators in hypoxia-induced metastasis. Whether hypoxia-induced microvesicles influence bone metastasis specifically remains unknown.
Since Paget’s seed and soil hypothesis described more than a century ago, we know that the tropism of tumor cells for a specific tissue is not random. The interaction between the receptor CXCR4 (C-X-C chemokine receptor type 4) expressed by tumor cells and its ligand CXCL12 present in colonized tissues illustrates this notion. CXCR4 belongs to a molecular signature associated with breast cancer metastasis to bone, and its overexpression in breast cancer cells promotes bone metastasis [63]. Bone is an environment particularly rich in CXCL12. It has been shown that cancer-associated fibroblasts produce CXCL12 that primes cancer cells for metastasis to the bone [64]. Interestingly, hypoxia and HIF1alpha induce the expression of both CXCL12 and CXCR4 in a variety of cell types [29, 65], indicating that the CXCL12/CXCR4 pathway may be important downstream of HIF signaling in promoting bone metastasis. The role of this pathway may not be specific to bone metastasis though, since other organs such as the lungs express high levels of CXCL12. Accordingly, inhibiting CXCR4 expression or blocking its function has been shown to inhibit lung metastasis in orthotopic cancer models [66, 67]. Another gene expressed by tumor cells that is associated with poor prognosis and bone metastases in cancer patients is osteopontin (OPN) [68, 69], which promotes tumor growth and metastasis in mouse models [70, 71]. OPN is involved at the primary site in tumor vascularization [72], the recruitment of BMDCs [73], and the transformation of normal fibroblasts into cancer-associated fibroblasts [74], thus promoting cancer progression. Interestingly, OPN expression is induced by hypoxia [72]. It would be therefore interesting to evaluate the possibility that OPN produced in primary tumors could mediate bone metastasis downstream of HIF.
Hypoxia in cancer cells at the primary tumor site has been reported to generally promote metastasis [16], including to bones [17, 19]. It is still unclear, however, whether some of the mechanisms mediated by HIF could induce specifically metastasis to the bone, but not to other tissues. At the opposite end of the metastatic process, the bone microenvironment also presents hypoxic regions. Although it has been proposed that combined treatments of mice with inhibitor of HIF1alpha and TGFbeta reduce bone metastasis through a dual action on tumor cells and on the bone microenvironment [19], the role of bone-specific hypoxia in bone metastasis has been addressed directly only recently and is presented below.
Role of the Hypoxic Bone Microenvironment in Bone Metastasis: Homing to the Bone
While the role of HIF signaling in tumor cells and in the tumor microenvironment at the primary tumor site has been relatively well described, the role of hypoxia that is restricted to the bone microenvironment and its influence in bone metastasis has been investigated only recently, and much is still unknown. Important gradients of hypoxia have been observed in the bone [75]. Although further studies are required to reliably measure oxygen tensions in long bones, available data indicate a range of 6 to 1% of O2, depending on the areas considered (reviewed in [76]). The specific organization of the vasculature of the long bones may be responsible for these important oxygen gradients.
The bone microenvironment comprises many different cell types (osteoblasts, osteoclasts, endothelial cells, adipocytes, MSCs, hematopoietic stem cell (HSCs) and other hematopoietic cells, etc.) that could affect individually or collectively bone metastasis. In fact, several crosstalks have been reported between osteoblasts, osteoclasts, endothelial and hematopoietic cells, and thus, affecting one cell type most likely affects indirectly the others. The endosteal surface below the growth plate presents low oxygenation levels. Endosteal arterioles found in this region are closely associated with skeletal progenitor cells, and invalidation of HIF1alpha in endothelial cells impairs bone formation [77]. Several studies have also demonstrated that HIF signaling in osteoblasts promotes simultaneously bone formation and bone angiogenesis [78,79,80]. Therefore, these data indicate that local hypoxia in the bone is responsible for the coupling of angiogenesis and osteogenesis, a process that could be mediated by osteoblast-derived VEGF [81]. Other genes activated by HIF signaling such as Cxcl12 and Cxcr4 could be involved in this coupling, since CXCL12-CXCR4 signaling in osteoblasts promotes bone formation [82] and this pathway has been reported to stimulate angiogenesis in other tissues. However, local hypoxia could also promote bone formation directly, independently of pro-angiogenic functions. Low oxygen tensions likely maintain the stemness and survival of MSCs and osteoblast progenitors (osteoprogenitors) [83], a mechanism that could explain why HIF signaling is required for bone formation. Consistent with these data, we observed that activation or suppression of HIF signaling in osterix-expressing cells increases or decreases the number of osteoprogenitors, respectively [84••].
Since tumor cells colonize tissues via blood vessels, one could assume that increasing bone mass and angiogenesis would increase bone metastasis. Consistent with this idea, a recent study showed that genetic inactivation of the beta-2 adrenergic receptor in osteoblasts leads to reduced VEGF production and to decreased bone colonization by breast cancer cells [85]. Our results also support this idea, since we showed that activation of HIF signaling in osteoprogenitor cells, which increases locally bone mass and angiogenesis, promotes bone metastasis [84••]. But the level of vascularization cannot alone explain everything, since some cancers do not disseminate to bones in spite of a high vascularization, and since other highly vascularized organs are less frequently colonized compared to bones. Hence, other factors independent of blood vessel numbers influence locally bone metastasis.
The ability of specific cancers to disseminate to specific tissues may depend on the relative levels of receptors and ligands expressed by the tumor cells and the targeted tissues, respectively. For instance, tumor cells from breast and prostate cancers but also melanoma present a high tropism for bone. These cells have in common that they highly express the receptors CXCR4 [86] and receptor activator of NF-kappaB (RANK) [87]. On the other hand, CXCL12 and RANK Ligand (RANKL), their respective ligands, are highly expressed in bones, and several publications suggested that these ligands act as chemoattractant of these tumor cells [88,89,90]. Of note, although robust data demonstrate that RANKL and CXCL12 act as chemoattractant in vitro, visualizing chemoattraction in a mouse skeleton is difficult, and our interpretation of the mechanisms of action of these cytokines in vivo is complicated due to their pleiotropic roles. In any event, it is interesting that the expression of both CXCL12 and RANKL is induced by hypoxia in various cell types [65, 87, 91,92,93,94,95]. In bones, we found that osteoprogenitor cells are highly hypoxic and express very high levels of CXCL12, in a HIF1alpha-dependent fashion [84••]. More importantly, we demonstrated that increased colonization of the bones by breast cancer cells, observed upon activation of HIF signaling in osteoprogenitors, involves CXCL12-CXCR4 signaling. Whether the hypoxic bone microenvironment favors bone metastasis through increased production of RANKL in osteoblast lineage cells remains unknown. Similarly, since hypoxia and HIF signaling promote the expression of key bone metastasis inducers such as LOX [54••], PLOD2 [47], RANKL [87, 88], OPN [72] and CXCL12 [29] in cancer cells, it would be interesting to assess whether the same factors could be also induced in the cells of the bone microenvironment.
Activation of HIF signaling in osteoblast lineage cells could also influence more directly the homing of tumor cells to the bone. Several publications showed that DTCs interact with osteoblasts. In prostate cancer, DTCs have been shown to home in the osteoblast niche and compete with local HSCs for the support provided by osteoblast lineage cells [90, 96]. In breast cancer, DTCs have been shown to disseminate near osteoblasts rather than near osteoclasts, and to form adherent junctions with them [97•]. When osteoblast-derived N-cadherin interacts with cancer-derived E-cadherin, this activates the mTOR pathway in cancer cells, promoting metastasis progression [97•]. We observed that breast cancer cells preferentially colonize regions in the bones enriched in osterix-positive osteoprogenitors that are highly hypoxic, and that express high levels of CXCL12 [84••]. While CXCL12 alone might explain this specific tropism for the hypoxic osteoprogenitors, it is possible that breast cancers cells interact more directly with hypoxic osteoprogenitors, independently of CXCL12. Since mTOR activates HIF signaling in tumor cells [12], these cellular interactions could lead to a particularly robust activation of HIF signaling in tumor cells induced concomitantly by hypoxia and mTOR, leading to a particularly aggressive metastatic progression.
Role of the Hypoxic Bone Microenvironment in Bone Metastasis: Metastatic Outgrowth and Dormancy
HIF signaling could also affect bone metastasis by modulating bone resorption. Bone resorption is the process by which osteoclasts degrade the bone matrix. In osteolytic bone metastasis, bone resorption promotes locally the growth of bone metastases by releasing, in the bone marrow, growth factors present in the bone matrix [98]. Hypoxia could stimulate bone resorption directly by stimulating osteoclast activity [99], or indirectly by increasing the pool of osteoblasts secreting osteoclastogenic factors such as IL6 and RANKL. Indeed, both IL6 and RANKL expression can be induced by HIF signaling [26, 69, 88] and are known to promote bone metastasis though activation of osteoclastogenesis that stimulates tumor cell growth locally [98, 100•]. On the other end, activation of HIF signaling in osteoblasts also strongly activates osteoprotegerin expression, the decoy receptor for RANKL that inhibits osteoclastogenesis [80]. More work is therefore needed to evaluate the impact of bone resorption downstream of HIF signaling in bone metastasis.
In contrast to osteolytic metastasis, in osteoblastic metastasis, tumor cells promote osteoblast differentiation and activity, which produce growth factors for cancer cells. Tumor cells synthetize factors like BMPs, Wnt, Endothelin 1 or PDGF, to promote osteoblast differentiation and the accumulation of newly formed bone [101,102,103]. In turn, osteoblast lineage cells produce TGFbeta, IGF and FGF that favor tumor cell survival and growth. Hypoxia could be involved in this process, as some of these factors have been reported to be induced by hypoxia and/or HIF signaling, such as BMP [104], Endothelin 1 [105], or PDGF [106].
Although much less studied than the mechanisms involved in the homing and the growth of DTCs in bones, the control of dormancy of tumors cells present in the bone marrow is essential, since it is for a large part responsible for metastatic relapse that can occur years after cancer therapy [42]. A recent publication unraveled the role of hypoxia in regulating breast cancer cell dormancy in the bone marrow [107••]. Leukemia inhibitory factor (LIF, an IL6 family member) is produced by osteoblast lineage cells and induces the dormancy and quiescence of DTCs upon binding to its receptor LIFR. Hypoxia exposure downregulates the signaling pathway downstream of LIFR, which relieves breast cancer cells from dormancy and induces their proliferation in bones [107••]. This study is concordant with the observation that HIF-induced urokinase-type plasminogen activator receptor (uPAR encoded by Plaur) inhibits dormancy [108, 109]. Hence, as opposed to the role of hypoxia at the primary tumor site that could favor dormancy, these studies propose that dormant DTCs in the bone could be reactivated by local hypoxia exposure. Interestingly, dormant breast cancer cells have been localized in proximity to cells expressing high levels of CXCL12 in the bone marrow [110]. Hence, HIF-induced CXCL12 expression in osteoprogenitor cells could also participate to the control of tumor cell dormancy in the skeleton.
Role of the Hypoxic Bone Microenvironment Beyond Bone Metastasis
A large part of the bone microenvironment is made of hematopoietic cells, and important crosstalks exist between these cells and that of the osteoblast lineage. Notably, osteoblasts control various aspects of hematopoiesis [111]. In particular, they have been shown to support the HSC niche [112]. Interestingly, hypoxia is detected in HSC niches [113, 114] and is important for HSC maintenance [115]. Moreover, activation of HIF2alpha signaling in the osteoblast lineage promotes red blood cells formation through increased production of erythropoietin (EPO) [78]. Consequently, hypoxia could locally affect bone metastasis by altering the anti-tumoral immune response, and/or tumor oxygenation and thus tumor growth, by altering the number of erythrocytes. Since hematopoietic cells are mobilized in the bloodstream, these effects mediated by HIF signaling in osteoblast lineage cells could also affect tumor progression outside the skeleton. Recent publications report that osteoblasts can promote the activation and the recruitment of immune-suppressive cells in mammary and lung adenocarcinomas, which leads to increased tumor growth [116••, 117••]. Importantly, we recently discovered that activation of HIF signaling in osteoprogenitors strikingly promotes breast cancer growth and dissemination outside the skeleton [84••]. We identified that this systemic effect is mediated at least in part by a direct communication between osteoblasts and breast cancer cells through CXCL12/CXCR4 signaling. However, additional mechanisms involved in the systemic control mediated by the activation of HIF signaling in osteoblast lineage cells may exist. It would be interesting for instance, to investigate whether local hypoxia in the bone marrow affects the immune response against tumors, in a systemic fashion.
Conclusions
It is generally admitted that the large majority of solid tumors are hypoxic, and that activation of HIF signaling promotes their growth and dissemination. However, the published data supporting this notion were obtained with a limited number of models, focused essentially on breast cancer, and on prostate cancer to a lesser extent. Whether the findings reported are valid in all types of cancers is unknown. Moreover, the majority of the conclusions drawn on metastasis are based on studies focused on lung metastasis. Only a few studies addressed the impact of HIF signaling specifically on tumor dissemination to bones, using intracardiac and orthotopic injections of tumor cell lines where HIF signaling was over-activated or suppressed. While these studies provided valuable information on tumor colonization of the bones, these experiments did not address the question of whether HIF signaling in primary tumors controls spontaneous metastasis to bones, in a spontaneous cancer model. Our lack of data on spontaneous bone metastasis results for a large part from the very limited number of spontaneous cancer models, and by the fact that in mice, unlike in humans, spontaneous bone metastasis seems a rare event that is particularly hard to detect.
The role of HIF signaling in the bone marrow in bone metastasis remained elusive until recently. The recent discoveries that HIF can reactivate dormant metastatic breast cancer cells that reside in bones, and that hypoxic osteoprogenitors promote the colonization of the skeleton by breast cancer cells and their metastatic outgrowth, emphasize the critical importance of tissue hypoxia in one of the organ most frequently colonized in cancer patients. But we also learned that the importance of tissue hypoxia in bones extends beyond the skeleton. These discoveries are important since they indicate that targeting HIF signaling specifically in bones might be a good strategy to treat cancer patients with or without bone metastases.
In spite of this recent significant progress in our understanding of the role of HIF signaling in bone metastasis, several questions remain. We still do not know whether hypoxia affects tumor dissemination to bones particularly, compared to other organs. We also do not know whether HIF-independent mechanisms activated by hypoxia contribute to bone metastasis. The relative importance of HIF2alpha versus HIF1alpha in this process is also not totally clear. Opposite roles have been often reported between HIF1alpha and HIF2alpha in cancer biology (reviewed in [118]), with HIF1alpha often identified as the important pro-tumorigenic factor. We observed that activation of HIF1alpha, but not HIF2alpha, in osteoprogenitor cells promotes systemic breast cancer growth and dissemination [84••]. However, this does not exclude a specific role of HIF2alpha in the control of bone metastasis, which may affect mechanisms and/or cell types that are different than those influenced by HIF1alpha.
Most of our knowledge of the role of HIF signaling in bone metastasis comes from animal models, and the clinical relevance of the majority of the experimental findings presented here still needs to be evaluated. Several clinical studies show that hypoxia and HIF expression in primary tumors are associated with metastasis and poor prognosis [8, 18]. Interestingly, HIF1alpha expression in primary breast tumors was associated with the presence of micrometastases in the bone marrow of patients [119]. However, whether activation of HIF signaling in primary tumors of cancer patients specifically influences bone metastasis remains unclear, due the limited clinical data available. Similarly, the clinical relevance of the role of the hypoxic bone microenvironment in bone metastasis observed in mice is unknown, due at least in part to the difficulty to measure accurately oxygen tension in bones in vivo.
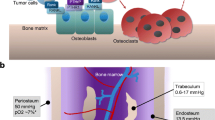
Even in animals models, the role of the tumor microenvironment is particularly complex to study at the primary tumor sites and in bones, since it involves many different cellular, molecular and structural components, but also because its spatial and temporal organization evolves continuously. In the bone microenvironment, like in primary tumors located outside the skeleton, the role of HIF signaling has been evaluated in a very limited number of cell types comprised in the tumor stroma. While some of the factors and cellular mechanisms triggered by hypoxia have emerged (Fig. 1), further studies are necessary and will very likely contribute to improve our understanding of bone metastasis, and may lead to new therapeutic strategies to limit tumor spread in patients.

HIF signaling controls bone metastasis in primary tumors and in the bone microenvironment. In breast cancer, HIF signaling instigates bi-directional signals between the breast and the skeleton that promote systemic tumor growth and dissemination. In mammary tumors, HIF induces the secretion of LOX, priming the bone microenvironment for tumor cell invasion. In bones, activation of HIF signaling in osteoprogenitors induces CXCL12 secretion that promotes mammary tumor growth and metastasis to bones and to other tissues. Tissue hypoxia in bones also alleviates tumor dormancy through repression of LIFR signaling. See text for more details. (Credit: this figure is original but the breast, blood vessel and bone diagrams were adapted from Servier Medical Art illustrations licensed under Creative Commons Attribution 3.0 Unported License)
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529(7586):298–306. This review article presents our current understanding of the general mechanisms involved in tissue colonization by tumor cells.
• Werb Z, Lu P. The role of stroma in tumor development. Cancer J. 2015;21(4):250–3. This review article presents an interesting overview of recent advances in our understanding of the influence of non-tumoral cells in cancer.
Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58(7):1408–16.
Vaupel P, Mayer A, Höckel M. Tumor hypoxia and malignant progression. Meth Enzymol. 2004;381:335–54.
Goldberg MA, Dunning SP, Bunn HF. Regulation of the erythropoietin gene: evidence that the oxygen sensor is a heme protein. Science. 1988;242(4884):1412–5.
Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88(13):5680–4.
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402.
Semenza GL. The hypoxic tumor microenvironment: a driving force for breast Cancer progression. Biochim Biophys Acta. 2016;1863(3):382–91.
Briggs KJ, Koivunen P, Cao S, Backus KM, Olenchock BA, Patel H, et al. Paracrine induction of HIF by glutamate in breast Cancer: EglN1 senses cysteine. Cell. 2016;166(1):126–39.
Kaelin WG. Cancer and altered metabolism: potential importance of hypoxia-inducible factor and 2-oxoglutarate-dependent dioxygenases. Cold Spring Harb Symp Quant Biol. 2011;76:335–45.
Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22(20):7004–14.
Agani F, Jiang BH. Oxygen-independent regulation of HIF-1: novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr Cancer Drug Targets. 2013;13(3):245–51.
Lin A, Li C, Xing Z, Hu Q, Liang K, Han L, et al. The LINK-A lncRNA activates normoxic HIF1α signalling in triple-negative breast cancer. Nat Cell Biol. 2016;18(2):213–24.
Goto Y, Zeng L, Yeom CJ, Zhu Y, Morinibu A, Shinomiya K, et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1α. Nat Commun. 2015;6:6153.
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408.
Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352(6282):175–80.
Hiraga T, Kizaka-Kondoh S, Hirota K, Hiraoka M, Yoneda T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007;67(9):4157–63.
Yang M-H, Wu M-Z, Chiou S-H, Chen P-M, Chang S-Y, Liu C-J, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10(3):295–305.
Dunn LK, Mohammad KS, Fournier PGJ, McKenna CR, Davis HW, Niewolna M, et al. Hypoxia and TGF-beta drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS One. 2009;4(9):e6896.
Xiang L, Gilkes DM, Chaturvedi P, Luo W, Hu H, Takano N, et al. Ganetespib blocks HIF-1 activity and inhibits tumor growth, vascularization, stem cell maintenance, invasion, and metastasis in orthotopic mouse models of triple-negative breast cancer. J Mol Med. 2014;92(2):151–64.
Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105(50):19579–86.
Charles N, Ozawa T, Squatrito M, Bleau A-M, Brennan CW, Hambardzumyan D, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010;6(2):141–52.
Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13(6):472–82.
Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci U S A. 2012;109(49):E3367–76.
Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. 2015;7(277):277ra30.
Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41(4):518–28.
Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A. 2013;110(43):17450–5.
Chaturvedi P, Gilkes DM, Takano N, Semenza GL. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc Natl Acad Sci U S A. 2014;111(20):E2120–9.
Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E, et al. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13(3):206–20.
Foster JG, Wong SCK, Sharp TV. The hypoxic tumor microenvironment: driving the tumorigenesis of non-small-cell lung cancer. Future Oncol. 2014;10(16):2659–74.
Lee Y-H, Bae HC, Noh KH, Song K-H, Ye S-k, Mao C-P, et al. Gain of HIF-1α under normoxia in cancer mediates immune adaptation through the AKT/ERK and VEGFA axes. Clin Cancer Res. 2015;21(6):1438–46.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48.
Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. 2016;126(10):3672–9.
Chaturvedi P, Gilkes DM, Wong CCL, Kshitiz, Luo W, Zhang H, et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Invest. 2013;123(1):189–205.
Dales J-P, Garcia S, Meunier-Carpentier S, Andrac-Meyer L, Haddad O, Lavaut M-N, et al. Overexpression of hypoxia-inducible factor HIF-1alpha predicts early relapse in breast cancer: retrospective study in a series of 745 patients. Int J Cancer. 2005;116(5):734–9.
Generali D, Berruti A, Brizzi MP, Campo L, Bonardi S, Wigfield S, et al. Hypoxia-inducible factor-1alpha expression predicts a poor response to primary chemoendocrine therapy and disease-free survival in primary human breast cancer. Clin Cancer Res. 2006;12(15):4562–8.
Schindl M, Schoppmann SF, Samonigg H, Hausmaninger H, Kwasny W, Gnant M, et al. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin Cancer Res. 2002;8(6):1831–7.
Jin Y, Wang H, Ma X, Liang X, Liu X, Wang Y. Clinicopathological characteristics of gynecological cancer associated with hypoxia-inducible factor 1α expression: a meta-analysis including 6,612 subjects. PLoS One. 2015;10(5):e0127229.
Yamamoto Y, Ibusuki M, Okumura Y, Kawasoe T, Kai K, Iyama K, et al. Hypoxia-inducible factor 1alpha is closely linked to an aggressive phenotype in breast cancer. Breast Cancer Res Treat. 2008;110(3):465–75.
Luan Y, Gao C, Miao Y, Li Y, Wang Z, Qiu X. Clinicopathological and prognostic significance of HIF-1α and HIF-2α expression in small cell lung cancer. Pathol Res Pract. 2013;209(3):184–9.
Wang HX, Qin C, Han FY, Wang XH, Li N. HIF-2α as a prognostic marker for breast cancer progression and patient survival. Genet Mol Res. 2014;13(2):2817–26.
Bragado P, Sosa MS, Keely P, Condeelis J, Aguirre-Ghiso JA. Microenvironments dictating tumor cell dormancy. Recent Results Cancer Res. 2012;195:25–39.
•• Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol. 2017;19(2):120–32. This article demonstrates that tissue hypoxia in primary tumors gives rise to a subpopulation of DTCs that remain dormant even when they no longer are exposed to hypoxia. These post-hypoxic dormant tumor cells could evade chemotherapy and lead to disease relapse.
Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci U S A. 2014;111(50):E5429–38.
Luo D, Wang J, Li J, Post M. Mouse snail is a target gene for HIF. Mol Cancer Res. 2011;9(2):234–45.
Gilkes DM, Xiang L, Lee SJ, Chaturvedi P, Hubbi ME, Wirtz D, et al. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signaling in breast cancer cells. Proc Natl Acad Sci U S A. 2014;111(3):E384–93.
Gilkes DM, Bajpai S, Wong CC, Chaturvedi P, Hubbi ME, Wirtz D, et al. Mol Cancer Res. 2013;11(5):456–66.
Pouysségur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441(7092):437–43.
Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XHF, Lee DJ, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156(5):1002–16.
Zhang H, Wong CCL, Wei H, Gilkes DM, Korangath P, Chaturvedi P, et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2012;31(14):1757–70.
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–7.
• Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017;168(4):670–91. This review article presents a good overview of the various biological mechanisms involve in the dissemination and the metastatic outgrowth of primary carcinomas.
Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15(1):35–44.
•• Cox TR, Rumney RMH, Schoof EM, Perryman L, Høye AM, Agrawal A, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature. 2015;522(7554):106–10. This article presents evidence that hypoxia induces primary breast cancer cells to secrete LOX, which circulates through blood to reach the skeleton, to induce osteolytic lesions by activating osteoaclasts prior to tumoral colonization of the bones. These pre-metastatic lesions facilitate bone metastasis.
•• Reynaud C, Ferreras L, Di Mauro P, Kan C, Croset M, Bonnelye E, et al. Lysyl oxidase is a strong determinant of tumor cell colonization in bone. Cancer Res. 2017;77(2):268–78. This article shows that LOX production by colorectal cancer cells promotes bone metastasis and osteolytic lesions by activating osteoclasts through LOX-driven secretion of IL8.
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816–26.
Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18(6):883–91.
• Hoshino A, Costa-Silva B, Shen T-L, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–35. This article shows that tumor cells that have a tropism for a specific tissue produce specific exosomes (carrying specific integrins) that selectively fuse with cells present in the targeted tissue to establish a pre-metastatic niche.
King HW, Michael MZ, Gleadle JM. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer. 2012;12:421.
Wang T, Gilkes DM, Takano N, Xiang L, Luo W, Bishop CJ, et al. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc Natl Acad Sci U S A. 2014;111(31):E3234–42.
Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood. 2014;124(25):3748–57.
Aga M, Bentz GL, Raffa S, Torrisi MR, Kondo S, Wakisaka N, et al. Exosomal HIF1α supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene. 2014;33(37):4613–22.
Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3(6):537–49.
Zhang XHF, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013;154(5):1060–73.
Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10(8):858–64.
Liang Z, Wu T, Lou H, Yu X, Taichman RS, Lau SK, et al. Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res. 2004;64(12):4302–8.
Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005;65(3):967–71.
Hou X, Wu X, Huang P, Zhan J, Zhou T, Ma Y, et al. Osteopontin is a useful predictor of bone metastasis and survival in patients with locally advanced nasopharyngeal carcinoma. Int J Cancer. 2015;137(7):1672–8.
Tuck AB, O'Malley FP, Singhal H, Harris JF, Tonkin KS, Kerkvliet N, et al. Osteopontin expression in a group of lymph node negative breast cancer patients. Int J Cancer. 1998;79(5):502–8.
Rodrigues LR, Teixeira JA, Schmitt FL, Paulsson M, Lindmark-Mänsson H. The role of osteopontin in tumor progression and metastasis in breast cancer. Cancer Epidemiol Biomark Prev. 2007;16(6):1087–97.
Shevde LA, Samant RS, Paik JC, Metge BJ, Chambers AF, Casey G, et al. Osteopontin knockdown suppresses tumorigenicity of human metastatic breast carcinoma, MDA-MB-435. Clin Exp Met. 2006;23(2):123–33.
Raja R, Kale S, Thorat D, Soundararajan G, Lohite K, Mane A, et al. Hypoxia-driven osteopontin contributes to breast tumor growth through modulation of HIF1α-mediated VEGF-dependent angiogenesis. Oncogene. 2014;33(16):2053–64.
McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133(6):994–1005.
Sharon Y, Raz Y, Cohen N, Ben-Shmuel A, Schwartz H, Geiger T, et al. Tumor-derived osteopontin reprograms normal mammary fibroblasts to promote inflammation and tumor growth in breast cancer. Cancer Res. 2015;75(6):963–73.
Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508(7495):269–73.
Johnson RW, Sowder ME, Giaccia AJ. Hypoxia and bone metastatic disease. Curr Osteoporos Rep. 2017;15(4):231–8.
Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507(7492):323–8.
Rankin Erinn B, Wu C, Khatri R, Wilson Tremika LS, Andersen R, Araldi E, et al. The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell. 2012;149(1):63–74.
Wang Y, Wan C, Deng L, Liu X, Cao X, Gilbert SR, et al. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Investig. 2007;117(6):1616–26.
Wu C, Rankin EB, Castellini L, Fernandez-Alcudia J, LaGory EL, Andersen R, et al. Oxygen-sensing PHDs regulate bone homeostasis through the modulation of osteoprotegerin. Genes Dev. 2015;29(8):817–31.
Maes C, Goossens S, Bartunkova S, Drogat B, Coenegrachts L, Stockmans I, et al. Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. EMBO J. 2010;29(2):424–41.
Zhu W, Liang G, Huang Z, Doty SB, Boskey AL. Conditional inactivation of the CXCR4 receptor in osteoprecursors reduces postnatal bone formation due to impaired osteoblast development. J Biol Chem. 2011;286(30):26794–805.
Stegen S, van Gastel N, Eelen G, Ghesquière B, D'Anna F, Thienpont B, et al. HIF-1α promotes glutamine-mediated redox homeostasis and glycogen-dependent bioenergetics to support Postimplantation bone cell survival. Cell Metab. 2016;23(2):265–79.
•• Devignes CS, Aslan Y, Brenot A, Devillers A, Schepers K, Fabre S, et al. HIF signaling in osteoblast-lineage cells promotes systemic breast cancer growth and metastasis in mice. Proc Natl Acad Sci U S A. 2018. This article demonstrates for the first time that osteoblast lineage cells directly promote breast cancer growth and metastasis outside the skeleton. This systemic pro-tumorigenic effect is mediated at last in part by CXCL12, which is secreted in large amounts by hypoxic osteoprogenitor cells upon activation of HIF signaling.
Mulcrone PL, Campbell JP, Clément-Demange L, Anbinder AL, Merkel AR, Brekken RA, et al. Skeletal colonization by breast Cancer cells is stimulated by an osteoblast and β2AR-dependent neo-Angiogenic switch. J Bone Miner Res. 2017;32(7):1442–54.
Muller A. Involvement of chemokine receptors in breast cancer metastasis. 2001;410.
Tang Z-N, Zhang F, Tang P, Qi X-W, Jiang J. Hypoxia induces RANK and RANKL expression by activating HIF-1α in breast cancer cells. Biochem Biophys Res Commun. 2011;408(3):411–6.
Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. 2006;440(7084):692–6.
Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62(6):1832–7.
Wang N, Docherty FE, Brown HK, Reeves KJ, Fowles ACM, Ottewell PD, et al. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: evidence from in vivo models. J Bone Miner Res. 2014;29(12):2688–96.
Dandajena TC, Ihnat MA, Disch B, Thorpe J, Currier GF. Hypoxia triggers a HIF-mediated differentiation of peripheral blood mononuclear cells into osteoclasts. Orthod Craniofac Res. 2012;15(1):1–9.
Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002;46(10):2587–97.
Martin SK, Diamond P, Williams SA, To LB, Peet DJ, Fujii N, et al. Hypoxia-inducible factor-2 is a novel regulator of aberrant CXCL12 expression in multiple myeloma plasma cells. Haematologica. 2010;95(5):776–84.
Park HJ, Baek KH, Lee HL, Kwon A, Hwang HR, Qadir AS, et al. Hypoxia inducible factor-1alpha directly induces the expression of receptor activator of nuclear factor-kappaB ligand in periodontal ligament fibroblasts. Mol Cells. 2011;31(6):573–8.
Zou D, Zhang Z, He J, Zhang K, Ye D, Han W, et al. Blood vessel formation in the tissue-engineered bone with the constitutively active form of HIF-1alpha mediated BMSCs. Biomaterials. 2012;33(7):2097–108.
Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121(4):1298–312.
• Wang H, Yu C, Gao X, Welte T, Muscarella Aaron M, Tian L, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015;27(2):193–210. This article presents one of the first demonstrations that osteoblast lineage cells directly promote breast cancer metastasis to bone. Cell adhesion between osteoblasts and tumor cells mediated by cadherins leads to the activation of mTOR signaling in tumor cells in bones, enhancing metastatic growth and osteolysis.
Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2(8):584–93.
Hulley PA, Bishop T, Vernet A, Schneider JE, Edwards JR, Athanasou NA, et al. Hypoxia-inducible factor 1-alpha does not regulate osteoclastogenesis but enhances bone resorption activity via prolyl-4-hydroxylase 2. J Pathol. 2017;242(3):322–33.
• Luo X, Fu Y, Loza AJ, Murali B, Leahy KM, Ruhland MK, et al. Stromal-initiated changes in the bone promote metastatic niche development. Cell Rep. 2016;14(1):82–92. This article shows that senescent osteoblasts establish metastatic niches by secreting IL6, which promotes locally osteoclastogenesis and bone metastasis.
Dai J, Hall CL, Escara-Wilke J, Mizokami A, Keller JM, Keller ET. Prostate cancer induces bone metastasis through Wnt-induced bone morphogenetic protein-dependent and independent mechanisms. Cancer Res. 2008;68(14):5785–94.
Ustach CV, Huang W, Conley-LaComb MK, Lin C-Y, Che M, Abrams J, et al. A novel signaling axis of matriptase/PDGF-D/ß-PDGFR in human prostate cancer. Cancer Res. 2010;70(23):9631–40.
Yin JJ, Mohammad KS, Käkönen SM, Harris S, Wu-Wong JR, Wessale JL, et al. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc Natl Acad Sci U S A. 2003;100(19):10954–9.
Wang H, Lindborg C, Lounev V, Kim J-H, McCarrick-Walmsley R, Xu M, et al. Cellular hypoxia promotes heterotopic ossification by amplifying BMP signaling. J Bone Miner Res. 2016;31(9):1652–65.
Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol Chem. 2001;276(16):12645–53.
ten Freyhaus H, Dagnell M, Leuchs M, Vantler M, Berghausen EM, Caglayan E, et al. Hypoxia enhances platelet-derived growth factor signaling in the pulmonary vasculature by down-regulation of protein tyrosine phosphatases. Am J Respir Crit Care Med. 2011;183(8):1092–102.
•• Johnson RW, Finger EC, Olcina MM, Vilalta M, Aguilera T, Miao Y, et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol. 2016;18(10):1078–89. This article reports that tissue hypoxia presents in the bone microenvronement releases disseminated breast cancer cells from dormancy, and induces them to proliferate and to form a bone metastasis by repressing LIFR-induced dormancy.
Mauro CD, Pesapane A, Formisano L, Rosa R, D'Amato V, Ciciola P, et al. Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Sci Rep. 2017;7(1):9388.
Vishnoi M, Peddibhotla S, Yin W, Scamardo AT, George GC, Hong DS, et al. The isolation and characterization of CTC subsets related to breast cancer dormancy. Sci Rep. 2015;5:17533.
Price TT, Burness ML, Sivan A, Warner MJ, Cheng R, Lee CH, et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci Transl Med. 2016;8(340):340ra73.
Panaroni C, Tzeng YS, Saeed H, Wu JY. Mesenchymal progenitors and the osteoblast lineage in bone marrow hematopoietic niches. Curr Osteoporos Rep. 2014;12(1):22–32.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–6.
Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. 2013;15(5):533–43.
Parmar K, Mauch P, Vergilio J-A, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci. 2007;104(13):5431–6.
Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391–402.
•• Engblom C, Pfirschke C, Zilionis R, Da Silva Martins J, Bos SA, Courties G, et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science. 2017;358(6367). This article shows that primary lung adenocarcinoma remotely activates bone formation and osteoblasts that mobilize a specific subset of pro-tumorigenic neutrophils in the circulation. These cells then colonize lung tumors promoting their growth. This works in one of the first to demonstrate that osteoblasts remotely promote tumor growth.
•• Rossnagl S, Altrock E, Sens C, Kraft S, Rau K, Milsom MD, et al. EDA-Fibronectin Originating from Osteoblasts Inhibits the Immune Response against Cancer. PLoS Biol. 2016;14(9):e1002562. This article provides the first demonstration that osteoblasts remotely promote tumor progression (in mouse models of melanoma and breast cancer) by mobilizing specific myeloid-derived cells that inhibit the immune response against tumors.
Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22.
Woelfle U, Cloos J, Sauter G, Riethdorf L, Janicke F, van Diest P, et al. Molecular signature associated with bone marrow micrometastasis in human breast cancer. Cancer Res. 2003;63(18):5679–84.
Acknowledgements
This manuscript was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM) and the Fondation ARC pour la Recherche Contre le Cancer (Project # R10081HS and C14007HS to S. Provot). The work performed by the authors cited here also received the support of the University of California San Francisco Academic Senate Committee on Research (Fund # 34935/500394 to S. Provot), and the Association le Cancer du Sein Parlons-en (Pink Ribbon award to S. Provot). C.S. Devignes was supported by the French Ministry of Research, and the Fondation ARC, and was recipient of the Société Française de Biologie des Tissus Minéralisés (SFBTM) award. The authors would like to acknowledge the investigators who generated the work highlighted in this review and to apologize for the published articles that were not cited due to space constraints.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Claire-Sophie Devignes, Yetki Aslan, and Sylvain Provot declare no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Molecular Biology of Bone Metastasis
Rights and permissions
About this article

Cite this article
Devignes, CS., Aslan, Y. & Provot, S. Signaling Pathways Underlying Bone Metastasis: Hypoxia Signaling in Bone Metastasis and Beyond. Curr Mol Bio Rep 4, 69–79 (2018). https://doi.org/10.1007/s40610-018-0090-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-018-0090-1