

Abstract
The advent of total-body positron emission tomography (PET) has vastly broadened the range of research and clinical applications of this powerful molecular imaging technology1. Such possibilities have accelerated progress in fluorine-18 (18F) radiochemistry with numerous methods available to 18F-label (hetero)arenes and alkanes2. However, access to 18F-difluoromethylated molecules in high molar activity is mostly an unsolved problem, despite the indispensability of the difluoromethyl group for pharmaceutical drug discovery3. Here we report a general solution by introducing carbene chemistry to the field of nuclear imaging with a [18F]difluorocarbene reagent capable of a myriad of 18F-difluoromethylation processes. In contrast to the tens of known difluorocarbene reagents, this 18F-reagent is carefully designed for facile accessibility, high molar activity and versatility. The issue of molar activity is solved using an assay examining the likelihood of isotopic dilution on variation of the electronics of the difluorocarbene precursor. Versatility is demonstrated with multiple [18F]difluorocarbene-based reactions including O–H, S–H and N–H insertions, and cross-couplings that harness the reactivity of ubiquitous functional groups such as (thio)phenols, N-heteroarenes and aryl boronic acids that are easy to install. The impact is illustrated with the labelling of highly complex and functionalized biologically relevant molecules and radiotracers.
Similar content being viewed by others

Main
Since the pioneering work of Curtius4 and Staudinger5, carbenes have had an important role in organic and organometallic chemistry6, and have found applications in biological7, medicinal8 and materials9 sciences. One area yet to embrace the synthetic power of carbenes in radiolabelled form is nuclear science, and more specifically, positron emission tomography (PET) (Fig. 1a). This molecular imaging technology, routinely used for clinical diagnosis and drug development, requires radiotracers for in vivo tracking of complex biological processes10. Radiolabelling is possible with a range of positron-emitting radionuclides, including fluorine-18 (18F), which displays outstanding properties (97% β+ decay, 109.8-min half-life and 635-KeV positron energy)11. A vital criterion for broad utility is high molar activity (Am), which is best achieved using cyclotron-produced [18F]fluoride12. Despite recent advances in 18F-radiochemistry, access to 18F-labelled polyfluoroalkylated molecules is mostly an unsolved problem, a considerable drawback considering the omnipresence of these motifs in drug discovery3,13. A substantial obstruction to progress in 18F-polyfluoroalkylation is low Am caused by 19F-fluoride leaching from the fluorinated precursors used in 18F-labelling. The most fruitful efforts have focused on 18F-trifluoromethylation with metal-mediated and radical strategies using [18F]fluoride14,15,16. Strikingly, molecules featuring a difluorinated motif found to be vital for drug efficacy are either not within reach as 18F-isotopologues, only accessible in prohibitively low Am or from precursors that require lengthy syntheses17,18,19,20,21,22. In a unique approach, a [18F]CF2H radical precursor was generated from [18F]fluoride, but the more nucleophilic character of •CF2H with respect to •CF3 limits its utility to a prohibitively narrow pool of heteroarenes23. With this current state of play, the distinct advantages of CF2H routinely embraced in drug discovery programmes3, including reduced lipophilicity compared with CF3 and hydrogen-bond donor ability, are not within reach for PET ligand discovery. Difluorocarbene chemistry can offer a general solution3,24. Tens of reagents have been invented that can be activated under various conditions enabling the released difluorocarbene (DFC) to participate in insertions25,26, cycloadditions24,27,28 and cross-coupling reactions29,30,31,32 leading to complex molecules substituted with (X)CF2H/R (X = Csp3, Csp2, O, N, S). In stark contrast, studies on [18F]difluorocarbene ([18F]DFC) are rare. In 1970, energetic 18F atoms from nuclear recoil were found to react with CF2H2 or CF4 to release [18F]DFC on substitution followed by elimination33. This study on the mechanism of atomic exchange reactions is neither practical nor suitable for PET ligand discovery. Here we describe the merits of a bespoke [18F]DFC reagent prepared in high Am. Its broad reactivity profile enabling O–H, S–H and N–H insertions, and cross-coupling reactions offers exciting opportunities in 18F-radiolabelling for PET ligand discovery and more generally for nuclear medicine.

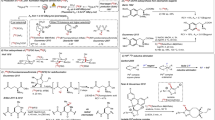
a, Development of the [18F]difluorocarbene reagent [18F]1 offering opportunities in PET radiotracer discovery. b, Initial studies. Two-step one-pot radiosynthesis of [18F]1b and subsequent 18F-difluoromethylation of [1,1′-biphenyl]-4-ol. c, Stepwise mechanism for difluorocarbene release from 1a. Carbene and nucleophilic reactivity of [18F]1b. dXY, interatomic distance between atoms X and Y; TS, transition state; Grel, relative Gibbs free energy; ΔGTS3ǂ, Gibbs free energy of activation for the DFC transfer from 1a to phenolate. Radiochemical yield (RCY) determined by radioHPLC. a1,1-Diphenyl-ethylene (0.10 mmol), NaOH (0.05 mmol), propylene carbonate (0.3 ml), 200 °C. bAcetophenone (0.04 mmol), lithium bis(trimethylsilyl)amide (0.10 mmol), tetrahydrofuran/1,3-dimethylimidazolidin-2-one (9:1), −78 °C, 20 min. cMg (0.80 mmol), N,N-dimethylformamide/acetic acid (9:1), room temperature, 20 min. d, 19F NMR fluoride leaching assay. Ratio [6]/[2] measured at 1 h (Supplementary Fig. 14).
Results and discussion
Initial experiments indicated that a strategy based on 19F/18F exchange of in situ-generated DFC had a poor prognosis, encouraging the implementation of an approach based on an 18F-labelled reagent (Supplementary Fig. 26)34. We ruled out volatile candidates (for example, HCF3 and ClCF2H) or salts (for example, ClF2CO2Na and Ph3P+CF2CO2−; where Ph is phenyl) that would be difficult to handle, purify or characterize, as well as reagents that preferentially require fluoride for DFC release to avoid isotopic dilution (for example, TMSCF2Br; where TMS is trimethylsilyl). Considering the demand for methods enabling direct 18F-difluoromethylation of oxygen, sulfur and nitrogen nucleophiles, we opted for a reagent releasing [18F]DFC under basic conditions to allow simultaneous activation of the heteroatom nucleophile for insertion reactions. Although ((difluoromethyl)sulfonyl)benzene (1a) is not used as a DFC precursor, it stood out as an attractive candidate for labelling. In 1960, Hine and Porter reported that 1a reacts with sodium methoxide in methanol to provide difluoromethyl ether at a rate faster than expected for an SN2 reaction, an observation suggesting a mechanism involving DFC formation35; no further studies ensued probably owing to the invention of more efficient DFC reagents than 1a. From a radiochemistry perspective, however, radiochemical yield does not necessarily correlate with chemical yield, and in our judgement, 1a could offer clear advantages over newer reagents (Supplementary Fig. 27, Supplementary Table 7). Specifically, [18F]1a could be within reach using cyclotron-produced [18F]fluoride by applying a protocol that minimizes radiosynthesis time and therefore decay, and critically offers opportunities to solve the Am problem by tuning the electronic properties of the aryl group. In initial experiments, we opted to label 1-(tert-butyl)-4-((difluoromethyl)sulfonyl)benzene (1b) considering volatility and precursor stability. Nucleophilic substitution of (bromofluoromethyl)-(4-(tert-butyl)phenyl)sulfane (2b) with [18F]fluoride followed by RuCl3/NaIO4 oxidation afforded [18F]1b isolated in 10% ± 2% (n = 5) non-decay corrected activity yield (AY) (Fig. 1b). Gratifyingly, the reaction of [18F]1b, a model phenol and KOH gave [18F]3 in 72% radiochemical yield (RCY), suggesting that [18F]DFC release and O–H insertion took place under these conditions. The radiosynthesis was automated on the cassette-based Trasis AllinOne platform, and [18F]1b was obtained in an overall synthesis time of 72 min in a non-decay corrected AY of up to 6% and Am of 10 ± 0 GBq µmol−1 (n = 2, decay corrected to the end of synthesis) from 148 GBq of starting activity (Supplementary Fig. 33). At this stage, further studies focused on the mechanism and improving the Am.
Quantum chemical studies, performed at the B2PLYP-D3/(ma)-def2-TZVPP//B3LYP-D3/def2-TZVPD level of theory with SMD (solvation model based on density) acetonitrile, gave insight into the mechanism of DFC release under basic conditions, its reactivity with a phenol and the effect of phenyl substitution of [18F]1 (Fig. 1c, Supplementary Table 8). DFC formation from 1a occurs by stepwise α-elimination. Initial C–H deprotonation by hydroxide is facile and exergonic by 24.9 kJ mol−1, whereas the subsequent C–S cleavage, which liberates free DFC, has a barrier of 76.3 kJ mol−1 and is endergonic by 44.5 kJ mol−1. The overall barrier for phenol difluoromethylation, in which C–O formation between free DFC and the phenolate anion is rate limiting, is 78.6 kJ mol−1. Although recombination of DFC and the phenyl sulfinate anion is kinetically competitive, this will occur reversibly, whereas C–O formation is highly exergonic by 81.7 kJ mol−1 and therefore irreversible. The possibility of a concerted SN2-like carbene transfer was considered; however, with an activation barrier of 112.2 kJ mol−1 (Supplementary Fig. 67), this can be rejected in favour of the stepwise process illustrated (Fig. 1c). A series of synthetically accessible difluoromethyl para-substituted aryl sulfones were examined computationally. In each case, the C–O formation step with phenolate defines the overall activation barrier, and is consistent with experimental studies (Supplementary Table 8); more electron-deficient substituents were computationally predicted to show greater reactivity. Each reagent considered showed both kinetic feasibility and thermodynamic irreversibility of C–O formation, suggesting flexibility with regard to the choice of the para-substituent for the optimization of Am. Experimentally, [18F]1b with 1,1-diphenylethylene and NaOH underwent [2 + 1] cycloaddition to afford the gem‐difluorinated cyclopropane product [18F]4 in 36% RCY, demonstrating that [18F]1b releases [18F]DFC under these conditions. We also prepared difluoromethylated alcohol [18F]5 by reacting acetophenone with [18F]1b under base activation followed by reductive cleavage of the sulfone group. These results demonstrated the divergent reactivity profile of [18F]1 that can serve either as a [18F]DFC precursor or as a surrogate of the [18F]CF2H anion (Fig. 1c).
Under labelling conditions, a plausible scenario to account for the low Am of [18F]1b is nucleophilic substitution of 2b with hydroxide, affording an α-fluorohemiacetal prone to 19F-fluoride elimination, which then competes for reaction with 2b (Supplementary Table 13). Computational studies predict that SN2 displacement is more facile with a hydroxide nucleophile than with fluoride, with standard activation barriers (that is, neglecting differences in concentration) of 62.5 kJ mol−1 and 92.8 kJ mol−1, respectively (Supplementary Fig. 69).
Experimentally, 19F/18F isotope exchange of sulfone 1b was not observed under the conditions applied for 18F-incorporation, but importantly, the reaction of 2b with K222/K2CO3 in the absence of external fluoride source gave (4-(tert-butyl)phenyl)(difluoromethyl)sulfane 6b, as evidenced by 19F NMR spectroscopy (Supplementary Fig. 14, Supplementary Table 1). As varying aryl substitution may influence the degree of fluoride leaching and offer a pathway to improve Am, five DFC precursors featuring H (2a), p-tBu (2b), p-NO2 (2c) p-Cl (2d) and p-OMe (2e) substitution were reacted with K2.2.2./K2CO3 in the absence of external fluoride. These all afforded the corresponding (aryl)(difluoromethyl)sulfanes (6), albeit in varying amounts (Fig. 1d). The results indicated that 6 was formed in increased amounts in the following order: Cl ≈ NO2 < tBu < H < OMe, highlighting that chloro or nitro para-substitution may increase Am by reducing 19F-fluoride leaching. Experiments performed in the presence of [18F]fluoride (135–148 GBq of starting activity) informed that [18F]1d was obtained in the highest Am (131 ± 29 GBq µmol−1, n = 2, decay corrected to the end of synthesis), a result corroborating our 19F NMR assay.
With [18F]1b and [18F]1d in hand, our priority objective was to develop a versatile route to 18F-labelled ArOCF2H, a motif increasingly encountered in medicinal chemistry3,36. At present, 18F-labelling requires over-engineered starting materials (ArOCHFCl or ArOCHFCO2H; where Ar is aryl) that are not readily accessible and afford [18F]ArOCF2H in prohibitively low Am (<1 GBq µmol−1)21,37. 18F-Difluoromethylation of alcohol precursors with [18F]DFC represents a direct approach to [18F]ArOCF2H, and complements 11C-methylation protocols routinely applied in radioligand design38,39. Building on our initial experiments with [1,1′-biphenyl]-4-ol and [18F]1b (Fig. 1b), we applied optimized reaction conditions consisting of reacting 0.20 mmol of [1,1′-biphenyl]-4-ol with [18F]1b or [18F]1d for 20 min at 80 °C under aqueous KOH conditions (acetonitrile (MeCN)/H2O = 6/1, v/v, 0.7 ml) to a range of phenols. A variety of functionalized electronically and sterically differentiated (hetero)aryl phenols found in pharmaceutical agents gave the desired labelled products. Specifically, the method tolerates alkoxy, ketone, alkyl, ester, halide, cyano, nitro, sulfonamide, aldehyde and basic amine functionalities ([18F]3, [18F]6–[18F]23 (27–88% RCY)) (Fig. 2). It is noteworthy that [18F]DFC inserted site-selectively into the phenolic O–H of 4-hydroxybenzaldehyde ([18F]15, 51%). A substrate with an ester prone to basic hydrolysis was labelled applying modified reaction conditions (NaH, N,N-dimethylformamide (DMF) at 140 °C) ([18F]23, 27% RCY). Even a challenging, less acidic benzylic alcohol (4-methoxyphenyl)methanol underwent [18F]DFC insertion, albeit with a lower RCY of 14% ([18F]24). Next, we investigated [18F]DFC insertions into S–H and N–H bonds. These 18F-labelling reactions lead to motifs that at present either require multi-step precursor synthesis, limiting the scope ([18F]SCF2H)22, or are not available in their 18F-labelled form ([18F]NCF2H). Electronically neutral, deficient and rich (hetero)aromatic thiophenols readily underwent [18F]DFC insertion, providing access to all [18F]SCF2H products ([18F]25–29, 70%–90% RCY). Benzimidazole was successfully subjected to N–H insertion under either aqueous (KOH; MeCN/H2O = 6/1), ([18F]30, 81% RCY) or anhydrous reaction conditions (NaH, DMF, 61% RCY). (1H-benzo[d]imidazol-2-yl)methanol underwent selective 18F-difluoromethylation at nitrogen affording [18F]31 (52% RCY). 1H-Indazole gave [18F]32 in good RCY (73%) (1:1 regioisomers). Similarly, 1H-benzo[d][1,2,3]triazole was a competent substrate ([18F]33, 71% RCY). Functional-group tolerance investigated with a robustness screening is broad (Supplementary Figs. 15–17). With this information, we considered the late-stage 18F-difluoromethylation of complex biologically active molecules (Fig. 3). The 18F isotopologue of the anti-inflammatory drug roflumilast [18F]35, along with eight [18F]OCF2H and [18F]NCF2H drug analogues (oestrone [18F]34, 2′-hydroxy-3-phenylpropiophenone [18F]36, chloroxine [18F]37, PF-03774076 [18F]38, triclabendazole [18F]39 and thiabendazole [18F]40) were successfully labelled with RCYs reaching 82%. Moreover, the [18F]OCF2H analogues of the PET radioligands P2X7R, PMB-protected PF-06809247, DASA-23, MPC-6827, DPA-714 and iloperidone were obtained in up to 98% RCY ([18F]41–[18F]46). As the proton source leading to OCF2H originates from the solvent (Supplementary Figs. 3, 4), [2H18F]OCF2H radiotracers ([2H18F]41, 84% RCY, 94% D; [2H18F]46, 74% RCY, 96% D; [2H18F]34, 64% RCY, 96% D) were all within reach. This is important because deuterium (D) incorporation is a common strategy to increase the metabolic stability of drugs and radiotracers40. For drug analogues (fenofibrate, 3-fluoro-5-[(2-methyl-1,3-thiazol-4-yl)ethynyl]benzonitrile (FMTEB), N-tert-butyloxycarbonyl(Boc)-fluoxetine, phenylalanine, flutamide and etoricoxib) that do not feature a phenol functionality, we developed a one-pot sequence from aryl boronic acids (Fig. 3b). Oxidation with urea hydrogen peroxide (0.20 mmol, 1.0 equiv.) in MeCN for 5 min followed by [18F]DFC O–H insertion led to [18F]47, [18F]48, [18F]49, [18F]50, [18F]51 and [18F]52 in good RCYs (22–86%). For fenofibrate featuring a C(sp2)–Cl bond, we developed a one-pot sequence consisting of borylation–oxidation–18F-difluoromethylation (Supplementary Fig. 45). With the knowledge that our 18F-difluoromethylation protocol is not detrimentally impacted by impurities, as evidenced by one-pot tandem procedures, a telescoped radiosynthesis of [18F]46 directly from [18F]fluoride was implemented to reduce the time required for labelling and purification. A one-pot 18F-fluorination–oxidation sequence to generate [18F]1b, followed by C18 filtration, 18F-difluoromethylation, semi-preparative high-performance liquid chromatography (HPLC) purification and reformulation afforded [18F]46 in 2% AY and >99% radiochemical purity (135 min total synthesis time) (Supplementary Fig. 50). This result is comparable to AY obtained for clinically relevant [18F]UCB-J prepared directly from [18F]fluoride41. An in vivo study using naive C57BL/6J mice was undertaken with FCH2CH2O-substituted [18F]DPA-714 and its OCF2H analogue [18F]4642,43. The study started with the radiosynthesis of [18F]46 (680 MBq, Am = 40 GBq μmol−1) using [18F]1d and the Trasis AllinOne platform. Extracts of mouse plasma and brain homogenates 5-min post-injection of [18F]46 were absent of metabolites based on radio-HPLC; this is in contrast to [18F]DPA-714 where radiolabelled metabolites were observed (Supplementary Figs. 62–65). These initial data corroborate expectations on the metabolic stability of OCF2H (ref. 44). To further demonstrate the utility of this [18F]DFC method in supporting (pre)clinical PET studies, [18F]46 was successfully used to image microglial activation in the striatum of a quinolinic acid lesion model of Huntington’s disease (Supplementary Fig. 66)45.

aReaction at 100 °C. bReaction in DMF (0.3 ml) at 140 °C with NaH (0.10 mmol) instead of KOH. cReaction in DMF (0.3 ml) at 60 °C with NaH (0.1 mmol) instead of KOH.

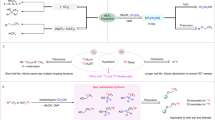
a, Scope of biologically relevant molecules and radioligands from phenols and N-heterocycles. Inset: 18F-labelling of an analogue of DPA-714 ([18F]46)(top left); coronal brain PET image of C57BL/6J mouse injected with [18F]46 to image microglial activation (top right); Trasis AllinOne automated radiosynthesis platform (bottom). b, Scope of [18F]OCF2H from boronic acids and aryl chlorides (Supplementary Figs. 44, 45). RCY determined by radioHPLC. Standard conditions: 0.2 mmol substrate, MeCN (0.6 ml), KOH(aq.) (0.1 ml, 25 w/w%). aReaction at 80 °C. bMeCN-d3, D2O, 100 °C (Supplementary Fig. 43). cReaction at 100 °C. dReaction in DMF (0.3 ml) at 140 °C with NaH (0.10 mmol) instead of KOH. eReaction on 0.10 mmol scale.
Next, we investigated the use of transition-metal complexes for the capture and transfer of [18F]DFC derived from [18F]1b and [18F]1d for site-selective aromatic 18F-difluoromethylation, an additional unsolved problem in 18F-radiochemistry46. Metal-DFC complexes known to convert aryl boronic acids into difluoromethylated arenes served as a starting point for investigations29,30,31,32. 1a is amenable to copper-mediated cross-coupling with aryl boronic acids leading to (phenylsulfonyl)difluoromethyl-substituted arenes, so the challenge was to favour the formation of 18F-difluoromethylated arene products from [18F]147. After extensive optimization studies on 4-biphenyl boronic acid (0.20 mmol) (Supplementary Fig. 46, Supplementary Table 6), [18F]4-(difluoromethyl)-1,1′-biphenyl ([18F]53) was formed in 45% RCY under basic conditions (40 µl KOH(aq.)) in the presence of Pd2dba3 (2.50 µmol, where dba is dibenzylideneacetone), Xantphos (7.50 µmol) and hydroquinone (50 µmol) in 1,4-dioxane (1.0 ml) at 130 °C. In the absence of Pd or hydroquinone, [18F]53 was not formed, or in substantially decreased RCY (Supplementary Table 6). In contrast to palladium cross-coupling reactions that must be performed under anaerobic conditions, our 18F protocol does not require the exclusion of moisture or air29,30,31,32. These optimized conditions were applied to various aryl boronic acids (Fig. 4), including those featuring heterocycles commonly found in drug-discovery pipelines, yielding [18F]56, [18F]58, [18F]60 and [18F]61 in up to 45% RCY. (6-Methoxypyridin-3-yl)boronic acid underwent site-selective 18F-difluoromethylation at the 3-position in 27% RCY ([18F]56), thereby complementing radical processes favouring the 2- and 4-positions23. Substrates presenting functional groups such as alkenes ((4-vinylphenyl)boronic acid) and alkynes ((3-cyano-5-((2-methylthiazol-4-yl)ethynyl)phenyl)boronic acid) yielded [18F]62 and [18F]64 in 41% and 24% RCY, respectively. Aryl boronic acids can therefore be used either for 18F-difluoromethylation or 18F-difluoromethoxylation. Fenofibrate ([18F]63 and [18F]47), FMTEB ([18F]64 and [18F]48), N-Boc-fluoxetine ([18F]65 and [18F]49) and protected phenylalanine ([18F]66 and [18F]50) were subjected to cross-coupling or tandem oxidation 18F-difluoromethylation conditions to afford either [18F]CF2H or [18F]OCF2H analogues from a common precursor (RCY = 20–86%).

[18F]DFC cross-coupling from (hetero)aryl boronic acids. RCY determined by radioHPLC. Reactions performed with 1,4-dioxane (0.3–1.0 ml) at 115–135 °C. a1,4-dioxane-d8 (1.0 ml), D2O, hydroquinone-d2, 120 °C (Supplementary Figs. 47, 56).
Using hydroquinone-d2 and 1,4-dioxane-d8, the [2H18F]CF2H analogue of fenofibrate [2H18F]63 was obtained in 15% RCY (90% D). Inductively coupled plasma mass spectrometry analysis of [18F]53 indicated a ruthenium and palladium content of 242.18 µg l−1 and 18.53 µg l respectively, which is below the threshold recommended by ICH Q3D(R1) guidelines for in-human injection48.
Conclusion
PET ligand discovery is at the forefront of molecular imaging and, similarly to pharmaceutical discovery, can immediately benefit from a diversity-oriented approach to radiolabelling. Although most practicing radiochemists are familiar with 18F-incorporation leading to aryl and alkyl fluorides, the ambition to consider 18F-polyfluoroalkyl substitution as a means to invent PET ligands has been tempered by the likelihood of low Am, and the discouraging requirement to synthesize over-engineered precursors only accessible after time-consuming multi-step syntheses. We now present [18F]difluorocarbene radiochemistry as a demonstration that the unique properties of the difluoromethyl group can now be exploited in PET ligand discovery programmes without compromising on Am and precursor availability. The simplicity of the protocol and the diverse range of molecules labelled in this study should encourage rapid adoption in PET centres that have access to cyclotron-produced [18F]fluoride, and more generally spark programmes to advance nuclear medicine imaging.
Data availability
Materials and methods, optimization studies, experimental procedures, mechanistic studies, 1H NMR, 13C NMR and 19F NMR spectra, and high-resolution mass spectrometry, infrared and HPLC data are available in the Supplementary Information.
References
Reardon, S. Whole-body PET scanner produces 3D images in seconds. Nature 570, 285–287 (2019).
Ajenjo, J., Destro, G., Cornelissen, B. & Gouverneur, V. Closing the gap between 19F and 18F chemistry. EJNMMI Radiopharm. Chem. 6, 33 (2021).
Sap, J. B. et al. Late-stage difluoromethylation: concepts, developments and perspective. Chem. Soc. Rev. 50, 8214–8247 (2021).
Buchner, E. & Curtius, T. Ueber die Einwirkung von Diazoessigäther auf aromatische Kohlenwasserstoffe. Ber. Dtsch. Chem. Ges. 18, 2377–2379 (1885).
Staudinger, H. & Kupfer, O. Über reaktionen des methylens. III. Diazomethan. Ber. Dtsch. Chem. Ges. 45, 501–509 (1912).
Hopkinson, M. N., Richter, C., Schedler, M. & Glorius, F. An overview of N-heterocyclic carbenes. Nature 510, 485–496 (2014).
Geri, J. B. et al. Microenvironment mapping via Dexter energy transfer on immune cells. Science 367, 1091–1097 (2020).
Marinelli, M., Santini, C. & Pellei, M. Recent advances in medicinal applications of coinage-metal (Cu and Ag) N-heterocyclic carbene complexes. Curr. Top. Med. Chem. 16, 2995–3017 (2016).
Smith, C. A. et al. N-heterocyclic carbenes in materials chemistry. Chem. Rev. 119, 4986–5056 (2019).
Campbell, M. G. et al. Bridging the gaps in 18F PET tracer development. Nat. Chem. 9, 1–3 (2017).
Deng, X. et al. Chemistry for positron emission tomography: recent advances in 11C‐, 18F‐, 13N‐, and 15O‐labeling reactions. Angew. Chem. Int. Ed. 58, 2580–2605 (2019).
McCluskey, S. P., Plisson, C., Rabiner, E. A. & Howes, O. Advances in CNS PET: the state-of-the-art for new imaging targets for pathophysiology and drug development. Eur. J. Nucl. Med. Mol. Imaging 47, 451–489 (2020).
Prchalová, E., Štěpánek, O., Smrček, S. & Kotora, M. Medicinal applications of perfluoroalkylated chain-containing compounds. Future Med. Chem. 6, 1201–1229 (2014).
Huiban, M. et al. A broadly applicable [18F]trifluoromethylation of aryl and heteroaryl iodides for PET imaging. Nat. Chem. 5, 941–944 (2013).
Levin, M. D. et al. A catalytic fluoride-rebound mechanism for C(sp3)–CF3 bond formation. Science 356, 1272–1276 (2017).
van der Born, D. et al. A universal procedure for the [18F]trifluoromethylation of aryl iodides and aryl boronic acids with highly improved specific activity. Angew. Chem. Int. Ed. 53, 11046–11050 (2014).
Mizuta, S. et al. Catalytic decarboxylative fluorination for the synthesis of tri-and difluoromethyl arenes. Org. Lett. 15, 2648–2651 (2013).
Verhoog, S. et al. Silver-mediated 18F-labeling of aryl–CF3 and aryl–CHF2 with 18F-fluoride. Synlett 27, 25–28 (2016).
Shi, H. et al. Synthesis of 18F‐difluoromethylarenes from aryl (pseudo) halides. Angew. Chem. Int. Ed. 55, 10786–10790 (2016).
Yuan, G. et al. Metal-free 18F-labeling of aryl–CF2H via nucleophilic radiofluorination and oxidative C–H activation. Chem. Commun. 53, 126–129 (2017).
Sap, J. B. et al. Synthesis of 18F-difluoromethylarenes using aryl boronic acids, ethyl bromofluoroacetate and [18F]fluoride. Chem. Sci. 10, 3237–3241 (2019).
Zhao, Q. et al. Radiosynthesis of [18F]arylSCF2H using aryl boronic acids, S-(chlorofluoromethyl) benzenesulfonothioate and [18F]fluoride. CCS Chem. 3, 1921–1928 (2021).
Trump, L. et al. Late‐stage 18F‐difluoromethyl labeling of N‐heteroaromatics with high molar activity for PET imaging. Angew. Chem. Int. Ed. 58, 13149–13154 (2019).
Ni, C. & Hu, J. Recent advances in the synthetic application of difluorocarbene. Synthesis 46, 842–863 (2014).
Fier, P. S. & Hartwig, J. F. Synthesis of difluoromethyl ethers with difluoromethyltriflate. Angew. Chem. Int. Ed. 52, 2092–2095 (2013).
Xie, Q. et al. Efficient difluoromethylation of alcohols using TMSCF2Br as a unique and practical difluorocarbene reagent under mild conditions. Angew. Chem. Int. Ed. 56, 3206–3210 (2017).
Birchall, J. M., Haszeldine, R. N. & Roberts, D. W. Cyclopropane chemistry. Part II. Cyclopropanes as sources of difluorocarbene. J. Chem. Soc. Perkin Trans. I 1071–1078 (1973).
Jia, Y., Yuan, Y., Huang, J., Jiang, Z. & Yang, Z. Synthesis of difluorinated heterocyclics through metal-free [8+1] and [4+1] cycloaddition of difluorocarbene. Org. Lett. 23, 2670–2675 (2021).
Feng, Z., Min, Q. & Zhang, X. Access to difluoromethylated arenes by Pd-catalyzed reaction of arylboronic acids with bromodifluoroacetate. Org. Lett. 18, 44–47 (2016).
Deng, X., Lin, J. & Xiao, J. Pd-catalyzed transfer of difluorocarbene. Org. Lett. 18, 4384–4387 (2016).
Feng, Z., Min, Q., Fu, X., An, L. & Zhang, X. Chlorodifluoromethane-triggered formation of difluoromethylated arenes catalysed by palladium. Nat. Chem. 9, 918–923 (2017).
Fu, X. et al. Controllable catalytic difluorocarbene transfer enables access to diversified fluoroalkylated arenes. Nat. Chem. 11, 948–956 (2019).
Smail, T. & Rowland, F. S. Insertion reactions of mono-and difluorocarbene with hydrogen halides. J. Phys. Chem. 74, 1866–1871 (1970).
Prakash, G. S. et al. Long‐lived trifluoromethanide anion: a key intermediate in nucleophilic trifluoromethylations. Angew. Chem. Int. Ed. 53, 11575–11578 (2014).
Hine, J. & Porter, J. J. The formation of difluoromethylene from difluoromethyl phenyl sulfone and sodium methoxide. J. Am. Chem. Soc. 82, 6178–6181 (1960).
Xing, L. et al. Fluorine in drug design: a case study with fluoroanisoles. ChemMedChem 10, 715–726 (2015).
Khotavivattana, T. et al. 18F‐labeling of aryl–SCF3, –OCF3 and –OCHF2 with [18F]fluoride. Angew. Chem. Int. Ed. 54, 9991–9995 (2015).
Dahl, K., Halldin, C. & Schou, M. New methodologies for the preparation of carbon-11 labeled radiopharmaceuticals. Clin. Transl. Imaging 5, 275–289 (2017).
Pipal, R. W. et al. Metallaphotoredox aryl and alkyl radiomethylation for PET ligand discovery. Nature 589, 542–547 (2021).
Mullard, A. Deuterated drugs draw heavier backing. Nat. Rev. Drug Discov. 15, 219–222 (2016).
Cai, Z. et al. Synthesis and in vivo evaluation of [18F]UCB-J for PET imaging of synaptic vesicle glycoprotein 2A (SV2A). Eur. J. Nucl. Med. Mol. Imaging 46, 1952–1965 (2019).
Zheng, J., Winkeler, A., Peyronneau, M.-A., Dollé, F. & Boisgard, R. Evaluation of PET imaging performance of the TSPO radioligand [18F]DPA-714 in mouse and rat models of cancer and inflammation. Mol. Imaging Biol. 18, 127–134 (2016).
Keller, T. et al. Radiosynthesis and preclinical evaluation of [18F]F-DPA, a novel pyrazolo[1,5a]pyrimidine acetamide TSPO radioligand, in healthy Sprague Dawley rats. Mol. Imaging Biol. 19, 736–745 (2017).
Kuchar, M. & Mamat, C. Methods to increase the metabolic stability of 18F-radiotracers. Molecules 20, 16186–16220 (2015).
Lelos, M. J. & Dunnett, S. B. Generating Excitotoxic Lesion Models of Huntington’s Disease 209–220 (Springer, 2018).
Zhou, W. et al. Transition-metal difluorocarbene complexes. Chem. Commun. 57, 9316–9329 (2021).
Li, X. et al. Copper-mediated aerobic (phenylsulfonyl)difluoromethylation of arylboronic acids with difluoromethyl phenyl sulfone. Chem. Commun. 52, 3657–3660 (2016).
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Quality guidelines. ICH https://www.ich.org/page/quality-guidelines (2019).
Acknowledgements
This research has received funding from the Engineering and Physical Sciences Research Council (EP/V013041/1, J.B.I.S.), Pfizer, Janssen, UCB, the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement number 721902 (C.F.M., S.M.H. and T.A.M.). J.F. is grateful to the Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship, generously supported by GlaxoSmithKline, MSD, Syngenta and Vertex. J.B.I.S. acknowledges financial support from an EPSRC Doctoral Prize (EP/T517811/1). R.S.P. acknowledges the RMACC Summit supercomputer at the University of Colorado Boulder and Colorado State University, the Extreme Science and Engineering Discovery Environment (XSEDE) through allocation TG-CHE180056, and support from the National Science Foundation (NSF CHE-1955876). We thank B. G. Davis, S. Verhoog and T. C. Wilson for comments, and T. Khotavivattana for preliminary experiments.
Author information
Authors and Affiliations
Contributions
J.B.I.S. and C.F.M. labelled the tBu-substituted difluorocarbene reagent and performed all insertions and cycloadditions with this reagent. J.B.I.S., C.F.M. and J.F. prepared the substrates and performed all automation experiments. J.B.I.S. and J.F. performed the cross-coupling reactions, one-pot procedures, the synthesis of the radiotracer for the imaging study, the radiosynthesis of all difluorocarbene reagents, and the experiments with the Cl-substituted difluorocarbene reagent, and developed the NMR assay to probe isotopic dilution. J.B.I.S. and N.J.W.S. performed preliminary studies for the radiosynthesis of the tBu-substituted difluorocarbene reagent. A.B.D. and R.S.P. performed and analysed the computational studies. T.A.M. and C.F.M. did an initial metabolic stability study. J.B.I.S., J.F., M.J.L. and S.J.P. performed all the in vivo experiments. S.M.H. prepared selected substrates. J.B.I.S., J.F. and V.G. conducted the revisions. J.B.I.S., R.S.P. and V.G. wrote the manuscript. All authors read and commented on the paper. V.G. conceived and supervised the project.
Corresponding author
Ethics declarations
Competing interests
C.G. is an employee of UCB Pharma. C.W.a.E. is an employee of Pfizer Inc. A patent application (no. GB2113561.1; Difluorocarbene radiosynthesis) has been filed, from which V.G., J.B.I.S., C.F.M., M.T., N.J.W.S., S.M.H. and A.A.T. may benefit from royalties. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks John Groves and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
This file contains: Materials and methods, Supplementary Text, Figs. 1–69, Tables 1–14, NMR spectra and References.
Rights and permissions
About this article

Cite this article
Sap, J.B.I., Meyer, C.F., Ford, J. et al. [18F]Difluorocarbene for positron emission tomography. Nature 606, 102–108 (2022). https://doi.org/10.1038/s41586-022-04669-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04669-2
- Springer Nature Limited
This article is cited by
-
Highlight selection of radiochemistry and radiopharmacy developments by editorial board
EJNMMI Radiopharmacy and Chemistry (2023)
-
Copper-catalysed difluorocarbene transfer enables modular synthesis
Nature Chemistry (2023)
-
Highlight selection of radiochemistry and radiopharmacy developments by editorial board
EJNMMI Radiopharmacy and Chemistry (2022)