

Abstract
Translation initiation defines the identity and quantity of a synthesized protein. The process is dysregulated in many human diseases1,2. A key commitment step is when the ribosomal subunits join at a translation start site on a messenger RNA to form a functional ribosome. Here, we combined single-molecule spectroscopy and structural methods using an in vitro reconstituted system to examine how the human ribosomal subunits join. Single-molecule fluorescence revealed when the universally conserved eukaryotic initiation factors eIF1A and eIF5B associate with and depart from initiation complexes. Guided by single-molecule dynamics, we visualized initiation complexes that contained both eIF1A and eIF5B using single-particle cryo-electron microscopy. The resulting structure revealed how eukaryote-specific contacts between the two proteins remodel the initiation complex to orient the initiator aminoacyl-tRNA in a conformation compatible with ribosomal subunit joining. Collectively, our findings provide a quantitative and architectural framework for the molecular choreography orchestrated by eIF1A and eIF5B during translation initiation in humans.
Similar content being viewed by others

Main
Human protein synthesis is initiated when ribosomal subunits identify a translation start site on messenger RNA (mRNA) and assemble a functional 80S ribosome. The multi-step process has been outlined biochemically and is mediated by numerous eukaryotic initiation factors (eIFs) and the initiator aminoacyl-tRNA3,4,5 (\({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\)). To begin, the 43S pre-initiation complex (43S PIC)—the small (40S) ribosomal subunit bound by eIF1, eIF1A, eIF3, eIF5 and the eIF2–GTP–\({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) complex—loads onto the mRNA, facilitated by eIF4 proteins bound near the N7-methylguanosine (m7G) cap6,7. Directional 5′ to 3′ movement of the now 48S initiation complex along the 5′ untranslated region scans for translation start sites in favourable sequence contexts8,9. A start codon (typically AUG) correctly placed at the ribosomal P site triggers remodelling of the complex from an ‘open’ scanning-permissive conformation to a ‘closed’ scanning-arrested one. The transition involves repositioning of the 40S head and \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\), release of eIF1 and the eIF2–GDP complex, and relocation of eIF59,10. To complete initiation, the GTPase eIF5B catalyses joining of the large (60S) ribosomal subunit to form the 80S ribosome11,12,13. Subsequent GTP hydrolysis-dependent departure of eIF5B enables protein synthesis to commence11,14.
What allows ribosomal subunits to join and form stable inter-subunit contacts remains unclear. One challenge is that the acceptor stem of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) must be slotted into the correct narrow channel on the incoming 60S subunit. Proper \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) orientation avoids steric clashes and poises its aminoacylated 3′ end for accommodation in the peptidyl transfer centre. We hypothesized that universally conserved eIF1A and eIF5B might have a central role in this process. eIF1A resides in the 40S ribosomal A site during initiation and mediates scanning and start codon recognition via its flexible N- and C-terminal tails8,9. Although the eIF5B-bound 48S initiation complex has not been visualized, structures exist for the eIF5B-bound 80S complex15,16,17. These snapshots suggest that eIF5B core domains (G, II and III) dock onto the 40S subunit outside of the A site. Domain IV of the protein extends inwards to latch onto the acceptor stem of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\). Complementary molecular and genetic studies further suggest that eukaryote-specific contacts between domain IV of eIF5B and the C-terminal tail of eIF1A facilitate 60S subunit joining18,19,20,21,22. Yet, whether these two factors coordinate to enable ribosomal subunit joining by guiding formation of inter-subunit contacts or by actively remodelling initiation complexes remains unknown.
To uncover how eIF5B and eIF1A mediate ribosomal subunit joining, we combined real-time single-molecule assays of human translation initiation with targeted structural analyses. We first performed in vitro reconstituted experiments in which fluorescently labelled ribosomal subunits, eIF1A and eIF5B were monitored directly throughout initiation using single-molecule spectroscopy. Our assays revealed a transient state in which both eIF1A and eIF5B are bound just prior to 60S subunit joining. Using single-particle cryo-electron microscopy (cryo-EM), we characterized structurally this hitherto elusive intermediate, which established the structural basis for eIF5B-catalysed ribosomal subunit joining.
Direct monitoring of translation initiation
We established a real-time single-molecule assay to monitor human translation initiation directly (Extended Data Fig. 1a,b). β-Globin mRNA was tethered to an imaging surface within zero-mode waveguides (ZMWs), which enable four-colour fluorescence detection in real time23 (Extended Data Fig. 1c). Saturating concentrations of eIF4 proteins (4A, 4B, 4E and 4G), ATP and GTP were then added to the tethered mRNA. Separately, the 43S PIC was pre-assembled and contained 40S subunits labelled with Cy3 on the N terminus of uS19. Upon start of data acquisition at 30 °C, a mixture with 5 nM 43S PIC (including 40S–Cy3), 1 µM eIF5B and 100 nM 60S subunits labelled with Cy5 on the C terminus of uL18 was added to the imaging surface. When excited by a 532 nm laser, appearance of Cy3 fluorescence indicated that the 40S subunit was loaded onto the mRNA. Subsequent appearance of Cy3(donor)–Cy5(acceptor) Förster resonance energy transfer (FRET) indicated successful joining of the 60S subunit24 (FRET efficiency (EFRET) = 0.65 ± 0.01) (Extended Data Fig. 1d–g). As expected, 40S subunit loading onto mRNA was dependent on the presence of eIF4 proteins (40S loading rate (kon,40S) = 0.049 ± 0.002 s−1 at 5 nM, equivalent to approximately 10 µM−1 s−1) (Extended Data Fig. 1h,i). Consistent with the low nanomolar equilibrium dissociation constants of the eIFs for the initiation complex25, the majority of loaded 43S PICs progressed to 60S subunit joining (60S joining rate (kon,60) = 0.033 ± 0.001 s−1 at 100 nM), which was dependent on the presence of eIF4 proteins and eIF5B (Extended Data Fig. 1h,i). The mean elapsed time from 40S subunit loading to 60S subunit joining that we observed (approximately 30 s) agreed well with initiation timescales obtained in cells26 (20–30 s).
eIF5B binds after eIF2 release
We next tracked fluorescently labelled eIF5B as it mediated 60S subunit joining. We labelled N-terminally truncated eIF5B on its N terminus with a Cy3.5 dye via a fused ybbR peptide tag, which was functional in bulk translation (Extended Data Fig. 2a,b). After the eIF4–mRNA complex was tethered in ZMWs, a mixture of the 43S PIC (10 nM) (labelled by 40S–Cy3), eIF5B–Cy3.5 and 60S–Cy5 subunits was added (Fig. 1a). Fluorescent signals were monitored in real time upon excitation by a 532 nm laser, with the presence of eIF5B on initiation complexes determined by direct excitation of the Cy3.5 dye. The predicted distance between eIF5B and 40S subunit labelling sites was incompatible with FRET16,17 (Extended Data Fig. 2c).

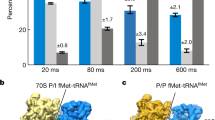
a, Schematic of the single-molecule experiment. The 43S PIC (10 nM, labelled via 40S–Cy3 subunits (green) and including eIFs 1, 1A, 3, 5 and eIF2–GTP–Met-tRNAiMet), 20 nM eIF5B–Cy3.5 (orange) and 100 nM 60S–Cy5 (red) subunits were added to m7G-capped β-globin mRNA tethered within ZMWs in the presence of saturating concentrations of eIFs 4A, 4B, 4G and 4E, and 1 mM ATP and GTP at 30 °C. Fluorescence data were acquired at 10 frames per second for 600 s with excitation via a 532 nm laser. b, Example single-molecule fluorescence data that depict sequential association of the 40S subunit (green), eIF5B (orange) and the 60S subunit (red). Dwell times correspond to eIF5B association (t1), 60S subunit joining (t2) and eIF5B departure times from the 80S complex (t3). The loss of the 60S–Cy5 fluorescence signal was owing to photobleaching of the dye, rather than departure of the ribosomal subunit. c, Density heat map (arbitrary units) of the normalized 40S–Cy3, eIF5B–Cy3.5, and 60S–Cy5 fluorescence intensities synchronized to joining of the 60S subunit (n = 136). d, Observed times for eIF5B association (t1), 60S subunit joining (t2) and eIF5B departure from the 80S complex (t3) at 20 nM eIF5B–Cy3.5 and 100 nM 60S–Cy5 at 30 °C (n = 136). In one experiment (grey, n = 150), eIF5 concentration was reduced 20-fold (from 50 nM to 2.5 nM) and eIF5B–Cy3.5 concentration was increased to 40 nM. The 43S PIC was present at 10 nM (via 40S–Cy3) in all experiments. The fast-phase rates are indicated, which corresponds to association (t1 and t2) or dissociation (t3) rates and were derived by fits to exponential functions (dashed lines). AU, arbitrary units.
We observed sequential association of the 40S subunit, eIF5B and the 60S subunit, which was followed by rapid departure of eIF5B (Fig. 1b,c and Extended Data Fig. 2d). Unlike transient eIF5B sampling previously observed on yeast 48S complexes14, human eIF5B stably associated in a concentration-independent manner with 48S complexes prior to 60S subunit joining (eIF5B association rate (kon,5B) ≈ 0.1 s−1 at 20 and 40 nM) (Fig. 1d and Extended Data Fig. 2e). The apparent concentration-independent association of eIF5B suggests the presence of a slow upstream step that limits its binding. Nevertheless, once eIF5B bound, the 60S subunit joined in a concentration- and temperature-dependent manner (kon,60S ≈ 0.14 ± 0.004 s−1 at 200 nM, 30 °C, equivalent to approximately 0.7 µM−1s−1) (Fig. 1d and Extended Data Fig. 2e,f). Formation of the 80S initiation complex triggered rapid and temperature-dependent departure of eIF5B (eIF5B dissociation rate (koff,5B) ≈ 1.2 ± 0.1 s−1 at 30 °C), which was prevented by substitution of the catalytic histidine to block GTP hydrolysis (koff,5B < 0.01 s−1 at 30 °C) (Fig. 1d and Extended Data Fig. 2e–g).
We hypothesized that the departure of eIF2 was needed for eIF5B to bind initiation complexes, since both proteins bind the acceptor stem of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{M}}{\rm{e}}{\rm{t}}}\). We therefore monitored eIF5B association with initiation complexes upon a 20-fold reduction in eIF5 concentration (from 50 nM to 2.5 nM), as eIF5 activates GTP hydrolysis by eIF2 to enable eIF2 departure. When eIF5 was limiting, the apparent eIF5B association rate was reduced by at least tenfold (kon,5B ≈ 0.009 s−1 at 40 nM) (Fig. 1d and Extended Data Fig. 2e). We reasoned that pre-equilibration of 48S initiation complexes with a saturating concentration of eIF5 would allow eIF2 release and accelerate eIF5B association (Extended Data Fig. 3a). Indeed, eIF5B stably associated approximately ninefold more rapidly and in a concentration-dependent manner with pre-equilibrated 48S complexes to mediate 60S subunit joining (kon,5B ≈ 20 µM−1 s−1) (Extended Data Fig. 3b–e). Conversely, addition of a limiting concentration of eIF5 (2.5 nM) simultaneously with labelled eIF5B to tethered 48S complexes equilibrated in the absence of eIF5 delayed eIF5B association nearly 50-fold (Extended Data Fig. 3c). eIF5B association also was nearly eliminated when departure of eIF2 was prevented by using the non-hydrolyzable GTP analogue GDPNP (Extended Data Fig. 3f). Our findings therefore suggest that eIF2 must dissociate from initiation complexes before eIF5B binds.
eIF1A and eIF5B co-occupy complexes
We next directly tracked eIF1A–Cy5, labelled via an S102C substitution25, throughout translation initiation. We added double-labelled 43S PICs (40S–Cy3 and eIF1A–Cy5), 1 µM unlabelled eIF5B and 200 nM 60S subunits labelled with Cy5.5 to eIF4–mRNA complexes tethered within ZMWs at 30 °C (Fig. 2a). Nearly all loaded 43S PICs began in a 40S (donor)-to-eIF1A (acceptor) FRET state (90 ± 4 %, 158 out of 175; EFRET = 0.42 ± 0.01), as predicted given the proximity of the labelling sites (approximately 50 Å) (Fig. 2b,c and Extended Data Fig. 4a–d). Most complexes were joined by the 60S subunit during the first eIF1A binding event (56 ± 7 %, 88 out of 158) (Extended Data Fig. 4b). Complexes with multiple eIF1A binding events also were competent for 60S subunit joining (87 out of 175; eIF1A reassociation rate (k1A,reass.) ≈ 1.5 ± 0.1 s−1 at 45 nM), which suggests the mRNA-loaded complexes can remodel dynamically (Extended Data Fig. 4c,e). Although eIF1A bound stably to the 48S complex (eIF1A dissociation rate (k1A,off) ≈ 0.075 ± 0.003 s−1), the protein departed within a few hundred milliseconds after 60S subunit joining (k1A,off > 10 s−1, mean lifetime < 0.2 s) (Fig. 2d and Extended Data Fig. 4f). eIF1A departure from the 80S complex was delayed at least tenfold (k1A,off ≈ 1.7 ± 0.1 s−1, mean lifetime = 3 ± 1.5 s) when GTP hydrolysis by eIF5B was prevented by using GDPNP (Fig. 2d and Extended Data Fig. 4f). Notably, multiple lines of evidence confirmed that fluorescently labelled eIF1A and eIF5B functionally substituted for the unlabelled proteins (Extended Data Fig. 4g,h). Thus, GTP hydrolysis by eIF5B accelerates eIF1A dissociation from 80S initiation complexes, and eIF1A release precedes eIF5B departure, consistent with findings in yeast19,20,27.

a, Schematic of the single-molecule experiment. The double-labelled 43S PIC (10 nM, labelled by 40S–Cy3 (green) and eIF1A–Cy5 (red)), 1 µM unlabelled eIF5B and 200 nM 60S–Cy5.5 (purple) subunit were added to β-globin mRNA tethered within ZMWs in the presence of saturating concentrations of of eIFs 4A, 4B, 4G and 4E, and 1 mM ATP and GTP at 30 °C. During imaging, eIF1A–Cy5 was present at 4.5-fold molar excess relative to the 40S subunit. Fluorescence data were acquired at 10 frames per second for 600 s with excitation via a 532 nm laser. b, Example single-molecule fluorescence data that depict recruitment of a double-labelled 43S PIC (40S–eIF1A FRET state) followed by 60S subunit joining (40S–60S FRET state) and rapid ejection of eIF1A from the 80S initiation complex. The full fluorescence trace is shown in Extended Data Fig. 4b. c, Density heat map (arbitrary units) of the normalized 40S–Cy3, eIF1A–Cy5 and 60S–Cy5.5 fluorescence intensities synchronized to joining of the 60S subunit (n = 175). d, Observed eIF1A lifetimes on the 80S initiation complexes in the presence of eIF5B–GTP (purple, n =175) or eIF5B–GDPNP (green, n = 151) or on the pre-equilibrated 48S initiation complex when eIF5B was present (dark grey, n = 589) or absent (light grey, n = 831). Fast-phase dissociation rates are indicated, derived or estimated from fits to double-exponential functions (dashed lines).
As suggested above, eIF1A and eIF5B reside simultaneously on initiation complexes when the 60S subunit joins. Addition of 20 nM eIF5B–Cy3.5 and 10 nM eIF1A–Cy5 to double-labelled 48S initiation complexes (40S–Cy3 and eIF1A–Cy5) tethered within ZMWs revealed complexes that contained both proteins (Extended Data Fig. 5a). The complexes produced FRET signals from both 40S–eIF1A and eIF5B–eIF1A interactions (Extended Data Fig. 5b,c). The signal from the eIF5B–eIF1A interaction is consistent with the proximity of eIF5B and eIF1A labelling sites predicted by crude structural comparisons (approximately 70 Å) (Extended Data Fig. 5b). Whereas reassociation rates were similar, eIF1A dissociated from 48S complexes about sevenfold more slowly when eIF5B was present relative to when eIF5B was absent (k1A,off ≈ 0.059 ± 0.002 versus 0.41 ± 0.01 s−1) (Fig. 2d and Extended Data Fig. 5d). Simultaneous addition of double-labelled 43S PICs (40S–Cy3 and eIF1A–Cy5), eIF5B–Cy3.5 and 60S–Cy5.5 subunits to tethered mRNA revealed that nearly all 60S subunit joining events occurred when eIF1A and eIF5B were both present (90 ± 10%) (Extended Data Fig. 5e–h).
Structure of the eIF1A–eIF5B initiation complex
To understand how the two proteins collaborate to mediate 60S subunit joining, we used our single-molecule findings to design a vitrification strategy that captured a 48S complex co-bound by eIF1A and eIF5B on cryo-EM grids. Immediately before plunge freezing, pre-assembled 48S complexes were incubated with eIF5B for 1 min at 30 °C in the presence of ATP and GTP. At this time point, the 48S complex should be saturated with both eIF1A and eIF5B, and the complex is competent for 60S subunit joining (Extended Data Figs. 2e and 3c). Vitrified samples were analysed by single-particle cryo-EM.
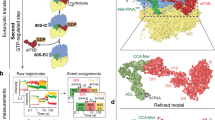
After data collection, image processing and in silico classifications, we identified a subset of around 190,000 particles that yielded a high-resolution map after refinement (overall resolution of approximately 3.2 Å) (Extended Data Fig. 6a, b and Extended Data Table 1). This subset represents around 84% of all 40S particles. Consistent with our prediction above, robust density was visible for the 40S subunit, an aminoacylated tRNA near the ribosomal P site, eIF1A in the A site and eIF5B (Fig. 3a). Peripheral regions of the map were heterogenous, such as the acceptor stem of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) and eIF5B domains located furthest from the 40S surface, namely domains G and IV. In these flexible areas, local resolution values were in the 4–6 Å range, with sufficient quality to place secondary structure elements unambiguously (Extended Data Fig. 6c–e). Density attributable to other eIFs—including eIF3—was absent from cryo-EM maps in this class, even at low map thresholds. The lack of density for eIF3 suggests that it may be highly dynamic or potentially has departed the initiation complex after eIF2 departure. A small subgroup of 40S particles (around 2,100 (0.65% of the dataset)) presented density ascribable to eIF3, but the reduced number of particles precluded further interpretation.

a, Post-processed density for the cryo-EM map (overall resolution 3.2 Å) of the eIF1A- and eIF5B-bound initiation complex, with 40S subunits coloured grey, \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) in green, eIF1A in red and eIF5B in blue. Right, view of the refined molecular model derived from the cryo-EM data centred around eIF1A, with GTP coloured yellow. Domains of eIF5B and regions of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) are indicated. ASL, anticodon stem loop; AS, acceptor stem; h, helix. The cryo-EM data and final atomic coordinates were deposited at the EMDB and PDB under accessions EMD-26067 and 7TQL, respectively. b, Left, zenithal view of the cryo-EM map with components coloured as in a. Centre, view of the molecular model along the acceptor stem axis of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\). Right, detailed view of the GTP-binding pocket of eIF5B. Y1063, which belongs to eIF5B domain III, collaborates with G-domain residues V640 and I664, part of the hydrophobic gate, to protect the γ-phosphate of GTP from hydrolysis. c, Corresponding views of eIF5B domain IV in the 40S state with eIF1A present (left) and after 60S joining and eIF1A departure (right). Departure of eIF1A allows for an approximately 30º rotation of eIF5B domain IV towards the 60S subunit, bringing the acceptor stem of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) closer to the peptidyl transfer centre. d, Enlarged view of the eIF5B–eIF1A interface. Terminal eIF5B α-helices h13 and h14 approach eIF1A, stabilizing weak interactions (more than 4 Å interacting distance) nucleated around eIF1A M50 and eIF5B residue I1219.
eIF5B density corresponds to the GTP-bound state. The core superdomain of eIF5B, comprising domains G, II and III, are present in the GTPase binding site on the 40S platform, which represents the largest interaction surface (approximately 820 Å2) in the complex (Fig. 3a). The G domain of eIF5B is exposed to solvent, with no direct 40S contacts. It is positioned via interactions with domain III and the base of helix 12. Consistent with a GTP-bound state, switch I of the G domain is ordered and density for the γ-phosphate of GTP is apparent (Fig. 3b and Extended Data Fig. 6e). The catalytic histidine is maintained away from the γ-phosphate by a steric barrier formed by residues I664 of switch I and V640 of the P loop (Fig. 3b, right). These two residues form the prototypical hydrophobic gate that must be pierced by the catalytic histidine to catalyse GTP hydrolysis. The gate is aided by Y1063, as its position at the base of helix 12 interferes with the trajectory the catalytic histidine must follow to reach the γ-phosphate28.
Domain IV of eIF5B directly interacts with \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) and eIF1A. Mediated by rigid helix 12 that extends away from the G-II-III superdomain, eIF5B domain IV is latched onto the acceptor stem of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\). Of note, relative to its position in 80S initiation complexes15,16,17, domain IV is rotated about 30 degrees along the helix 12 axis towards the ribosomal A site (Fig. 3c and Supplementary Video 1). This rotation positions the two eukaryote-specific, C-terminal α-helices of eIF5B (helices 13 and 14) to contact eIF1A directly, which is positioned identical to earlier initiation intermediates29,30,31,32,33,34,35,36,37,38,39 (Extended Data Fig. 7a,b). The interaction interface is relatively small (approximately 150 Å2) and nucleated around a cluster of hydrophobic residues (M50 of eIF1A and I1219 of eIF5B) (Fig. 3d). Whereas the flexible N-terminal tail of eIF1A is disordered, we observed additional density proximal to W1207 on helix 14 of eIF5B that may correspond to I141 of the flexible eIF1A C-terminal tail (Extended Data Fig. 7a). Such contacts are consistent with NMR studies on the two proteins in isolation40,41.
The rotation of eIF5B domain IV induced by eIF1A reorients the \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) configuration to be compatible with 60S subunit joining. We compared our model to two previous structures of eIF2-bound intermediates—scanning-competent ‘open’37 or post-scanning ‘closed’ complexes36—and to elongation-competent 80S ribosomes42 (Extended Data Fig. 7b). In our eIF5B-bound complex, the 40S subunit head conformation is similar to both the post-scanning ‘closed’ and elongation-competent conformations (Extended Data Fig. 7c). By contrast, the \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) configuration most closely resembles an elongation state; the ASL at the 40S P site is nearly identical to the final elongation state, and the acceptor stem and elbow region are almost perpendicular to the ASL (Extended Data Fig. 7d). In this position and mediated by eIF1A, eIF5B domain IV and the acceptor stem are aligned to slot directly into the approximately 35 Å channel formed by the central protuberance and helix 69 of the incoming 60S subunit (Fig. 4a,b). Thus, our structural analyses suggest that the 40S–eIF1A–eIF5B–acceptor-stem network of interactions are needed to funnel multiple conformations of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) towards a position compatible with 60S subunit joining.

a,b, Structural models that depict how eIF1A and eIF5B domain IV orient the acceptor stem of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) to slot into the channel formed by the central protuberance (CP) and helix 69 (h69) of the incoming 60S subunit (a); as it joins, the 60S subunit triggers GTP hydrolysis by eIF5B, which accelerates eIF1A departure by h69, probably by facilitating back-rotation of the 40S subunit from a semi-rotated state to a classical non-rotated state (b) (see Discussion). c, Analysis of eIF5B mutants in single-molecule assays. Top, views of the eIF5B-bound 48S complex that depict the location of R1105 and R1174, which contact \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\), and R1188, Q1189, R1199, E1218 and I1219, which form an interface that contacts eIF1A. Centre left, observed lifetimes of the wild-type (WT) and indicated mutant eIF5B proteins on the 48S complex at 20 nM at 30 °C. n = 156 (WT), 251 (RR/AA) and 196 (RQREI/ERERR). Centre right, observed 60S joining times, quantified relative to 40S loading, when the indicated eIF5B protein was present at 20 nM at 30 °C. n = 133 (WT), 123 (RR/AA) and 93 (RQREI/ERERR). Bottom, summary of eIF5B dissociation rates from and 60S subunit association rates with the 48S initiation complex (only fast-phase rates are shown; see Extended Data Fig. 8g,i for all rates). d, Proposed model for how human eIF1A and eIF5B collaborate to orient \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) to catalyse ribosomal subunit joining, consistent with the single-molecule dynamics and structures. PTC, peptidyl transfer centre.
We used our single-molecule assays to assess the role of residues in eIF5B that our cryo-EM-derived model suggested mediate contacts with \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) or eIF1A in translation initiation. Contacts with the \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) acceptor stem were disrupted by substitution of conserved R1105 and R1174 with alanine (RR/AA) (Fig. 4c and Extended Data Fig. 8a,b). Contacts with eIF1A were disrupted either by truncation of eIF5B prior to helix 14 (Δ−h14) or five targeted substitutions of residues in helices 13 and 14 that invert surface contacts with eIF1A (RQREI/ERERR) (Fig. 4c and Extended Data Fig. 8a,b). After the eIF4–mRNA complex was tethered in ZMWs, a mixture of the 43S PIC (labelled with 40S–Cy3), wild-type or mutant eIF5B–Cy3.5 and 60S–Cy5 was added, and fluorescent signals were monitored in real time (Extended Data Fig. 8c–e). Relative to the wild-type protein, the three mutant eIF5B proteins were much more likely (5- to 7-fold) to sample the 48S complex (eIF5B dissociation rate (k5B,off) ≈ 2–4 s−1) (Fig. 4c and Extended Data Fig. 8f,g). The destabilized mutant eIF5B proteins reduced the efficiency of 60S subunit joining, and when the 60S did join, it occurred at least 2- to 3-fold more slowly than with the wild-type protein (Fig. 4c and Extended Data Fig. 8h,i). Thus, the eIF5B–eIF1A–40S–\({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) interactions stabilize and remodel the 48S initiation complex to allow 60S subunit joining.
Discussion
Our single-molecule and structural results suggest a model for how eIF1A and eIF5B mediate human ribosomal subunit joining (Fig. 4d and Supplementary Video 2). The eIF1A–40S subunit complex, presumably with the other components of the 43S PIC, loads onto the mRNA. eIF1A remains stably bound in the A site throughout scanning and start codon recognition, consistent with its key roles in those steps. Once eIF2 departs, eIF5B stably binds and forms the 40S late initiation complex (40S LIC). The delay from 40S subunit loading until eIF5B association (about 10 s) therefore suggests an upper bound for the integrated timescale of scanning and start codon recognition on β-globin mRNA. Stable eIF5B association with the 40S LIC probably provides the molecular basis for how eIF5B prevents reassociation43 of eIF2–GDP with \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\). Furthermore, relative to yeast14, the enhanced stability of human eIF5B on the 40S LIC provides insights needed to understand how increased eIF5B abundance drives aberrant translation in lung adenocarcinomas44.
For eIF5B to bind stably to the 40S LIC, domain IV of the protein must contact both eIF1A and \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\). Loss of contacts with either led to a 100-fold decrease in eIF5B lifetime on the complex and inhibited 60S subunit joining. Thus, multiple relatively weak interactions probably stabilize this conformationally flexible GTPase on the initiation complex in its active form. Previous structural analyses of the protein in isolation or bound to eIF1A have suggested that eIF5B adopts both extended and compact conformations, with domain IV either extended away from or adjacent to the G–II–III superdomain40,41,45,46,47. In the context of initiation, contacts with eIF1A may enhance or stabilize extension of domain IV away from the G-II-III superdomain of eIF5B, as the compact conformation would clash sterically with eIF1A in the ribosomal A site.
Eukaryote-specific contacts between eIF1A and eIF5B reconfigure the \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) acceptor stem, driven by rotation of eIF5B domain IV along the helix 12 axis. This surprising rotation—rather than a rigid, swing-like movement predicted by studies in prokaryotes48 and archaea45—funnels the complex into a conformation that slots directly into the channel formed by the central protuberance and helix 69 of the 60S subunit. Formation of the 80S ribosome triggers hydrolysis of GTP by eIF5B to accelerate ejection of eIF1A by helix 69 of the 60S subunit (within approximately 100 ms), probably by accelerating back-rotation of the 40S subunit from a semi-rotated state to a classical non-rotated state, similar to prokaryotes49,50,51. Indeed, inter-subunit back-rotation moves the apical loop of helix 69 by around 16 Å further into the ribosomal A site, where eIF1A resides. eIF1A departure frees eIF5B domain IV and the acceptor stem to rotate approximately 30° towards the peptidyl transfer centre. The rotation poises the aminoacylated end of the tRNA for its final accommodation upon eIF5B departure, which occurs within another few hundred milliseconds. Dynamic rotation of eIF5B domain IV mediated by eIF1A therefore plays a key role in the late steps of translation initiation.
Collectively, our findings suggest that the eIF1A–eIF5B–\({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) network of interactions serves as a final fidelity checkpoint to ensure that initiation complexes are competent for the 60S subunit to join. They also demonstrate that eIF5B, mediated by contacts with eIF1A, actively remodels the 40S LIC to facilitate 60S subunit joining, rather than serving simply as a chaperone of inter-ribosomal subunit contacts. Almost immediately after formation of the 80S initiation complex, GTP hydrolysis by eIF5B catalyses a series of events that clears the ribosomal A site and positions the aminoacylated end of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) in the peptidyl transfer centre of the ribosome, which commits the ribosome to synthesize a protein.
Methods
eIF5B cloning
N-terminally truncated (587–1220) eIF5B with an N-terminal ybbR-tag and relevant mutants were purchased as geneblocks from IDT and cloned into a vector purchased from the UC Berkeley QB3 MacroLab (vector 1B) using their standard protocol. Contacts with \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) were disrupted by R1105A and R1174A substitution. Contacts with eIF1A were disrupted by truncation of eIF5B at Q1203 (Δ−helix 14) or five substitutions that inverted properties along the interaction interface (R1188E, Q1189R, R1199E, E1218R and I1219R). All sequences were verified by Sanger sequencing. The resulting plasmids encoded eIF5B(587–1220) tagged on the N terminus with a 6-histidine tag fused to maltose-binding protein (MBP) followed by a TEV protease cleavage site, a short NA linker, and the ybbR peptide tag (NH2-6His–MBP–TEV–NA–ybbR–eIF5B-COOH).
β-globin mRNA
A synthetic DNA was purchased from IDT that contained a 5′ flanking sequence (pUC19 backbone sequence), a T7 promoter (TAATACGACTCACTATAG), the human β-globin transcript 1 (NM_000518.5, 628 nts), 8 non-templated adenosines, and a 3′ flanking sequence (pUC19 backbone sequence). Linear DNA templates were amplified using NEB Phusion polymerase and a 5′ primer (TGACCATGATTACGCCAAGC) that annealed upstream of the T7 promoter and a 3′ primer (TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTttgcaatgaaaataaatgttttttattagg) that annealed to the 3′ end of β-globin transcript and encoded a poly(A)30 tail. The mRNA was in vitro transcribed using the T7 Megascript kit (Invitrogen, AM1334) and standard conditions. Transcribed RNAs were purified using the GeneJET RNA Purification Kit (ThermoFisher, K0732). The 3′ terminus was biotinylated by potassium periodate oxidation followed by reaction with biotin-hydrazide, as described52. The 5′ terminus was capped with a type I 7-methylguanosine cap (m7G) using the Vaccinia Capping System (NEB, M2080S) and 2′-O-methyltransferase (NEB, M0366), following the one-step protocol. 5′-capped and 3′-biotinylated β-globin mRNA was purified using the GeneJET RNA Purification Kit and stored at −20 °C until use.
Purification and fluorescent labelling of ybbR-tagged eIF5B
All wild-type and mutant ybbR-eIF5B expression plasmids were transformed into Rosetta2 cells purchased from the UC Berkeley QB3 MacroLab and grown overnight at 37 °C on LB agar plates supplemented with 50 µg ml−1 kanamycin. Liquid cultures of single colonies were grown to OD600 of 0.5 at 37 °C in LB supplemented with kanamycin. Cultures were shifted to 18 °C for 30 min, 0.5 mM IPTG was added, and cultures were grown for 16–20 h at 18 °C. Cells were collected by centrifugation at 5,000g for 15 min at 4 °C in a Fiberlite F9 rotor (ThermoFisher, 13456093). Cells were lysed by sonication in lysis buffer (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 10% (v/v) glycerol, 20 mM imidazole, and 5 mM β-mercaptoethanol), and lysates were cleared by centrifugation at 38,000g for 30 min at 4 °C in a Fiberlite F21 rotor followed by filtration through a 0.22 µm syringe filter. Clarified lysate was loaded onto a Ni-NTA gravity flow column equilibrated in lysis buffer, washed with 20 column volumes (CV) of lysis buffer, 20 CV of wash buffer (20 mM Tris-HCl pH 8.0, 1 M NaCl, 10% (v/v) glycerol, 40 mM imidazole, and 5 mM β-mercaptoethanol), and 10 CV of lysis buffer. Recombinant proteins were eluted with five sequential CV of elution buffer (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 10% (v/v) glycerol, 300 mM imidazole, and 5 mM β-mercaptoethanol). Fractions with recombinant protein were identified by SDS–PAGE analysis. The relevant fractions were dialysed overnight at 4 °C into TEV cleavage buffer (50 mM HEPES-KOH pH 7.5, 200 mM NaCl, 10% (v/v) glycerol, and 1 mM DTT) in the presence of excess TEV protease. Fluorescent labelling via the ybbR tag was performed essentially as described52,53. In brief, ~10 µM ybbR-eIF5B was supplemented with 10 mM MgCl2 and incubated at 37 °C for 90 min in the presence of 2–4 µM Sfp synthase enzyme and 20 µM of Cy3.5–CoA substrate. Free dye was removed via purification over 10DG-desalting columns (Bio-Rad, 7322010) equilibrated in TEV cleavage buffer supplemented with 20 mM imidazole. TEV protease, Sfp synthase, and the cleaved 6His tag were removed via a subtractive Ni-NTA gravity column equilibrated in TEV buffer, with the flow-through collected. Labelled proteins were purified using size-exclusion chromatography (SEC) on a Superdex 75 or 200 column (23 ml) equilibrated in SEC storage buffer (20 mM HEPES-KOH pH 7.5, 150 mM KOAc, 10% (v/v) glycerol, and 1 mM DTT). Fractions containing eIF5B proteins were concentrated using a 30 kD MWCO Amicon Ultra centrifugal filter, aliquoted, flash frozen on liquid N2, and stored at −80 °C.
In vitro extract-based translation assays
The nanoluciferase GAPDH reporter mRNA was prepared as described24. Standard HeLa cell-free translation (ThermoFisher, 88882) reactions were programmed with a final mRNA concentration of 100 nM and 840 nM of the indicated eIF5B protein. An equal volume of eIF5B SEC (storage) buffer (without protein) was added to a control reaction to account for buffer effects on IVT activity. IVT reactions were incubated at 37 °C for 45 min and then immediately transferred to an ice water bath and diluted 1:1 with cold Glo Lysis Buffer (Promega, E2661). All samples were brought to room temperature and mixed with a 1:1 volume of nGlow solution (Promega, N1110). Samples (90% of total volume) were loaded into non-adjacent wells of a 384-well plate. Sample luminescence was measured 11 min post nGlow solution addition using a BioTek Neo2 multi-mode plate reader (25 °C, 114LUM1537 filter, gain of 135). Luminescence signal was monitored for an additional 30 min at 3 min intervals to verify luminesce signal of all samples decayed at the same rate. Prism8 (Graphpad) was used for all analyses.
Purification of human eIFs, \({{\bf{Met-tRNA}}}_{{\bf{i}}}^{{\bf{M}}{\bf{e}}{\bf{t}}}\) and ribosomal subunits
Recombinant eIF154, eIF1A54, eIF3j54, eIF4AI54,55, eIF4E55,56 and eIF557 proteins were purified as described. Recombinant eIF4B was purified essentially as described54,55, except cells were lysed solely by sonication and a subtractive Ni-NTA step was added prior to ion-exchange chromatography (heparin). Coding sequence for eIF4G residues 165–1599 was inserted into 6His-tagged pFASTBAC1 vector and purified similar to a described protocol56, with the following modifications: 15 µM leupeptin was added to growth media 24 h before collection, cells were lysed by sonication, and a subtractive Ni-NTA step was added, which was followed by ion-exchange chromatography (heparin) and concentration into SEC buffer with 300 mM KOAc. Native human eIF2 and eIF3 (lacking 3j) were purified as described54,58. Human \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) was transcribed in vitro and aminoacylated as described24. eIF1A(S102C) was purified and fluorescently labelled with Cy5 dye as described25. Human 40S and 60S ribosomal subunits were purified from the appropriately edited HEK293T cell lines and labelled with fluorescent dyes as described24,52. Cell line identities were confirmed by PCR assays, Sanger sequencing, western blotting, and fluorescence gels (after labelling), as described24. Cells were not tested for mycoplasma contamination.
ZMW-based single-molecule spectroscopy
All real-time imaging was conducted using a modified Pacific Biosciences RSII DNA sequencer and the Maggie software (v. 2.3.0.3.154799), which were described previously23. All experiments were performed at 30 °C (unless otherwise noted) using a 532 nm excitation laser at 0.32 µW/µm2, which directly excited Cy3 and Cy3.5 dyes. Cy5 and Cy5.5 dyes were excited via FRET as indicated. Four-colour fluorescence emission (Cy3, Cy3.5, Cy5 and Cy5.5) was detected at 10 frames per second for 600 s. ZMW chips were purchased from Pacific Biosciences, which were passivated by reaction with polyvinylphosphonic acid to form a covalent Al-polyphosphonate coating59. Prior to imaging, all ZMW chips were washed with 0.2% Tween-20 and TP50 buffer (50 mM Tris-OAc pH 7.5, 100 mM KCl). Washed chips were coated with neutravidin by a 5 min incubation with 1 µM neutravidin diluted in TP50 buffer supplemented with 0.7 mg ml−1 UltraPure BSA and 1.3 µM of pre-annealed DNA blocking oligonucleotides (CGTTTACACGTGGGGTCCCAAGCACGCGGCTACTAGATCACGGCTCAGCT) and (AGCTGAGCCGTGATCTAGTAGCCGCGTGCTTGGGACCCCACGTGTAAACG). The imaging surface then was washed with TP50 buffer at least four times.
Real-time initiation single-molecule assays
For all single-molecule assays, the ‘initiation reaction buffer’ was: 20 mM HEPES-KOH, pH 7.3, 70 mM KOAc, 2.5 mM Mg(OAc)2, 0.25 mM spermidine, 0.2 mg ml−1 creatine phosphokinase, 1 mM ATP•Mg(OAc)2, and 1 mM GTP•Mg(OAc)2. The ‘imaging buffer’ was the initiation reaction buffer supplemented with casein (62.5 µg ml−1) and an oxygen scavenging system60: 2 mM TSY, 2 mM protocatechuic acid and 0.06 U µl−1 protocatechuate-3,4-dioxygenase.
To prepare the eIF2–GTP–\({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) ternary complex, 3.3 µM eIF2 was incubated in initiation reaction buffer (excluding ATP•Mg(OAc)2) for 10 min at 37 °C to saturate eIF2 with GTP. The eIF2–GTP complex then was incubated with 2.3 µM \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) for 5 min at 37 °C to form ternary complex. In a few instances, GTP was replaced with a non-hydrolyzable analogue (GDPNP) during formation of ternary complex, which prevents GTP hydrolysis and subsequent departure of eIF2 from the initiation complex.
To prepare the 43S PIC, 1 µM eIF1, 1 µM eIF1A, 500 nM ternary complex (by eIF2), 1.2 µM eIF5, 400 nM eIF3, 1.2 µM eIF3j and 240 nM 40S–Cy3 subunits were incubated for 5 min at 37 °C in initiation reaction buffer. In experiments with double-labelled 43S PICs (40S–Cy3, eIF1A–Cy5), unlabelled eIF1A was substituted for an equal concentration (by Cy5) of eIF1A(S102C)–Cy5.
In all full real-time single-molecule initiation assays, β-globin mRNA with a 5′ m7G cap, poly(A)30 tail, and 3'-terminal biotin moiety was tethered to a prepared and neutravidin-coated ZMW imaging surface. Immediately after tethering and washing the surface with imaging buffer, a mixture of 2 µM eIF4A, 440 nM eIF4B, 260 nM eIF4G, and 320 nM eIF4E in 20 µl of imaging buffer was added to the surface. The surface and reaction mixture were incubated at room temperature for 5-10 min as the instrument initialized. At the start of data acquisition, a 20 µl mixture of 10-20 nM 43S PIC (by 40S–Cy3), 2 µM eIF5B, and 200-400 nM 60S–Cy5/5.5 subunits in imaging buffer was added to the surface. In experiments with fluorescently labelled eIF5B, unlabelled eIF5B was replaced with 40-80 nM eIF5B–Cy3.5. Importantly, addition of this second reaction mixture to the surface doubled the total reaction volume (to 40 µl, since 20 µl with eIF4 proteins was already present); thus, final concentrations of all components during data collection are halved from the values reported above. Final concentrations for labelled components in particular figures are reported in the legends and text.
In a few instances, the 48S initiation complex was pre-equilibrated and tethered to the imaging surface. To do this, 90 nM 43S PIC (by 40S–Cy3), 90 nM of β-globin mRNA, 2 µM eIF4A, 1.1 µM eIF4B, 650 nM eIF4G, and 800 nM eIF4E was incubated for 15 min at 37 °C in initiation reaction buffer. The resulting complex was diluted 1:12.5, immediately added to a prepared and neutravidin-coated ZMW imaging surface, and incubated for 10 min at room temperature to tether the complex to the surface. After washing the surface with imaging buffer, a 20 µl mixture of 1 µM eIF1A, 2 µM eIF4A, 440 nM eIF4B, 260 nM eIF4G, and 320 nM eIF4E in 20 µl of imaging buffer was added to the surface. At the start of data acquisition, a 20 µl mixture of 40–80 nM eIF5B ± 200 nM 60S–Cy5 subunits in imaging buffer was added to the surface. In experiments with labelled eIF1A, a 20 µl mixture with 20 nM eIF1A–Cy5 and 40 nM eIF5B–Cy3.5 was added to the imaging surface with a tethered and double-labelled 48S PIC (in which unlabelled eIF1A was replaced with labelled eIF1A). As above, final concentration of all components during data collection are halved from the reported values.
To probe the role of GTP hydrolysis by eIF5B, the GTP in the imaging buffer was replaced with an equal concentration of the non-hydrolyzable analogue GDPNP. In this experimental setup, ternary complex would remain in the GTP-bound state, as GTP dissociates very slowly from the ternary complex61,62.
Data analysis
Experimental movies that captured fluorescence intensities over time were processed using MATLAB R2018-b as described previously23,24,52. In all analyses, ZMWs with the desired fluorescence signals were identified by filtering for the desired signals.
To determine EFRET, fluorescence data from individual ZMWs with the desired fluorescence signals (for example, 40S–60S FRET) were background corrected using SPARTAN 3.7.063, with single molecules indicated by single-step photobleaching events of the donor fluorophore. FRET on and off states were assigned automatically using vbFRET (version june10)64, which were visually inspected and manually corrected as needed. An EFRET threshold of 0.1 was used in all experiments. EFRET was defined as:
where ID and IA represent fluorescence intensities of the donor and acceptor fluorophores. EFRET values observed across all events and molecules were binned (50 bins, −0.2 to 1.2) and fit to gaussian functions to determine the mean and standard deviations.
To determine association and dissociation kinetics, binding events of individual components (for example, 40S, eIF5B and 60S) were assigned manually based on the appearance and disappearance of the respective fluorescence signals. The observed times for an event to occur from 100–200 individual molecules (that is, ZMWs) were used to calculate cumulative probability functions of the observed data (cdfcalc, MATLAB), which were fit to single- or double-exponential functions in MATLAB (cftool, non-linear least squares method) as described23,65. All derived association rates, median association times, lifetimes, and the number of molecules examined are reported in the relevant figures and legends. The exponential function was defined as:
where t is time (in s), b and c are rates, and d is an adjustment factor. If a parameter yielded a phase that represented less than 10% of the population, only a single phase was used to derive the respective rate and reported (that is, single-exponential function).
Statistical analyses
To calculate errors for the efficiency of a given binding event, bootstrap analyses (n = 10,000) were performed to calculate 95% confidence intervals for the observed proportions using R (4.1.2) and RStudio (Ghost Orchid release) (mosaic library, rflip approach). To calculate errors for median association times and lifetimes, bootstrap analyses (n = 10,000) were performed to calculate 95% confidence intervals of the observed median using MATLAB (bootstrp and bootci functions). Reported errors for derived rates represent 95% confidence intervals yielded from fits to linear, single-exponential or double-exponential functions, as indicated.
Formation of the eIF5B-bound 48S initiation complex for cryo-EM analysis
The eIF2–GTP–\({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) ternary complex was formed as above. To prepare the 43S PIC, 1.5 µM eIF1, 1.5 µM eIF1A, 1 µM ternary complex (by eIF2), 1.5 µM eIF5, 800 nM eIF3, and 600 nM 40S–Cy3 subunits were incubated for 5 min at 37 °C in initiation reaction buffer. The 43S PIC was incubated on ice. In parallel, 200-mesh Quantifoil R2/1 grids (Electron Microscopy Sciences, Q250AR1) were glow-discharged for 25 s in a PELCO EasiGlow glow discharger (Ted Pella, conditions: negative charge, 15 mA, 0.4 mBar, 25 s). After grids were prepared, 300 nM 43S PIC (by 40S), 500 nM of β-globin mRNA, 1 µM eIF4A, 1 µM eIF4B, 650 nM eIF4G, and 800 nM eIF4E were incubated for 3 min at 37 °C in initiation reaction buffer. Immediately after, 1 µM eIF5B was added and the resulting reaction mixture was incubated for ~55 s at 30 °C. A 3 µl sample from the mixture was applied to each grid at 21 °C and 95% humidity. The total time elapsed after eIF5B addition until application to the grid was ~60 s, and the final concentration of 40S subunit was 250 nM. The sample was vitrified by plunging into liquid ethane after 2.5 s blotting using a Leica EM GP (Leica Microsystems) plunger.
Cryo-EM data collection, image processing, and structure determination
Vitrified grids were screened for ice thickness and intactness on a Glacios microscope (ThermoFisher) equipped with a Gatan K2 Summit direct electron detector. High-resolution data were collected on a Titan Krios equipped with a BioQuantum-K3 imaging system operated in super-resolution mode with pixel size 0.42 Å per pixel and fluence of 20 e A−2 s−1 using SerialEM software (version 3.9beta5). The defocus range was set between −0.5 and −2 μm. Image processing was done using Relion3.166 with ctf estimation via a wrapper to CtfFind467. After 2 times binning in motion correction and ctf estimation, a Laplacian autopicking job identified 2 million particles in the dataset. Initial particle extraction was performed with a binned size of 8 and a 2D classification job (200 classes, tau fudge 4 and the subset option deactivated) was used to discard bad particles or poorly aligned ones. The selected particles were subjected to a 3D refinement job using as initial model a volume obtained by stochastic gradient descent. After 3D refinement, particles were re-extracted at bin 8 with the recentre option activated. Next, a 3D classification job using a soft mask around the factors and the tRNA and without alignment (20 classes, tau-fudge parameter set to 3) found a major population with clear features for the tRNA and the factors. Particles belonging to this class (~190,000) were selected, re-extracted to bin 2 and re-refined at this pixel size. This reconstruction reached ~4 Å resolution which was further improved by refinement of the ctf parameters and Bayesian polishing of the beam induced particle movements. The final subset of ‘shiny’ particles was re-refined and reached an overall resolution of 3.2 Å after post-processing. Map exploration revealed features compatible with the reported resolution, like clear side chain density, especially for tyrosine, phenylalanine, leucine and tryptophan residues. Final post-processed maps were used for model building in Coot (v. 0.8.9.1)68. Good general density, especially for bulky residues, allowed proper tracing of the polypeptide of human eIF5B and eIF1A. Modelling of eIF5B was aided by the AlphaFold prediction69, which domain wise, fit the experimental maps accurately. Only minor manual, rigid-body adjustments of individual domains were necessary to fully account for the experimental density prior to stereochemical model refinement. The final model was refined in real space with Phenix (v. 1.14) imposing secondary structure and Ramachandran restraints70. This refined model was checked in Coot, corrected in problematic areas, and re-refined in the reciprocal space with Refmac (v. 5.8), using secondary structure restraints computed in ProSmart (v. 0.8)71. The quality of the final model was monitored by Molprobity (v. 4.5)72. Pymol (v. 2.4) and ChimeraX-1.373 were used for analysis of the structure and figure making.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Code availability
All custom codes used in this study to process and analyse single-molecule data are available publicly under an open-source license at: https://github.com/puglisilab/Lapointe-2022-Nature.
References
Tahmasebi, S., Khoutorsky, A., Mathews, M. B. & Sonenberg, N. Translation deregulation in human disease. Nat. Rev. Mol. Cell Biol. 19, 791–807 (2018).
Robichaud, N., Sonenberg, N., Ruggero, D. & Schneider, R. J. Translational control in cancer. Cold Spring Harb. Perspect. Biol. 11, a032896 (2019).
Pain, V. M. Initiation of protein synthesis in eukaryotic cells. Eur. J. Biochem. 236, 747–771 (1996).
Merrick, W. C. & Pavitt, G. D. Protein synthesis initiation in eukaryotic cells. Cold Spring Harb. Perspect. Biol. 10, a033092 (2018).
Hershey, J. W. B., Sonenberg, N. & Mathews, M. B. Principles of translational control. Cold Spring Harb. Perspect. Biol. 11, a032607 (2019).
Aitken, C. E. & Lorsch, J. R. A mechanistic overview of translation initiation in eukaryotes. Nat. Struct. Mol. Biol. 19, 568–576 (2012).
Sokabe, M. & Fraser, C. S. Toward a kinetic understanding of eukaryotic translation. Cold Spring Harb. Perspect. Biol. 11, a032706 (2019).
Hinnebusch, A. G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 83, 779–812 (2014).
Hinnebusch, A. G. Structural Insights into the mechanism of scanning and start codon recognition in eukaryotic translation initiation. Trends Biochem. Sci. 42, 589–611 (2017).
Hashem, Y. & Frank, J. The jigsaw puzzle of mRNA translation initiation in eukaryotes: a decade of structures unraveling the mechanics of the process. Annu. Rev. Biophys. 47, 125–151 (2018).
Pestova, T. V. et al. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature 403, 332–335 (2000).
Lee, J. H. et al. Initiation factor eIF5B catalyzes second GTP-dependent step in eukaryotic translation initiation. Proc. Natl Acad. Sci. USA 99, 16689–16694 (2002).
Shin, B. S. et al. Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell 111, 1015–1025 (2002).
Wang, J. et al. eIF5B gates the transition from translation initiation to elongation. Nature 573, 605–608 (2019).
Fernández, I. S. et al. Molecular architecture of a eukaryotic translational initiation complex. Science 342, 1240585 (2013).
Wang, J. et al. Structural basis for the transition from translation initiation to elongation by an 80S–eIF5B complex. Nat. Commun. 11, 5003 (2020).
Yamamoto, H. et al. Structure of the mammalian 80S initiation complex with initiation factor 5B on HCV-IRES RNA. Nat. Struct. Mol. Biol. 21, 721–727 (2014).
Choi, S. K. et al. Physical and functional interaction between the eukaryotic orthologs of prokaryotic translation initiation factors IF1 and IF2. Mol. Cell. Biol. 20, 7183–7191 (2000).
Olsen, D. S. Domains of eIF1A that mediate binding to eIF2, eIF3 and eIF5B and promote ternary complex recruitment in vivo. EMBO J. 22, 193–204 (2003).
Acker, M. G., Shin, B.-S., Dever, T. E. & Lorsch, J. R. Interaction between eukaryotic initiation factors 1A and 5B is required for efficient ribosomal subunit joining. J. Biol. Chem. 281, 8469–8475 (2006).
Fringer, J. M., Acker, M. G., Fekete, C. A., Lorsch, J. R. & Dever, T. E. Coupled release of eukaryotic translation initiation factors 5B and 1A from 80S ribosomes following subunit joining. Mol. Cell. Biol. 27, 2384–2397 (2007).
Pisareva, V. P. & Pisarev, A. V. eIF5 and eIF5B together stimulate 48S initiation complex formation during ribosomal scanning. Nucleic Acids Res. 42, 12052–12069 (2014).
Chen, J. et al. High-throughput platform for real-time monitoring of biological processes by multicolor single-molecule fluorescence. Proc. Natl Acad. Sci. USA 111, 664–669 (2014).
Lapointe, C. P. et al. Dynamic competition between SARS-CoV-2 NSP1 and mRNA on the human ribosome inhibits translation initiation. Proc. Natl Acad. Sci. USA 118, e2017715118 (2021).
Sokabe, M. & Fraser, C. S. Human eukaryotic initiation factor 2 (eIF2)–GTP–Met–tRNAi ternary complex and eIF3 stabilize the 43 S preinitiation complex. J. Biol. Chem. 289, 31827–31836 (2014).
Koch, A., Aguilera, L., Morisaki, T., Munsky, B. & Stasevich, T. J. Quantifying the dynamics of IRES and cap translation with single-molecule resolution in live cells. Nat. Struct. Mol. Biol. 27, 1095–1104 (2020).
Acker, M. G. et al. Kinetic analysis of late steps of eukaryotic translation initiation. J. Mol. Biol. 385, 491–506 (2009).
Huang, B. Y. & Fernández, I. S. Long-range interdomain communications in eIF5B regulate GTP hydrolysis and translation initiation. Proc. Natl Acad. Sci. USA 117, 1429–1437 (2020).
Passmore, L. A. et al. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol. Cell 26, 41–50 (2007).
Hashem, Y. et al. Structure of the mammalian ribosomal 43S preinitiation complex bound to the scanning factor DHX29. Cell 153, 1108–1119 (2013).
Lomakin, I. B. & Steitz, T. A. The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature 500, 307–311 (2013).
Weisser, M., Voigts-Hoffmann, F., Rabl, J., Leibundgut, M. & Ban, N. The crystal structure of the eukaryotic 40S ribosomal subunit in complex with eIF1 and eIF1A. Nat. Struct. Mol. Biol. 20, 1015–1017 (2013).
Hussain, T. et al. Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell 159, 597–607 (2014).
Des Georges, A. et al. Structure of mammalian eIF3 in the context of the 43S preinitiation complex. Nature 525, 491–495 (2015).
Simonetti, A. et al. eIF3 peripheral subunits rearrangement after mRNA binding and start-codon recognition. Mol. Cell 63, 206–217 (2016).
Llácer, J. L. et al. Translational initiation factor eIF5 replaces eIF1 on the 40S ribosomal subunit to promote start-codon recognition. eLife 7, e39273 (2018).
Brito Querido, J. et al. Structure of a human 48S translational initiation complex. Science 369, 1220–1227 (2020).
Simonetti, A., Guca, E., Bochler, A., Kuhn, L. & Hashem, Y. Structural insights into the mammalian late-stage initiation complexes. Cell Rep. 31, 107497–107497 (2020).
Kratzat, H. et al. A structural inventory of native ribosomal ABCE1–43S pre-initiation complexes. EMBO J. 40, e105179 (2021).
Nag, N. et al. eIF1A/eIF5B interaction network and its functions in translation initiation complex assembly and remodeling. Nucleic Acids Res. 44, 7441–7456 (2016) .
Marintchev, A., Kolupaeva, V. G., Pestova, T. V. & Wagner, G. Mapping the binding interface between human eukaryotic initiation factors 1A and 5B: A new interaction between old partners. Proc. Natl Acad. Sci. USA 100, 1535–1540 (2003).
Shao, S. et al. Decoding mammalian ribosome-mrna states by translational GTPase complexes. Cell 167, 1229–1240.e1215 (2016).
Pisarev, A. V. et al. Specific functional interactions of nucleotides at key −3 and +4 positions flanking the initiation codon with components of the mammalian 48S translation initiation complex. Genes Dev. 20, 624–636 (2006).
Suresh, S. et al. eIF5B drives integrated stress response-dependent translation of PD-L1 in lung cancer. Nat. Cancer 1, 533–545 (2020).
Roll-Mecak, A., Cao, C., Dever, T. E. & Burley, S. K. X-ray structures of the universal translation initiation factor IF2/eIF5B. Cell 103, 781–792 (2000).
Kuhle, B. & Ficner, R. eIF5B employs a novel domain release mechanism to catalyze ribosomal subunit joining. EMBO J. 33, 1177–1191 (2014).
Zheng, A. et al. X-ray structures of eIF5B and the eIF5B–eIF1A complex: the conformational flexibility of eIF5B is restricted on the ribosome by interaction with eIF1A. Acta Crystallogr. D 70, 3090–3098 (2014).
Kaledhonkar, S. et al. Late steps in bacterial translation initiation visualized using time-resolved cryo-EM. Nature 570, 400–404 (2019).
Marshall, R. A., Aitken, C. E. & Puglisi, J. D. GTP hydrolysis by IF2 guides progression of the ribosome into elongation. Mol. Cell 35, 37–47 (2009).
Ling, C. & Ermolenko, D. N. Initiation factor 2 stabilizes the ribosome in a semirotated conformation. Proc. Natl Acad. Sci. USA 112, 15874–15879 (2015).
Sprink, T. et al. Structures of ribosome-bound initiation factor 2 reveal the mechanism of subunit association. Sci. Adv. 2, e1501502 (2016).
Johnson, A. G. et al. RACK1 on and off the ribosome. RNA 25, 881–895 (2019).
Yin, J., Lin, A. J., Golan, D. E. & Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc. 1, 280–285 (2006).
Fraser, C. S., Berry, K. E., Hershey, J. W. B. & Doudna, J. A. eIF3j is located in the decoding center of the human 40S ribosomal subunit. Mol. Cell 26, 811–819 (2007).
Özeş, A. R., Feoktistova, K., Avanzino, B. C. & Fraser, C. S. Duplex unwinding and ATPase activities of the DEAD-box helicase eIF4A are coupled by eIF4G and eIF4B. J. Mol. Biol. 412, 674–687 (2011).
Feoktistova, K., Tuvshintogs, E., Do, A. & Fraser, C. S. Human eIF4E promotes mRNA restructuring by stimulating eIF4A helicase activity. Proc. Natl Acad. Sci. USA 110, 13339–13344 (2013).
Sokabe, M., Fraser, C. S. & Hershey, J. W. The human translation initiation multi-factor complex promotes methionyl-tRNAi binding to the 40S ribosomal subunit. Nucleic Acids Res. 40, 905–913 (2012).
Damoc, E. et al. Structural characterization of the human eukaryotic initiation factor 3 protein complex by mass spectrometry. Mol. Cell. Proteomics 6, 1135–1146 (2007).
Korlach, J. et al. Selective aluminum passivation for targeted immobilization of single DNA polymerase molecules in zero-mode waveguide nanostructures. Proc. Natl Acad. Sci. USA 105, 1176–1181 (2008).
Aitken, C. E., Marshall, R. A. & Puglisi, J. D. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 94, 1826–1835 (2008).
Erickson, F. L. & Hannig, E. M. Ligand interactions with eukaryotic translation initiation factor 2: role of the gamma-subunit. EMBO J. 15, 6311–6320 (1996).
Kapp, L. D. & Lorsch, J. R. GTP-dependent recognition of the methionine moiety on initiator tRNA by translation factor eIF2. J. Mol. Biol. 335, 923–936 (2004).
Juette, M. F. et al. Single-molecule imaging of non-equilibrium molecular ensembles on the millisecond timescale. Nat. Methods 13, 341–344 (2016).
Bronson, J. E., Fei, J., Hofman, J. M., Gonzalez, R. L. & Wiggins, C. H. Learning rates and states from biophysical time series: a Bayesian approach to model selection and single-molecule FRET data. Biophys. J. 97, 3196–3205 (2009).
Lawson, M. R. et al. Mechanisms that ensure speed and fidelity in eukaryotic translation termination. Science 373, 876–882 (2021).
Zivanov, J., Nakane, T. & Scheres, S. H. W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in. IUCrJ 7, 253–267 (2020).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Casañal, A., Lohkamp, B. & Emsley, P. Current developments in Coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci. 29, 1069–1078 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018).
Nicholls, R. A., Long, F. & Murshudov, G. N. Low-resolution refinement tools in REFMAC5. Acta Crystallogr. D 68, 404–417 (2012).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Acknowledgements
We thank M. Lawson, J. Carette and other members of the Puglisi and Carette laboratories for helpful guidance, discussions and feedback; and P. Sarnow and the Sarnow laboratory for sharing cell culture equipment. Some of this work was performed at the Stanford-SLAC Cryo-EM Facilities, supported by Stanford University, SLAC and the National Institutes of Health S10 Instrumentation Programs. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. C.P.L. was a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-#2321-18); C.A. was supported by a Stanford Bio-X Fellowship; and J.W. was supported by a postdoctoral scholarship from the Knut and Alice Wallenberg Foundation (KAW 2015.0406). This work was funded, in part, by the National Institutes of Health (GM011378 and AG064690 to J.D.P.; GM092927 to C.S.F.; and K99GM144678 to C.P.L.) and by the Intramural Research Program of the National Institutes of Health (to T.E.D.).
Author information
Authors and Affiliations
Contributions
Conceptualization: C.P.L., I.S.F. and J.D.P. Methodology: C.P.L., R.G., M.S., C.A., J.W., N.V., B.S.-S., E.M., T.E.D., C.S.F., I.S.F. and J.D.P. Resources: C.P.L., R.G., M.S., C.A., N.V. and C.S.F. Investigation: C.P.L., R.G., E.M. and I.S.F. Visualization: C.P.L., I.S.F. and J.D.P. Funding acquisition: T.E.D., C.S.F. and J.D.P. Project administration: T.E.D., C.S.F., I.S.F. and J.D.P. Supervision: T.E.D., C.S.F., I.S.F. and J.D.P. Writing, original draft: C.P.L., I.S.F. and J.D.P. Writing, review and editing: C.P.L., R.G., M.S., C.A., J.W., T.E.D., C.S.F., I.S.F. and J.D.P.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Katrin Karbstein, Assen Marintchev and Meni Wanunu for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Real-time monitoring of human ribosomal subunit recruitment to β-globin mRNA.
a. Human 40S and 60S ribosomal subunits were labeled with fluorescent dyes on the N-terminus of uS19 or C-terminus of uL18, respectively. These labeling positions yield an inter-subunit FRET signal upon formation of translation-competent 80S ribosomes. The structural model was obtained from PDB: 4UG0. Of note, the first nine N-terminal amino acids of uS19 and last two C-terminal acids of uL18 were unresolved in the structure, which also does not account for the fused ybbR tags (11 amino acids). Thus, the reported distance between the 40S and 60S labeling sites is only an approximation. b. A real-time single molecule fluorescence assay using zero-mode waveguides (ZMWs) on a custom PacBio RSII DNA sequencer. Components of interest are tethered to the imaging surface within individual ZMWs, and reaction components are added directly to the surface. Time-resolved, real-time 4-color fluorescence emission was monitored across ~150,000 ZMWs after excitation with a 532 nm laser. The order and time elapsed between relevant fluorescence signals (e.g., green & orange) were determined in ~100–200 ZMWs with the desired signals. Rates of association and dissociation were determined using probability based statistical models; cumulative distribution functions of the observed times were calculated and subsequently fit to exponential functions to yield association or dissociation rates, as appropriate. c. Schematic of a real-time single-molecule assay to monitor ribosomal subunit joining on tethered β-globin mRNA (the ‘tethered mRNA’ setup). β-globin mRNA with a 5' m7G cap, poly(A)30 tail, and 3'-terminal biotin moiety was tethered to the neutravidin-coated ZMW imaging surface. Immediately after tethering, eIFs 4A, 4B, 4G, and 4E were added. After start of data acquisition and excitation via the 532 nm laser at 30 °C, the 43S PIC (5 nM via the labeled 40S-Cy3), unlabeled eIF5B (1 µM), 60S-Cy5 subunits (100 nM), and 1 mM of ATP and GTP were added. Unlabeled 43S PIC components were present at \(\ge \) 1.5-fold excess during each step of the experiment. Fluorescence data were acquired for 600 s. d. Cartoon schematic of theoretical single-molecule fluorescence data where 40S and 60S subunits were recruited to an mRNA to form 80S ribosomes. The 40S subunit association time (t40S) was defined as the time elapsed from addition of the 43S PIC until appearance of the Cy3 signal. The 60S subunit association time (t60S) was defined as the time elapsed from 40S subunit association until appearance of the 40S(Cy3,donor)-60S(Cy5,acceptor) FRET signal. e. Example single-molecule fluorescence trace with FRET efficiency (EFRET) plot. Recruitment of the 40S subunit (as the 43S PIC) was indicated by a burst of Cy3 (green) fluorescence intensity. 60S subunit joining was indicated by appearance of the 40S-60S FRET signal. f. Density maps of normalized 40S-Cy3 and 60S-Cy5 fluorescence intensities post-synced to 60S subunit joining (n = 128). As in the individual trace shown in panel D, joining of the 60S subunit led to an anti-correlated decrease in Cy3 and increase in Cy5 signals, indicative of intersubunit FRET when the 80S ribosome formed. g. Histogram and single gaussian function fit (line) of the observed EFRET in the real-time initiation assay. The mean EFRET (µ) was 0.65 ± 0.1 and the standard deviation (σ) was 0.17 ± 0.1 (n = 168), consistent with structural predictions for translation-competent 80S ribosomes. h. Table of 40S loading and 60S joining efficiencies (left) and a plot of observed 40S association times (right) in the indicated conditions. 40S subunit loading efficiency (80% labeling efficiency in all experiments) was defined as the fraction of 1,000 analyzed ZMWs with at least one stable (> 10 s) 40S subunit association event. The concentration of 40S subunits (5 nM) was optimized to yield a single recruitment event per ZMW, which is most probable when < 30% of ZMWs contain a recruitment event, as predicted by Poisson distribution statistics. 60S subunit joining efficiency (with errors propagated from 95% CIs) was defined the fraction of recruited 40S subunits (300 analyzed) with a 60S subunit joining event (indicated by Cy3-Cy5 FRET), which was normalized to account for relative 60S subunit labeling efficiencies (35 or 80% labeled). 40S association time (t40S) was defined as above (n = 135, 128, 131, 155, and 137, from left to right), and the median observed times are represented by the orange lines. i. Cumulative probability plot of 40S and 60S subunit association times and the fits to single-exponential functions, which yielded apparent association rates of kon,40S ≈ 0.049 ± 0.002 s−1 and kon,60S ≈ 0.033 ± 0.001 s−1 when added at 5 nM and 100 nM (final concentration), respectively (n = 128).
Extended Data Fig. 2 Real-time analysis of eIF5B-mediated ribosomal subunit joining in humans.
a. Fluorescence scans of SDS-PAGE gels that analyzed purified and fluorescently-labeled eIF5B proteins. ‘WT’ corresponds to the wild-type protein with the N-terminal domain truncated (residues 1-586 removed) and an N-terminal ybbR peptide tag (11 amino acids). ‘H706E’ corresponds to the GTPase deficient version of the protein. Each protein was analyzed by SDS-PAGE twice. The full scans of the cropped gel are available in Supplementary Figure 1. b. Plot of the relative light units (RLU) from nanoLuciferase in vitro translation assays where the indicated eIF5B proteins were supplemented in the extract (840 nM final concentration). Three replicates (n = 3) were completed and are overlayed on the bar plots. Error bars represent standard error of the mean. c. Structural model of the yeast 80S initiation complex bound by eIF5B (PDB: 6WOO16) used to predict the potential distance between the human 40S subunit (ybbR tag fused to N-term of uS19) and eIF5B (ybbR tag fused to residue 587) labeling sites. The relatively long predicted distance (>130 Å) precluded detection using FRET. eIF5B therefore was monitored via direct excitation and emission in all experiments. d. Theoretical real-time single-molecule fluorescence data from a ZMW with sequential association of the 40S subunit (green), eIF5B (orange), and 60S subunit (red), which was followed by departure of eIF5B from the newly-formed 80S initiation complex. The dwell times were defined as follows: between appearance of 40S and eIF5B signals, as the eIF5B association time (t1); between appearance of eIF5B and 60S signal, indicated by 40S-60S FRET, as 60S joining time (t2); and between appearance of 60S until loss of eIF5B, as eIF5B departure time (t3). e. Cumulative probability plots of the observed times at the indicated concentrations for eIF5B and 60S subunits at 30 °C. In all experiments, the 43S PIC was present at 10 nM (final concentration, via 40S-Cy3). WT and H706E indicate whether wild-type or catalytically-inactive eIF5B-Cy3.5 were present, respectively. Lines represent fits of observed data to single- or double-exponential functions, which yielded the indicated association or dissociation rates. All errors represent 95% confidence intervals (C.I.). The number of molecules analyzed were: 136 (black), 148 (purple), 134 (blue), 146 (red), and 150 (gray). f. Cumulative probability (top) and Eyring (bottom) plots of observed 60S joining and eIF5B departure times at 20, 25, 30, and 35 °C. These data were obtained using tethered, pre-equilibrated 48S initiation complexes (see, Extended Figure 3a for more details on the experiment setup) to provide sharper focus on eIF5B dynamics and 60S subunit joining. eIF5B-Cy3.5 and 60S-Cy5 subunits were present at 20 nM and 100 nM, respectively. Lines on the cumulative probability plots represent fits of the observed data to single or double-exponential functions, which yielded the indicated rates (errors represent 95% C.I.). Eyring plots (bottom) were modeled via linear regression analyses, which yielded the indicated enthalpies and entropies of activation. The number of molecules analyzed were: 116 (20 °C), 109 (25 °C), 120 (30 °C), and 44 (35 °C). g. Example single-molecule fluorescence data (top) and density plots (bottom) from the tethered mRNA setup when catalytically-inactive eIF5B (H706E) was examined at 30 °C. 40S-Cy3, eIF5B(H706E)-Cy3.5, and 60S-Cy5 subunits were present at 10 nM, 20 nM, and 100 nM, respectively. eIF5B(H706E) was unable to depart the 80S initiation complex in the presence of 1 mM GTP.
Extended Data Fig. 3 eIF5B associated more rapidly with pre-equilibrated 48S initiation complexes.
a. Schematic of an alternative experimental setup (‘tethered 48S’) used to examine eIF5B association with pre-equilibrated 48S initiation complexes. In this setup, a pre-formed 48S initiation complex at equilibrium on the β-globin mRNA was tethered to the ZMW imaging surface in the presence of 1 mM ATP and GTP. After removal of untethered components, data acquisition began via excitation with a 532 nm laser, and (final concentrations) 20 nM eIF5B-Cy3.5, 100 nM 60S-Cy5 subunits, 1 µM eIF1A, and 1 mM ATP and GTP were added. ZMWs with tethered 48S PICs were identified by the initial presence of 40S-Cy3 fluorescence signal (green). b. Example single-molecule fluorescence data from a tethered 48S PIC experiment where eIF5B association led to 60S subunit joining and rapid departure of eIF5B from the 80S initiation complex at 30 °C. The dwells were defined as in the ‘tethered mRNA’ experimental setup (see, Extended Data Fig. 2d). c. Comparison of observed eIF5B association times (t1) in tethered mRNA versus tethered 48S PIC experimental setups at 30 °C. The pre-equilibration of the 48S PIC prior to tethering allowed the slow upstream step to proceed, which enabled rapid and concentration-dependent eIF5B association. Fits of the observed association times are represented by the lines, which yielded the indicated rates (n = 172 and 111 for 20 & 40 nM, respectively). In one experiment (magenta, n = 113), the 48S PIC was formed and pre-equilibrated in the absence of eIF5, and then 2.5 nM eIF5 was added simultaneously with labeled eIF5B and 60S subunits to the tethered 48S PIC. The ‘tethered mRNA’ data were replotted from Extended Data Fig. 2e to facilitate comparisons. All errors represent 95% C.I. d. Example single-molecule fluorescence data from the tethered 48S complex experimental setup where wild-type eIF5B-Cy3.5 (20 nM) was added to pre-equilibrated 48S initiation complxes either in the absence of 60S subunits (top) or when 60S subunits (100 nM) were present but GTP was replaced with 1 mM GDPNP in free solution (bottom). e. Plot of the total lifetime of wild-type eIF5B-Cy3.5 on tethered 48S complexes in the indicated conditions. In the presence of 60S subunits and 1 mM GTP, eIF5B remained on initiation complexes for a total of ~15 s (t2 + t3). The total lifetime of eIF5B was lengthened approximately 34-fold to ~510 s when GTP was replaced with 1 mM of the non-hydrolyzable analog, GDPNP. eIF5B total lifetime also was lengthened dramatically (to ~ 130 s) when 60S subunits were omitted and 1 mM GTP was present. f. Table of 60S subunit joining and eIF5B association efficiencies when eIF2–\({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\)–GTP or eIF2–\({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\)–GDPNP were present at 30 °C. 60S subunit joining efficiency was quantified in the tethered mRNA experimental setup with 1 µM unlabeled eIF5B present. The 95% CI of the observed efficiencies are indicated, with 200 or 204 40S loading events or 48S complexes analyzed in each experiment, respectively. Observed efficiencies were corrected to account for the 40S and 60S subunit labeling efficiencies, which were 80% and 38%, respectively, with errors propagated from the observed 95% CIs. eIF5B association efficiency (45% labeled) was quantified in the tethered 48S PIC setup (to facilitate analyses) in the absence of 60S subunits.
Extended Data Fig. 4 eIF1A resides on initiation complexes until the 60S subunit joins.
a. Structural model that depicts the proximity of eIF1A and 40S subunit labeling sites. eIF1A was fluorescently labeled via an S102C substitution followed by reaction with Cy5-maleimide dye, which yields a fully-active protein in translation assays, as described previously25. 40S subunits were labeled with Cy3 on the N-terminus of uS19, via a fused ybbR tag (11 amino acid), as above. The relative proximity of the two labeling sites suggested the presence of eIF1A on the 40S subunit could be monitored via FRET. Of note, the first three N-terminal amino acids of uS19 were unresolved in the structure, which also does not account for the fused ybbR tags (11 amino acids). Thus, the reported distance between the 40S and eIF1A labeling sites is only an approximation. b, c. Theoretical (top) and example (middle, bottom) real-time single-molecule fluorescence data from a ZMW where doubly labeled 43S PICs (10 nM via 40S-Cy3; eIF1A-Cy5), 1 µM unlabeled eIF5B, and 200 nM 60S-Cy5.5 subunits were added to tethered eIF4ABGE-mRNA complexes at 30 °C. During imaging, eIF1A-Cy5 was present at 4.5-fold molar excess relative to 40S subunits. Key association and dissociation events are labeled on the theoretical plot. The lifetime of eIF1A on the 48S PIC (t1A,48S) was defined as the duration of individual 40S(Cy3,donor)-eIF1A(Cy5,acceptor) FRET events. In these experiments, 60S subunit joining was indicated by appearance of 40S(Cy3,donor)-60S(Cy5.5,acceptor) FRET. eIF1A lifetime on the 80S complex (t1A,80S) was defined as the time elapsed from 60S subunit joining until eIF1A departure (loss of 40S-eIF1A FRET). Panel B depicts an mRNA where eIF1A was co-recruited with the 40S subunit (signal begins in 40S-eIF1A FRET state) and that initial, co-recruited eIF1A protein was present until the 60S subunit joined. Panel C depicts a complex where multiple eIF1A binding events occurred prior to 60S subunit joining. In both cases, eIF1A departed either concomitantly (left) with or within a few hundred milliseconds (right) after 60S subunit joining. d. Plot of FRET efficiency (EFRET) distributions for 40S(Cy3,donor)-eIF1A(Cy5,acceptor) FRET. ‘All’ represents all observed 40S-eIF1A FRET events (n = 433), ‘60S joined’ represents events that overlapped with 60S subunit joining (n = 175), and ‘No 60S joining’ represents events that did not overlap with 60S subunit joining (n = 258). The lines represent fits of the observed data to single gaussian functions, which yielded the indicated means (µ) and standard deviations (σ). e. Cumulative probability plot of observed eIF1A reassociation times with the 48S initiation complex prior to 60S subunit joining at 30 °C (n = 258). eIF1A-Cy5 was present at ~45 nM in free solution. The line represents a fit of the observed data to a double-exponential function, which yielded the indicated rates. All errors represent 95% C.I. f. Cumulative probability plot of observed eIF1A lifetimes on either the 48S or 80S initiation complexes when either eIF5B-GTP or eIF5B-GDPNP was present at 30 °C. The line represents a fit of the observed data to either single- or double-exponential functions, which yielded the indicated rates. All errors represent 95% C.I. g. Table of 60S subunit joining efficiency when eIF1A-Cy5 was present in or eIF1A was absent from initiation reactions. The 95% CI of the observed efficiencies are indicated, with 500 40S loading events analyzed in each experiment. Observed efficiencies were corrected to account for the 40S and 60S subunit labeling efficiencies, which were 80% and 80%, respectively, with errors propagated from the observed 95% CIs. h. Cumulative probability plot of observed 60S subunit joining times at 30 °C when either 40 nM eIF5B-Cy3.5 (gold, n = 134) or 45 nM eIF1A-Cy5 (slate, n = 175) was present. These findings further confirmed that fluorescently-labeled eIF1A and eIF5B are fully active, as the proteins yielded identical rates of 60S subunit joining as to when the unlabeled version is present (unlabeled eIF1A was present in the eIF5B-Cy3.5 experiment, and vice versa). Moreover, the rare instances where the 60S subunit joined when eIF1A was absent from the reaction (reported in panel H) occurred 4-fold slower (grey, n = 100). The lines represent fits of the observed data to single-exponential functions, which yielded the indicated rates. All errors represent 95% C.I.
Extended Data Fig. 5 eIF1A and eIF5B reside simultaneously on initiation complexes when the 60S subunit joins.
a. Schematic of single-molecule experiments that examined eIF1A and eIF5B dynamics on the 48S initiation complex at equilibrium. Pre-formed 48S initiation complexes on β-globin mRNA were tethered at equilibrium within ZMWs in the presence of 1 mM ATP and GTP. After removal of untethered components, data acquisition began via excitation with a 532 nm laser, and an imaging mix that contained (final concentrations) 10 nM eIF1A-Cy5 (red) and 20 nM eIF5B-Cy3.5 (orange) was present at 30 °C. ZMWs with tethered 48S PICs were identified by the initial presence of 40S-Cy3 fluorescence signal (green). b. Rudimentary structural comparison where a low-resolution model of human eIF5B (PDB: 4UJC) was docked onto a high-resolution model of a mammalian 48S PIC post-recognition of the start codon (PDB: 6YAL). From these crude analyses, eIF1A was predicted to be within FRET distance (< 80 Å) of the N-terminus of truncated eIF5B, consistent with the eIF5B(Cy3.5, donor)-eIF1A(Cy5,acceptor) FRET signal observed in our single-molecule assays. c. Example single-molecule fluorescence data that depicts either: top, a complex with 40S(Cy3,donor)-eIF1A(Cy5,acceptor) FRET in the absence of eIF5B signal (‘eIF5B absent’); bottom, a complex with both 40S(Cy3,donor)-eIF1A(Cy5,acceptor) and eIF5B(Cy3.5,donor)-eIF1A(Cy5,acceptor) FRET, (‘eIF5B present’). Focused analyses were conducted on both forms of the 48S PIC to derive kinetic parameters. d. Cumulative probability plots of observed eIF1A reassociation times with (left) or eIF1A lifetimes on (right) the 48S initiation complex at 30 °C at equilibrium. eIF1A-Cy5 was present at 10 nM. The lines represent fits of the observed data to double-exponential functions, which yielded the indicated rates. All errors represent 95% C.I. 831 and 589 eIF1A binding events were analyzed when eIF5B was present or absent, respectively. e. Schematic of the four-color single-molecule experiment. The doubly labeled 43S PIC (10 nM by 40S-Cy3; eIF1A-Cy5), 40 nM eIF5B-Cy3.5, and 200 nM 60S-Cy5.5 subunit were added to b-globin mRNA tethered within ZMWs in the presence of saturating concentrations of eIFs 4A, 4B, 4G, and 4E, and 1 mM ATP and GTP at 30 °C. During imaging, eIF1A-Cy5 was present at 4.5-fold molar excess relative to the 40S subunit. Fluorescence data were acquired for 600 s with excitation via the 532 nm laser. The potential FRET signals are indicated in the box. f, g. Theoretical (panel F) and example (panel G) four-color single-molecule fluorescence data from a ZMW where a loaded doubly labeled 43S PIC (40S-Cy3, green; eIF1A-Cy5, red) was bound by eIF5B-Cy3.5 (orange), which was followed by 60S-Cy5.5 (purple) subunit joining. The 43S PIC was recruited in a 40S(Cy3, donor)-eIF1A(Cy5, acceptor) FRET state. Once eIF5B bound, the eIF1A-Cy5 signal increased due to eIF5B(Cy3.5, donor)-eIF1A(Cy5, acceptor) FRET. Joining of the 60S subunit was indicated by appearance of Cy5.5 fluorescence signal due to 40S(Cy3, donor)-60S(Cy5.5, acceptor) FRET. Departure of eIF1A-Cy5 and eIF5B-Cy3.5 was indicated by loss of Cy5 and Cy3.5 fluorescence signals, respectively. In panel G, the full experimental window is depicted on the left, and the middle and right panels represent zoomed views of the indicated time windows. Given bleed through across the four fluorescent channels, the fluorescence signals in each channel were made transparent before relevant events for presentation here. h. Quantification of eIF1A and eIF5B occupancy upon 60S subunit joining in the four-color single-molecule experiment. The potentially convoluted FRET signals and fluorescence bleed through among channels and factors precluded rigorous kinetic analyses, as exact frames for association and dissociation were extremely challenging to assign. However, the experiment did allow the presence of eIF1A and eIF5B upon 60S subunit joining to be quantified unmistakably. On a large majority of 48S initiation complexes (183/271), eIF1A-Cy5 signal was present when the 60S subunit joined, which corresponded to an estimated eIF1A occupancy of about 85 ± 5 % after correction for eIF1A labeling efficiency (~80 %). Of those eIF1A-bound 48S complexes, nearly half (88/183) also contained eIF5B-Cy3.5 signal when the 60S subunit joined, which indicated that about 90 ± 10 % of the 48S PICs contained both eIF1A and eIF5B, after correction for eIF5B labeling efficiency (~45%). eIF1A preceded eIF5B association on nearly all (79/88; 90 ± 20 %) 48S complexes that contained both labeled proteins when the 60S subunit joined.
Extended Data Fig. 6 Fourier Shell Correlation curves, local resolution, particle distribution plot and representative cryo-EM density examples.
a. Fourier Shell Correlation (FSC) curves computed for independently refined half-maps of the final subgroup of particles before masking (blue) and after masking (black). The red curve corresponds to FSC curve of phase-randomized structure factors beyond 7Å. b. Model versus map FSC (black). Red and blue curves correspond to a model-map overfitting validation test performed for the final model. A per-atom, random distortion of 0.5Å was introduced in the model which was subsequentially refined against half-map 1 only. FSC between the distorted and refined model against half-map 1 (blue) and half-map 2 (red, not included in the refinement) are nearly identical. c. Unsharpened map colored according to local resolution calculations with the resolution scale indicated on the left and a zoomed view for \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\)/eIF1A/eIF5B on the right. d. Eulerian angular distribution for the final refined set of particles in a mollweide spherical projection. e. Several views of the final post-processed cryo-EM density used for model building and refinement. The areas of the map to which the different views belong are indicated.
Extended Data Fig. 7 eIF5B, through contacts with eIF1A, reorients \({{\bf{Met-tRNA}}}_{{\bf{i}}}^{{\bf{M}}{\bf{e}}{\bf{t}}}\) conformation in initiation complexes.
a. Post-processed cryo-EM density with eIF1A colored red, \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) green, and eIF5B blue. While nearly the entire flexible C-terminal tail of eIF1A was disordered, extra density proximal to W1207 of eIF5B was observed, which likely corresponds to I141 in the eIF1A C-terminal tail. b. Position of eIF1A and \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) in scanning, post-scanning, and eIF5B-bound initiation complexes. eIF1A remains bound in the ribosomal A site throughout initiation, until the 60S subunit joins. c. Structural models of the 40S subunit (with ribosomal proteins omitted) that indicate movement of the 18S rRNA in the indicated states relative to the final position in the elongation-competent 80S ribosome. In both the post-scanning and eIF5B-bound states, the 40S head region resembles the conformation in the 80S ribosome. d. Comparison of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) conformations in scanning-competent (orange), post-scanning (grey), and eIF5B-bound 48S (green) complexes. eIF5B reorients the elbow and acceptor stem region of \({\text{Met-tRNA}}_{{\rm{i}}}^{{\rm{Met}}}\) to avoid steric clashes (black arrowheads) with the incoming 60S subunit.
Extended Data Fig. 8 eIF5B contacts with eIF1A and \({{\bf{Met-tRNA}}}_{{\bf{i}}}^{{\bf{M}}{\bf{e}}{\bf{t}}}\) mediate 60S subunit joining.
a. Alignments of human and yeast eIF5B protein sequences at the indicated regions. b. Fluorescence scans of a SDS-PAGE gel that analyzed the indicated purified and fluorescently-labeled eIF5B proteins. All proteins had the N-terminal domain removed (residues 1-586) and contained an N-terminal ybbR peptide tag (11 amino acids). Each protein was analyzed by SDS-PAGE twice. The full scans of the cropped gel are available in Supplementary Figure 1. c. Schematic of the single-molecule experiment to assess the role of the indicated residues in eIF5B. The 43S PIC (10 nM via 40S-Cy3 subunits, green), wild-type or mutant eIF5B-Cy3.5 (orange, at 20 or 40 nM), and 100 nM 60S-Cy5 (red) subunit were added to β-globin mRNA tethered within ZMWs in the presence of saturating concentrations of eIF4ABGE and 1 mM ATP and GTP at 30 °C. Fluorescence data were acquired for 600 s with excitation via the 532 nm laser. d. Theoretical real-time single-molecule fluorescence data from a ZMW with sequential association of the 40S subunit (green), eIF5B (orange), and 60S subunit (red), which was followed by departure of eIF5B from the newly-formed 80S initiation complex. In this experiment, the lifetimes of all eIF5B binding events that began prior to 60S subunit joining were analyzed. Events that concluded prior to 60S subunit joining were classified as ‘samples’, and events that concluded after the 60S subunit joined were classified as ‘productive’. To facilitate cross-comparisons in these experiments, the 60S subunit joining time was defined as the time elapsed from 40S subunit loading (appearance of green fluorescence) until 60S subunit joining, as indicated by 40S(Cy3, donor)-to-60S(Cy5, acceptor) FRET. e. Example single-molecule fluorescence data obtained when the indicated eIF5B proteins were present. The left plots represent the fluorescence intensities throughout the entire 600 s data collection window, and the right are zoomed views of events of interest. f. Fraction of observed eIF5B binding events on the 48S complex that were samples or productive, defined as in panel D. g. Cumulative probability plots of observed eIF5B lifetimes on the 48S complex when the indicated eIF5B protein was present at the indicated concentration (20 or 40 nM). The lines represent fits of the observed data to double-exponential functions, which yielded the indicated rates. All errors represent 95% C.I. The number of binding events analyzed (n) are indicated in the figure. h. Table of the 60S subunit joining efficiency in ZMWs that contained at least one 40S subunit loading event when the indicated eIF5B proteins were present at 40 nM. The 95% CI of the observed efficiencies are indicated, with 1,000 40S loading events analyzed in each experiment. Relative efficiencies were determined relative to the wild-type protein, with errors propagated from the 95% CIs. i. Cumulative probability plots of observed 60S joining times on the 48S complex when the indicated eIF5B protein was present at the indicated concentration (20 or 40 nM). The lines represent fits of the observed data to exponential functions, which yielded the indicated rates. All errors represent 95% C.I. The number of binding events analyzed (n) are indicated in the figure.
Supplementary information
Supplementary Information
This file contains Supplementary Fig. 1 and legends for Supplementary Videos 1 and 2.
Rights and permissions
About this article

Cite this article
Lapointe, C.P., Grosely, R., Sokabe, M. et al. eIF5B and eIF1A reorient initiator tRNA to allow ribosomal subunit joining. Nature 607, 185–190 (2022). https://doi.org/10.1038/s41586-022-04858-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04858-z
- Springer Nature Limited
This article is cited by
-
Genetic investigations on singleton school aged children reveal novel variants and new candidate genes for hearing loss
Scientific Reports (2024)
-
Fluorescence resonance energy transfer at the single-molecule level
Nature Reviews Methods Primers (2024)
-
The molecular basis of translation initiation and its regulation in eukaryotes
Nature Reviews Molecular Cell Biology (2024)
-
Human tumor suppressor protein Pdcd4 binds at the mRNA entry channel in the 40S small ribosomal subunit
Nature Communications (2024)
-
Structural basis of LRPPRC–SLIRP-dependent translation by the mitoribosome
Nature Structural & Molecular Biology (2024)