
Abstract
Phedimus latiovalifolius (Y.N.Lee) D.C.Son & H.J.Kim is exclusively distributed in the high mountains in the Korean Peninsula, mainly along the Baekdudaegan mountain range. Despite its morphological and distributional distinction from other Phedimus Raf. species, its taxonomic identity and phylogenetic relationship with congeneric species remain unclear. This study employs genotyping-by-sequencing-derived genome-wide single nucleotide polymorphisms to establish the monophyly of P. latiovalifolius and its relationship with closely related species. Genetic diversity and population differentiation of P. latiovalifolius are also assessed to provide baseline genetic information for future conservation and management strategies. Our phylogenetic analyses robustly demonstrate the monophyletic nature of P. latiovalifolius, with P. aizoon (L.) ‘t Hart identified as its closest sister lineage. There is no genetic evidence supporting a hybrid origin of P. latiovalifolius from P. aizoon involving either P. ellacombeanus (Praeger) ‘t Hart or P. kamtschaticus (Fisch.) ‘t Hart. Population genetic analyses reveal two major groups within P. latiovalifolius. A higher genetic variation is observed in P. ellacombeanus than in the congeneric species. Notably, most of the genetic variation exists within P. latiovalifolius populations. Given its distribution and the potential role of Baekdudaegan as an East Asian Pleistocene refugia, P. latiovalifolius could be considered rare and endemic, persisting in the refugium across glacial/interglacial cycles.
Similar content being viewed by others
Introduction
The genus Phedimus Raf.1 encompasses a group of succulent stonecrop plants within the family Crassulaceae, comprising approximately 20 species globally. Traditionally, Phedimus was classified under the broad genus Sedum L., however, this genus segregated from Sedum, because the species within Phedimus are distinguished by flattened leaves featuring serrate or crenate margins and longitudinally costate or sub-smooth testa. In contrast, Sedum species exhibit terete or semiterete leaves with entire margins and reticulate or papillate-reticulate testa2,3,4. Molecular phylogenetic studies consistently support the monophyly of Phedimus and its separation from Sedum sensu stricto3,4,5,6,7. Within the genus Phedimus, two subgeneric groups are recognized: Phedimus and Aizoon (L.K.A. Koch ex Schönland) Ohba & Turland5,6. Subgenus Phedimus comprises approximately five species with purple or white petals occurring in the Eurasian regions from the Aegean to South Persia and North Caucasus. These species are mainly diploid, except for P. spurius (M.Bieb.) ‘t Hart, exhibiting a simple descending dysploidy series with base chromosome numbers (x) ranging from x = 7, 6, to 52,8. In contrast, subgenus Aizoon includes between 12 and 15 species with yellow petals ranging from East Europe in the South Urals to the Far East. Aizoon species have a base chromosome number of x = 8, with extensive polyploidy and aneuploidy reported9,10,11,12.
The subgenus Aizoon exhibits distinct morphological and karyological characteristics, yet the differentiation of its component species poses challenges due to the uniformity of floral traits and significant variability in vegetative features complicated further by extensive polyploidy and aneuploidy. This has led to intricate issues in defining species and understanding interspecific relationships9,10,11,12,13. In the context of Korea, species recognition within subgenus Aizoon has been inconsistent among authors, ranging from one14 to six15 species. Presently, eight species are recognized in Korea, constituting nearly half of the 15 species in subgenus Aizoon. This includes the recent recognition of P. latiovalifolius16, previously misreported as P. latiovalifolium17. The identified species are P. aizoon, P. ellacombeanus, P. kamtschaticus, P. middendorffianus (Maxim.) 't Hart, P. takesimensis (Nakai) 't Hart, P. zokuriensis (Nakai) 't Hart, and P. daeamensis T.Y.Choi & D.C.Son. Notably, five of these species are narrow endemics with limited geographical distributions: the evergreen creeping P. takesimensis on Ulleung Island, P. zokuriensis and P. latiovalifolius in central Korea, P. daeamensis in Mt. Daeam (Gangwon-do Province), and P. ellacombeanus in southern Korea. In contrast, P. aizoon and P. kamtschaticus are widely distributed throughout the Korean Peninsula, as well as in Russia, China, Mongolia, and Japan. P. middendorffianus, found in northern Korea, exhibits a broader distribution throughout Russia, China, and Japan.
Phedimus latiovalifolius is exclusively found in an elevation of approximately 1000 m above sea level (a.s.l.) within the narrow confines of the east-central Korean Peninsula. This region is part of the main mountain range and watershed crest line known as the Baekdudaegan (hereafter referred to as the 'BDDG'). The BDDG, which contains 14 subsidiary mountain ranges, runs through almost the entire peninsula for over 1400 km (701 km within South Korea), stretching from Mt. Baekdu (alt. 2744 m) to the north to Mt. Jiri (alt. 1915 m) to the south18. Although it occupies only approximately 2.6% of the total land and 4% of the total forest area of the South Korean Peninsula, the BDDG is a well-known biodiversity hotspot with high species richness and endemism, harboring approximately one-third (ca. 1326 species in the South Korean part of this mountain range) of the total flora of the Korean Peninsula. Of nearly 1300 species, 108 native to the BDDG are endemic to the Korean Peninsula, and 56 rare species are listed around the BDDG19.
Initially described as Sedum latiovalifolium Y. Lee, P. latiovalifolius was identified on Geumdaebong Peak on Mt. Taebaek, Korea, at an altitude of around 1300 m a.s.l. The distinctiveness of P. latiovalifolius was recognized by Lee (1992)16, who highlighted its unique features such as the broadly ovate leaves forming a rosette on the terminal stem (Fig. 1) and its restricted distribution to Geumdaebong (Figs. 1A–C and 2), although he suggested its alliance to Sedum ellacombeanum (≡ current P. ellacombeanus). Lee20 later proposed a potential hybrid origin hypothesis involving Sedum aizoon (≡ P. aizoon) and S. ellacombeanum (≡ P. ellacombeanus) or S. kamtschaticum (≡ P. kamtschaticus), as P. aizoon and P. kamtschaticus coexist with P. latiovalifolius on Geumdaebong Peak. While P. latiovalifolius is limited to elevations above 1000 a.s.l. within a narrow range in Korea, its putative parental species, P. aizoon, and P. kamtschaticus, exhibit wide altitudinal and geographical ranges in East Asia21,22.

Photographs of the Phedimus latiovalifolius found in the type locality, Geumdaebong Peak (A-C; 1345 m a.s.l.), and two other localities, Mt. Dosol (D; 1029 m a.s.l.) and Mt. Seorak (E and F; 1010 m a.s.l.). Photo credit: Seung-Chul Kim.

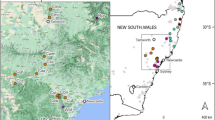
Geographic distributions of the Phedimus latiovalifolius populations and sympatric Phedimus species. Populations are coded by different icons and colors representing the sampling localities, species, and sizes (see Table 1 for population codes).
Once considered synonymous with P. ellacombeanus (Praeger) 't Hart8, P. latiovalifolius was only recently recognized as a distinct Korean endemic species. Morphometric analysis of Phedimus species in Korea did not initially identify P. latiovalifolius as a distinct species, associating it with P. kamtschaticus in the UPGMA phenogram23. P. kamtschaticus displayed varied morphological characteristics with a wide range of geographic and ecological distribution clustered with few other Phedimus species (P. zokuriensis, P. ellacombeanus, and P. takesimensis). P. latiovalifolius revealed the morphological closeness to the southern population of P. kamtschaticus, while it was distantly placed from P. aizoon. Yoo and Park24 refuted the hybrid origin of P. latiovalifolius between P. aizoon and P. kamtschaticus based on morphology (18 characters) and isozyme data (ten isozyme loci). P. latiovalifolius neither clustered with its putative parental species, P. aizoon or P. kamtschaticus in phenetic analysis, nor shared the high-frequency marker alleles with them in isozyme data. Recently, Moon and Jang25 reported that P. latiovalifolius, P. takesimensis, and P. middendorffianus should be recognized as distinct taxa based on their morphological characteristics. Molecular phylogenetic studies on interspecific relationships within the subgenus Aizoon species in Korea have encountered challenges in establishing robust phylogenetic relationships26,27. For instance, Seo et al. (2020)26 conducted a phylogenetic analysis of five Korean taxa (P. takesimensis, P. aizoon, P. kamtschaticus, P. ellacombeanus, and P. latiovalifolius) to assess the anagenetic speciation of P. takesimensis on Ulleung Island. However, the overall lack of resolution and support, along with insufficient sampling, prevented the determination of the closest sister species to P. latiovalifolius. Recently, Kim et al. (2023)27 explored overall species relationships within the subgenus Aizoon using complete plastomes and nrDNA ITS sequences. Despite these efforts, neither the monophyly of P. latiovalifolius nor the phylogenetic relationships within Aizoon were adequately addressed, attributed to insufficient sampling and/or limited tree resolution and node support.
The main goal of this study was to investigate the monophyly of P. latiovalifolius and determine its origin by inferring its phylogenetic relationships with closely related sympatric congeneric species. This study included the populations of the putative parental species, P. aizoon, P. kamtschaticus, and P. ellacombeanus as well as the populations of P. latiovalifolius to test its hybrid origin hypothesis suggested by Lee20 (See Fig. 2 and Table 1 for sampling details). In addition, we estimated the genetic diversity and population differentiation of P. latiovalifolius and other Phedimus species, utilizing 6642 genotyping-by-sequencing (GBS)-derived single nucleotide polymorphisms (SNPs) at the genome-wide level. GBS involves restriction enzymes for genome complexity reduction and next-generation sequencing (NGS) for SNP discovery and genotyping28. GBS method is suitable for population, taxonomic, and phylogenetic studies29,30, offering a cost-effective, high-throughput sequencing method for various species, including apple, barley, Brassica, maize, rice, wheat, and chickpea31,32,33,34,35,36,37. The findings from this study provide essential genetic information for establishing conservation and management strategies for highly vulnerable mountain species affected by climate change in Korea.
Results
Phylogenetic trees derived from SNPs of GBS analysis
Phylogenetic analyses were conducted based on 6642 SNPs obtained from GBS analysis to elucidate the relationships among 101 accessions of P. latiovalifolius and other Phedimus species. The analyses employed ML IQ-TREE (Fig. 3) and SVDQuartets (Fig. 4) trees. P. latiovalifolius emerged as the sole monophyletic species with robust bootstrap support (100% BS for each tree), while the species identified as P. aizoon, P. kamtschaticus, and P. ellacombeanus were not monophyletic. Both ML and SVDQuartets trees indicated P. aizoon as the sister species to P. latiovalifolius, although P. aizoon was not monophyletic. In the ML tree, the paraphyletic Geumdaebong Peak population (PA_KDB), inclusive of the three accessions of PA_JBS, was identified as the sister to P. latiovalifolius (100% BS). The SVDQuartets tree suggested a similar relationship with 99% BS. Further examination within P. latiovalifolius revealed, albeit weakly supported, two major lineages in both phylogenetic trees (except several accessions placed outside of two major lineages on ML tree). The first lineage (BS < 50% on ML and 73% BS on SVDQuartets trees) primarily comprised the Geumdae-bong Peak populations (PL_KDB and PL_KDBS). The second lineage included the Mt. Seorak population, coupled with the Manhang-jae Pass population (86% BS on the ML tree and BS < 50% on the SVDQuartets tree). The SVDQuartets tree (Fig. 4) suggested that the Mt. Dosol (PL_DSS) and Daegwallyeong Pass (PL_DGR) populations were embedded within the clade of PL_KDB and PL_KDBS.

Maximum likelihood (ML) tree produced by IQ-TREE and visualized by the Interactive Tree of Life (iTOL) based on the GBS-derived 6642 SNPs for 101 accessions of P. latiovalifolius and sympatric Phedimus species. Numbers on branches are bootstrap support (BS) values of > 50% with 1000 bootstrap replicates.

SVDQuartets bootstrap consensus tree generated PAUP and visualized by the Interactive Tree of Life (iTOL) based on the GBS-derived 6642 SNPs for 101 accessions of P. latiovalifolius and sympatric Phedimus species. Numbers on branches are bootstrap support (BS) values of > 50% with 100 bootstrap replicates.
Genetic diversity and analysis of molecular variation
The genetic diversity of P. latiovalifolius, excluding two small populations (sample size of ≤ 3), was assessed using STACKS (Table 2). Expected heterozygosity ranged from 0.2073 (PL_MHJ) to 0.2309 (PL_KDBS), with a mean of 0.2190. Nucleotide diversity (π) varied, with the PL_SRK population displaying the highest (0.2397) and PL_MHJ the lowest (0.2192), averaging at 0.2296. For comparative analysis with congeneric species, two populations of P. aizoon from Gangwon-do Province exhibited the lowest mean expected heterozygosity (0.1783) and π (0.1906). Conversely, two populations of P. ellacombeanus from the southern Korean Peninsula displayed the highest expected heterozygosity (0.2214) and nucleotide diversity (0.2351).
Within the mountainous endemic species, P. latiovalifolius, majority of the variation (86%) was observed within populations, while the remaining variation (14%) was distributed among populations (Table 3). Similar levels of genetic variation were found in other Phedimus species; P. aizoon (within-population variation of 80.4%, among population variation of 19.6%), and P. ellacombeanus (80.2%, 19.8%). Across the three species, variations among species, among populations, and within populations were 20%, 20.7%, and 59.3%, respectively. Pairwise genetic differentiation (Fst) was notable between all pairs of P. latiovalifolius and other Phedimus species, ranging from 0.151 (PL_MHJ and PA_KDB) to 0.286 (PL_KDBS and PE_WJB) (Table 4). Within P. latiovalifolius populations, pairwise genetic differentiation ranged from 0.012 (PL_SRK and PL_MHJ) to 0.075 (PL_MHJ and PL_KDBS). Overall, P. aizoon and P. ellacombeanus populations showed comparable levels of intraspecific genetic differentiation (0.096 and 0.065, respectively). Considering both intra- and interspecific genetic differentiation, it is apparent that all populations of P. latiovalifolius were genetically differentiated from congeneric species.
Population structure and gene flow
The PCA results based on 6642 SNP loci elucidated the genetic relationships within P. latiovalifolius populations and their connections with closely related Phedimus species (Fig. 5). P. latiovalifolius exhibited distinctiveness from all other congeneric species, forming an independent cluster on PCA plot. On the other hand, all the other congeneric species were overlapped and clustered together. Within P. latiovalifolius, two discernible genetic clusters emerged. The first cluster comprised populations from Geumdaebong Peak South (PL_KDBS), Geumdaebong (PL_KDB), Mt. Dosol (PL_DSS), and Daegwanryong Pass (PL_DGR), excluding one accession of PL_KDB. The second cluster encompassed two geographically separate populations: Mt. Seorak (PL_SRK) and Manhangjae Pass (PL_MHJ). For P. aizoon, two populations (excluding several accessions of PA_JBS) appeared isolated from other congeneric species, namely P. kamtschaticus, P. ellacombeanus, and P. latiovalifolius. All but one population (PK_SDR) of P. kamtschaticus, and the geographically separated coastal P. ellacombeanus, grouped together. The northernmost population of P. kamtschaticus (PK_SDR) exhibited some distance from the P. kamtschaticus and P. ellacombeanus clusters. The Mantel test revealed no significant correlations between geographic and genetic distances for the six populations of P. latifovalifolius (r = 0.1, p = 0.85) (Fig. 6). The demographic history of P. latifovalifolius inferred from folded SNP frequency spectra (SFS) indicated a bottleneck in its effective population size between 100 and 1000 thousand years ago (kya) based on 95% confidence interval, with a bottom approximately 200 kya (Fig. 7). The effective populations of P. latifovalifolius appeared as maintained quite stable since a recovery from the bottleneck, approx. 100 kya.

Score plot for principal component analysis (PCA) for 101 accessions of P. latiovalifolius and sympatric Phedimus species based on the GBS-derived 6642 SNPs.

Relationship between geographic distance (km) and genetic distances (Nei’s Gst) among the populations of P. latiovalifolius.

Inferred demographic history of P. latiovalifolius based on 54 individuals. Orange line: median of 200 inferences based on subsampling. Dark grey lines: 12.5% and 87.5% confidence interval of the inference. Light grey lines: 2.5% and 97.5% confidence interval of the inference.
The population genetic structure was assessed by individual ancestries of 101 accessions of P. latiovalifolius (six populations) and closely related Phedimus species (two populations of P. aizoon, two of P. ellacombeanus, and three of P. kamtschaticus) using ADMIXTURE V1.3.038. Although the optimal K value was determined as K = 3, reflecting the lowest cross-validation error (Supplementary Fig. S1), bar plots of the Q estimates for K = 2, 3, 4, and 5 are presented for comparison (Fig. 8). The partitioned bar plots for all K values reveal distinct non-admixture genetic assignment patterns for P. latiovalifolius populations, while the populations of all other Phedimus species shared the same or admixed genetic clusters in common. Within P. latiovalifolius, two genetic clusters were identified at K = 3, aligning with ML and SVDQuartet trees (Figs. 3 and 4) and PCA results (Fig. 5): one comprising two geographically separated populations (PL_MHJ and PL_SRK), and the other including the remaining populations (PL_DGR, PL_DSS, PL_KDB, and PL_KDBS) except one accession from PL_KDB (PL626204). At K = 3, P. ellacombeanus, two populations of P. kamtschaticus (PK_CRS and PK_KDB), and several accessions of P. aizoon (PA_JBS) shared the same non-admixture genetic assignment. P. aizoon (all PA_KDB accessions and three PA_JBS accessions) and P. kamtschaticus (all PK_SDR accessions) were identified as admixtures of the two genetic clusters. Genetic structure analysis did not differentiate P. kamtschaticus from P. aizoon or P. ellacombeanus.

Stacked bar plots of Q estimates of ancestry fractions using the best K (3) and comparative K (2, 4, and 5) values based on the GBS-derived SNPs.
The best TreeMix graph was chosen as M2 (two migration edges) with the highest value for Δm (the second-order rate of change in likelihood across values of m) in optM, explaining 99.46% variances (Supplementary Fig. S2). The threshold (99.8%) recommended by Pickrell and Pritchard39 for the proportion of explained variance was not achieved for all tested migration events from 0 to 10. The M2 TreeMix graph displayed a topology similar as a species tree (SVDQuartet tree; Fig. 4); i.e., the monophyly of P. latiovalifolius with two major lineages, a close relationship between P. latiovalifolius and P. aizoon (specifically PA_KDB population), and that the remaining species of P. aizoon, P. kamtschaticus, and P. ellacombeanus were intermixed (Fig. 9). Treemix results indicated two gene flow events between P. latiovalifolius and either P. aizoon (PA_KDB population) or P. kamtschaticus (PK_SDR population), corresponding to the population genetic structure analyzed by ADMIXTURE (Fig. 8). The direction of gene flow was PL_KDBS of P. latiovalifolius into PK_SDR of P. kamtschaticus and PA_KDB of P. aizoon into PL_DSS of P. latiovalifolius, which did not support the hypothesis of hybrid origin of P. latiovalifolius between P. aizoon and P. kamtschaticus. Furthermore, we found no indication of any gene flow events, consistent with the hybrid origin hypothesis, either when the number of migration edges in the model increased (Supplementary Fig. S3) or when two migration edges were iterated for ten replicates (Supplementary Fig. S4). None of the possible three- and four-population tests (f3 statistics/f4 statistics) implemented in TreeMix supported gene flow between P. aizoon and P. kamtschaticus involving the hybrid origin of P. latiovalifolius. All significantly negative f3 statistics (Z-score < − 3) as well as negative or positive f4 statistics (Significant Z-score >|3|) did not infer that P. latiovalifolius was the product of admixture between hypothesized parents in its hybrid origin (Supplementary Tables S1, S2, and S3).

A Maximum-likelihood (ML) tree with optimal two migration events (M2) inferred with TreeMix. The scale bar shows 10 times the average standard error of entries in the sample covariance matrix. Two estimated migration events are represented by an arrow and are colored according to their migration weight. B Residual fit plotted from the ML tree in (A). Residuals above zero represent populations that are more closely related to each other and thus are candidates for admixture events.
Discussion
Origin, genetic diversity, and population structure of P. latiovalifolius
Based on the GBS-derived genome-wide SNPs, this study successfully confirmed the monophyly of P. latiovalifolius and determined its sister lineage using concatenation-based ML and coalescent-based SVDQuartets analyses. All 54 accessions of P. latiovalifolius from the six populations (PL_DSS, PL_DGR, PL_MHJ, PL_SRK, PL_KDB, and PL_KDBS) formed a 100% BS-supported clade in both phylogenetic trees (Figs. 3 and 4). Initially, P. latiovalifolius was described as a distinct Phedimus species based on morphological differences in the broadly ovate leaves of the terminal stem arranged in a rosette form, as well as its restricted distribution to Geumdaebong Peak, Mount Taebaek, in Gangwon-do Province. Such morphological and geographical distinctions are now supported by molecular evidence based on its monophyly and genetic differentiation from three representative congeneric species (P. aizoon, P. kamtschaticus, and P. ellacombeanus). Genetic differentiation of P. latiovalifolius was further corroborated unambiguously by genetic structure and PCA analyses (Fig. 5 and 8), as well as pairwise genetic differentiation based on Fst (Table 4). P. latiovalifolius demonstrated the non-admixture genetic assignment patterns of exclusive genetic clusters distinct from other Phedimus species on ADMIXTURE bar plot. PCA score plot also displayed an independent placement of P. latiovalifolius apart from the intermixed cluster of other congeneric species. Thus, this study further confirmed the monophyly of P. latiovalifolius, which was preliminarily suggested based on the limited chloroplast and nrDNA ITS noncoding sequences26,27.
In terms of phylogenetic relationships among closely related Phedimus species, the monophyletic P. latiovalifolius shared the most recent common ancestor with P. aizoon in both ML and SVD Quartets trees, although P. aizoon was not monophyletic. Particularly, P. latiovalifolius was closest to the Geumdaebong Peak population of P. aizoon (PA_KDB), the type locality of P. latifolvalifolius, in the SVD Quartets tree (Fig. 4; 99% BS support). In the ML tree (Fig. 3; 100% BS support), it was also closest to Geumdaebong Peak population (PA_KDB), but inclusive few accessions of the Mt. Jangbyeong (PA_JBS) population of P. aizoon. The nrDNA ITS tree, based on broad sampling, identified several distinct geographical lineages of P. aizoon, suggesting that one geographical lineage in the Korean Peninsula might have been involved in the origin of P. latifovalifolius during the Late Miocene26,27. P. aizoon is a highly polymorphic species with extensive polyploid and aneuploid series that often intergrades with the sympatric, highly variable P. kamtschaticus11,13. The ploidy level of P. latiovalifolius and the specific geographical lineage of P. aizoon that contributes to P. latiovalifolius require further investigation based on broad sampling to fully understand the origin of the high mountain endemic to the Korean Peninsula.
The current results do not support the hybrid origin of P. latiovalifolius between P. aizoon and P. ellacombeanus or between P. kamtschaticus and P. ellacombeanus20, corroborating the previous findings based on morphological and allozyme data24. The M2 TreeMix graph displayed two potential gene flow events between P. latiovalifolius and either P. aizoon (PA_KDB population) or P. kamtschaticus (PK_SDR population) (Fig. 9). The direction of gene flow was PL_KDBS of P. latiovalifolius into PK_SDR of P. kamtschaticus and PA_KDB of P. aizoon into PL_DSS of P. latiovalifolius without any indication of gene flow events consistent with the hybrid origin hypothesis, either when the number of migration edges in the model increased (Supplementary Fig. S3) or when two migration edges were iterated for ten replicates (Supplementary Fig. S4). None of the three- and four-population tests (f3 statistics/f4 statistics) implemented in TreeMix inferred that P. latiovalifolius was the product of admixture involving the hybrid origin of P. latiovalifolius (Supplementary Tables S1, S2, and S3).
Korean Baedudaegan Mountains and a glacial refugium for boreal and temperate species
Phedimus latiovalifolius, now confirmed as a Korean endemic species, occurs disjunctively in narrow ranges of several adjacent high mountains (within the main mountain range and watershed crest line known as the Baekdudaegan, 'BDDG') in the east-central Korean Peninsula. The BDDG is a well-known biodiversity hotspot, harboring high species richness and endemism. Several phylogeographic and population genetic studies have suggested that the BDDG serves as a biodiversity reservoir, and this is associated with its role as a glacial refugium, presenting relevant genetic evidence from several plant species found in the BDDG area18,40,41,42,43,44. In the case of a large assemblage of boreal and temperate plants, it has been suggested that the BDDG should be regarded as glacial refugia in addition to other well-known East Asian Pleistocene refugia (e.g., the Hengduan Mountains, the Nanling Mountains, or the central China Mountains) 45,46,47.
Chung et al. (2017)41 conducted a review of the genetic evidence supporting the role of the BDDG as a glacial refugium for several Korean species (e.g., Kalopanax septemlobus Koidz.48, Pinus koraiensis Siebold & Zucc.49, Quercus variabilis Blume50, Veratrum album ssp. oxysepalum (Turcz.) Hultén51, and 16 other species40). These studies generally revealed the expected genetic pattern, with plants persisting in refugia, consistent with the "southern richness to northern purity" paradigm52 and the basic Expansion–Contraction model (EC model) of Quaternary demography53 explaining the spatial genetic consequences with lower genetic diversity in postglacially colonized northern regions comparing to southern glacial refugia throughout the repeated cycles of latitudinal range shifts. They also exhibited relatively high levels of intrapopulation genetic diversity, ancestral haplotypes, and/or significant numbers of unique haplotypes/alleles. Paleoecological data further supported these genetic studies, indicating that the BDDG sustained a mixture of boreal and temperate forests during the Last Glacial Maximum (LGM).
Phedimus latiovalifolius is distributed disjunctively on several adjacent high mountains in the central BDDG. Given its geographically disjunctive distributional pattern and the potential role of the BDDG as an East Asian Pleistocene refugium, P. latiovalifolius could be considered as a rare, endemic species that persisted in the BDDG refugium through altitudinal shifts throughout glacial/interglacial cycles. The levels of genetic diversity and differentiation in P. latiovalifolius can be compared to those of other plant species mainly occurring in the BDDG41. Although a direct comparison between different types of molecular data based on allozymes41 and genome-wide SNPs by GBS (current study) is not feasible, it seems that the herbaceous perennial P. latiovalifolius (mean He = 0.219) exhibits higher genetic diversity than endemic plants (mean He = 0.063), narrowly distributed plants (mean He = 0.105), and short-lived herbaceous perennials (mean He = 0.096). An allozyme study based on nine populations of P. latiovalifolius sampled from its type locality (Geumdaebong Peak) found a similar level of mean heterozygosity (He = 0.26)24 as the current GBS-based study. Additionally, the same study suggested that narrowly distributed P. latiovalifolius in the BDDG contained a similar level of genetic diversity as more widely distributed and sampled congeners, such as P. aizoon (He = 0.255) and P. kamtschaticus (He = 0.257) (see also He = 0.203 for P. kamtschaticus in earlier Chung et al.’s study54). Therefore, the high genetic diversity of P. latiovalifolius appears to align with the general pattern found in other BDDG plant species. However, the population differentiation measured by Fst for P. latiovalifolius (mean Fst = 0.047) suggests much lower differentiation than that of narrowly distributed (Fst or Gst = 0.242) or short-lived herbaceous perennials (Fst or Gst = 0.233). Thus, the very low population differentiation found in P. latiovalifolius is exceptional for plant species in the BDDG, which typically show low to moderate among-population genetic variability (mean Fst = 0.175). However, this statement might be somewhat misleading, as Fst and Gst range widely, from 0.027 (herbaceous perennial Adenophora grandiflora Nakai; Campanulaceae) to 0.627 (herbaceous perennial Leontice microrryncha S.Moore; Berberidaceae) (see Table 1 of Chung et al., 201740,41). Genetic differentiation greatly depends on breeding systems, pollination and dispersal modes, demographic history, ecological attributes, and many other factors55,56,57. Given the short dispersal distances of seeds via splash rain seed dispersal mechanisms typically found in the genus and family58,59,60, the low level of population differentiation in P. latiovalifolius was unexpected and contrasted with P. takesimensis on Ulleung Island26. We hypothesize that the two geographically disjunct patterns among populations in P. latiovalifolius, corroborated by PCA and genetic structure analyses, could be formed by postglacial contraction (i.e., vicariance resulting from climate-related population fragmentation and local extinction during interglacial periods) rather than by long-distance dispersal. The overall genetic patterns of plant species restricted to the BDDG can be further synthesized based on comparative phylogeographic and population genetic studies, including the diverse life history traits of plant species.
Given its levels of genetic diversity in comparison to more widely occurring congeneric species, P. latiovalifolius appears less likely to go extinct due to genetic factors for conservation perspectives. Furthermore, following a demographic expansion that began 100,000 years ago, the estimated effective population size (Ne) change revealed a stable size for P. latiovalifolius (Fig. 7). However, the abundance of P. latiovalifolius along both sides of hiking trails may negatively affect pollinator visitation and harm plants that support species establishment and growth. In addition, hiking trails may promote the introduction of non-native plant species into natural areas, thereby facilitating competition with P. latiovalifolius and other native plant species. The over-harvesting of this succulent species with high ornamental values in rock gardens and succulent gardens could be an additional threat. The long-term survival of P. latiovalifolius may be negatively impacted by the ongoing climate change, which is expected to result in an upward shifting of the species due to rising temperatures and altered precipitation patterns. For the conservation of P. latiovalifolius, we suggest securing seeds or fragments of plants from two geographically disjunct groups of populations as ex situ strategies. In addition, it is recommended to conduct demographic monitoring of current populations concurrently with ex situ conservation strategies.
In conclusion, we determined the taxonomic identity of P. latiovalifolius by providing extensive genetic evidence for its monophyly based on the genome-wide SNPs derived by GBS. We conducted a large-scale population genetic and phylogenetic analyses of six populations of P. latiovalifolius and compared them to representative populations of other sympatric Phedimus species: P. kamtschaticus, P. aizoon, and P. ellacombeanus. The monophyly of P. latiovalifolius was strongly demonstrated, and its genetic distinctiveness was further unambiguously corroborated by genetic structure, PCA, and genetic differentiation analyses. P. latiovalifolius has been confirmed as a Korean-endemic species that occurs disjunctively in several adjacent high-altitude mountains located within the BDDG on the Korean Peninsula. It can be regarded as a rare endemic species that has persisted in the BDDG refugium throughout glacial/interglacial cycles through altitudinal shifts. Furthermore, the populations of P. latiovalifolius showed a geographically disjunct pattern, presumably formed by post-glacial range contraction rather than by recent long-distance dispersal. Regarding the origin of P. latiovalifolius, the phylogenetic relationships among Phedimus species did not reveal any genetic evidence for the potential hybrid hypothesis previously proposed. Although it displayed genetic closeness to P. aizoon, P. latiovalifolius was not phylogenetically closely related to its purported parent species, without any indication of the admixture product or any gene flow events responsible for its hybrid origin.
Materials and methods
Plant materials
To detect GBS-derived genome-wide SNPs, we sampled 145 wild accessions of Phedimus latiovalifolius (85 accessions), P. aizoon (20), P. ellacombeanus (20), and P. kamtschaticus (20) (Fig. 2 and Table 1). P. latiovalifolius was collected from six populations of Daegwallyeong Pass (population code PL_DGR; 952 m), Geumdaebong Peak, Mt. Taebaek (PL_KDB; 1345 m) (Fig. 1 A-C), the south from Geumdaebong Peak, Mt. Taebaek (PL_KDBS; 1311 m), Manhangjae Pass, Mt. Hambaek (PL_MHJ; 1297 m), Mt. Dosol (PL_DSS; 1029 m) (Fig. 1 D), and Mt. Seorak (PL_SRK; 1010 m) (Fig. 1E,F) in mountainous areas of central Korea. These six populations represented the entire range of P. latiovalifolius populations in South Korea. Two sympatric species, P. aizoon and P. kamtschaticus were collected from the overlapped distribution areas with P. latiovalifolius; two populations of P. aizoon from Mt. Jangbyeong (PA_JBS; 703 m) and Geumdaebong Peak, Mt. Taebaek (PA_KDB; 1345 m), and three populations of P. kamtschaticus from Sottongnyeong (PK_SDR; 98 m), Mt. Cheongryang (PK_CRS; 200 m), and Geumdaebong Peak, Mt. Taebaek (PK_KDB; 1345 m). Two populations of P. ellacombeanus were collected from Wujebong Peak (PE_WJB; 92 m) and Egari (PE_EGR; 21 m) on the southern seashore area of the Korean Peninsula. As Phedimus species are neither highly threatened nor legally protected, permits were not required to be collected. All samples were collected from wild populations on the Korean Peninsula and one representative voucher specimen per each population were deposited at the Ha Eun Herbarium (SKK), Sungkyunkwan University, Korea (see the voucher information in Table 1). Experimental research and field studies on wild plants, including the collection of plant materials, complied Sungkyunkwan University and Korean guidelines and legislation. Species identification was performed by Seung-Chul Kim, an expert in Korean Phedimus species. In this study, we complied with the IUCN Policy Statement on Research Involving Species at Risk of Extinction and the Convention on the Trade in Endangered Species of Wild Fauna and Flora.
DNA isolation, GBS Library construction, sequencing, and variants calling
Total genomic DNA was extracted from silica gel-dried leaves using a DNeasy Plant Mini kit (Qiagen, Valencia, CA, United States) following the manufacturer’s instructions. The extracted DNAs were submitted to Seeders Inc. (Daejeon, Korea) for GBS library construction. Libraries were prepared by the restriction enzyme digestion of DNA with ApeK1 (GCWGC), followed by ligation of barcoded adapters according to a protocol modified by Elshire et al.61. The pooled libraries, each containing either 96 or 49 samples, were sequenced on an Illumina HiSeq X system (Illumina, San Diego, CA, USA) with an average 151 bp length for paired-end reads. Two sequencing runs were conducted consecutively. Sequence data were deposited in the NCBI Sequence Read Archive (SRA BioProject number PRJNA1037601, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1037601).
Raw reads were demultiplexed using barcode sequences associated with each sample to generate separate FASTQ files for all 145 samples. Adapter sequences were removed using Cutadapt v1.8.362, and the demultiplexed reads were trimmed using DynamicTrim (phred score ≥ 20) and LengthSort (short read length ≥ 25 bp) in SolexaQA v1.1363. Poor-quality sequences with Phred quality scores below Q = 20 (or an error probability of 0.05) were removed, and short read lengths of < 25 bases were discarded.
Variant calling was conducted by the DeNovoGBS function implemented in NGSEPcore_4.1.064. Default parameters were used to discover and genotype Single Nucleotide Variants (SNVs): K-mer length 31, the maximum number of read clusters 2,000,000, maximum value allowed for a base quality score, 30; and minimum variant quality, 40. The obtained SNVs were filtered using VCFTOOLS v0.1.1665; the applied parameters were mac (Minor Allele Count) 3, min-meanDP (minimum mean depth values) 3, minQ (minimum Quality value) 30, max-missing (maximum missing genotypes) 0.5, hwe (sites for Hardy–Weinberg Equilibrium) 0.01, and min-r2 (minimum squared correlation coefficient between genotypes) 0.8. A total of 44 samples with extensive missing data (> 50%) were removed to maintain optimal dataset quality. The SNP data matrix, including 6642 SNPs shared by all 13 populations (-r 0.75 and -p 13 applied), was generated by the program ‘populations’ in STACKS v1.4866 in multiple output formats of variant call format (VCF), STRUCTURE, and Phylip. PLINK v1.90b6.2167 was used to convert the SNP data into binary PLINK (.bed; contains genotype information) and ordinary PLINK (.ped, containing variant formation) files of 101 accessions of P. latiovalifolius and closely related Phedimus species for application in population genetics analyses.
Phylogenetic and population genetic analyses
To determine the phylogenetic relationships between P. latiovalifolius and other Phedimus species in Korea, we conducted maximum likelihood (ML) analyses based on genome-wide 6642 SNPs in 101 samples using W-IQ-TREE68,69. Based on the Kim et al.’s nrDNA ITS tree27, P. kamtschaticus sampled from Sottongnyeong, Gangwon-do Province (PK_SDR), was used as the outgroup. Ultrafast bootstrap support (BS) was calculated from 1000 bootstrap replicates to determine clade robustness70. The best-fit substitution models were checked according to the Bayesian information criterion using ModelFinder71 implemented in IQ-TREE. Additionally, a species-partitioned SVDQuartets bootstrap consensus tree72 was generated using the default setting of 100,000 random quartets with the QFM quartet tree search algorithm and bootstrapping of 100 replicates in PAUP 4.0a16972,73. ML IQ-TREE and SVDQuartets phylogenetic trees were visualized using the online tool, the Interactive Tree of Life (iTOL) v674.
The summary statistics of genetic diversity were presented by ‘populations’ in STACKS v1.48. Two populations of P. latiovalifolius, PL_DSS, and PL_DGR, were excluded due to a sample size of ≤ 3. In addition, the diversity statistics for three populations of P. kamtschaticus, PK_SDR, PK_CRS, and PK_KDB, were excluded because of their small population sizes (5, 3, and 4, respectively). Thus, 83 accessions were analyzed for genetic diversity, AMOVA, and Fst. Analyses of molecular variance (AMOVA)75 and pairwise genetic differentiation between and among populations of P. latiovalifolius and congeneric Phedimus species were performed using GENODIVE ver. 3.576. The pairwise correlation between geographic distance and genetic distance (Nei’s Gst) among populations of P. latiovalifolius was calculated using Pearson’s correlation coefficient. The demographic history of P. latiovalifolius was inferred from folded SFSs using Stairway Plot 277. The applied parameters were 6.075 × 10–9 per locus per year for mutation rate (u), 10 years for generation time (g), and 200 inferences that were used for the closely related genus, Rhodiola L. (Crassuraceae)78, as no data were found for Phedimus.
PLINK was used to estimate the population genetic structure and differentiation of the mountainous endemic P. latiovalifolius and congeneric Phedimus species. The eigenvalues and eigenvectors were calculated using principal component analysis (PCA) to examine the genetic similarities and relationships between individuals, and a PCA plot was drawn using the R statistical software (R 4.0.2). Population genetic structure was estimated by the maximum likelihood estimation of individual ancestries using ADMIXTURE ver. 1.3.038. The best K value (the number of populations assumed for which the model had the best predictive accuracy) was chosen based on a low cross-validation error compared with other K values. Stacked bar plots of Q estimates of ancestry fractions were generated for target K values (best K and comparative K values) in R.
We used TreeMix v1.1239 to infer the admixture network among the populations of P. latiovalifolius and the congeneric Phedimus species. Based on the allele frequency of the SNP dataset, TreeMix first builds a Maximum Likelihood estimate of populations, including branch lengths (proportional to the amount of genetic drift between populations), and allows for migration between populations. Model fit was assessed by the degree to which migration edges (signifying gene flow between populations) reduced the residual genetic covariance among populations. To infer the best model, we tested models containing approximately 0 ~ 10 migration edges with ten replicates each. The dataset was filtered by removing the sites with missing data using VCFTOOLS v0.1.1665, and subsampled and bootstrapped with 80% of SNPs. The parameters of -global, -seed $RANDOM, and “-bootstrap -k 30” were used to run TreeMix to build ML trees by resampling blocks of 30 SNPs to ensure independency between blocks. Based on the output files produced by TreeMix, the OptM package79 in R was used to compare the model fit across models of different migration edges and replicates and to estimate the optimal number of migration edges. The R script “plotting_funcs.R” (https://github.com/joepickrell/pophistory-tutorial/blob/master/example2/plotting_funcs.R, accessed on February 16, 2023) was used to visualize the TreeMix graphs with migration results. The dataset was grouped by species and population to calculate all possible f3 (C, A, B) and f4 (A, B, C, D) statistics80 implemented in TreeMix to provide an analysis for comparison with the population admixture.
Data availability
The raw read sequences generated for this study can be found in the Sequence Read Archive (SRA) at the National Center for Biotechnology Information (NCBI) under the BioProject number PRJNA1037601 (Submission ID SUB13962522) with the title of “101 Phedimus GBS”.
References
Rafinesque, C.S. No. VI. Art. 5. Museum of Natural Sciences. in The American Monthly Magazine and Critical Review Vol. 1 (ed. Biglow H.) 438 (Mercein, 1817).
Thiede, J. & Eggli, U. Crassulaceae in The families and genera of vascular plants, Vol. 9. (ed. Kubitzki, K.) 83–118. (Springer, 2007), https://doi.org/10.1007/978-3-540-32219-1_12
Ohba, H., Bartholomew, B. M., Turland, N. J. & Fu, K. New combinations in Phedimus (Crassulaceae). Novon 10, 400–402 (2000).
't Hart, H. Infrafamilial and generic classification of the Crassulaceae in Evolution and systematics of the Crassulaceae (eds. 't Hart, H. & Eggli, U.) 159–172 (Backhuys, Leiden, 1995).
Mayuzumi, S. & Ohba, H. The phylogenetic position of eastern Asian Sedoideae (Crassulaceae) inferred from chloroplast and nuclear DNA sequences. Syst. Bot. 29, 587–598 (2004).
Gontcharova, S. B., Artyukova, E. V. & Gontcharov, A. A. Phylogenetic relationships among members of the subfamily Sedeae (Crassulaceae) inferred from the ITS region sequences of nuclear rDNA. Russ. J. Genet. 42, 654–661 (2006).
Gontcharova, S. B. & Gontcharov, A. A. Molecular phylogeny and systematics of flowering plants of the family Crassulaceae DC. Mol. Biol. 43, 794–803 (2009).
't Hart, H. & Bleij, B. Phedimus. in Illustrated handbook of succulent plants: Crassulaceae (ed. Eggli, U.) 196–203 (Springer, 2003).
Uhl, C. H. & Moran, R. Chromosomes of Crassulaceae from Japan and South Korea. Cytologia 37, 59–81 (1972).
Amano, M. Biosystematic study of Sedum L. subgenus Aizoon (Crassulaceae). I. Cytological and morphological variations of Sedum aizoon L. var. floribundum Nakai. Bot. Mag. Tokyo 103, 67–85 (1990).
Amano, M. & Ohba, H. Biosystematic study of Sedum L. subgenus Aizoon (Crassulaceae). II. Chromosome numbers of Japanese Sedum aizoon var. aizoon. Bot. Mag. Tokyo 105, 431–441 (1992).
Chung, G. Y., Choi, M. J., Nam, B. M. & Choi, H. J. Chromosome numbers of 36 vascular plants in South Korea. J. Asia-Pac. Biodivers. 13, 504–510 (2020).
Chung, Y. H. & Kim, J. H. A taxonomic study of Sedum section Aizoon in Korea. Korean J. Pl. Taxon. 19, 189–227 (1989).
Lee, T.B. Illustrated flora of Korea. 404–408 (Hyangmunsa, 1980).
Oh, S. Y. The phytogeographical studies of family Crassulaceae in Korea. Research Rev. Kyungpook Nat. Univ. 39, 123–159 (1985).
Lee, Y. N. New taxa on Korean flora (4). Korean J. Pl. Taxon. 22, 7–11 (1992).
Son, D. C., Kim, H.-J., Moon, A.-R., Jang, C.-G. & Chang, K. S. A new combination in Phedimus (Crassulaceae), with neotypification of Sedum latifovalifolium. Phytotaxa 278, 294–296 (2016).
Chung, M. Y., López-Pujol, J. & Chung, M. G. Is the Baekdudaegan “the Southern Appalachians of the East”? A comparison between these mountain systems, focusing on their role as glacial refugia. Korean J. Pl. Taxon. 46, 337–347 (2016).
Korea Forest Service. Baek-du-daegan. To Go Beyond the Korean Peninsula and Become Northeast Asia’s Core Ecosystem. (2016).
Lee, T.B. A natural hybrid of the genus Sedum. (S. aizokamtschatica hyb. Nov.). Natural Plant 50, 5–6 (2000). (In Korean)
Fu, K.J., Ohba, H. & Gilbert, M.G. Crassulaceae in Flora of China. (eds. Wu, Z.Y. & Raven, P.H.) 202–268 (Missouri Botanical Garden Press, 2001).
Ohba, H. Crassulaceae in Flora of Japan. (eds. Iwatsuki, K., Boufford, D.E. & Ohba, H.) 139–152 (Heibonsha Ltd., 2002).
Lee, K.-B., Yoo, Y.-G. & Park, K.-R. Morphological relationships of Korean species of Sedum L. subgenus Aizoon (Crassulaceae). Korean J. Plant Tax. 33, 1–15 (2003).
Yoo, Y.-G. & Park, K.-R. A test of the hybrid origin of Korean endemic Sedum latiovalifolium (Crassulaceae). Korean J. Pl. Taxon. 46, 378–391 (2016).
Moon, A. R. & Jang, C. G. Taxonomic study of genus Sedum and Phedimus (Crassulaceae) in Korea based on external morphology. Korean J. Plant Tax. 33, 116–129 (2020).
Seo, H.-S., Kim, S.-H. & Kim, S.-C. Chloroplast DNA insights into the phylogenetic position and anagenetic speciation of Phedimus takesimensis (Crassulaceae) on Ulleung and Dokdo Islands. Korea. PLoS One 15, e0239734 (2020).
Kim, Y. et al. Plastome-based backbone phylogeny of East Asian Phedimus (Subgenus Aizoon: Crassulaceae), with special emphasis on Korean endemics. Front. Plant Sci. 14, 823 (2023).
Davey, J. W. et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 12, 499–510 (2011).
Favre, F., Jourda, C., Besse, P. & Charron, C. Genotyping-by-Sequencing technology in plant taxonomy and phylogeny. Methods Mol. Biol. 2222, 167–178 (2021).
De Luca, D., Del Guacchio, E., Cennamo, P., Paino, L. & Caputo, P. Genotyping-by-sequencing provides new genetic and taxonomic insights in the critical group of Centaurea tenorei. Front. Plant Sci. 14, 1130889 (2023).
Gardner, K. M. et al. Fast and cost-effective genetic mapping in apple using next-generation sequencing. G3 Genes Genomes Genet. 4, 1681–1687 (2014).
Liu, H. et al. An evaluation of genotyping by sequencing (GBS) to map the Breviaristatum-e (ari-e) locus in cultivated barley. BMC Genom. 15, 1–1 (2014).
Bird, K. A. et al. Population structure and phylogenetic relationships in a diverse panel of Brassica rapa L. Front. Plant Sci. 8, 321 (2017).
Su, C. et al. High density linkage map construction and mapping of yield trait QTLs in maize (Zea mays) using the genotyping-by-sequencing (GBS) technology. Front. Plant Sci. 8, 706 (2017).
Spindel, J. et al. Bridging the genotyping gap: Using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor. Appl. Genet. 126, 2699–2716 (2013).
Hyun, D. Y. et al. Genotyping-by-sequencing derived single nucleotide polymorphisms provide the first well-resolved phylogeny for the genus Triticum (Poaceae). Front. Plant Sci. 11, 688 (2020).
Verma, S. et al. High-density linkage map construction and mapping of seed trait QTLs in chickpea (Cicer arietinum L.) using Genotyping-by-Sequencing (GBS). Sci. Rep. 5, 1–4 (2015).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012).
Chung, M. Y., López-Pujol, J., Lee, Y. M., Oh, S. H. & Chung, M. G. Clonal and genetic structure of Iris odaesanensis and Iris rossii (Iridaceae): Insights of the Baekdudaegan Mountains as a glacial refugium for boreal and temperate plants. Plant Syst. Evol. 301, 1397–1409 (2015).
Chung, M. Y., López-Pujol, J. & Chung, M. G. The role of the Baekdudaegan (Korean Peninsula) as a major glacial refugium for plant species: A priority for conservation. Biol. Conserv. 206, 236–248. https://doi.org/10.1016/j.biocon.2016.11.040 (2017).
Chung, M. Y. et al. Comparison of genetic variation between northern and southern populations of Lilium cernuum (Liliaceae): implications for Pleistocene refugia. PLoS One 13, e0190520. https://doi.org/10.1371/journal.pone.0190520 (2018).
Chung, M. Y. et al. Patterns of genetic diversity in rare and common orchids focusing on the Korean Peninsula: Implications for conservation. Bot. Rev. 84, 1–25 (2018).
Lee, S.-J. et al. Phylogeography of the Asian lesser white-toothed shrew, Crocidura shantungensis, in East Asia: Role of the Korean Peninsula as refugium for small mammals. Genetica 146, 211–226. https://doi.org/10.1007/s10709-018-0014-2 (2018).
López-Pujol, J., Zhang, F.-M., Sun, H.-Q., Ying, T.-S. & Ge, S. Centres of plant endemism in China: Places for survival or for speciation?. J. Biogeogr. 38, 1267–1280 (2011).
Qiu, Y.-X., Fu, C.-X. & Comes, H. P. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 59, 225–244 (2011).
Tian, H. Z. et al. Genetic diversity in the endangered terrestrial orchid Cypripedium japonicum in East Asia: Insights into population history and implications for conservation. Sci. Rep. 8, 1–13 (2018).
Sakaguchi, S. et al. Climate oscillation during the quaternary associated with landscape heterogeneity promoted allopatric lineage divergence of a temperate tree Kalopanax septemlobus (Araliaceae) in East Asia. Mol. Ecol. 21, 3823–3838 (2012).
Kim, Z. S., Hwang, J. W., Lee, S. W., Yang, C. & Gorovoy, P. G. Genetic variation of Korean pine (Pinus koraiensis Sieb. Et Zucc.) at allozyme and RAPD markers in Korea, China and Russia. Silvae Genet. 54, 235–246 (2005).
Chen, D. et al. Phylogeography of Quercus variabilis based on chloroplast DNA sequence in East Asia: Multiple glacial refugia and mainland-migrated island populations. PLoS One 7, e47268 (2012).
Kikuchi, R., Pak, J.-H., Takahashi, H. & Maki, M. Disjunct distribution of chloroplast DNA haplotypes in the understory perennial Veratrum album ssp. oxysepalum (Melanthiaceae) in Japan as a result of ancient introgression. New Phytol. 188, 879–891. https://doi.org/10.1111/j.1469-8137.2010.03398.x (2010).
Hewitt, G. The genetic legacy of the quaternary ice ages. Nature 405, 907–913 (2000).
Provan, J. & Bennett, K. D. Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 23, 564–571 (2008).
Chung, M. Y., López-Pujol, J. & Chung, M. G. Comparative genetic structure between Sedum ussuriense and S. kamtschaticum (Crassulaceae), two stonecrops co-occurring on rocky cliffs. Am. J. Bot. 101, 946–956 (2014).
Hamrick, J.L. & Godt, M.J.W. Allozyme diversity in plant species in Plant Population Genetics. Breeding and Genetic Resources (eds. Brown, A.H.D., Clegg, M.T. & Weir, B.S.) 43–63 (Sinauer Associates, 1989).
Holsinger, K. E. & Weir, B. S. Genetics in geographically structured populations: Defining, estimating and interpreting FST. Nature Genet. 10, 639–650 (2009).
Ballesteros-Mejia, L., Lima, N. E., Lima-Ribeiro, M. S. & Collevatti, R. G. Pollination mode and mating system explain patterns in genetic differentiation in Neotropical plants. PLoS One 11, e0158660 (2016).
Gibbs, R.D. Botany: An evolutionary approach. (The Blakiston Company, 1950).
Brodie, H. J. The splash-cup dispersal mechanism in plants. Can. J. Bot. 29, 224–234 (1951).
Nakanishi, H. Splash seed dispersal by raindrops. Ecol Res. 17, 663–671 (2002).
Elshire, R. J. et al. A robust, simple genotyping-by sequencing (GBS) approach for high diversity species. PLoS One 6, e19379. https://doi.org/10.1371/journal.pone.0019379 (2011).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. https://doi.org/10.14806/ej.17.1.200 (2011).
Cox, M. P., Peterson, D. A. & Biggs, P. J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 11, 485. https://doi.org/10.1186/1471-2105-11-485 (2010).
Parra-Salazar, A., Gomez, J., Lozano-Arce, D., Reyes-Herrera, P. H. & Duitama, J. Robust and efficient software for reference-free genomic diversity analysis of genotyping-by-sequencing data on diploid and polyploid species. Mol. Ecol. Resour. 22, 439–454 (2022).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics. 27, 2156–2158 (2011).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K., von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Kubatko, L. S. & Degnan, J. H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 56, 17–24. https://doi.org/10.1080/10635150601146041 (2007).
Swofford, D.L. Version 4.0a169; Phylogenetic Analysis Using Parsimony (PAUP). (2020). http://phylosolutions.com/paup-test/
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Excoffier, L., Smouse, P. E. & Quattro, J. M. Analysis of molecular variance inferred from metric distances among DNA haplotypes - application to human mitochondrial-DNA restriction data. Genetics 131, 479–491 (1992).
Meirmans, P. G. GENODIVE version 3.0: Easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol. Ecol. Resour. 20, 1126–1131 (2020).
Liu, X. & Fu, Y. X. Stairway Plot 2: Demographic history inference with folded SNP frequency spectra. Genome Biol. 21, 280 (2020).
You, J. et al. Comparative phylogeography study reveals introgression and incomplete lineage sorting during rapid diversification of Rhodiola. Ann. Bot. 129, 185–200 (2022).
Fitak, R. R. OptM: Estimating the optimal number of migration edges on population trees using Treemix. Biol. Methods Protoc. 6, bpab017 (2021).
Reich, D., Thangaraj, K., Patterson, N., Price, A. L. & Singh, L. Reconstructing Indian population history. Nature 461, 489–494 (2009).
Acknowledgements
This research was funded in part by the National Institute of Biological Resources (NIBR, grant number 2020NIBR202005201), under the program “A Study on Plant Resources on DNA Sequences Utilizing Next Generation Sequencing Technique (3rd year).”
Author information
Authors and Affiliations
Contributions
M-SC, YK, and S-CK designed the experiments and M-SC, YK, S-HK, JYY, and S-CK collected the samples. M-SC, YK, J-HJ, JYY, and S-HK performed the experiments and analyzed the data. M-SC drafted the manuscript, and S-CK revised it. All the authors have approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cho, MS., Kim, Y., Kim, SH. et al. Phylogenetic relationships and genetic diversity of the Korean endemic Phedimus latiovalifolius (Crassulaceae) and its close relatives. Sci Rep 14, 16255 (2024). https://doi.org/10.1038/s41598-024-63272-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-63272-9
- Springer Nature Limited