
Abstract
Many peninsulas in the temperate zone played an important role as refugia of various flora and fauna, and the southern Korean Peninsula also served as a refugium for many small mammals in East Asia during the Pleistocene. The Asian lesser white-toothed shrew, Crocidura shantungensis, is a widely distributed species in East Asia, and is an appropriate model organism for exploring the role of the Korean Peninsula as a refugium of small mammals. Here, we investigated phylogenetic relationships and genetic diversity based on the entire sequence of the mitochondrial cytochrome b gene (1140 bp). A Bayesian tree for 98 haplotypes detected in 228 C. shantungensis specimens from East Asia revealed the presence of three major groups with at least 5 subgroups. Most haplotypes were distributed according to their geographic proximity. Pairwise FST’s and analysis of molecular variance (AMOVA) revealed a high degree of genetic differentiation and variance among regions as well as among populations within region, implying little gene flow among local populations. Genetic evidence from South Korean islands, Jeju-do Island of South Korea, and Taiwan leads us to reject the hypothesis of recent population expansion. We observed unique island-type genetic characteristics consistent with geographic isolation and resultant genetic drift. Phylogeographic inference, together with estimates of genetic differentiation and diversity, suggest that the southern most part the Korean Peninsula, including offshore islands, played an important role as a refugium for C. shantungensis during the Pleistocene. However, the presence of several refugia on the mainland of northeast Asia is also proposed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Climatic oscillations, such as the extensive environmental alterations that occurred during Pleistocene glacial–interglacial cycles, have changed the demographic and distributional patterns of species (Hewitt 2003, 2004). The action of glaciers during the Pleistocene period is one of the major formative causes underlying the genetic structuring of many vertebrate taxa in the northern hemisphere (Gómez and Lunt 2006; Hewitt 2000; Schmitt 2007; Shafer et al. 2010; Taberlet et al. 1998). Populations in refugia that formed during the glacial age would have been isolated and the species’ gene pool subdivided into distinct lineages (Hewitt 1996, 2000). Peninsulas are major refugia for many animal species (Tomasik and Cook 2005; Weiss and Ferrand 2007). Several peninsulas along the Mediterranean Sea, such as the Iberian, Italian, and Balkan, have played an especially important role as refugia of animals, including the ocellated lizard (Lacerta lepida; Miraldo et al. 2011), Italian treefrog (Hyla intermedia; Canestrelli et al. 2007), and Martino’s vole (Dinaromys bogdanovi; Krystufek et al. 2007).
Phylogeographic studies in East Asia have demonstrated the role of the Korean Peninsula as a refugium of various fauna and flora. East Asia experienced strong climatic oscillations during the glacial and interglacial periods although no large glaciers occurred after the Quaternary (Shi 2006; Zheng et al. 2003). The general point of view is that during the LGM period, the temperature declined by about 5–11 °C compared to the current climate of the Asian continent. After LGM, several small glacial periods were spaced, but the temperature of the entire Asian continent became warmer. As a result, the range of vegetation in East Asia has repeatedly experienced retractions and expansion (Yu et al. 2000). A study of the deciduous Oriental oak (Quercus variabilis), a broad-leaved tree, suggested that it had retreated to several refugia during the Pleistocene, including the southern part of the Korean Peninsula (Chen et al. 2012). Also during the Pleistocene glaciations, the eastern monsoon region and lower elevations of the southwestern plateau in China and the Korean Peninsula became refugia for black-spotted frog populations (Pelophylax nigromaculata) (Zhang et al. 2008). Phylogeographic studies also suggested the Korean Peninsula serves as a refugium for several mammals. Kim et al. (2013) showed a lack of gene flow between raccoon dogs in South Korea and Japan, and their phylogeographic pattern confirmed that the Korean Peninsula is the southernmost refugium. The Siberian chipmunk (Tamias sibiricus) has three distinct clades in South Korea, and two refugia may have existed in South Korea and east Russia-northeast China during the Pleistocene (Lee et al. 2008).
The genus Crocidura, containing the white-toothed shrew, is among the largest mammalian genera, with about 150–180 species. This genus is widely distributed in the Palaearctic, Oriental, and Afrotropical regions (Churchfield 1990; Corbet and Hill 1991; Hutterer 2005; Motokawa et al. 2003; Nowak 1999; Wójcik and Wolsan 1998). Approximately 8–10 species of crocidurine shrews inhabit in East Asia (Ohdachi et al. 2004). Three species, C. shantungensis, C. dsinezumi and C. lasiura, are present in the Korean Peninsula, and the first two are found on Jeju-do Island (Han et al. 2002; Iwasa et al. 2001; Motokawa et al. 2005; Won and Smith 1999). The Asian lesser white-toothed shrew (C. shantungensis Miller 1901) is endemic to East Asia including Korea, Russia, China, Taiwan, and Mongolia (Hutterer 2005). It is the smallest species of white-toothed shrew in South Korea and is widely distributed on the South Korean mainland and southern peripheral islands (Yoon et al. 2004). Taxonomically, Asian lesser white-toothed shrew has been considered synonymous with C. suaveolens in Europe, but was reported to be a separate species based on morphological (Jiang and Hoffmann 2001; Motokawa et al. 2003) and molecular genetic (Dubey et al. 2006; Ohdachi et al. 2004) studies.
The phylogeography of C. shantungensis in East Asia is poorly understood and information on intraspecific relationships is scarce. Phylogenetic studies of seven species of Crocidura in East Asia and two species (C. shantungensis and C. dsinezumi) on Jeju-do Island demonstrated that populations of C. shantungensis on the island and the East Asian mainland are genetically differentiated (Han et al. 2002; Ohdachi et al. 2004). Bannikova et al. (2009) established the distribution of Crocidura in Mongolia and Buryatia regions. Preliminary research by Koh et al. (2013) on genetic divergence of C. shantungensis in South Korea, adjacent islands and in continental East Asia indicated that the population on Jeju-do Island is genetically different from those on the mainland.
The Asian lesser white-toothed shrew is a good model organism for phylogeographic studies on refugia formation of small mammals endemic to East Asia. This species is abundant with a wide geographic distribution in the Korean Peninsula and adjacent countries in East Asia. It has a short generation time as well as limited dispersal capacity. In particular, the species’ high adaptability and reproductive rate can facilitate rapid evolution in response to topography and climatic changes.
In this study, we examined genetic diversity and the phylogeographic relationship of Asia lesser white-toothed shrews sampled from various localities of East Asia, i.e. South Korea, China, Russia, Mongolia, and Taiwan, by comparing the entire mitochondrial cytochrome b gene sequence. Our goals were to evaluate the present status of genetic diversity and differentiation of the Asia lesser white-toothed shrew in East Asia, and to clarify its phylogeographic relationships and evolutionary history in this region. We also discussed the possible formation of refugia of this species in East Asia during the glacial period.
Materials and methods
Sample collection
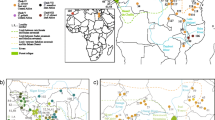
A total of 228 specimens of Asian lesser white-tooth shrew, Crocidura shantungensis, were collected from several localities in East Asia (Fig. 1; Table 1). Tissue samples were stored in 70% ethanol at − 20 °C until DNA extraction. All samples were legally collected and deposited into the Conservation Genome Resource Bank for Korean Wildlife (CGRB). The procedures involving animal samples followed the guidelines by Seoul National University Institutional Animal Care and Use Committee (SNUIACUC). We included sequences of mtDNA cytochrome b gene of C. shantungensis from Russia (10) and Mongolia (3) from GenBank for the analyses (Accession number: AB077080—AB077082, AB077278, EU742584, EU742593, KF144160—KF144163 from Russia; EU7442589—EU7442591 from Mongolia). The sequences generated in this study were deposited in GenBank (Accession nos. MF136304—MF136385, and MF152782).

Sampling localities for Asian lesser white-toothed shrew (C. shantungensis) based on mitochondrial cytochrome b sequences. Haplogroup 1: Subgroup A (KOM, KOI), Subgroup B (TAI), all the rest, Subgroup C (KOM, KOI, CHS, and RAM); Haplogroup 2: KOJ; Basal group: KOI. CHS Shandong in China, RAM Russia and Mongolia, TAI Taiwan, KOM South Korean mainland, KOI South Korean islands, KOJ Jeju-do Island in South Korea
Molecular techniques
Whole genomic DNA was isolated using the Qiagen DNeasy Blood & Tissue Kit (Qiagen, USA) according to the manufacturer’s instructions. The entire cytochrome b gene (1,140 bp) was amplified using a set of universal primers, L14724 5′-CGAAGCTTGATATGAAAAACCATCGTTG-3′ and H15915 5′-AACTGCAGTCAT CTCCGGTTTACAAGAC-3′ (Irwin et al. 1991). PCR mixtures were prepared in 30 µL volume containing 20–100 ng template DNA, 3 µL of 10× rTaq reaction buffer, 1.5 mM MgCl2, 100 µM each dNTPs, 10 pmol each primer, and 1 Unit of Takara rTaqTM polymerase (Takara, Japan). PCR amplification was conducted with initial denaturation at 94 °C for 4 min, followed by 35 cycles at 94 °C for 45 s, 44 °C for 60 s and 72 °C for 90 s, with a final extension at 72 °C for 7 min using a Takara PCR machine. 2 µL of PCR product were checked on a 1% agarose gel. The aforementioned primers and additional internal primers designed for this study, insec_F (5′-GATAAAGCTACTCTAACCCG-3′) and insec_R (5′-GGATGGAAAGGAATTTTGTC-3′), were used for sequencing. All samples were directly sequenced on an Applied Biosystems 3730 XL DNA sequencer according to the manufacturer’s instructions (NICEM, South Korea).
Genetic diversity and phylogenetic analyses
The sequences were determined, modified and subjected to multiple alignments using Geneious Pro v 5.3 (http://www.geneious.com, Kearse et al. 2012). The number of polymorphic sites were calculated, and the genetic diversity of each sample population was evaluated with respect to haplotype (h) and nucleotide (π) diversity using DnaSP v 5.10 (Librado and Rozas 2009). A pairwise distance matrix between populations was calculated using the Kimura 2-parameter model (Kimura 1980) implemented in MEGA v 7 (Kumar et al. 2016).
Phylogenetic trees to investigate evolutionary relationships were reconstructed using Bayesian inference (BI). Four individuals of Crocidura suaveolens (GenBank: EU742610—EU742613) were used as the outgroup. The best appropriate model of sequence evolution for Bayesian trees was the Hasegawa Kishino–Yano-model (HKY) with Gamma distribution (+ G), which was selected by Bayesian information criterion (BIC), as implemented in jModelTest v 2.1 (Darriba et al. 2012). BI and Bayesian posterior probabilities (BPP) were assessed by MrBayes v 3.2 (Ronquist and Huelsenbeck 2003). BI employed four simultaneous Monte Carlo Markov chains (one cold and three heated) with 2,000,000 generations and sampled every 100 generations. The first 25% of the data points were discarded as burn-in. The consensus tree was illustrated using FigTree v 1.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Population structure and demographic analyses
Haplotype relationships of C. shantungensis were constructed by a median-joining network (Bandelt et al. 1999) using the program Network v 4.6 (http://www.fluxus-engineering.com). Genetic differentiation among populations (pairwise FST’s) and an analysis of molecular variance (AMOVA) within and among geographic samples of six groups [Shandong in China (CHS); Russia and Mongolia (RAM); Taiwan (TAI); South Korean mainland (KOM); South Korean islands (KOI); Jeju-do Island in South Korea (KOJ)] were calculated using the Arlequin v 3.5 (Excoffier and Lischer 2010). Buryatia in Russia, Primorsky Krai in Russia, and Mongolia were distant from one another, but were small samples so they were combined as a single location (Russia and Mongolia, RAM) for analysis. Most of the population analyses were based on the above six groups. Arlequin v 3.5 was further used to evaluate historical demographic expansion using Tajima’s D (Tajima 1989) and Fu’s F S (Fu 1997). Mismatch distribution for each group was examined using Harpending’s raggedness index (Harpending et al. 1993) implemented in Arlequin v 3.5. This analysis quantifies smoothness of the observed mismatch distribution, and a non-significant result indicates population growth (Harpending 1994). We also used the Bayesian skyline plot (BSP) to estimate the demographic history of each lineages (clades) using the program BEAST v 1.8.4 (Drummond and Rambaut 2007) and we used the calculated a substitution rate from Esselstyn and Brown (2009). TRACER v 1.6 (Rambaut et al. 2014) was used to reconstruct the demographic history through time.
Divergence time within the C. shantungensis was inferred using a Bayesian analysis implemented with BEAST v 1.8.4 (Drummond and Rambaut 2007) between the major lineage. This analysis used the Yule speciation model and relaxed uncorrelated lognormal clock with sequences partitioned into 1st + 2nd and 3rd codon positions with reference to Esselstyn and Brown (2009). Analyses were initiated with an UPGMA starting tree and run for 2 × 107 generations with trees and parameters sampled every 2000 generations. We calibrated analyses with the oldest known fossil Crocidura (5.03 Mya ago; Butler 1998). Analyses were repeated twice using different random seeds to test for convergence. The estimated values and associated effective sample size (ESS) for each model parameter were viewed in TRACER v 1.6 (Rambaut et al. 2014). The maximum clade credibility tree was produced in TreeAnnotator v 1.8.4 in BEAST (Drummond and Rambaut 2007). We also calculated divergence time (T) according to the equation proposed by Graur and Li (2000): T = K/(2r), where r is the substitution rate. This method was assessed in the Bayesian phylogenetic tree, calibrated using prior estimates of substitution rate: substitution rate obtained from the oldest known fossil data of Crocidura (5.03 Mya ago; Butler 1998). Sequence divergence (K, substitutions/site) was derived from the mean value of P-distance between clusters, using Mega v 7 (Kumar et al. 2016).
Results
Genetic diversity of C. shantungensis in East Asia
A total of 98 haplotypes were detected among the full sequences of mitochondrial cytochrome b gene from 241 C. shantungensis individuals from East Asia. There were 156 nucleotide substitutions, with 99 parsimony-informative sites and 57 singletons. No insertions or deletions were observed. Base frequencies for A, C, G and T, respectively were 0.2994, 0.2473, 0.1240 and 0.3293. Haplotype distribution for each location is shown in Table 1. C. shantungensis haplotypes were not shared between most locations, and samples from most South Korean islands exhibited unique haplotypes. Only a single haplotype, Hap95, was shared between South Korean mainland (KOM) and South Korean islands (KOI). Although several haplotypes from KOM, Shandong in China (CHS) and Russia and Mongolia (RAM) were clustered together in the tree and network (Figs. 2, 3), they were not shared between these regions.

Bayesian (MCMC) haplotype tree of Asian lesser white-toothed shrew (C. shantungensis) based on mtDNA cyt-b gene (1,140 bp) sequences. Hasegawa Kishino–Yano-model (HKY) + Gamma distributed (+ G) model was implemented. Bayesian posterior probability is shown for branches with > 50% support. Haplogroup 1: Subgroup A (KOM, KOI), Subgroup B(TAI), all the rest, Subgroup C (KOM, KOI, CHS, and RAM); Haplogroup 2: KOJ; Basal group: KOI. CHS Shandong in China, RAM Russia and Mongolia, TAI Taiwan, KOM South Korean mainland, KOI South Korean islands, KOJ Jeju-do Island in South Korea

Median-joining network showing the distribution of 98 haplotypes from C. shantungensis cytochrome b gene. The size of the circles is proportional to the number of individuals
Overall haplotype diversity (h) and nucleotide diversity (π) across the six regions were 0.972 and 1.127%, respectively (Table 2). The lowest level of nucleotide diversity was found in Jeju-do Island in South Korea (KOJ 0.192%), followed by Russia and Mongolia (RAM 0.335%), Taiwan (TAI 0.396%), South Korean mainland (KOM 0.577%), Shandong in China (CHS 0.593%), and South Korean islands (KOI 1.144%). Pairwise genetic distances between C. shantungensis populations ranged from 0.005 in CHS vs. RAM to 0.019 in KOJ vs. TAI, and the mean genetic distance within group ranged from 0.002 in KOJ to 0.012 in KOI (Table 3). Pairwise FST’s ranged from 0.116 to 0.824, with the highest value between RAM and KOJ.
Genetic relationship of C. shantungensis in east Asia
The Bayesian tree based on the Hasegawa Kishino–Yano-model (HKY) + Gamma distributed (+ G) genetic distances revealed three main haplogroups with several minor subgroups (Fig. 2). Haplogroup 1 mainly consisted of haplotypes from Shandong in China (CHS), Russia and Mongolia (RAM), Taiwan (TAI), South Korean mainland (KOM), and South Korean islands (KOI). Haplogroup 2 included haplotypes from only Jeju-do Island in South Korea (KOJ), and Basal group was composed of haplotypes from KOI. Only one individual (Hap95) from KOI, collected on the southern coast of South Korea (Haenam-gun, Jeolla Province, Korea), belonged to Basal group. Haplogroup 1 was further subdivided into at least three subgroups, mostly clustering based on geographic range: Subgroup A, from KOM and KOI; Subgroup B, from TAI; and all the rest Subgroup C, from the broad expanse of geographic regions in central and northeast Asia, i.e., CHS, RAM, KOM, and KOI. The median-joining network tree (Fig. 3) showed genealogical groupings similar to the results from phylogenetic tree (Fig. 2). Subgroup A of the main Haplogroup 1, which was formed by samples from the Korean mainland and islands (KOM, KOI) and central and northeast Asia (CHS, RAM), revealed a star-like shape in the center of the median-joining network. In contrast, Haplogroup 1_Subgroup B, Haplogroup 2, and Basal group were formed by samples mainly from islands in Korea (KOJ and KOI) and Taiwan (TAI), occupying peripheral positions of the network.
Genetic structure of C. shantungensis in east Asia
In general, pairwise FST’s, as a measure of genetic differentiation between populations, were high and significant for most comparisons. The lowest FST (0.116) was detected between samples from Russia and Mongolia (RAM) and South Korean islands (KOI), and the highest (0.824) between RAM and Jeju-do Island in South Korea (KOJ) (Table 3). Although samples from Shandong in China (CHS) and Russia and Mongolia (RAM) are geographically distant from South Korean mainland (KOM) and KOI, pairwise FST values were lower than those involving KOJ. (0.122 in CHS vs. RAM; 0.245 in CHS vs. KOM, 0.142 in CHS vs. KOI, 0.233 in RAM vs. KOM, 0.116 in RAM vs. KOI, 0.737 in KOM vs. KOJ, 0.480 in KOI vs. KOJ) Analysis of molecular variance (AMOVA) was performed according to the following hierarchical settings: Set 1, three populations consisting of Haplogroup 1 (CHS, RAM, TAI, KOM, and KOI), Haplogroup 2 (KOJ), and Basal group (KOI); Set 2, six populations based on regional proximity; Set 3: the three populations from South Korea (KOM, KOJ, KOI) (Table 4). Results showed that Asian lesser white-toothed shrew groups were significantly differentiated for all three hierarchical settings. In Set 1, 58.72% of genetic variation in C. shantungensis mtDNA was accounted for among groups, and 29.07% among populations within groups, whereas in Sets 2 and 3, respectively, 27.45 and 21.87% of genetic variation was accounted for among regions, and 49.86, and 54.41% among populations within regions.
Demographic history
Two neutrality tests, Tajima’s D and Fu’s F S , revealed negative values for all samples most groups of C. shantungensis, except Fu’s F S value for South Korean islands (KOI), showing a recent demographic growth for C. shantungensis from most regions (Table 5). Samples from KOM showed significantly negative values in both tests supporting a model of demographic expansion. Values for Shandong in China (CHS), Russia and Mongolia (RAM), and Jeju-do Island in South Korea (KOJ) were negative but non-significant values in both tests [CHS (D = 1.188, p = 0.131 and F S = − 2.324, p = 0.062); RAM (D = − 1.088, p = 0.139 and F S = − 1.478, p = 0.209); KOJ (D = − 0.638, p = 0.278 and F S = − 1.291, p = 0.212)]. TAI samples generated a significantly negative value in Tajima’s D test, but was not significant in Fu’s F S test (Tajima’s D test: D = − 1.676, p = 0.023; Fu’s F S test: F S = − 1.305, p = 0.358). The values for KOI were negative for Tajima’s D test and positive for Fu’s F S test, but not significant in either.
The mismatch distribution confirmed that not only the entire sample of C. shantungensis but also each regional group was multimodal (Fig. 4). This was expected as effects of sampling from multiple locations. The Harpending’s Raggedness index (r) was not significant for CHS, RAM, and KOM, whereas r was significant for samples from TAI, KOI, and KOJ. Caution is required when interpreting the mismatch distribution analysis in this study since demographic analyses can be heavily affected by group sub-structuring (e.g. more genetic lineages within a group, as in this case of KOI and KOM).

Mismatch distribution of each regional population under the sudden expansion model. Mismatch distributions based on pairwise site differences between sequences. CHS Shandong in China, RAM Russia and Mongolia, TAI Taiwan, KOM South Korean mainland, KOI South Korean islands, KOJ Jeju-do Island in South Korea
The divergence time using a Bayesian analysis between C. shantungensis and C. suaveolens was approximately 1.88 Mya (95% CI 0.80–3.02 Mya). Haplogroup 1 and Haplogroup 2 (KOJ) were separated from Basal group during the period of 1.16 Mya (95% CI 0.43–1.199 Mya). Haplogroup 1 including Shandong in China (CHS), Russia and Mongolia (RAM), Taiwan (TAI), South Korean mainland (KOM), and South Korean islands (KOI) was separated from Haplogroup 2 (KOJ) during the period of 1.03 Mya (95% CI 0.44–1.76 Mya). The percentage sequence divergence and estimated divergence time among haplogroups represented in the Bayesian phylogenetic tree are shown in Table 6. Based on mutation rates of r = 26.1 (Essenlstyn and Brown 2009), the divergence time (T) between C. shantungensis and C. suaveolens was dated to approximately 1.59 Mya (Table 6). The most recent divergence was between subgroups A and C, at 134 Kya. Divergence time between subgroups A and B (Taiwan) was 230 Kya, and subgroup B (Taiwan) and subgroup C (islands in southern part of South Korea) was 192 Kya. The mainland of South Korea (KOM) and islands in of South Korea (KOI) were separated from Jeju-do island in South Korea (KOJ) during the period of 307 Kya. Our Bayesian skyline plots showed that the effective population sizes of each lineage except for RAM marginally increased around 100,000–900,000 years ago (Fig. 5). RAM did not show clearly the population expansion across time.

Bayesian Skyline Plots. The Y-axis indicates the effective population size while X-axis indicates time from the present (left) to the past (right). The solid line is the median estimate and the purple area represents the standard error. CHS Shandong in China, RAM Russia and Mongolia, TAI Taiwan, KOM South Korean mainland, KOI South Korean islands, KOJ Jeju-do Island in South Korea
Discussion
Genetic diversity and phylogeographic relationships
Nucleotide diversity across all C. shantungensis samples (1.127%; within-group range of 0.192% − 1.144%) is comparable with the ranges (0.4–1.2%) observed for Eurasian shrews (C. suaveolens group) (Dubey et al. 2006). Unlike the continental populations of C. shantungensis, a reduced level of genetic diversity was observed in samples from several islands including Taiwan, and Jeju-do Island in South Korea. We ascribe this phenomenon to the typical pattern caused by peripheral isolation, where relatively low levels of genetic diversity are expected from founder effects and subsequent genetic drift after migration from a source area into the islands. In addition, little or no gene flow between island and mainland populations should enhance genetic differentiation in such regions. Lack of shared haplotypes between Jeju-do Island and the South Korea mainland, and unique haplogroups found in Jeju-do Island suggest that migration of Asian lesser white-toothed shrew to the latter could have taken place only during periods of glaciation when a land bridge formed between the island and continent. A similar pattern of distribution of mtDNA haplotypes was recently reported for several widely distributed mammal species, including the Siberian roe deer, Caproulus pygargus (Lee et al. 2016), European badger, Meles meles (Frantz et al. 2014), and raccoon dogs, Nyctereutes procyonoides (Kim et al. 2013). C. shantungensis populations between the offshore islands and mainland of South Korea belonged to the same haplogroup and showed relatively low level of genetic differentiation, suggesting possible ongoing gene flow between these regions. On the other hand, lack of shared haplotypes (with one exception of haplotype 95) between these regions may also imply limited gene flow between them. However, because this contradictory interpretation could be caused by effects of small sample size, analysis for more samples from these regions could address this question.
The levels of genetic divergence (0.5–1.9%) between geographic samples of Asian lesser white-toothed shrew are similar to those of other small mammals: 0.8–1.9% for Siberian chipmunk, Tamias sibiricus (Lee et al. 2008), 0.0–0.9% for Korean field mouse, Apodemus peninsulae, and 0.5–1.2% for striped field mouse, A. agrarius (Sakka et al. 2010).
Phylogeographic relationships based on the Bayesian tree (Fig. 2) suggest two monophyletic haplogroups (Haplogroups 1 and 2) and a remaining non-monophyletic group (Basal group). The non-monophyletic group (Basal group), represented only by C. shantungensis from the offshore islands in the southern part of South Korea (KOI) was basal to the other samples, where Haplogroups 1 and 2 shared a common ancestor. Subgroups within Haplogroup 1 seem to have diverged similarly from each other. Interestingly, CHS, RAM, and KOM in Haplogroup 1 occupy the center of the star-like shape in the median-joining network (Fig. 3), implying a recent population expansion in these regions. Together, our results strongly suggest that Asian lesser white-toothed shrew spread from the continent of East Asia into the periphery of the species’ geographic range and the islands.
Population demographic and evolutionary history
As a whole, the samples of Asian lesser white-toothed shrew used in this study generated significantly negative values in Tajima’s D and Fu’s FS neutrality test (Table 5), and non-significant values of Harpending’s Raggedness index (r). The multimodal mismatch distribution in total seems to reflect the combined effects of different geographic groups. Results generally revealed an apparent geographic effect on the genetic relationships between sample locations.
All of the population demography analyses, such as the neutrality test with a significant negative value (D = − 1.941, p < 0.005 and F S = − 24.450, p < 0.005), a unimodal shape of the mismatch distribution, and a non-significant Harpending’s Raggedness index (r = 0.007, p value = 0.690), provide evidence of population expansion in the main Haplogroup 1 (Avise 2000). Shandong of China (CHS) and Russia and Mongolia (RAM) had non-significant negative neutrality values, but r was non-significant. The number of samples in each group was small and they did not form a clade in the tree. So, we analyzed the combination of CHS and RAM. The result was evidence that C. shantungensis in Central-Northeast Asia experienced a population expansion: significant negative values (D = − 1.963, p < 0.01 and F S = − 6.420, p < 0.01) in the neutrality tests, a unimodal shape of the mismatch distribution, and a non-significant Harpending’s Raggedness index (r = 0.063, p value = 0.100).
There are at least three distinct genetic groups of Asian lesser white-toothed shrew on the Korean Peninsula (Table 4, set 3), owing to the peripheral isolation of the island populations during the glacial and interglacial periods. Samples from the South Korean mainland (KOM) showed a significant negative value in two neutrality tests, a non-significant r value, and the multimodal mismatch distribution, supporting a hypothesis of recent population demographic growth in this region.
In the case of the geographically divided Jeju-do Island in South Korea (KOJ), negative but non-significant neutrality test values, and a significant Harpending’s Raggedness index suggest that this population has not experienced recent population growth. Our results indicate that populations on Jeju-do Island in South Korea (KOJ, Haplotype 2) and the offshore islands (KOI) diverged about 307 Kya, and KOJ and basal group diverged 249 Kya in Graur and Li (2000)’s equation. KOJ and basal group diverged 0.44–1.76 Mya in Bayesian approach implemented in BEAST. Motokawa et al. (2003) and Kim et al. (2015) showed that there are morphological differences between C. shantungensis in South Korea, Jeju-do island, and Taiwan, and that populations in Tsushima (Japan) or Jeju were separated from the Korean Peninsula during the Pleistocene. Kartavtseva and Park (2010) proposed that the Jeju-do Island population has been isolated for a long time and the genetic variation has accumulated on the Y chromosome. These results are consistent with our findings for the mtDNA cyt-b gene. The relatively low level of genetic diversity (h = 0.809 and π = 0.192%) found on Jeju-do Island in South Korea (KOJ) implies that its C. shantungensis population may have undergone a prolonged bottleneck after migration into the island (Avise 2000).
Basal group comprises most of the Asian lesser white-toothed shrews South Korean islands (KOI), and is basal to the other haplogroups, implying animals from these southern offshore islands share the ancestral polymorphism of C. shantungensis in East Asia. Moreover, samples from the islands do not share haplotypes with the Korea mainland, except for one sample from Haenam-gun, and each island is composed of unique haplotypes without sharing haplotypes with other islands. The high level of genetic diversity found in the KOI population can be explained as a sampling effect caused by pooling specimens of heterogeneous genetic makeup. Tajima’s D values were not significantly negative, and Fu’s F S (= 2.558) were positive, indicating a recent population bottleneck or some form of balancing selection.
Interestingly, the population on Ulleung-do Island, positioned far away from the Korea mainland, showed few haplotypes despite a large sample size, and these haplotypes belong to the main Haplogroup 1 that was composed mainly of samples from the mainland. Unlike Jeju-do Island, Ulleung-do Island is a volcanic island that was not connected to the continent during the Pleistocene (Chung and Yang 1999). Thus, the presence of animals on the island is best explained by anthropogenic introduction, and that the low level of genetic diversity is the result of founder effects, and is maintained by genetic drift. A similar explanation may hold for other remote islands, such as Gageo-do Island, Geomun-do Island, Oeyeon-do Island, and Hong-do Island which belong to the same haplogroup as mainland Korea.
The Taiwan Strait, about 200 km wide and 50 m deep on average, is an effective barrier isolating China and Taiwan by blocking the movement and gene flow of fauna distributed on both sides (Jang-Liaw et al. 2008; Tzeng 1986). However, the Chinese continent and Taiwan have been connected via land bridge formation more than once due to changes in sea-level during the Pleistocene. These changes in sea level could provide opportunities for animals to migrate between mainland China and Taiwan via land bridge formation (Emery et al. 1971; Yang 1991). We found a lack of shared haplotypes, formation of distinct haplogroups, and a high degree of genetic differentiation between samples from these two regions. Divergence time suggests that the C. shantungensis population from Taiwan was separated from China about 192 Kya in Graur and Li (2000)’s equation and 0.26–1.21 Mya in Bayesian approach. Therefore, it is presumed that the Asian lesser white-toothed shrew in Taiwan has undergone independent evolution since migration from the continent during the Pleistocene. A similar high level of genetic differentiation in these regions has been reported for several vertebrates, such as the Taiwanese mole shrew, Anourosorex Yamashinai (Yuan et al. 2006), reptiles such as the Taiwanese bamboo viper, Trimeresurus stejnegeri (Creer et al. 2002), amphibians such as the fanged dicroglossine frog, Limnonectes fujianensis (Jang-Liaw and Chou 2011), and freshwater fishes such as the Taiwan shoveljaw carp, Varicorhinus barbatulus (Wang et al. 2004).
The Asian lesser white-toothed shrew in Taiwan could be further divided geographically into three minor groups: northern Taipei, central Taichung, and southern Pingtung. Of these three, the Taichung population seems to be ancestral to the other two based on the Bayesian tree and network analyses (Figs. 2, 3). The estimated divergence time of C. shantungensis within Taiwan is 57–134 Kya in Graur and Li (2000)’s equation and 7–598 Kya in Bayesian approach. Likewise, some vertebrates in Taiwan diverged during the middle and last Pleistocene, such as L. fujianensis 270–660 Kya (Jang-Liaw and Chou 2011), A. yamashinai 630–710 Kya (Yuan et al. 2006), and Buergeria robusta 35–50 Kya (Lin et al. 2012).
Divergence time
Our genetic evidence suggests that divergence time of regional C. shantungensis populations in East Asia probably occurred during the Pleistocene epoch of the Quaternary Period. Pleistocene climatic oscillations have played a major role in shaping the present geographic distribution and genetic structure of temperate species (Avise 2000; Hewitt 2000). Although much of the Korean Peninsula and China were not covered by ice sheets during the Ice Age (Qiu et al. 2011; Zhou et al. 2004), it is probable that the cold and dry climate (Harrison et al. 2001) played a role in the decline and isolation of small mammal populations (Sakka et al. 2010; Zhang et al. 2008) resulting in different genetic groups through allopatric differentiation. This scenario is similar to that generally proposed to explain the genetic structure of many organisms in other regions, such as Europe and North America (Avise 2000).
Our results showed not only the genetic differences between the Korean Peninsula and continental China–Russia, but also unique genetic features among the several islands off the southern coast of the Korean Peninsula, Jeju Island, and Taiwan. The divergence period of C. shantungensis in East Asia dates to the middle Pleistocene (130–773 Kya; Head and Gibbard 2015) at 134–364 Kya [when using the equation of Graur and Li (2000)]. This result is similar to others for small mammals such as in A. agaricus, Russia-South Korea vs. Central Asian which diverged 175–192 Kya, and Taiwan vs. China which diverged 450–500 Kya (Sakka et al. 2010). The divergence time for C. shantungensis in China and Taiwan is estimated as 192–397 Kya, a little later than A. agaricus, but after the second major ice age of the Pleistocene.
Formation and implications of refugia of asian lesser white-toothed shrew on the korean Peninsula
The Asian lesser white-toothed shrew inhabits a wide variety of habitats, but the species is vulnerable to cold weather. It is found mainly in the lowlands, such as grasslands, broadleaf forest, river banks, foothills, around streams, and cultivated lands, rather than in the highlands (Abe 2005; Ohdachi et al. 2009; Yoon et al. 2004). Therefore, it is assumed that several refugia must have existed that protected inhabitants against the cold of the Pleistocene Ice Age. Dubey et al. (2006) suggested that the white-toothed shrews in the northern part of East Asia would have gone extinct when the first major glaciations took place at the beginning of the Pleistocene (1.84 Mya), except for C. shantungensis in the potential refugium of the Shandong region on the Pacific coast of China. The high level of genetic diversity observed in Shandong in China (CHS) in this study supports this hypothesis.
In addition, measures of genetic diversity were higher in the South Korean mainland (KOM) compared with other regions of East Asia. The larger number of haplotypes and haplogroups, as well as the distribution of haplotypes, implies the presence of relatively large gene pools in mainland South Korea. Notably, the Asian lesser white-toothed shrew population from the offshore islands of southern Korea are characterized by a high level of genetic diversity and heterogeneous haplotypes. Indeed, most islands retain unique haplotypes as well as the island-type haplogroup (Basal group), but haplotypes of some islands belong to the main haplogroup (Basal group) characteristic of the Korea mainland. Since these islands were connected with mainland Korea during the Pleistocene, it is presumed that they likely represent the southern limit of refugia on the Korean Peninsula. The Korean Peninsula, therefore, could have been an important refugium during the Pleistocene where genetic diversity and heterogeneous genetic features were maintained.
We also propose that C. shantungensis has undergone rapid and independent regional evolution, which is facilitated by short-dispersal behavior, small home range (male: 163.48 m2; Female: 95.46 m2 by Lin et al. 2009), and short generation time. Indeed, pairwise FST’s (Table 3) and AMOVA (Table 4) showed conspicuous genetic differences across the species’ geographic ranges. In general, high levels of genetic differentiation were detected among most of the geographic samples of C. shantungensis, implying a low-level of gene flow between regions. These behavioral and genetic features of C. shantungensis could have led to the formation of multiple large and/or small refugia in the species’ geographic range during the Pleistocene. The Korean Peninsula and part of the China–Russia region have played important roles as refugia for several small rodents (Tamias sibiricu, Apodemus peninsulae, and A. agrarius) during the Pleistocene (Lee et al. 2008; Sakka et al. 2010). Kim et al. (2013), in a recent study of raccoon dog, proposed that there were at least two refugia on the mainland of north-eastern Asia during the glacial Age. Taken together, it is concluded that the Korean Peninsula has played an important role in providing refugia for small mammals during the Pleistocene. Moreover, the peculiar geographic position of the Korean Peninsula may have promoted peripheral evolution of small mammal species throughout glaciations.
In future studies, fine-scale sampling within East Asia, including the offshore islands of Korea and the use of additional markers such as microsatellites, will help develop a more complete picture of the phylogenetic relationships and fine mapping of refugia formation for C. shantungensis in East Asia.
References
Abe H (2005) A guide to the mammals of Japan. Tokai University Press, Hadano
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge
Bandelt H-J, Forster P, Rohl A (1999) Median_Joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Bannikova AA, Sheftel BI, Lebedev VS et al (2009) Crocidura shantungensis, a new species for Mongolia and Buryatia. Doklady Biol Sci 424:68–71
Butler PM (1998) Fossil history of shrews in Africa. In: Wójcik JM, Wolsan M (eds) Evolution of Shrews. Mammal Research Institute, Polish Academy of Sciences, Bialowieza, pp 121–132
Canestrelli D, Cimmaruta R, Nascetti G (2007) Phylogeography and historical demography of the Italian treefrog, Hyla intermedia, reveals multiple refugia, population expansions and secondary contacts within peninsular Italy. Mol Ecol 16:4808–4821
Chen D, Zhang X, Kang H et al (2012) Phylogeography of Quercus variabilis based on chloroplast DNA sequence in East Asia: multiple glacial refugia and mainland-migrated island populations. PLoS One 7:e47268
Chung K-H, Yang H-J (1999) A study on the fauna and speciation of the Cheju Island and Ulrung Dageletin Korea. J Kyonggi Basic Sci 12:189–200
Churchfield S (1990) The natural history of shrews. Christopher Helm, A&C Black, London
Corbet GB, Hill JE (1991) A world list of mammalian species, 3rd edn. Natural History Museum Publications & Oxford University Press, London
Creer S, Malhotra A, Thorpe RS, Chou WH (2002) Multiple causation of phylogeographical pattern as revealed by nested clade analysis of the bamboo viper (Trimeresurus stejnegeri) within Taiwan. Mol Ecol 10:1967–1981
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and high- performance computing. Nat Methods 9:772
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. https://doi.org/10.1186/1471-2148-7-214
Dubey S, Zaitsev M, Cosson J-F et al (2006) Pliocene and Pleistocene diversification and multiple refugia in a Eurasian shrew (Crocidura suaveolens group). Mol Phylogenet Evol 38:635–647. https://doi.org/10.1016/j.ympev.2005.11.005
Emery KO, Nino H, Sullivan B (1971) Post-pleistocene levels of the East China Sea. Woods Hole Oceanographic Institute Press, Massachusetts
Esselstyn JA, Brown RM (2009) The role of repeated sea-level fluctuations in the generation of shrew (Soricidae: Crocidura) diversity in the Philippine Archipelago. Mol Phylogenet Evol 53:171–181. https://doi.org/10.1016/j.ympev.2009.05.034
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Frantz AC, McDevitt AD, Pope LC et al (2014) Revisiting the phylogeography and demography of European badgers (Meles meles) based on broad sampling, multiple markers and simulations. Heredity 113:443–453. https://doi.org/10.1038/hdy.2014.45
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Gómez A, Lunt DH (2006) Refugia within refugia: patterns of phylogeographic concordance in the Iberian Peninsula. In: Phylogeography of Southern European Refugia. Springer, London
Graur D, Li W-H (2000) Fundamentals of molecular evolution, 2nd edn. Sinauer Associates, Inc., Sunderland
Han S, Iwasa MA, Ohdachi SD et al (2002) Molecular phylogeny of Crocidura shrews in northeastern Asia: a special reference to specimens on Cheju Island, South Korea. Acta Theriol 47:369–379
Harpending HC (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol 66:591–600
Harpending HC, Sherry ST, Rogers AR, Stoneking M (1993) The genetic structure of ancient human populations. Curr Anthropol 34:483–496
Harrison SP, Yu G, Takahara H, Prentice IC (2001) Diversity of temperate plants in east Asia. Nature 413:129–130
Head MJ, Gibbard PL (2015) Formal subdivision of the quaternary system/period: past, present, and future. Quatern Int 383:4–35
Hewitt GM (1996) Some genetic consequences of ice ages, and their role, in divergence and speciation. Biol J Lin Soc 58:247–276
Hewitt GM (2000) The genetic legacy of the quaternary ice ages. Nature 405:907–913
Hewitt GM (2003) Ice ages: their impact on species distributions and evolution. In: Evolution on planet Earth. Academic press, New York
Hewitt GM (2004) Genetic consequences of climatic oscillations in the Quaternary. R Soc 359:183–195. https://doi.org/10.1098/rstb.2003.1388
Hutterer R (2005) Mammal species of the world: a taxonomic and geographic reference, 3rd edn. Johns Hopkins University Press, Baltimore
Irwin DM, Kocher TD, Wilson AC (1991) Evolution of the cytochrome b gene of mammals. J Mol Evol 32:128–144
Iwasa MA, Ohdachi S, Han S-H et al (2001) Karyotype and RFLP of the nuclear rDNA of Crocidura sp. on Cheju Island, South Korea (Mammalia, Insectivora). Mammalia 65:451–460. https://doi.org/10.1515/mamm.2001.65.4.451
Jang-Liaw N-H, Chou W-H (2011) Phylogeography of the fanged dicroglossine frog, Limnonectes fujianensis (Anura, Ranidae), in Taiwan. Zoolog Sci 28:254–263. https://doi.org/10.2108/zsj.28.254
Jang-Liaw N-H, Lee T-H, Chou W-H (2008) Phylogeography of Sylvirana latouchii (Anura, Ranidae) in Taiwan. Zool Sci 25:68–79. https://doi.org/10.2108/zsj.25.68
Jiang X, Hoffmann RS (2001) A revision of the white-toothed shrews (Crocidura) of Southern China. J Mammal 82:1059–1079
Kartavtseva IV, Park I-S (2010) Y chromosome peculiarities and chromosomal G- and C-staining in Crocidura shantungensis Miller, 1901 (Soricomorpha: Soricidae). Comp Cytogenet 4:67–71
Kearse M, Moir R, Wilson A et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kim S-I, Park S-K, Lee H et al (2013) Phylogeography of Korean raccoon dogs: implications of peripheral isolation of a forest mammal in East Asia. J Zool 290:225–235. https://doi.org/10.1111/jzo.12031
Kim T, Park S, Kim Y et al (2015) Characteristics of external and cranial morphological characters of asian lesser white-toothed shrew (Crocidura shantungensis). Korean Soc Environ Biol 33:441–449
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. https://doi.org/10.1007/BF01731581
Koh HS, Kartavtseva IV, Lee BK et al (2013) A preliminary study on genetic divergence of the Asian lesser white-toothed shrew Crocidura shantungensis (mammalia: Soricomorpha) in mainland Korea, adjacent islands and continental east Asia: cytochrome b sequence analysis. Russ J Theriol 12:71–78
Krystufek B, Buzan EV, Hutchinson WF, Hänfling B (2007) Phylogeography of the rare Balkan endemic Martino’s vole, Dinaromys bogdanovi, reveals strong differentiation within the western Balkan Peninsula. Mol Ecol 16:1221–1232
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Lee M-Y, Lissovsky AA, Park S et al (2008) Mitochondrial cytochrome B sequence variations and population structure of Siberian Chipmunk (Tamias Sibiricus) in Northeastern Asia and population substructure in South Korea. Mol Cells 26:566–575
Lee YS, Markov N, Argunov A et al (2016) Genetic diversity and phylogeography of Siberian roe deer, Caproulus pygargus, in central and peripheral populations. Ecol Evol. https://doi.org/10.1002/ece3.2458
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lin T-T, You E-M, Lin YK (2009) Social and genetic mating systems of the Asian lesser white-toothed shrew, Crocidura shantungensis, in Taiwan. J Mammal 90:1370–1380. https://doi.org/10.1644/08-MAMM-A-346R1.1
Lin HD, Chen YR, Lin SM (2012) Strict consistency between genetic and topographic landscapes of the brown tree frog (Buergeria robusta) in Taiwan. Mol Phylogenet Evol 62:251–262. https://doi.org/10.1016/j.ympev.2011.09.022
Miller SG (1901) Descriptions of three new Asiatic shrews. Biol Soc Wash 14:157–159
Miraldo A, Hewitt GM, Paulo OS, Emerson BC (2011) Phylogeography and demographic history of Lacerta lepida in the Iberian Peninsula: multiple refugia, range expansions and secondary contact zones. BMC Evol Biol. https://doi.org/10.1186/1471-2148-11-170
Motokawa M, Lin L-K, Harada M, Hattori S (2003) Morphometric geographic variation in the Asian lesser white-toothed shrew Crocidura shantungensis (Mammalia, Insectivora) in East Asia. Zoolog Sci 20:789–795
Motokawa M, Yu H-T, Harada M (2005) Diversification of the white-toothed shrews of the genus Crocidura (Insectivora: Soricidae) in East and Southeast Asia. Mammal Study 30:S53–S64
Nowak RM (1999) Walker’s mammals of the world, 6th edn. Johns Hopkins University Press, Baltimore
Ohdachi SD, Iwasa MA, Nesterenko VA et al (2004) Molecular phylogenetics of crocidura shrews (insectivora) in East and Central Asia. J Mammal 85:396–403
Ohdachi SD, Ishibashi Y, Iwasa MA et al (2009) The wild mammals of Japan. Shoukadoh Book Sellers and Mammalogical Society of Japan, Kyoto
Qiu Y, Fu C, Peter H (2011) molecular phylogenetics and evolution plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of quaternary climate and environmental change in the world’s most diverse temperate flora. Mol Phylogenet Evol 59:225–244
Rambaut A, Suchard MA, Xie D, Drummond AJ (2014) Tracer v 1.6. http://beast.bio.ed.ac.uk/Tracer. Accessed 1 May 2018
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. https://doi.org/10.1093/bioinformatics/btg180
Sakka H, QUÉRÉ JP, Kartavtseva I et al (2010) Comparative phylogeography of four Apodemus species (Mammalia: Rodentia) in the Asian Far East: evidence of Quaternary climatic changes in their genetic structure. Biol J Lin Soc 100:797–821
Schmitt T (2007) Molecular biogeography of Europe: pleistocene cycles and postglacial trends. Front Zool 4:11. https://doi.org/10.1186/1742-9994-4-11
Shafer ABA, Cullingham CI, Côté SD, Coltman DW (2010) Of glaciers and refugia: a decade of study sheds new light on the phylogeography of northwestern North America. Mol Ecol 19:4589–4621
Shi YF (2006) The quaternary glaciations and environmental change in China (Hardbac ks). Hebei Science Technology Press, Shijiazhuang, pp 65–101
Taberlet P, Fumagalli L, Wust-Saucy A-G, Cosson J-F (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464
Tajima F (1989) The effect of change in population size on DNA polymorphism. Genetics 123:597–601
Tomasik E, Cook J (2005) Mitochondrial phylogeography and conservation genetics of wolverine (Gulo Gulo) of Northwestern North America. J Mammal 86:386–396. https://doi.org/10.1644/BER-121.1
Tzeng C (1986) Distribution of the freshwater fishes of Taiwan. Taiwan Museum 39:127–146
Wang JP, Lin HD, Huang S et al (2004) Phylogeography of Varicorhinus barbatulus (Cyprinidae) in Taiwan based on nucleotide variation of mtDNA and allozymes. Mol Phylogenet Evol 31:1143–1156
Weiss S, Ferrand N (2007) Phylogeography of southern european refugia: evolutionary perspectives on the origins and conservation of European biodiversity. Springer, Dordrecht
Wójcik JM, Wolsan M (1998) Evolution of shrews. Mammal Research Institute, Polish Academy of Sciences, Bialowieza
Won C, Smith KG (1999) History and current status of mammals of the Korean Peninsula. Mammal Review 29:3–33
Yang Z (1991) Evolution of eastern shelf of China in quaternary and its environmental consequences. In: Liang M, Zhang J (eds) Correlation of onshore and offshore quaternary in China. Science Press, Beijing, pp 1–22
Yoon M-H, Han S-H, Oh H-S, Kim JG (2004) The mammals of Korea. Dongbangmedia, Seoul
Yu G, Chen X, N i J, Cheddadi R et al (2000) Palaeovegetation of China: a pollen data-based synthesis for the mid-Holocene and last glacial maximum. J Biogeogr 27:635–664
Yuan SL, Lin LK, Oshida T (2006) Phylogeography of the mole-shrew (Anourosorex yamashinai) in Taiwan: implications of interglacial refugia in a high-elevation small mammal. Mol Ecol 15:2119–2130. https://doi.org/10.1111/j.1365-294X.2006.02875.x
Zhang H, Yan J, Zhang G, Zhou K (2008) Phylogeography and demographic history of chinese black-spotted frog populations (Pelophylax nigromaculata): evidence for Independent refugia expansion and secondary contact. BMC Evol Biol 8:21. https://doi.org/10.1186/1471-2148-8-21
Zheng YQ, Yu G, Wang SM et al (2003) Simulations of LGM climate of East Asia by regional climate model. Sci China 46:753–764
Zhou SZ, Jijun L, Zhang SQ et al (2004) Quaternary glaciations in China. Dev Quat Sci 2:105–113. https://doi.org/10.1016/S1571-0866(04)80116-7
Acknowledgements
We gratefully acknowledge Dr. Thomas W. Sappington, USDA-ARS, for his critical comments and edits on this manuscript. We would like to express our deep gratitude to all cooperators who donated the Asian lesser white-toothed shrew samples for this study including Conservation Genome Resource Bank for Korean Wildlife, National Institute of Biological Resources, Korea National Institute of Health, Tunghai University, National Taiwan University, and Shandong University at Weihai. This study was supported by a Grant (2010–0025751) from the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology, and partially supported by the Research Institute for Veterinary Science, Seoul National University. Also, this study was supported by the Major International (Regional) Joint Research Project of National Sciences Foundation of China (NSFC, 31110103910).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Lee, SJ., Lee, MY., Lin, LK. et al. Phylogeography of the Asian lesser white-toothed shrew, Crocidura shantungensis, in East Asia: role of the Korean Peninsula as refugium for small mammals. Genetica 146, 211–226 (2018). https://doi.org/10.1007/s10709-018-0014-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-018-0014-2