

Abstract
Translation initiation determines both the quantity and identity of the protein that is encoded in an mRNA by establishing the reading frame for protein synthesis. In eukaryotic cells, numerous translation initiation factors prepare ribosomes for polypeptide synthesis; however, the underlying dynamics of this process remain unclear1,2. A central question is how eukaryotic ribosomes transition from translation initiation to elongation. Here we use in vitro single-molecule fluorescence microscopy approaches in a purified yeast Saccharomyces cerevisiae translation system to monitor directly, in real time, the pathways of late translation initiation and the transition to elongation. This transition was slower in our eukaryotic system than that reported for Escherichia coli3,4,5. The slow entry to elongation was defined by a long residence time of eukaryotic initiation factor 5B (eIF5B) on the 80S ribosome after the joining of individual ribosomal subunits—a process that is catalysed by this universally conserved initiation factor. Inhibition of the GTPase activity of eIF5B after the joining of ribosomal subunits prevented the dissociation of eIF5B from the 80S complex, thereby preventing elongation. Our findings illustrate how the dissociation of eIF5B serves as a kinetic checkpoint for the transition from initiation to elongation, and how its release may be governed by a change in the conformation of the ribosome complex that triggers GTP hydrolysis.
Similar content being viewed by others
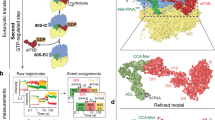
Main
Despite decades of study, the transition from translation initiation to elongation in eukaryotes remains poorly understood. During translation initiation, the 40S ribosome subunit in complex with initiation factors is recruited to a 7-methylguanosine (m7G)-capped mRNA and scans the 5′ untranslated region until the correct AUG start codon is recognized (termed ‘scanning’), at which point a post-scanning 48S pre-initiation complex (PIC) is formed1,2. The subsequent joining of the 60S subunit to this 48S PIC is catalysed by the universally conserved factor eIF5B, independently of its GTPase activity6,7,8,9. The conformation of yeast eIF5B when bound to GTP is remodelled after association with a ribosome, and this conformational change enables eIF5B to contact directly both Met-tRNAMeti and the GTPase activation centre on the 60S subunit10,11,12. These contacts are probably essential for triggering the hydrolysis of GTP and the subsequent release of eIF5B–GDP, which allows the ribosomal A site to accept the first elongator aminoacyl-tRNA (aa-tRNA) and enter the elongation phase12. Bypassing the hydrolysis of GTP by eIF5B with a mutation that weakens the ribosome-binding affinity of the factor increases the frequency of the first AUG codon in an mRNA being bypassed (‘leaky scanning’) in vivo8—which implies that the timing of the dissociation of eIF5B from the 80S complex and the subsequent entry into elongation is critical to start-site selection. The steps that govern this transition—and their timescales—have not been elucidated in eukaryotes because the complex, multi-step nature of the transition challenges traditional biochemical approaches.
Here we overcome these limitations by using single-molecule fluorescence microscopy to observe translation initiation directly, and thereby elucidate the dynamics and pathways that are involved in the assembly of the 80S complex and subsequent acceptance of the first elongator aa-tRNA. We first reconstituted an active yeast translation system from purified ribosomes and core eukaryotic initiation and elongation factors (Extended Data Fig. 1) that is suitable for single-molecule analyses13. We then applied this system to study the assembly of the 80S complex in real time using a zero-mode waveguide (ZMW)-based instrumentation platform14, which is capable of multi-colour single-molecule fluorescence imaging at high concentrations of fluorescently labelled ligands.
Given that we observed efficient and stable formation of the 48S PIC on an uncapped, unstructured model mRNA that has been well-characterized previously9 (Fig. 1a, Extended Data Fig. 2a), we formed and immobilized single 48S PICs (containing the cyanine 3-labelled 40S subunit, Cy3–40S) within ZMWs and delivered Cy5–60S along with the other required factors (Extended Data Fig. 3a). We observed efficient joining of the 60S subunit on single 48S PICs—around 90% (n = 267 out of 300) of the 48S PICs bound to Cy5–60S to form the 80S complex (Extended Data Fig. 3b, c). This required eIF5B; in the absence of eIF5B, no 80S formation was observed in the 15-min time window that was used for imaging (n = 500); of note, yeast that lack eIF5B are able to survive (albeit with a severe growth defect8) despite the critical role of this factor, which may be because the presence of polyamines can lead to functional subunit joining in the absence of eIF5B (Extended Data Fig. 3d, e). The association rate of 60S subunits was comparable to that from previous bulk measurements9 (Extended Data Fig. 3c). When we labelled the first elongator Phe-tRNAPhe with Cy3.5 and included this in the delivery mix as a ternary complex with eukaryotic elongation factor 1A (eEF1A) and GTP (Cy3.5–Phe-TC) (Fig. 1b), around 80% (n = 164 out of 200) of the 60S joining events (signalled by single-molecule fluorescence resonance energy transfer, smFRET) were followed by a subsequent Cy3.5–Phe-TC arrival event (Fig. 1c). This association of aa-tRNA with the ribosomal A site was codon- and tRNA-specific and led to further elongation (Extended Data Fig. 4). These results demonstrate that real-time 80S assembly occurs in the correct reading frame and that our single-molecule system for translation initiation is fully active.

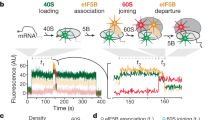
a, mRNA constructs used in single-molecule assays (Extended Data Fig. 5d). All of the mRNAs contain a UUC Phe codon after the AUG start codon and are biotinylated at their 3′-ends. p, phosphate group; nt, nucleotide. b, Experimental setup for single-molecule assays. 48S PICs containing Cy3–40S, Met-tRNAMeti and the 3′-end-biotinylated mRNA of interest were immobilized in ZMWs in the presence of the required initiation factors (eIFs). Experiments were started by illuminating ZMWs with a green laser and delivering Cy5–60S, Cy3.5–Phe-TC and initiation factors. c, Example experimental trace (bottom) and schematic (top) of the molecular events along the reaction coordinate. IC, initiation complex; EC, elongation complex. d, The dwell times between the 60S joining and the arrival of Phe-TC to the A site were measured and fitted to single-exponential distributions to estimate the average time, Δt, of the transition from initiation to elongation. For bars from bottom to top: n = 118, n = 130, n = 130, n = 121, n = 189, n = 159, n = 164 and n = 136 molecules. Error bars show 95% confidence intervals.
To determine an initial rate for the transition from initiation to elongation, we measured the dwell times between 60S joining and the arrival of Cy3.5–Phe-TC, and determined the average time of this transition (Δt) (Fig. 1c). The Δt value was 30.2 ± 1.0 s (n = 147) (± 95% confidence interval) when assayed under the same conditions as were used in previous bulk kinetic analyses of yeast translation initiation9 (26 °C and buffer containing 3 mM free Mg2+). This is substantially longer than that in bacterial translation, for which the Δt value was reported to be approximately 1 s at 20 °C3,4,5. For direct comparisons with the bacterial rates, we also determined the Δt value at 20 °C (92.2 ± 2.5 s) (Fig. 1d). The Δt value remained similar after addition of 150 nM eIF3 and 200 nM eEF3 (and 1 mM ATP:Mg2+) or addition of 500 nM hypusine-modified eIF5A15—indicating that the transition from initiation to elongation is independent of these factors (Extended Data Fig. 5a). Changing the concentration of Cy3.5–Phe-TC did not notably alter the Δt value (Extended Data Fig. 5b). Thus, the rate of the transition is limited by a step that occurs before binding of the first aa-tRNA to the A site.
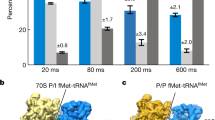
We next questioned whether the slow transition was a result of the m7G-cap-independence of initiation on the model mRNA (that is, the lack of the m7G cap on the model mRNA and eIF4F proteins in the reaction). We formed 48S PICs on RPL41A or RPL30 mRNAs that were completely capped with m7G (cap-RPL41A or cap-RPL30, respectively) (Fig. 1a, Methods) in the presence of eIF4F, eIF4B and eIF3 proteins and performed the same experiments as above. The 48S PICs readily formed in a cap-dependent manner (Extended Data Fig. 2b), and cryo-electron microscopy (cryo-EM) analyses of the complexes showed that they were in the post-scanning, mRNA-channel-‘closed’ state16 (Extended Data Fig. 2c–e). Thus, the 48S PIC that was central to our single-molecule assays is an authentic and on-pathway complex. However, the Δt values at 20 °C were 81.4 ± 2.5 s and 65.7 ± 3.4 s for cap-RPL41A and cap-RPL30, respectively—similar to the Δt value that was measured on the model mRNA (Fig. 1d). Increasing the Cy3.5–Phe-TC concentration did not markedly change the Δt value (Extended Data Fig. 5c), which further demonstrates that the rate-limiting step occurs before aa-tRNA binding to the A site. Our findings suggest that the slow transition to elongation may be a general feature of yeast translation.
We next tested whether the sequence context near the start codon affected entry into the elongation phase of translation. When the optimal yeast Kozak sequence17 was inserted upstream of the AUG codon in the model mRNA (designated as model mRNA-Kozak), the Δt value was reduced by around threefold to 34.1 ± 1.3 s at 20 °C (Fig. 1a, d). This effect was largely abolished by an A to U substitution at the −3 position (Extended Data Fig. 5d), consistent with the critical role of that residue within the Kozak sequence18. We have thus uncovered a role of the Kozak sequence in the control of this transition. However, even the fastest rate we observed at 20 °C was still more than 30-fold slower than that observed in E. coli3,4,5. To understand the energetics of the rate-limiting step in the eukaryotic transition, we increased the reaction temperature to 30 °C (which is within the optimal temperature range for growth of S. cerevisiae19), and found that Δt depended on temperature across all assayed mRNAs (Fig. 1d, Extended Data Fig. 5d). At 30 °C, the Δt value decreased by approximately 4–5-fold compared to the value at 20 °C for the model, cap-RPL41A and cap-RPL30 mRNAs, and by around 2.5-fold for the model mRNA-Kozak.
The temperature dependence of the Δt value suggested that slow conformational rearrangements and/or factor dissociation that are predominantly driven by changes in enthalpy limited the transition. We hypothesized that dissociation of eIF5B was the rate-limiting step, given that its binding site on the ribosome overlaps with that of the elongator aa-tRNAs12. Previous kinetic analyses of translation in rabbit reticulocyte lysate suggest that GTP hydrolysis by an initiation factor limits the rate at which the first peptide bond is formed after the joining of ribosomal subunits20. This GTP hydrolysis could plausibly be that by eIF5B. If eIF5B indeed gates the transition to elongation, we would expect its lifetime on the 80S complex to be similar to the Δt values that we measured above.
To test this hypothesis, we fluorescently labelled eIF5B (Extended Data Fig. 6a) and tracked the protein throughout the reaction pathways using a similar experimental scheme as above. A Cy5.5 dye was attached to N-terminally truncated eIF5B (remaining residues 396–1002) through a ybbR tag fused to the N terminus. Note that this truncated eIF5B is the standard construct in the eukaryotic translation field6,9,21,22; it was used in all of our measurements above that involved eIF5B (Extended Data Fig. 6b) and neither the tag nor the dye affected the function of eIF5B (Extended Data Fig. 6c, d). After immobilizing pre-formed Cy3–48S PICs in ZMWs, we co-delivered Cy5.5–eIF5B, Cy5–60S and Cy3.5–Phe-TC to the immobilized complexes in the presence of other required factors. With the model mRNA and direct illumination of all fluorophores, we observed transient sampling events of Cy5.5–eIF5B to the 48S PIC before 60S joining (mean lifetime = 0.59 ± 0.06 s, n = 903 events; Extended Data Fig. 7a), which is consistent with the dynamic interaction of eIF5B with the 48S PIC that has been suggested by previous measurements with ensemble fluorescence approaches9. After the joining of Cy5–60S to form the 80S complex (as shown by the smFRET signal), the average lifetime of Cy5.5–eIF5B on the ribosome was prolonged and nearly equal to Δt (Fig. 2a–c, Extended Data Fig. 7a). The Δt value measured here matched that measured with unlabelled eIF5B, which confirms that labelling did not affect eIF5B function (Extended Data Fig. 6e). Furthermore, dissociation of Cy5.5–eIF5B always preceded the binding of Cy3.5–Phe-TC to the A site of the 80S ribosome, which occurred very rapidly (within 1 s) after eIF5B departure (Fig. 2a, d). Similar results were observed for all mRNAs at both 20 °C and 30 °C (Fig. 2c). Notably, the Δt value was not reduced by using full-length eIF5B, which indicates that the lack of the non-conserved N-terminal region8,10 of the protein did not interfere with the transition rate (Extended Data Fig. 6b, f, g). Thus, the presence of eIF5B on the de novo-assembled 80S ribosome limits the rate of the transition from initiation to elongation.

a, b, Sample trace (a) and schematic (b) showing the correlation of Cy5.5–eIF5B occupancy on the ribosomal complex with Cy5–60S joining and the arrival of Cy3.5–Phe-TC. c, For all assayed mRNAs, the Δt value, average eIF5B lifetime on 80S and average tRNA arrival time after eIF5B departure were estimated by fitting the dwell times to single-exponential distributions (Extended Data Fig. 9g). From bottom to top for each group of three bars: n = 141, n = 164, n = 133, n = 131 and n = 134 molecules. Error bars show 95% confidence intervals. d, Contour plots of the departure of Cy5.5–eIF5B (bottom; normalized Cy5.5 fluorescence intensity (norm. Cy5.5 int.) changing from 1 to 0) and the arrival of Cy3.5–Phe-TC (top; normalized Cy3.5 intensity (norm. Cy3.5 int.) changing from 0 to 1) in experiments performed with model mRNA in the presence of GTP. Plots were generated by superimposing all the analysed fluorescence traces such that the time point of Cy3.5–Phe-TC arrival was set at 0. Contours are plotted from tan (lowest population) to red (highest population); n = 134. e, The same types of average dwell times as in c were determined for experiments performed with the model mRNA in the presence of 3 mM free Mg2+ at 20 °C. Experiments were performed either in the presence of GTP with unlabelled wild-type (WT) eIF5B (n = 164; data taken from Fig. 1d), Cy5.5-labelled wild-type eIF5B (n = 134; data taken from c), unlabelled eIF5B(H505Y) (n = 119) or Cy5.5-labelled eIF5B(H505Y) (n = 80); or separately in the presence of GDPNP with unlabelled eIF5B(H505Y) (n = 80) (Extended Data Fig. 9h).
We hypothesize that the rate of GTP hydrolysis by eIF5B defines its residence time on the 80S complex. The Δt value that was measured here on the model mRNA was similar to the mean time of GTP hydrolysis by eIF5B during initiation on the same type of model mRNA (approximately 30.2 s versus 28 s at 26 °C)21. Notably, inclusion of a non-hydrolysable GTP analogue (GDPNP) trapped eIF5B on the 80S complex (average Cy5.5–eIF5B lifetime on the 80S was around 850 s and was limited by photobleaching and imaging time; n = 105), and prevented the association of aa-tRNA with the ribosomal A site (Extended Data Fig. 7b, c). To further investigate this finding, we used two previously reported mutant versions of eIF5B that exhibit disrupted GTPase activity8. In the first of these mutants (eIF5B(T439A)), tRNA association with the A site was inhibited (only 17 out of 131 assembled 80S ribosomes underwent A-site tRNA association) and the Δt value increased to around 370 s at 20 °C (Extended Data Fig. 8). By contrast, in the second mutant (eIF5B(H505Y)), which also has weakened affinity for the ribosome, the Δt value decreased by about 15-fold (n = 119) compared to wild-type eIF5B (Fig. 2e, Extended Data Fig. 8). The H505Y mutation instead reduced the occupancy time of eIF5B on the ribosome compared to wild type independent of GTP hydrolysis (Fig. 2e). Thus, eIF5B departure is indeed controlled by GTP hydrolysis.
The marked dependence of the eIF5B departure rate from the 80S complex on temperature and on the concentration of free Mg2+ (Extended Data Fig. 9) suggests that the GTP hydrolysis activity of eIF5B is sensitive to the conformation of the ribosome. We therefore applied cryo-EM, guided by our single-molecule kinetics, to analyse the conformations of on-pathway ribosomal complexes that were assembled in the presence of wild-type eIF5B and GTP. We added eIF5B–GTP and 60S subunits to assembled 48S PICs (on the model mRNA) (Fig. 1a) and allowed the reaction to proceed at about 20 °C for 45 s (from mixing to grid plunge-freezing) (Methods), which corresponds to a pre-steady-state time point according to our single-molecule measurements (Extended Data Fig. 10a, Methods). Two distinct classes of 80S complexes were resolved: (1) the eIF5B-bound 80S initiation complex; and (2) the 80S elongation complex (Extended Data Fig. 10c, d). The two states were populated in agreement with predictions from our single-molecule kinetics (Extended Data Fig. 10a, b). Similar to that of the reported 80S complex bound to eIF5B–GDPCP12, the eIF5B-bound 80S initiation complex (class 1) adopts a semi-rotated inter-subunit conformation, with the Met-tRNAMeti in the 80S peptidyl/initiation configuration. Moreover, eIF1A density was absent in this complex. Its dissociation therefore occurs before the departure of eIF5B and is not the rate-limiting step9. After dissociation of eIF5B, the 80S initiation complex matured to the 80S elongation complex (class 2) in a non-rotated-state, with Met-tRNAMeti shifted to the 80S peptidyl/peptidyl configuration—poising the ribosome for rapid association of tRNA with the A site. Together, we propose that the irreversible hydrolysis of GTP by eIF5B and its subsequent dissociation serves as a final kinetic quality-control checkpoint of the assembly of the 80S complex before entry into the energy-expensive elongation phase.
In summary, we have demonstrated that in S. cerevisiae (and probably in other eukaryotic systems20), eIF5B gates the transition from translation initiation to elongation (Fig. 3). The shortest transition time that we measured in m7G-cap-dependent translation was approximately 8 s (in the presence of 2 mM free Mg2+ at 30 °C) (Extended Data Fig. 9b)—a timescale that is relevant to the estimated rates of translation initiation in yeast in vivo (which vary from several seconds to minutes at 30 °C)23,24. The transition to elongation is probably gated by the hydrolysis of GTP by eIF5B, as shown by the correspondence between the rate of GTP hydrolysis21 and the occupancy time of eIF5B on the 80S complex, and by our observation that the 80S complex bound to eIF5B (probably with GTP) is in a similar conformation to that observed for non-hydrolysable GTP12. We speculate that the long lifetime of eIF5B-bound 80S complexes could partially account for the enrichment of ribosome-protected mRNA fragments at the start codon that has been observed in ribosome profiling experiments in eukaryotic systems25,26,27. The relatively slow transition between translation initiation and elongation in the eukaryotic system compared with the bacterial counterpart provides a mechanism to control the density of ribosomes in the early part of the mRNA open reading frame23,28 to prevent ribosome collisions29,30, and a final kinetic checkpoint for accurate start-codon selection. Consistent with the latter proposal, disruption of GTPase activity results in a higher propensity for leaky scanning in vivo8, and the residence time of eIF5B on the 80S is strongly dependent on the sequence context that surrounds the start codon, with non-optimal Kozak sequences leading to longer residence times. Subtle conformational signalling—perhaps through the initiator tRNA—may modulate the final conformation of the 80S–eIF5B complex for rapid hydrolysis of GTP and subsequent formation of an elongation-competent 80S complex. Finally, we expect that there are many kinetic checkpoints in the early stages of eukaryotic translation. The application of similar single-molecule approaches will unveil the complicated dynamics that underlie mRNA selection, scanning and start-codon selection and the mechanisms by which they are regulated1,2.

eIF5B catalyses the joining of the 60S subunit to the 48S PIC to form the 80S initiation complex (80S IC). Dissociation of eIF5B from the 80S initiation complex requires the hydrolysis of GTP, and this plausibly leads to a change in the conformation of eIF5B that lowers its affinity for the 80S. Thus, the dissociation of eIF5B from the 80S initiation complex gates the transition to elongation, which is marked by the binding of an elongator aa-tRNA to the 80S elongation complex (80S EC). The effects of free Mg2+ concentration, temperature and the sequence context that surrounds the start codon on the rate of the transition indicate that conformational rearrangements may have key roles in governing the rate of eIF5B dissociation, probably by controlling its GTPase activity.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
The strategy for labelling with a ybbR tag
The ybbR-tag labelling strategy has previously been established31. In brief, 4′-phosphopantetheinyl transferase (SFP synthase) specifically transfers a CoA conjugate to a serine residue in the ybbR tag (an 11-amino-acid peptide sequence), resulting in the covalent linking of the CoA conjugate to the ybbR tag. The SFP synthase that was used in this study was purified as previously described31, and the Cy3-like dye analogue CoA 547 was purchased from New England BioLabs (S9349). Following the published protocol, we synthesized CoA-Cy5.5 dye by reacting CoA trilithium salt (Sigma-Aldrich, C3019-25MG; 0.52 mg, 0.66 μmol, 0.75 equivalent (equiv.)) with sulfo-cyanine5.5 maleimide (Lumiprobe, 17380; 1 mg, 0.88 μmol, 1 equiv.) at room temperature for 2 h in a 1-ml volume of 100 mM potassium phosphate pH 7.05 and 10% DMSO. This was followed by quenching the excess amount of maleimide with 8 mM DTT at room temperature for 10 min, which enables the product to be used without the need of further purification.
Unlabelled and labelled tRNAs and yeast ribosomal subunits
Native yeast Met-tRNAMeti was purchased from tRNA Probes, LLC (MI-60) and E. coli Phe-tRNAPhe and Lys-tRNALys labelled with Cy3.5 or Cy5 were previously characterized32. Unlabelled yeast ribosomal subunits were purified by sucrose density-gradient centrifugation as previously described13. Cy3–40S was prepared by the ybbR-tag labelling method as described in the next two sections below.
Genetically tagging the 40S subunit at uS19 with a ybbR tag
The yeast uS19 (RPS15) strain JD1346 (his3Δ1; leu2Δ0; ura3Δ0; YOL040c::KanMX4; pFL38(Pgal::RPS15; URA3)) and plasmid pJD1055 (pRPS15-HIS3) were provided by J. D. Dinman33. A ybbR-tag sequence (GATTCTCTTGAATTTATTGCTAGTAAGCTTGCG)31 was inserted at the N-terminal end of the uS19 sequence in the plasmid pJD1055, resulting in the plasmid pJFW1. This was introduced into yeast JD1346 using a standard lithium acetate yeast-transformation protocol34 followed by a 5′-FOA plasmid shuffle35, to obtain the yeast strain yJFW11 in which all the 40S ribosomal subunits were tagged. For ybbR-uS19-40S purification, the yeast strain yJFW11 was streaked onto a selection plate containing synthetic complete–His (SC-His) medium. A single colony was used to inoculate 5 ml of fresh SC-His liquid medium, and the culture was grown at 30 °C overnight with shaking at 200 r.p.m. The next morning, the stationary-phase yeast culture was used to inoculate a 1.8-l culture of YPAD medium with a 1:4,000 dilution for large-scale growth until an optical density at 600 nm (OD600 nm) of around 0.6 was reached, before collecting cells by fast centrifugation at 4 °C and 7,808g in a Fibrelite F9-6x1000 LEX Fixed Angle Rotor (Thermo Fisher Scientific) for 12 min. The cell pellet was flash-frozen in liquid nitrogen before storage at −80 °C for further ribosome purification.
Labelling of ybbR-uS19-40S
For labelling of the 40S subunit through the ybbR tag, a 300-μl labelling reaction was prepared by mixing 1 μM 40S, 2 μM SFP synthase and 10 μM CoA-547 dye in a buffer containing 50 mM HEPES-KOH pH 7.5, 1 mM DTT and 10 mM MgCl2. The reaction was incubated at 30 °C for 30 min. To purify the labelled 40S from free dye, 150 μl of the reaction mixture was loaded on top of a 750-μl sucrose cushion (30 mM HEPES-KOH pH 7.5, 2 mM DTT, 100 mM KOAc, 5 mM Mg(OAc)2 and 0.5 M sucrose) for ultracentrifugation at 351,955g at 4 °C for 90 min in a TLA100.2 rotor. The pellet containing labelled 40S was washed once with and then resuspended in the ribosome storage buffer (50 mM HEPES-KOH pH 7.5, 2 mM DTT, 100 mM KOAc, 3 mM Mg(OAc)2, 0.25 M sucrose) and stored at −80 °C after flash-freezing with liquid nitrogen.
Labelling of 60S
The Cy5-labelled SNAP-tagged 60S was prepared as previously described and characterized32. The Cy5.5-labelled ybbR-tagged 60S was prepared in the same way as described above for the Cy3–40S. For yeast genetics, the pRS315 plasmid used for the SNAP-tagged 60S yeast strain was mutated to carry the native uL18 (RPL5) gene sequence with a ybbR-tag sequence fused in frame at the C terminus before the stop codon. The new plasmid was transformed into JWY3733 strain (MATα; ura3-52; trp1Δ101; leu2Δ1; ade1; his3Δ200; rpl1::TRP1+ pRS316-RPL5) and plated onto SC-Leu plates, followed by screening for the loss of the pRS316-RPL5 on SC-Leu plates containing 5′-FOA. The resulting strain (MATα; ura3-52; trp1Δ101; leu2Δ1; ade1; his3Δ200; rpl1::TRP1+ pRS315-RPL5-ybbR) expresses uL18-ybbR as the sole source of the protein. The ybbR ribosome labelling efficiencies were generally around 75% based on absorbance measurements.
Purification of protein factors
Published strains, constructs and protocols were used for the purification of yeast eIF2, eIF3, eIF4E, eIF5, eIF5A, eIF5B (residues 396–1002), eEF1A, eEF1Bα, eEF2 and eEF313,15,32,36.
Purification of eIF1
The SUI1 gene was cloned into a pET28 vector such that the overexpressed protein had the following sequence: MGSSHHHHHHMENLYFQG–native eIF1, wherein the underlined sequence is a tobacco etch virus (TEV) protease cleavage site that cleaves between the Q and the G and this cleavage site is followed by the native eIF1 sequence. The final purified protein sequence is therefore the native eIF1 sequence, preceded by a Gly residue.The plasmid was transformed into Rosetta2(DE3) E. coli cells. Protein overexpression was performed at an OD600 nm of around 0.6 by induction with 1 mM IPTG at 30 °C for 4 h. Cells were pelleted and stored at −80 °C. The pellet was rapidly thawed and suspended in 50 mM potassium phosphate (pH 7.5), 500 mM KCl, 10 mM imidazole, 5 mM β-mercaptoethanol (βME), 10% (v/v) glycerol and 1 mM phenylmethylsulfonyl fluoride (PMSF). The cells were lysed by sonication and the lysate was clarified by centrifugation at 41,656g for 30 min at 4 °C in a F21-8x50y rotor (Thermo Fisher Scientific). The clarified lysate was loaded onto a 2-ml Ni-NTA column and washed with 50 ml wash buffer (50 mM potassium phosphate (pH 7.5), 300 mM KCl, 20 mM imidazole, 5 mM βME, 10% (v/v) glycerol). Histidine (His)-tagged protein was eluted with wash buffer containing 250 mM imidazole. The eluted protein was diluted five times in volume by addition of 20 mM HEPES-KOH pH 7.5, 10% (v/v) glycerol and 2 mM DTT, and loaded to a 5-ml HiTrap Heparin HP column for further purification as previously described13. The eluate was treated with TEV protease overnight on ice to cleave off the 6×His tag and eIF1 was recovered by flowing through a Ni-column to remove the free tag and TEV protease. The final eIF1 protein was purified on a HiLoad 26/600 Superdex 200-pg gel filtration column in storage buffer (20 mM HEPES-KOH pH 7.5, 100 mM KOAc, 10% (v/v) glycerol, 2 mM DTT), flash-frozen with liquid nitrogen and stored at −80 °C.
eIF1A
The TIF11 gene was cloned into a pET28 vector such that the overexpressed protein had the following sequence: MGSSHHHHHH MENLYFQG–native eIF1A, and the final purified protein sequence is the native eIF1A sequence preceded by a Gly residue. The protein overexpression and purification were performed in exactly the same way as for eIF1.
eIF4A
The TIF1 gene was cloned into a pET28 vector such that the overexpressed protein had the following sequence: MGSSHHHHHH MENLYFQG–native eIF4A, and the final purified protein sequence is the native eIF4A sequence preceded by a Gly residue. The protein overexpression and Ni-NTA purification were performed in the same way as for eIF1. After Ni-NTA purification, the protein was treated with TEV protease overnight to cleave off the 6×His tag, while dialysing at 4 °C against a buffer containing 50 mM potassium phosphate (pH 7.5), 300 mM NaCl and 5 mM βME. The eIF4A protein was recovered by flowing through a Ni-column to remove the cleaved 6×His tag and TEV protease. The final protein was purified on a HiLoad 26/600 Superdex 200-pg gel filtration column in storage buffer (20 mM HEPES-KOH pH 7.5, 100 mM KOAc, 2 mM DTT), flash-frozen with liquid nitrogen and stored at −80 °C.
eIF4B
The TIF3 gene was cloned into a pET22b vector such that the overexpressed protein had the following sequence: native eIF4B–ENLYFQGAAAELHHHHHH, and the final purified protein sequence is the native eIF4B sequence followed by ENLYFQ. The protein overexpression and purification were performed in exactly the same way as for eIF4A.
eIF4G
The TIF4631 gene was cloned into the multiple-cloning site 1 of a pETDuet-1 vector such that the overexpressed protein had the following sequence: native eIF4G-ENLYFQGHHHHHH, and the final purified protein sequence is the native eIF4G sequence followed by ENLYFQ. On the same plasmid, the native eIF4E gene (CDC33) was cloned into the multiple-cloning site 2, which has been proven to facilitate the overexpression of the full-length eIF4G in E. coli37. The protein overexpression and purification were performed in the same way as for eIF4A, except that 500 mM KCl was included in all the buffers used for the Ni-column purification. The final protein contained both eIF4G and wild-type eIF4E and the full-length eIF4G migrates slower than expected on SDS–PAGE for its molecular weight, which has previously been shown37,38,39. The full-length nature of eIF4G was further verified by Edman degradation and mass spectrometry.
eIF5B variants
When applicable, a ybbR-tag sequence was introduced into the pMGAHisTev5B plasmid13 after the TEV cleavage site, with or without a T439A or H505Y mutation, by site-directed mutagenesis. The protein was overexpressed and purified following the previously described protocol13. For full-length eIF5B, we started with the plasmid pC100540, deleted the Flag-tag sequence, inserted in a 8×His tag and a TEV cleavage site before the native eIF5B sequence, and cloned into the multiple-cloning site 1 of the pETDuet-1 vector. Proteins were overexpressed in Rosetta2(DE3) cells as described for eIF1 and the purification was performed as previously described13. To label the ybbR-tagged proteins with Cy5.5, 10 μM protein was incubated with 2 μM SFP synthase and 15 μM CoA-Cy5.5 dye in a buffer containing 50 mM HEPES-KOH pH 7.5, 1 mM DTT and 10 mM MgCl2 at room temperature for 30 min. The reaction mixture was loaded onto a Ni-NTA column equilibrated with the same buffer to remove His-tagged SFP synthase, and the flow-through was passed through a 10-DG desalting column to remove free dye from the protein. The final protein was purified on a Superdex 75 10/300 GL column in storage buffer (20 mM HEPES-KOH pH 7.5, 100 mM KOAc, 10% glycerol, 2 mM DTT) and stored at −80 °C. The ribosome-dependent GTPase activity was determined as previously described using the malachite green assay32.
mRNAs
All the mRNAs were prepared by T7 RNA polymerase run-off transcription. The RNA transcript sequences are as follows (with the start and stop codons underlined): model mRNA: GG(UC)8UAUGUUCAAAUAA(UC)16; model mRNA-Kozak: GG(UC)8UAAAAAAAUGUUCAAAUAA(UC)16; model mRNA-Kozak_-3U: GG(UC)8UAAAUAAAUGUUCAAAUAA(UC)16; RPL41A: GGAGACCACAUCGAUUCAAUCGAAAUGUUCGCCAAGUGGAGAAAGAAGAGAACUAGAAGACUUAAGAGAAAGAGACGGAAGGUGAGAGCCAGAUCCAAAUAA; RPL30: GGAAACAGACCGGAGUGUUUAAGAACCUACAGCUUAUUCAAUUAAUCAAUAUACGCAGAGAUGUUCCCAGUUAAAUCCCAAGAAUCUAUCAACCAAAAGUUGGCUUUGGUUAUCAAGUCUGGUAAGUACACCUUAGGUUACAAGUCCACUGUCAAGUCUUUGAGACAAGGUAAGUCUAAGUUGAUCAUCAUUGCCGCUAACACUCCAGUUUUGAGAAAGUCCGAAUUGGAAUAUUACGCUAUGUUGUCCAAGACUAAGGUCUACUACUUCCAAGGUGGUAACAACGAAUUGGGUACUGCUGUCGGUAAGUUAUUCAGAGUCGGUGUUGUCUCUAUUUUGGAAGCUGGUGACUCUGAUAUCUUGACCACCUUGGCUUAAAUAAGGUAAGUUCAAACGAUUU.
For the model mRNA, model mRNA-Kozak and model mRNA-Kozak_-3U, 3′-end biotinylation or Cy5-labelling was performed using periodate chemistry followed by hydrazide reaction as previously described41. The RNA was purified from the free hydrazide on a Superdex 75 10/300 GL column in storage buffer (20 mM HEPES-KOH pH 7.5 and 100 mM KOAc) and stored at −80 °C. The hydrazide analogues used were EZ-Link Biotin-LC-Hydrazide (Thermo Fisher Scientific, 21340) and cyanine5 hydrazide (Lumiprobe, 13070).
To obtain 3′-end-biotinylated or Cy3-labelled, fully m7G-capped RPL41A mRNA, the RNA transcript was: (1) ligated to a DNA oligonucleotide (5′pd(CT)12-biotin-3′ or 5′pd(CT)6-Cy3-d(CT)6-3′) (TriLink Biotechnologies) and gel purified; (2) m7G-capped using the Vaccinia capping system (New England BioLabs, M2080S); and (3) treated with RNA 5′polyphosphatase (Epicentre, RP8092H) and terminator 5′-phosphate-dependent exonuclease (Epicentre, TER51020) to remove uncapped RNA, followed by phenol and chloroform extraction and ethanol precipitation.
The 3′-end-biotinylated, fully m7G-capped RPL30 mRNA was prepared by: (1) 3′-end biotinylation using the periodate chemistry and hydrazide reaction41; and (2) m7G-capping and uncapped-RNA depleting as described above for RPL41A mRNA.
Native gel-shift analyses
The formation of pre-initiation and initiation complexes was monitored by 4% native PAGE analyses using protocols as previously described13. Samples were prepared in 1× Recon buffer (30 mM HEPES-KOH pH 7.5, 100 mM KOAc, 3 mM Mg(OAc)2). A ternary complex was prepared by incubating 3.9 μM eIF2 with 1 mM GTP:Mg2+ at 30 °C for 10 min, followed by addition of 2.7 μM Met-tRNAMeti and incubation at 30 °C for another 5 min.
For reactions with the model mRNA (Extended Data Fig. 2a), the 48S PIC was formed by mixing a final concentration of 1 μM eIF1, 1 μM eIF1A, threefold-diluted ternary complex (final 1.3 μM eIF2 and 0.9 μM Met-tRNAMeti), 50 nM Cy3–40S and 2 μM model mRNA–Cy5, and incubating at 30 °C for 15 min; the 80S PIC was formed by addition of a final concentration of 1 μM eIF5, 1 μM eIF5B and 200 nM 60S to the 48S PIC and further incubation at 30 °C for 5 min. Typically, 10 μl of each sample was mixed with 2 μl of native gel dye (50% sucrose, 0.02% bromophenol blue in 1× Recon buffer) and loaded into the 4% native PAGE gel for electrophoresis13; this was followed by scanning the gel for the Cy3 and Cy5 fluorescence with a Typhoon scanner.
For reactions with the cap-RPL41A mRNA (Extended Data Fig. 2b), the samples were prepared similarly to above, except 1 mM ATP:Mg2+ was supplemented into the reaction buffer, the cap-RPL41A–Cy3 mRNA was at 100 nM, 40S was unlabelled and, when applicable, 1 μM eIF4A, 1 μM eIF4B, 400 nM eIF4G, 200 nM eIF4E and 200 nM eIF3 were added.
Single-molecule inter-subunit FRET experiments using a total internal reflection fluorescence microscope
The ribosomal inter-subunit FRET signal was characterized on a previously described42,43 home-built, prism-based total internal reflection fluorescence microscope. The 80S complexes were assembled using Cy3–40S and Cy5–60S on the model mRNA–biotin in the same way as for the native gel shift assays. The 80S complexes were immobilized on a neutravidin-derivatized quartz slide according to reported protocols32,43. Unbound components were washed from the slide with 1× Recon buffer supplemented with 1 mM GTP:Mg2+ and an oxygen-scavenging system (containing 2.5 mM PCA (protocatechuic acid), 2.5 mM TSY and 2× PCD (protocatechuate-3,4-dioxygenase), purchased from Pacific Bioscience). Cy3 fluorophores were excited with a diode-pumped solid-state 532-nm laser at 1 kW cm−2. Fluorescence intensity trajectories were recorded at 10 frames per second with a total of 600 frames collected per movie by using the MetaMorph imaging software (Molecular Devices). Analysis of fluorescence intensity trajectories was performed in MATLAB using custom scripts42. Baseline correction of raw fluorescence traces and Cy3 signal bleed-through into the Cy5 channel were corrected on a trace-by-trace basis. The FRET states were assigned with a hidden Markov model-based approach as previously described and visually corrected42.
Real-time single-molecule experiments on the ZMW instrument
All the experiments were performed in the 1× Recon buffer supplemented with 1 mM GTP:Mg2+ (and 1 mM ATP:Mg2+ for m7G-capped mRNAs) unless otherwise specified. A ZMW-based PacBio RSII instrumentation14 was used for all the real-time measurements at 20 °C, 26 °C or 30 °C. For experiments performed with the model mRNA, model mRNA-Kozak and model mRNA-Kozak_-3U, in general, immediately before each experiment, the 48S PIC was formed as described for the native gel shift assays using final concentrations of 80 nM Cy3–40S, 500 nM biotinylated mRNA, 1 μM eIF1, 1 μM eIF1A, 300 nM eIF5, 1.3 μM eIF2 and 0.9 μM Met-tRNAMeti. In parallel, a ZMW chip was treated with 0.2% (w/v) Tween20 at room temperature for 10 min followed by treatment with neutravidin at room temperature for 5 min, which bound to the biotin-PEG (polyethylene glycol) on the chip. After immobilizing the 48S PIC via the biotin on the mRNA onto the neutravidin-coated surface, the chip was washed twice using an imaging buffer containing the oxygen-scavenging system and 62.5 μg ml−1 casein to remove unbound complexes, and loaded onto the RSII instrument in the presence of 20 μl of the imaging buffer. At the same time, a delivery mixture containing 200 nM Cy5-60S, 1 μM eIF5, eIF5B (2 μM unlabelled or 40 nM Cy5.5-labelled), Cy3.5–Phe-TC (pre-formed by incubating 8 μM eEF1A, 10 μM eEF1Bα with 1 mM GTP:Mg2+ at room temperature for 5 min, followed by the addition of 0.8 μM Cy3.5–Phe-tRNAPhe and another incubation at room temperature for 5 min; diluted eightfold in the delivery mixture for final 50 nM ternary complex or fourfold for final 100 nM ternary complex), oxygen-scavenging system and 62.5 μg ml−1 casein was prepared and loaded onto the instrument. For each experiment, the instrument delivered 20 μl of the delivery mixture to the chip at the beginning of the experiment, and recorded a 15-min movie at 10 frames per second, with 0.32 μW μm−2 of 532-nm laser illumination (for unlabelled eIF5B experiments), or 0.6 μW μm−2 of 532-nm laser and 0.1 μW μm−2 of 642-nm laser dual illumination (for Cy5.5-labelled eIF5B experiments). For the elongation assay with tRNA–tRNA smFRET, both the imaging buffer and the delivery mixture were supplemented with 1 mM ATP, and the delivery mixture contained 115 nM Cy5.5–60S, 1 μM eIF5, 2 μM eIF5A, 2 μM eIF5B, 1 μM eEF2, 400 nM eEF3, 100 nM Cy3.5–Phe-TC and 100 nM Cy5–Lys-TC (pre-formed separately by incubating 8 μM eEF1A, 10 μM eEF1Bα with 1 mM GTP:Mg2+ at room temperature for 5 min, followed by the addition of 0.8 μM Cy3.5–Phe-tRNAPhe or Cy5–Lys-tRNALys and another incubation at room temperature for 5 min), oxygen-scavenging system and 62.5 μg ml−1 casein. The imaging was performed at 20 °C with 0.32 μW μm−2 of 532-nm laser illumination.
Experiments with the cap-RPL41A and cap-RPL30 mRNAs were performed in a similar manner, except that 1 μM eIF4A, 1 μM eIF4B, 0.67 μM eIF4E, 300 nM eIF4G and 200 nM eIF3 were also included in the second incubation step for the 48S PIC formation, and 300 nM eIF3 was also added to the delivery mixture. When titrating free Mg2+ concentrations, the corresponding amount of Mg(OAc)2 was used in both the imaging buffer and the delivery mixture. When additional protein factors were added to each experiment, they were added to the delivery mixture in the denoted amounts. For experiments performed with GDPNP, the 48S PIC was formed in the presence of GTP as described above. After immobilization, the imaging buffer used contained 0.2 mM GTP; no GTP was added in the delivery mixture and 6 mM GDPNP:Mg2+ was added instead.
The resulting experimental movies were analysed using custom MATLAB scripts, as previously described14. In brief, fluorescence traces from the ZMWs were filtered on the basis of the presence of the fluorophores of interests at different time points (the signal from immobilized 48S PICs was expected to be present at the beginning of the movie, whereas signals from fluorophores attached to the 60S, eIF5B and tRNA were expected not to be). The different arrival and departure events were manually assigned based on the appearance and disappearance of the corresponding fluorescent signals.
Cryo-EM analysis of the 48S PIC formed with cap-RPL30 mRNA
The 48S PIC sample for the cryo-EM analysis was prepared in the same way as that for the real-time single-molecule experiments in the presence of ATP, GTP and 3 mM free Mg2+. After the incubation at 30 °C, 30 μl of the sample was diluted with 100 μl 1× Recon buffer and then concentrated to 30 μl using a Vivaspin 500 concentrator (100, 000 MWCO PES, Sartorius, VS0141) by centrifugation at 6,973g for 5 min at 4 °C. This step mimics the washing step in our single-molecule experiments for removing unbound components. A 3 μl sample was applied to each 25-s glow-discharged (in a PELCO easiGlow glow discharger, Ted Pella, Inc.) 200-mesh Quantifoil R2/1 gold grid (Electron Microscopy Sciences, Q250AR1) at 21 °C and 95% humidity. The grid was blotted for 2 s before plunging into liquid ethane using a Leica EM GP (Leica Microsystems). Automated data acquisition was performed using SerialEM44 on a Tecnai F20 microscope operated at 200 kV at a dose rate of 10 e− per pixel per s using a defocus range of −2.0 to −4.0 μm. Eighty movie-mode images with 10-s exposure time and 50 frames for each were recorded on a Gatan K2 Summit direct electron detector camera in a counting mode at a nominal magnification of 14,500 (yielding a pixel size of 2.53 Å on the specimen level). The drift correction was performed using MotionCor245 and the contrast transfer function (CTF) estimation was performed using CTFFIND-4.146. The images with bad CTF fitting were excluded and 78 images were selected for further data processing. The initial particle-picking was performed using EMAN247 and the saved box files were imported into RELION-3.0-beta-248 for the first 2D classification to generate some good 2D class averages as a new template for automatic particle-picking in RELION. The 3D initial model was built using RELION. After the first six-class 3D classification, 11,478 particles within one class from a total of 17,654 good particles were selected for the 3D auto-refine to obtain a map at 9.0-Å resolution (not shown). Then a soft mask around the Met-tRNAMeti region was created to perform the particle subtraction followed by the focused classification (without image alignment) into two classes: with Met-tRNAMeti and without Met-tRNAMeti. Finally, 4,930 particles were selected for the final 3D auto-refine to reconstruct the map at 9.9-Å resolution. The scanning-competent open (EMD 3049) or scanning-incompetent closed (EMD 3048) 48S PIC structures were low-pass-filtered to the same resolution (9.9 Å) and scaled to the same pixel size using the command relion_image_handler in RELION for visual comparisons in Chimera (UCSF) (see Extended Data Fig. 2c–e).
Cryo-EM analysis of the on-pathway 80S complexes during initiation and the transition to elongation
To probe the conformational states of the 80S complexes along the native initiation and transition to elongation pathways, we decided to analyse the sample corresponding to the reaction time point 45 s (under pre-steady-state conditions), when the 80S initiation complex should account for around 64.5% (and the 80S elongation complex account for around 35.5%; see Supplementary Note) of the total 80S complexes assembled on the model mRNA at 20 °C and 3 mM Mg2+ in the presence of wild-type eIF5B and GTP. Also, to ease the post-acquisition particle-classification procedure, we decided to use excess 48S PICs to 60S, such that free 60S particles would be minimized. To this end, we assembled 48S PICs in the same way as in the single-molecule experiments with the model mRNA at 3 mM free Mg2+, and the only difference was that the concentration of the unlabelled 40S was 300 nM (assuming 100% complex assembly, the 48S PIC concentration should be 300 nM). In parallel, an initiation mixture containing 150 nM unlabelled 60S, 1 μM eIF5 and 2 μM eIF5B was prepared. After mixing equal volumes of the 48S PIC with the initiation mixture, the final concentrations were 150 nM 48S PIC, 75 nM 60S and 1 μM eIF5B. At 20 °C under these conditions, we would expect the reaction kinetics to be similar to that of the single-molecule experiments described above.
While the 48S PIC and initiation mixtures were kept on ice, the 200-mesh R2/1 Quantifoil grids were glow-discharged for 25 s. When the Leica EM GP system was ready for grid plunging, 5 μl of the 48S PIC mixture was mixed with 5 μl of the initiation mixture, and incubated at about 20 °C for 30 s before approximately 3 μl of the sample was applied to each grid. The grid was immediately blotted for 2 s using a filter paper before being plunged into liquid ethane with 95% chamber humidity at about 21 °C (resulting in a reaction time from mixing to grid-freezing at 45 s). Automated data acquisition was done using SerialEM44 on a Tecnai F20 microscope operated at 200 kV at a dose rate of 10 e− per pixel per s using a defocus range of −0.7 to −2.5 μm. A total of 226 movie-mode images with 10-s exposure time and 50 frames for each were recorded on a Gatan K2 Summit direct electron detector camera in a counting mode at a nominal magnification of 14,500 (yielding a pixel size of 2.53 Å on the specimen level). The drift correction was performed using MotionCor245 and the CTF estimation was done using CTFFIND-4.146. The images with bad CTF fitting, too much drift or that were over-focused were excluded, and eventually 134 images were selected for further 2D and 3D classifications. The initial particle-picking was performed using EMAN247 and the saved box files were imported into RELION-3.0-beta-248 for the first 2D classification, to generate some good 2D class averages as a new template for better 80S particle-picking in RELION. The 3D initial model of the 80S complex was built using RELION. The following 2D and 3D classifications were used to eliminate junk or bad particles, or even free 40S or 60S particles. In total, 25,144 80S particles were selected for the 3D auto-refine to obtain the map at 7.8-Å resolution (not shown). The eIF5B Protein Data Bank (PDB) model was selected from the PDB file 4V8Z12 and fitted into the 7.8-Å 80S map. A zone with radius 6 around the fitted eIF5B PDB model was generated using Chimera. Then a soft mask around the eIF5B density was made using mask creation in RELION. The particle subtraction was done with this soft mask to perform the focused 3D classification (without image alignment) into two classes of 80S: 17,602 particles with eIF5B bound (class 1, 80S initiation complex; Extended Data Fig. 10c) and 7,542 without eIF5B (class 2, 80S elongation complex; Extended Data Fig. 10d). Finally 3D auto-refine reconstructed the 80S initiation complex map at 8.4-Å resolution and the 80S elongation complex map at 10.1-Å resolution.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Source data for figures are available as accompanying Source Data files. The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) server under the accession codes EMD-20324 (48S PIC), EMD-20325 (80S initiation complex) and EMD-20326 (80S elongation complex). Single-molecule fluorescence-trace data are available upon request.
Code availability
The MATLAB scripts used in this study are available at: https://drive.google.com/drive/u/1/folders/1-yKC0Nui7T7yoJScCuODPhCT4G52emLj
References
Hinnebusch, A. G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 83, 779–812 (2014).
Sokabe, M. & Fraser, C. S. Toward a kinetic understanding of eukaryotic translation. Cold Spring Harb. Perspect. Biol. 11, a032706 (2019).
Kaledhonkar, S. et al. Late steps in bacterial translation initiation visualized using time-resolved cryo-EM. Nature 570, 400–404 (2019).
Tsai, A. et al. Heterogeneous pathways and timing of factor departure during translation initiation. Nature 487, 390–393 (2012).
Goyal, A., Belardinelli, R., Maracci, C., Milón, P. & Rodnina, M. V. Directional transition from initiation to elongation in bacterial translation. Nucleic Acids Res. 43, 10700–10712 (2015).
Pestova, T. V. et al. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature 403, 332–335 (2000).
Lee, J. H. et al. Initiation factor eIF5B catalyzes second GTP-dependent step in eukaryotic translation initiation. Proc. Natl Acad. Sci. USA 99, 16689–16694 (2002).
Shin, B. S. et al. Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell 111, 1015–1025 (2002).
Acker, M. G. et al. Kinetic analysis of late steps of eukaryotic translation initiation. J. Mol. Biol. 385, 491–506 (2009).
Roll-Mecak, A., Cao, C., Dever, T. E. & Burley, S. K. X-ray structures of the universal translation initiation factor IF2/eIF5B: conformational changes on GDP and GTP binding. Cell 103, 781–792 (2000).
Kuhle, B. & Ficner, R. eIF5B employs a novel domain release mechanism to catalyze ribosomal subunit joining. EMBO J. 33, 1177–1191 (2014).
Fernández, I. S. et al. Molecular architecture of a eukaryotic translational initiation complex. Science 342, 1240585 (2013).
Acker, M. G., Kolitz, S. E., Mitchell, S. F., Nanda, J. S. & Lorsch, J. R. Reconstitution of yeast translation initiation. Methods Enzymol. 430, 111–145 (2007).
Chen, J. et al. High-throughput platform for real-time monitoring of biological processes by multicolor single-molecule fluorescence. Proc. Natl Acad. Sci. USA 111, 664–669 (2014).
Gutierrez, E. et al. eIF5A promotes translation of polyproline motifs. Mol. Cell 51, 35–45 (2013).
Llácer, J. L. et al. Conformational differences between open and closed states of the eukaryotic translation initiation complex. Mol. Cell 59, 399–412 (2015).
Hamilton, R., Watanabe, C. K. & de Boer, H. A. Compilation and comparison of the sequence context around the AUG startcodons in Saccharomyces cerevisiae mRNAs. Nucleic Acids Res. 15, 3581–3593 (1987).
Li, J. J., Chew, G. L. & Biggin, M. D. Quantitating translational control: mRNA abundance-dependent and independent contributions and the mRNA sequences that specify them. Nucleic Acids Res. 45, 11821–11836 (2017).
Walsh, R. M. & Martin, P. A. Growth of Saccharomyces cerevisiae and Saccharomyces uvarum in a temperature gradient incubator. J. Inst. Brew. 83, 169–172 (1977).
Lorsch, J. R. & Herschlag, D. Kinetic dissection of fundamental processes of eukaryotic translation initiation in vitro. EMBO J. 18, 6705–6717 (1999).
Acker, M. G., Shin, B. S., Dever, T. E. & Lorsch, J. R. Interaction between eukaryotic initiation factors 1A and 5B is required for efficient ribosomal subunit joining. J. Biol. Chem. 281, 8469–8475 (2006).
Pisareva, V. P. & Pisarev, A. V. eIF5 and eIF5B together stimulate 48S initiation complex formation during ribosomal scanning. Nucleic Acids Res. 42, 12052–12069 (2014).
Shah, P., Ding, Y., Niemczyk, M., Kudla, G. & Plotkin, J. B. Rate-limiting steps in yeast protein translation. Cell 153, 1589–1601 (2013).
Chu, D. et al. Translation elongation can control translation initiation on eukaryotic mRNAs. EMBO J. 33, 21–34 (2014).
Schuller, A. P., Wu, C. C. C., Dever, T. E., Buskirk, A. R. & Green, R. eIF5A functions globally in translation elongation and termination. Mol. Cell 66, 194–205 (2017).
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. S. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009).
Heyer, E. E. & Moore, M. J. Redefining the translational status of 80S monosomes. Cell 164, 757–769 (2016).
Ingolia, N. T. et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 8, 1365–1379 (2014).
Juszkiewicz, S. et al. ZNF598 Is a quality control sensor of collided ribosomes. Mol. Cell 72, 469–481 (2018).
Ikeuchi, K. et al. Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J. 38, e100276 (2019).
Yin, J., Lin, A. J., Golan, D. E. & Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc. 1, 280–285 (2006).
Petrov, A., Grosely, R., Chen, J., O’Leary, S. E. & Puglisi, J. D. Multiple parallel pathways of translation initiation on the CrPV IRES. Mol. Cell 62, 92–103 (2016).
Bowen, A. M. et al. Ribosomal protein uS19 mutants reveal its role in coordinating ribosome structure and function. Translation 3, e1117703 (2015).
Ito, H., Fukuda, Y., Murata, K. & Kimura, A. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153, 163–168 (1983).
Maniatis, T., Fritsch, E. F. & Sambrook, J. Molecular Cloning: a Laboratory Manual (Cold Spring Harbor Laboratory Press, 1982).
O’Leary, S. E., Petrov, A., Chen, J. & Puglisi, J. D. Dynamic recognition of the mRNA cap by Saccharomyces cerevisiae eIF4E. Structure 21, 2197–2207 (2013).
Mitchell, S. F. et al. The 5′-7-methylguanosine cap on eukaryotic mRNAs serves both to stimulate canonical translation initiation and to block an alternative pathway. Mol. Cell 39, 950–962 (2010).
Algire, M. A. et al. Development and characterization of a reconstituted yeast translation initiation system. RNA 8, 382–397 (2002).
Goyer, C. et al. TIF4631 and TIF4632: two yeast genes encoding the high-molecular-weight subunits of the cap-binding protein complex (eukaryotic initiation factor 4F) contain an RNA recognition motif-like sequence and carry out an essential function. Mol. Cell. Biol. 13, 4860–4874 (1993).
Choi, S. K. et al. Physical and functional interaction between the eukaryotic orthologs of prokaryotic translation initiation factors IF1 and IF2. Mol. Cell. Biol. 20, 7183–7191 (2000).
Rinaldi, A. J., Suddala, K. C. & Walter, N. G. Native purification and labeling of RNA for single molecule fluorescence studies. Methods Mol. Biol. 1240, 63–95 (2015).
Marshall, R. A., Dorywalska, M. & Puglisi, J. D. Irreversible chemical steps control intersubunit dynamics during translation. Proc. Natl Acad. Sci. USA 105, 15364–15369 (2008).
Blanchard, S. C., Kim, H. D., Gonzalez, R. L. Jr, Puglisi, J. D. & Chu, S. tRNA dynamics on the ribosome during translation. Proc. Natl Acad. Sci. USA 101, 12893–12898 (2004).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Shin, B. S. et al. Structural integrity of α-helix H12 in translation initiation factor eIF5B is critical for 80S complex stability. RNA 17, 687–696 (2011).
Munro, J. B., Sanbonmatsu, K. Y., Spahn, C. M. T. & Blanchard, S. C. Navigating the ribosome’s metastable energy landscape. Trends Biochem. Sci. 34, 390–400 (2009).
Acknowledgements
We are grateful to J. Lorsch, T. Dever, J. Dinman, J. Yin and C. Aitken for sharing constructs, strains and protocols; C. Sitron and O. Brandman for sharing equipment and knowledge for use of their Freezer/Mill cryogenic mill for yeast lysis; the Stanford PAN facility for mass spectrometry analyses of proteins; and members of the Puglisi laboratory for discussion and input. This work was supported by the US National Institutes of Health (NIH) grants GM113078 and AI047365 to J.D.P.; a Knut and Alice Wallenberg Foundation postdoctoral scholarship (KAW 2015.0406) to J.W.; a National Science Foundation Graduate Research Fellowship (DGE-114747) to A.G.J.; a Damon Runyon Fellowship funded by the Damon Runyon Cancer Research Foundation (DRG-2321-18) to C.P.L.; a Stanford Bio-X fellowship to J.C.; and a Stanford Interdisciplinary Graduate Fellowship and NIH Molecular Biophysics Training Grant T32-GM008294 to A.P.
Author information
Authors and Affiliations
Contributions
J.W. performed all the biochemical and single-molecule experiments; J.W. analysed the data with help from J.C., A.P. and A.N.P.; J.W. prepared the samples, D.-H.C. and J.W. acquired and D.-H.C. processed the cryo-EM data; J.W. and J.D.P. conceived the project with input from A.G.J., C.P.L and A.N.P.; and J.W., A.G.J., C.P.L., J.C., D.-H.C. and J.D.P. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 A reconstituted yeast translation system with fluorescently labelled ribosomes for inter-subunit smFRET.
a, The labelling of the yeast 60S ribosomal subunit with Cy5 by means of a SNAP tag fused to the uL18 protein and the reaction with a SNAP-647 dye have previously been established32. b, In this work, we engineered a yeast strain in which all of the 40S subunits carried the N-terminal ybbR-tagged uS19 protein. After purification, the 40S was labelled by SFP synthase with CoA-547 at the serine residue (in bold and underlined) of the ybbR tag, resulting in the Cy3–40S. c, The estimated distance between the 2 ribosomal labelling sites is within 50 Å of that from published yeast 80S 3D structures (g). The ribosome model was created in PyMOL, using data from PDB 4V8Z12. d, Total internal reflection fluorescence microscopy (TIRFM) experimental setup to characterize the inter-subunit FRET signal. 80S complexes were assembled from Cy3–40S and Cy5–60S on the model mRNA (Fig. 1a) in the presence of the required factors (Methods), and were immobilized on a quartz slide used for TIRFM imaging with green laser illumination. e, f, Sample TIRFM experimental trace (e) and the inter-subunit smFRET efficiency histogram (f; fitted with a single-Gaussian distribution, with a mean FRET efficiency of 0.89 ± 0.15 s.e.m.); n = 107 molecules. g, Estimated distances between the two labelling sites on the ribosomal subunits from a few examples of published yeast 80S structures in different functional states, and the expected FRET efficiencies based on a Förster radius (R0) of 54 Å for the Cy3–Cy5 FRET pair. h, Representative SDS–PAGE analysis of the purified core eukaryotic initiation factors (blue numbering) and elongation factors (red numbering) that were used for the reconstitution of the translation system. Each component was analysed at least three times with similar results.
Extended Data Fig. 2 Native gel shift assays and cryo-EM analysis showing active translation initiation with our purified yeast translation system.
a, Representative gel showing initiation on the model mRNA. A merged view of Cy5 (red) and Cy3 (blue) scans of the same gel is shown. For gel source data, see Supplementary Fig. 1. The model mRNA was labelled with Cy5 and 40S was labelled with Cy3. Addition of Cy3–40S, Met-tRNAMeti:eIF2:GTP (Met-TC), eIF1 and eIF1A to the model mRNA–Cy5 resulted in the formation of a distinct 48S PIC band. Further addition of eIF5, eIF5B and 60S led to the formation of the 80S band. The experiment was repeated three times with similar results. b, Representative gel showing initiation on the cap-RPL41A mRNA. For gel source data, see Supplementary Fig. 1. The cap-RPL41A mRNA was labelled with Cy3, and other components were unlabelled. The gel was scanned for Cy3 fluorescence. Various mRNA and protein complexes were formed in the absence of 40S. After adding 40S to the mixture, a distinct 48S PIC band was formed and further addition of eIF5B and 60S shifted this band to the 80S band. The apparent electrophoretic mobility of the 48S PIC that formed with the cap-RPL41A mRNA differs from that of the 48S PIC that formed with the model mRNA; this is probably a result of the different charges or hydrodynamic radius of the complex brought about by the capped mRNA. Both 48S PIC and 80S formation were very inefficient when the cap-binding eIF4F (initiation factors 4A, 4E and 4G) and eIF4B proteins were omitted from the reaction, demonstrating the dependence of initiation on the cap when the full set of initiation factors were added. The experiment was repeated three times with similar results. c–e, Using our regime for assembly of the 48S PIC, we would expect the 48S PIC to be in the post-scanning state, with the initiation factors that are required during the scanning process potentially dissociated from the complex. A cryo-EM map at 9.9 Å resolution was obtained for the 48S PIC that formed on the cap-RPL30 mRNA (grey). This map was compared with previously reported 48S structures16 in the scanning-competent, mRNA-channel-open configuration (c) (EMD 3049; yellow), or scanning-incompetent, mRNA-channel-closed configuration (d) (EMD 3048; cyan) (Methods). The cap-RPL30–48S PIC was assembled in the same way as for our single-molecule experiments, and the comparisons showed that it resembles the post-scanning closed state, with the Met-tRNAMeti positioned in the P-site (e). The modelled Met-tRNAMeti in the EMD 3048 structure, PDB 3JAP, is shown in red.
Extended Data Fig. 3 Real-time 80S assembly on the model mRNA at the single-molecule level.
a, smFRET assay for subunit joining in ZMWs. 48S PICs were formed by incubating Cy3–40S, Met-TC, model mRNA–biotin, eIF1, eIF1A and eIF5 at 30 °C for 15 min before immobilization in the ZMWs. After washing away free components, the experiment was started with green laser illumination and delivery of Cy5–60S, eIF5 and eIF5B. The reaction was performed in the 1× Recon buffer supplemented with 1 mM GTP:Mg2+ at 20 °C. b, Example experimental trace showing real-time observation of Cy5–60S joining to immobilized Cy3–48S PIC to form the 80S complex, which is identified by the appearance of the smFRET signal. Single photobleaching events are denoted. Similar results were obtained from three independent experiments. c, The cumulative probability distribution of the dwell times for 60S joining was fitted to a double-exponential equation, resulting in a fast phase rate of about 0.22 s−1 with about 46% amplitude, and a slow phase rate of about 0.03 s−1 with about 54% amplitude (n = 178). The kinetics are comparable to previous bulk measurements of the same reaction under similar conditions9 (about 77% fast phase with a rate of about 0.076 s−1; about 23% slow phase with a rate of 0.019 s−1). d, e, Spermidine-driven initiation in the absence of eIF5B. The cumulative probability distributions of the dwell times for 60S joining (d) and the transition to elongation (e) from experiments performed in the presence or absence of 3 mM spermidine and/or 1 µM eIF5B at 20 °C with the model mRNA were fitted to a double-exponential (d) or a single-exponential (e) equation. The estimated average fast and slow phase rates (kfast and kslow) and amplitudes (Afast and Aslow) of 60S joining (from d) and Δt values (from e) with the 95% confidence intervals are shown in the inset table in e. n = 232 (for addition of spermidine and eIF5B), n = 117 (for addition of spermidine without eIF5B) and n = 164 (for addition of eIF5B without spermidine). Notably, the Δt value is small in the presence of only spermidine—probably owing to the lack of the rate-limiting eIF5B dissociation step. Consistently, this Δt value falls in the same range of the average tRNA arrival times after eIF5B departure in those experiments that were performed with labelled eIF5B (Fig. 2).
Extended Data Fig. 4 Single-molecule assays demonstrate that the association of the first aa-tRNA to the A site is codon- and tRNA-specific and can lead to further elongation.
a, Experimental setup (left) and sample fluorescence trace (right) for the single-molecule assay to assess the tRNA-binding specificity of the A site. 48S PICs that contain Cy3–40S, Met-tRNAMeti and the 3′-end-biotyinlated model mRNA (encoding Met-Phe-Lys-stop) were immobilized in ZMWs in the presence of the required initiation factors. Experiments were started by illuminating ZMWs with a green laser and delivering Cy5–60S, Cy3.5–Lys-tRNALys:eEF1A:GTP ternary complex (Cy3.5–Lys-TC) and initiation factors. No Cy3.5–Lys-tRNALys-binding events were observed in the 15-min imaging window, demonstrating that the aa-tRNA association with the A site was codon- and tRNA-specific (n = 150). b–e, The elongation competence of the 80S complex is scored by smFRET between tRNAs (that is, (Cy3.5)tRNAPhe–(Cy5)tRNALys) after initiation and elongation to the second elongation codon in the model mRNA. To study the tRNA–tRNA smFRET in the context of ribosomal inter-subunit smFRET, we decided to use Cy3 and Cy5.5 to label the 40S and 60S subunits, respectively. Therefore, we engineered the yeast 60S to carry a ybbR tag at the C terminus of uL18 and labelled the 60S with Cy5.5–CoA by SFP synthase (b). c, The different tag and label did not notably affect the kinetics of the transition to elongation (red curve, Δt = 95.4 ± 2.6 s (mean ± 95% confidence interval); n = 146; model mRNA, 20 °C and 3 mM free Mg2+) compared with the original Cy5–SNAP-tagged 60S (black curve, Δt = 92.2 ± 2.5 s (mean ± 95% confidence interval); n = 164). d, Sample fluorescence trace from the experiments in which Cy5.5–60S, Cy3.5–Phe-TC, Cy5–Lys-TC, eEF2, eEF3:ATP, eIF5A and other required initiation factors were delivered to immobilized 48S PICs containing Cy3–40S, Met-tRNAMeti and the 3′-end-biotyinlated model mRNA, and illuminated with a green laser at 20 °C. Out of n = 152 molecules that showed the sequential 60S and Cy3.5–Phe-tRNA association events, n = 113 molecules showed the subsequent Cy3.5–tRNAPhe to Cy5–tRNALys FRET signal. e, The distribution of the dwell times between the appearance of the Cy3.5 and Cy5 signals was fitted to a single-exponential equation, and the average time was 142 ± 8 s (mean ± 95% confidence interval; n = 113). Thus, 74% of the first aa-tRNA associations with the A site yielded the elongation to the next codon.
Extended Data Fig. 5 Comparisons of the kinetics of the transition from initiation to elongation under various conditions.
a–c, Transition dwell times (open circles) were fitted to single-exponential distributions to estimate the average dwell times (that is, Δt values) (grey bars; error bars in black represent the 95% confidence intervals). a, b, Comparison of Δt values in experiments performed with the model mRNA at 20 °C when no extra factors were added (data taken from Fig. 1d; n = 164), or in the presence of eIF3 and eEF3 (n = 130) or with addition of eIF5A (n = 143) (a); or when the concentration of Cy3.5–Phe-TC was 50 nM (n = 221) or 100 nM (data taken from Fig. 1d; n = 164) (b). c, Similar comparison for experiments performed with the cap-RPL30 mRNA at 20 °C when the concentration of Cy3.5–Phe-TC was 100 nM (n = 118) or 200 nM (n = 132). d, The Δt values compared across all assayed mRNAs at 20 °C and 30 °C (Fig. 1a, d). In the model mRNA-Kozak_-3U, the A at the −3 position of the optimal Kozak sequence was mutated to U, which largely abolished the Kozak sequence effect. From bottom to top for each group: n = 118, n = 130, n = 130, n = 121, n = 149, n = 161, n = 189, n = 159, n = 164 and n = 136.
Extended Data Fig. 6 Neither truncation nor fluorescent labelling of eIF5B perturbs its function.
a, Fluorescent labelling of eIF5B with Cy5.5 by means of a ybbR tag at the N-terminal end, which is distal from the ribosomal subunit labels and hence not expected to interfere with the inter-subunit smFRET. The ribosome model was created in PyMOL, using data from PDB 4V8Z12. b, A N-terminally truncated version of eIF5B (eIF5B-Trunc) was used in most of our assays, as in other reported reconstituted, purified yeast translation assays8,9,11,12,13,14,15,21,25. Previous reports failed to purify the full-length protein and have demonstrated that the truncated protein supported initiation in vitro and in vivo8,9. c, We tagged eIF5B-Trunc at the N terminus with a ybbR tag and labelled the protein with a CoA–Cy5.5 dye using SFP synthase. A representative gel is shown, which was first scanned for Cy5.5 fluorescence (right) and subsequently stained with Coomassie blue (left) following SDS–PAGE analysis of ybbR–eIF5B-Trunc after labelling with and without SFP synthase. The experiment was repeated three times with similar results. d, The GTPase activity of Cy5.5–eIF5B-Trunc was not perturbed by the labelling. Multiple-turnover GTP hydrolysis was performed in 50 mM HEPES-KOH pH 7.5, 10 mM Mg(OAc)2 and 100 mM KOAc at 30 °C 30 min before quenching with malachite green assay solution. Where applicable, concentrations were: GTP, 100 µM; eIF5B-Trunc, 2.5 µM; Cy5.5–eIF5B-Trunc, 2.5 µM; 40S and 60S, 0.2 µM each. The GTP-only group was used as a negative control and the values were normalized to 0. Bars represent mean, and error bars indicate standard deviations of three biological replicates (individual data points are indicated with open circles). e, The dwell times (open circles) between 60S arrival and the arrival of Phe-TC to the A site were fitted to single-exponential distributions for experiments performed with Cy5.5–eIF5B (related to Fig. 2c) versus those with unlabelled eIF5B (related to Fig. 1d), and at 20 °C or 30 °C. Error bars (in black) represent the 95% confidence intervals of the average dwell times (Δt values). From bottom to top for each group: n = 141, n = 159, n = 164, n = 118, n = 133, n = 130, n = 131, n = 189, n = 134 and n = 164. f, Despite it being reported previously that purification of recombinant yeast full-length eIF5B (eIF5B-FL) cannot be achieved49, we were able to recombinantly express and purify it, as shown by a 12% SDS–PAGE gel analysis. The experiment was repeated three times with similar results. g, Use of full-length eIF5B in our assay did not lead to faster transition to elongation in experiments performed with the cap-RPL30 mRNA at 3 mM free Mg2+ and 20 °C. Error bars (in black) represent the 95% confidence intervals of the average dwell times (Δt values, grey bars) from fitting of the dwell times (open circles) to single-exponential distributions. n = 118 (related to Fig. 1d) (left) and n = 205 (right).
Extended Data Fig. 7 Use of a non-hydrolysable GTP analogue, GDPNP, traps eIF5B on the 80S and prevents the transition to elongation.
a, Sample trace from experiments performed with GTP (n = 134). The grey highlighted part of the trace is shown in Fig. 2a as a magnified view. b, Sample trace from experiments performed with non-hydrolysable GDPNP (n = 105), in which eIF5B was trapped on the 80S after 60S joining and no tRNA binding to the A site was observed. Black dashed boxes show Cy5.5 events that are transient eIF5B sampling events to the 48S PIC before 60S joining. c, The cumulative probability distributions of the lifetimes of Cy5.5–eIF5B on 80S in experiments performed with the model mRNA at 3 mM Mg2+ and 20 °C in the presence of GTP (n = 134; related to Fig. 2d) or GDPNP (n = 105). In the presence of GTP, the mean lifetime was 51.9 s ( ± 1.8 s, 95% confidence interval from fitting to a single-exponential equation); in the presence of GDPNP, the mean lifetime was 848 s ( ± 19 s, 95% confidence interval from fitting to a linear equation).
Extended Data Fig. 8 The effects of eIF5B mutations on the kinetics of 60S joining and the transition to elongation.
a, b, The cumulative probability distributions of the dwell times for 60S joining (a) and the transition to elongation (b) from experiments performed with unlabelled wild-type eIF5B (n = 164), the eIF5B(T439A) mutant (n = 131) or the eIF5B(H505Y) mutant (n = 119) and the model mRNA at 20 °C in the presence of 3 mM Mg2+ and 1 mM GTP. c, The estimated average fast and slow phase rates (kfast and kslow) and amplitudes (Afast and Aslow) of 60S joining (from a) and Δt values (from b) with the 95% confidence intervals from fitting the distributions to a double-exponential (wild-type eIF5B and eIF5B(H505Y)) or a single-exponential (eIF5B(T439A)) equation for the 60S joining; or to a single-exponential equation for the transition dwell times. ∗In the case of eIF5B(T439A), out of n = 131 molecules showing 60S joining signal, only n = 17 molecules showed the subsequent signal of aa-tRNA association with the A site. Thus, the eIF5B(T439A) mutant is defective in catalysing subunit joining (consistent with bulk measurement9), and inhibits tRNA association with the A site.
Extended Data Fig. 9 Free Mg2+ concentration modulates 60S joining and the transition to elongation.
a, The dwell times for the transition from initiation to elongation (open circles) were fitted to single-exponential distributions to estimate the average transition dwell times (Δt values, with 95% confidence intervals, red) from experiments performed with unlabelled eIF5B and cap-RPL30 mRNA or model mRNA-Kozak at 20 °C in the presence of 1–10 mM Mg2+ (data for 3 mM Mg2+ were taken from Fig. 1d). For cap-RPL30, unstable 80S formation was observed at 1 mM Mg2+ (f) and thus no Δt values were obtained. From bottom to top for each group: n = 108, n = 189, n = 124, n = 150, n = 144, n = 118 and n = 195. b, The dwell times for the transition from initiation to elongation (open circles) were fitted to single-exponential distributions to estimate the average transition dwell times (Δt values, with 95% confidence intervals, red) from experiments performed with unlabelled eIF5B and cap-RPL30 or model mRNA-Kozak, under the indicated conditions. From left to right for each group: n = 195, n = 152, n = 118, n = 130, n = 150, n = 132, n = 189 and n = 159. c, The dwell times (open circles) for the transition from initiation to elongation, eIF5B lifetimes on 80S, and tRNA arrival times after eIF5B departure were fitted to single-exponential distributions to estimate the average dwell times (with 95% confidence intervals, black). Experiments were performed with cap-RPL30 mRNA at 3 mM free Mg2+ and 20 °C in the presence of unlabelled eIF5B (n = 195; data taken from Fig. 1d) or Cy5.5–eIF5B (n = 164; data taken from Fig. 2c); or at 2 mM free Mg2+ and 30 °C in the presence of unlabelled eIF5B (n = 152; data taken from b) or Cy5.5–eIF5B (n = 150). d, The dwell times for 60S joining (open circles) from the same experiments as in a were fitted to single-exponential distributions (for model mRNA-Kozak) or double- exponential distributions (for cap-RPL30; the average times for the fast phase were plotted here) to estimate the average dwell time (with 95% confidence intervals, red). e, The cumulative probability distribution of the 60S joining times, showing the compromised rate of 60S joining in experiments that were performed with the model mRNA-Kozak at 1 mM Mg2+ and 20 °C as in a (n = 150). However, we still observed that the arrival of tRNA to the A site occurred readily after 80S formation. The kinetics of 60S joining under these reaction conditions were not fitted well by single- or double-exponential distributions and therefore no average time was deduced for the bar plot in d. f, Sample trace from experiments performed with cap-RPL30 mRNA at 1 mM Mg2+ and 20 °C as described in a (n = 200). g, h, Dot plots for Fig. 2c, e, showing the single-exponential distributions of the dwell times, with the average dwell times (with 95% confidence interval) in black. Notes: (1) increasing free Mg2+ concentrations increased the Δt values (that is, higher Mg2+ concentration favours the eIF5B-bound semi-rotated 80S conformation (Extended Data Fig. 10)). This seems contrary to the known effects of free Mg2+ concentrations on the conformation of the bacterial pre-translocation 70S elongation complex50: lower Mg2+ concentration favours the rotated state and higher Mg2+ concentration favours the non-rotated state. The apparent discrepancy might be a result of the different compositions of the complexes: in our case, the semi-rotated state of the 80S contains an acylated Met-tRNAMeti in the P site and a protein factor eIF5B bound in the A site; whereas in the pre-translocation 70S complex, the P-site tRNA is deacylated and a peptidyl-tRNA is bound in the A site. The differences in the energy landscape between these two types of complexes can also be explained by the observation that there are no apparent fluctuations between semi-rotated and non-rotated 80S conformations with eIF5B bound, whereas there are frequent fluctuations between rotated and non-rotated 70S conformations in the pre-translocation complexes. (2) The different magnitudes of dependence of Δt values on temperature (Fig. 1d) and Mg2+ concentration for different mRNAs suggest that there are mRNA sequence-context differences in the thermodynamics that govern the transition from initiation to elongation. Similarly, the minimal Mg2+ concentrations required for stable 80S assembly on different mRNAs are different.
Extended Data Fig. 10 Cryo-EM analyses of the on-pathway 80S complexes during initiation and the transition to elongation.
a, Cumulative probability distributions of the dwell times for 60S joining to 48S PIC (that is, the formation of the 80S complex, which is shown in black and designated as ‘total 80S’) and the subsequent association of aa-tRNA with the A site (corresponding to the Δt value, which is shown in blue and designated as ‘80S EC’) were from the single-molecule experiment with the model mRNA and wild-type eIF5B at 20 °C and 3 mM free Mg2+ (n = 164). The red curve corresponds to the evolution of the fraction of the eIF5B-bound 80S complex (‘80S IC’) over time, simulated with the kinetics of the other two curves. The dashed lines indicate that at time point 45 s, around 64.5% of the 80S complexes should be in the 80S initiation complex state under these conditions. b, The population sizes of the two classes of 80S complexes that were observed by cryo-EM (80S IC, 17,602 particles, 70%; 80S EC, 7,542 particles, 30%) were in agreement with predictions from our single-molecule kinetics at time point 45 s. c–f, The compositional and conformational differences between the 80S initiation and elongation complexes. g. Summary of cryo-EM data collection and processing for the 48S PIC (Extended Data Fig. 2c–e), 80S initiation complex (c) and 80S elongation complex (d) maps.
Supplementary information
Supplementary Information
This document contains the Supplementary Note and Supplementary Figure 1.
Rights and permissions
About this article

Cite this article
Wang, J., Johnson, A.G., Lapointe, C.P. et al. eIF5B gates the transition from translation initiation to elongation. Nature 573, 605–608 (2019). https://doi.org/10.1038/s41586-019-1561-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1561-0
- Springer Nature Limited
This article is cited by
-
The molecular basis of translation initiation and its regulation in eukaryotes
Nature Reviews Molecular Cell Biology (2024)
-
Start codon-associated ribosomal frameshifting mediates nutrient stress adaptation
Nature Structural & Molecular Biology (2023)
-
Single-molecule visualization of mRNA circularization during translation
Experimental & Molecular Medicine (2023)
-
Principles, challenges, and advances in ribosome profiling: from bulk to low-input and single-cell analysis
Advanced Biotechnology (2023)
-
eIF5B and eIF1A reorient initiator tRNA to allow ribosomal subunit joining
Nature (2022)