

Abstract
MFSD2A is a sodium-dependent lysophosphatidylcholine symporter that is responsible for the uptake of docosahexaenoic acid into the brain1,2, which is crucial for the development and performance of the brain3. Mutations that affect MFSD2A cause microcephaly syndromes4,5. The ability of MFSD2A to transport lipid is also a key mechanism that underlies its function as an inhibitor of transcytosis to regulate the blood–brain barrier6,7. Thus, MFSD2A represents an attractive target for modulating the permeability of the blood–brain barrier for drug delivery. Here we report the cryo-electron microscopy structure of mouse MFSD2A. Our structure defines the architecture of this important transporter, reveals its unique extracellular domain and uncovers its substrate-binding cavity. The structure—together with our functional studies and molecular dynamics simulations—identifies a conserved sodium-binding site, reveals a potential lipid entry pathway and helps to rationalize MFSD2A mutations that underlie microcephaly syndromes. These results shed light on the critical lipid transport function of MFSD2A and provide a framework to aid in the design of specific modulators for therapeutic purposes.
Similar content being viewed by others
Main
The blood–brain barrier is indispensable for normal brain function8. Historically, the restricted permeability of the brain vasculature has been attributed to specialized tight junctions between endothelial cells that prohibit the passage of water-soluble molecules9,10. Recent evidence shows that endothelial cells of the central nervous system also actively inhibit transcytosis to ensure the integrity of the blood–brain barrier, and full barrier integrity requires restriction of both paracellular and transcellular leakage. MFSD2A is a blood–brain-barrier-specific lipid transporter and a key inhibitor of transcytosis6,7,11. Mice that lack the lipid transport function of MFSD2A have blood–brain barrier leakage owing to upregulated transcytosis, without apparent tight junction disruption6,7,11. Mechanistically, MFSD2A-translocated phospholipids inhibit the formation of caveolae, suppressing transcytosis7,12. Thus, MFSD2A is a promising target for manipulating the permeability of the blood–brain barrier to facilitate drug delivery into the central nervous system7.
As a lipid transporter1,2, MFSD2A is unique among mammalian members of the major facilitator superfamily (MFS), which typically transport soluble substrates13. Moreover, MFSD2A shares limited sequence similarity with MFS transporters with known structures. The lack of a reliable structural model impedes a mechanistic understanding of MFSD2A. For example, it is unclear how MFSD2A mediates lipid transport and how the transport is coupled to sodium.
Here we report the structure of mouse MFSD2A by single-particle cryo-electron microscopy (cryo-EM). The structure, together with functional characterizations and molecular dynamics simulations, reveals the architecture of the transporter and provides a blueprint to understand lipid translocation and sodium-dependent transport.
Structural determination
To elucidate the structure and transport mechanism of MFSD2A, we used the extensively characterized mouse orthologue6,7,12, which has optimal biochemical properties and shares 84% sequence identity and 90% similarity with human MFSD2A (Extended Data Fig. 1). HEK293S cells that overexpress wild-type mouse MFSD2A showed robust uptake of TopFluor lysophosphatidylcholine (LPC) (a known substrate of MFSD2A1,2) as compared to control cells (Fig. 1a). Substituting D96, a residue that is critical for transport1, substantially reduced uptake. These results confirm that mouse MFSD2A is an LPC transporter with properties comparable to those of human MFSD2A.

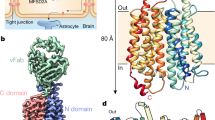
a, Uptake activities of mouse MFSD2A. WT, wild type. b, Cryo-EM map of MFSD2A, coloured by N- (cyan) and C- (green) domains. c, MFSD2A architecture. Ribbon representations are viewed from extracellular side (top) and membrane (bottom). N- and C-domains are coloured in cyan and green, respectively. d. Slab view of MFSD2A showing partially outward-open conformation, viewed from membrane (left) or extracellular side (right). Surface is coloured by electrostatic potential (red, −5 kT e−1; blue, 5 kT e−1). e, Uptake activities for disulfide-breaking mutations. f, Residues lining cavity bottom. Residues are shown as sticks. g, Uptake activity of MFSD2A variants with point mutations at cavity-lining residues. Uptake activities are normalized to that of the wild type (mean ± s.e.m., n = 6 biologically independent experiments for MFSD2A variants in a, e, g). P values from one-way analysis of variance (ANOVA) followed by Tukey’s post hoc multiple comparison test are indicated in a, e, g.
We identified a fortuitous loss-of-function point mutation, Q67H (Extended Data Fig. 2), that—on the basis of its location near the typical extracellular gate area in MFS—presumably arrests the transporter in an outward-facing conformation. We reasoned that this might reduce conformational heterogeneity and selected this variant for structural studies. Because MFSD2A is small (59 kDa) with few features outside the micelle, we used a single-chain variable fragment that binds MFSD2A as a fiducial marker to aid particle alignment. This strategy resulted in a 3.5 Å-resolution map (Fig. 1b, Extended Data Fig. 3), showing clear side-chain densities (Extended Data Fig. 4).
Overall structure
MFSD2A adopts a canonical MFS fold with structurally related N-terminal domain (hereafter, N-domain; transmembrane helix (TM)1 to TM6) and C-terminal domain (hereafter, C-domain; TM7 to TM12) (Fig. 1c, Extended Data Fig. 5). Our structure captures MFSD2A in an outward-facing conformation with a cavity at the interface of the N- and C-domains (Fig. 1d). One unique feature of MFSD2A is its extracellular domain, which is primarily formed by an elongated, ordered loop (extracellular loop (EL)3) between TM5 and TM6 and a loop between TM11 and TM12 (EL6) (Fig. 1c). EL3 reaches across the interface of the N- and C-domains and interacts with EL6 and EL4 (between TM7 and TM8). A conserved disulfide bridge forms between C216 on EL3 and C464 on EL6, providing covalent linkage (Fig. 1c). The extracellular domain may potentially constrain relative movement between the N- and C-domains. Mutating C216 or C464 to alanine substantially reduced transport (Fig. 1e), suggesting an important role of the extracellular domain stabilized by the disulfide bridge. Intracellularly, the N- and C-domains are connected by a long linker between TM6 and TM7 (Extended Data Fig. 6) that interacts with both domains and might stabilize the outward-open conformation. After the last transmembrane helix, a short helix reaches from the C-domain to the domain interface on the intracellular surface and may stabilize the intracellular closed conformation. Unlike sugar transporters within the MFS superfamily14, no helical bundle domain forms on the intracellular side of MFSD2A.
Translocation pathway
In the outward-facing conformation, the solvent-accessible cavity (which traverses around half of the membrane) is surrounded mainly by TM1, TM2, TM4, TM5, TM7, TM8, TM10 and TM11 (Fig. 1c), which probably form a substrate translocation pathway. The bottom half of the cavity has an overall negative electrostatic surface that becomes more neutral near the mouth of the cavity (Fig. 1d). This electrostatic distribution can accommodate the amphipathic nature of LPC such that its positively charged headgroup settles in the base of the cavity and its hydrophobic tail points to the extracellular side. To assess the functional roles of the residues near the bottom of the cavity, we performed mutagenesis studies (Fig. 1f, g). Majority alanine substitutions either abolished (R190A or H166A) or substantially reduced (S309A, F403A, W407A, E194A or F305A) transport activity—except for T439A, which showed little apparent effect. These results indicate important roles of the central pocket residues on both N- and C-domains. Potentially, highly conserved charged residues near the bottom of the cavity (such as H166) might directly interact with the zwitterionic phosphatidylcholine headgroups of LPC.
Lateral entry and lipid-binding site
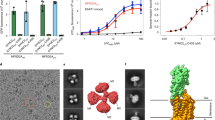
In the extracellular half of the membrane, the N- and C-domains of MFSD2A interact only loosely, which leaves lateral V-shaped openings to the membrane between TM5 and TM8 on one side and between TM2 and TM11 on the other (Fig. 1d). Extra lipid-like density is wedged in both openings (Fig. 2a, b, d) with the stronger density between TM5 and TM8 (TM5–TM8), indicating more stable interactions. Although the local resolution is insufficient to unambiguously resolve the lipid identity, LPC or a LPC-like or -containing molecule can be reasonably placed into the density between TM5 and TM8 (Fig. 2b). In this case, the LPC headgroup points to the centre of the cavity; its C-terminal acyl chain extends out parallel to the transmembrane helices, matching the lipid bilayer environment. The density between TM2 and TM11 (TM2–TM11) shows some flatness features (Fig. 2d); its identity is less clear. We probed the importance of residues constituting the potential lipid-binding sites through mutagenesis (Fig. 2c). N335 on TM8 lies at the entrance of the lateral opening near the kink of the tentatively modelled LPC. Mutating N335 substantially reduced transport, consistent with its potential role in lipid docking. The T202 sidechain on TM5 points towards the lipid density. Mutation to a bulky residue (T202F), but not to an alanine, substantially impaired transport, perhaps by sterically hindering lipid binding. By contrast, mutating Q334 on TM8 or Q207 on TM5—the side chains of which point away from the lipid density—only modestly affected transport. Likewise, mutating residues near the TM2–TM11 lipid density (F64A, S82A, R89A, T451A, F86A, D455A or R461A) had modest or negligible effects. Thus, the TM5–TM8 lateral opening is probably functionally more important than the TM2–TM11 opening. We propose that the TM5–TM8 lateral opening constitutes an LPC binding site en route to the central cavity, facilitating LPC diffusion into or from the membrane. It remains unclear whether the TM2–TM11 lateral opening might also be involved in lipid entry or exit.

a, MFSD2A cryo-EM map. Densities attributed to lipids at TM2–TM11 and TM5–TM8 lateral openings are coloured in yellow and purple, respectively. b, Zoomed-in view of lipid density between TM5 and TM8. Densities attributed to lipid are shown as purple mesh. LPC is tentatively modelled in the density. c, Uptake activities of MFSD2A variants with point mutations in lateral opening regions surrounding lipid density. Uptake activities are normalized to that of the wild type (mean ± s.e.m., n = 6 biologically independent experiments except N335A, n = 15 biologically independent experiments). P values from one-way ANOVA followed by Tukey’s post hoc multiple comparison test are indicated on bar chart. d, Zoomed-in view of lipid density between TM2 and TM11. Densities attributed to lipid are shown as gold mesh.
Sodium-binding site
The LPC transport activity of MFSD2A depends on sodium1,2. Within the MFS superfamily, the bacterial melibiose transporter MelB is a well-characterized Na+-coupled symporter15,16. Several key residues in the Na+-binding site of MelB are conserved in MFSD2A; the equivalent region of MFSD2A probably also forms a Na+-binding site (Fig. 3a). In particular, the side-chain oxygens of D92, D96, T95 and T163 form a pocket that is well-positioned to coordinate sodium at the centre. In this region of the cryo-EM map, extra density above background levels (Fig. 3b) might be compatible with Na+, given that 150 mM NaCl was included in the sample. However, the resolution is insufficient to assign or place the ion unambiguously.

a, Proposed Na+-binding site. Residues in the binding site are shown as sticks. b, Density map around the proposed Na+-binding site, shown as blue mesh. The proposed position of Na+ is represented as a yellow circle. c, Sodium positions from molecular dynamics simulations superimposed with the MFSD2A cryo-EM structure. Na+ ions bound at the Na1 site are shown as purple spheres, Na+ bound at the Na2 site as orange spheres (Methods) and all other Na+ as grey spheres. The initial Na+ position proposed on the basis of the cryo-EM data is shown as a yellow circle. d, Coordination of sodium bound at the Na1 site (purple sphere) in a representative simulation frame. e, Uptake activities of MFSD2A variants with point mutations in Na+-binding sites. Uptake activities are normalized to that of the wild type (mean ± s.e.m., n = 6 biologically independent experiments except for T95A, n = 9 biologically independent experiments; wild type, D96A and control are the same as in Fig. 1a). P values from one-way ANOVA followed by Tukey’s post hoc multiple comparison test are indicated on bar chart.
To validate the proposed sodium-binding site and elucidate sodium coordination, we performed all-atom molecular dynamics simulations of MFSD2A. In all 13 of the independent simulations (1.2 μs each) with a sodium ion placed in the putative binding pocket, the sodium quickly shifted about 2 Å towards TM2, forming polar interactions with D92, D96, T95 and T163. The sodium remained bound at this location (which we designate Na1) for the remainder of each simulation (Fig. 3c, d, Extended Data Fig. 7a). In four of five simulations initiated with no sodium present in the proposed binding pocket, a sodium ion from the extracellular solution diffused through the central cavity to spontaneously and stably bind at Na1, with coordination distances and geometry that are typical of sodium binding17.
In many simulations, a sodium ion also bound at a second site (which we designate Na2) about 4 Å from Na1, further from TM2 and closer to the extracellular solvent (Fig. 3c, Extended Data Fig. 7b–d). The sodium at Na2 forms salt bridges with D92 and E159, with water molecules providing additional coordination. Sodium ions often bind simultaneously at Na1 and Na2. Because both sites are located beside a large solvent-exposed cavity, sodium follows various pathways to reach them. After entering the cavity, sodium typically interacts briefly with Y55, Q56, E194, T198 and/or E316—in no particular order—before binding to Na1 or Na2. At Na2, sodium forms only two direct polar interactions with the protein (compared to six at Na1), resulting in weak and transient binding. Thus, we propose that Na1, which shows stable sodium binding, is the key sodium-binding site for the transport cycle.
We performed mutagenesis studies to probe the functional importance of the Na+-binding sites (Fig. 3e). For Na1, substituting alanine at D96 abolished transport, corroborating previous findings1 and consistent with strong coordination by both oxygens of the carboxylate group of D96 for Na+ at Na1. Mutating D92 or T95 also substantially impaired uptake, consistent with roles in coordinating Na+. Substituting alanine for T163 had little effect, whereas substituting a longer side chain (methionine) that potentially interferes with Na+ binding abolished uptake. In control experiments, mutating Q56 (which faces the central cavity) had little effect. Together, these results suggest that Na1 is functionally important and corroborate several critical residues found in human MFSD2A2. For Na2, mutating E159 only modestly affected activity. Thus, Na2 is probably not involved directly in coupled transport of Na+ and lipid. Sequence conservation analysis of MFSD2A and its close relative MFSD2B—a Na+-independent sphingosine 1-phosphate transporter18,19—revealed that D92 is conserved in MFSD2A but is substituted by glycine in MFSD2B, consistent with the differing Na+ requirements. Notably, K440 is next to Na1, where it forms electrostatic interactions with D96 and D92, stabilizing closely spaced carboxylate groups and thus helping to uphold Na1. Mutating K440 abolished transport (Fig. 3e). Likewise, mutating Y55 substantially reduced uptake. Y55 is within distance to interact with K440, which might further stabilize Na1. In a previous knock-in mouse study, the D96A substitution was shown to abolish lipid transport by MFSD2A and caused blood–brain barrier leakage7, recapitulating the MFSD2A-knockout phenotype. This highlights the functional importance of the sodium-binding site and corroborates the link between MFSD2A transport and permeability of the blood–brain barrier.
Disease-causing mutations
Several loss-of-function mutations of MFSD2A underlie lethal or progressive microcephaly syndromes4,5,20,21,22,23. For instance, T159M or S166L substitutions (corresponding to T163M and S170L in mouse, respectively) underlie a lethal microcephaly syndrome4,20. Our structure reveals that T163 is part of Na1. The substitution of T163 with methionine thus probably disrupts Na+ binding and impedes transport (Fig. 3d, e). S170 is below the base of the cavity at the interface of the N- and C- domains (Extended Data Fig. 8a), forms a hydrogen bond with R190 and closely contacts W407. S170 contributes to formation of the central binding pocket and possibly to closing of the intracellular gate. As expected, an S170L substitution reduced uptake close to background levels (Extended Data Fig. 8c). A third substitution, S339L (corresponding to S343L in mouse)5, may affect the helical bend of TM8 (Extended Data Fig. 8b), thereby affecting conformational transitions. The S343L substitution substantially reduced activity (Extended Data Fig. 8c), consistent with decreased LPC uptake in patients with a microcephaly syndrome5. A fourth substitution, T198M (corresponding to T202M in mouse)20,21, may interfere with substrate entry, as T202 lines the TM5–TM8 lateral opening (Fig. 2b). The T202M substitution impaired transport. Several mutations probably affect protein structure or folding. Three proline point mutations (P402H, P493L and P164T, which correspond to mouse P406H, P497L and P168T, respectively) are linked to microcephaly20,22,23. Given the unique properties of proline, these mutations probably cause conformational changes or destabilize the structure. In addition, an R326H/V250F double mutation (corresponding to R330H/A254F in mouse) underlies microcephaly20. Given that A254F, but not R330H, affected uptake (Extended Data Fig. 8c), the A254F substitution is probably responsible. Mutating A254 to a bulky residue presumably interferes with the packing interaction between TM3 and TM6, affecting structural stability or folding. These results corroborate that our structure of mouse MFSD2A provides a faithful model for interpreting function and disease-causing mutations of human MFSD2A.
Discussion
MFS transporters are thought to operate by a rocker–switch model24. Following a similar mechanism, opening or closing of the lateral openings of MFSD2A is expected to accompany opening or closing of the central cavity to the solution as the N- and C-domains rock during the transport cycle. For a Na+-coupled transporter, a central question is how Na+ is coupled to transport. Our structural analyses, molecular dynamics simulations and functional studies revealed a stable and functionally important Na+-binding site (Na1) at a strategic position close to the helical kink of TM2, which is encompassed by the Na+-binding residues D92 and D96. As the kink of TM2 gives rise to the V-shaped lateral opening between TM2 and TM11, Na+ binding might be linked directly to the opening or closing of the lateral opening, thus coupling Na+ to the state transition between outward- and inward-open conformations. In addition, the interaction between K440 in the C-domain and D92 and D96 in the N-domain stabilizes the Na+-binding site. This also potentially links Na+ binding and conformational state transition that involves relative rocking between the N- and C-domains. Moreover, the Na+-binding sites are near the base of the central cavity, where the LPC headgroup presumably binds. Thus, binding of LPC or sodium might induce local conformational changes, affecting binding of the other substrate and yielding transport coupling.
To our knowledge, the structure of MFSD2A represents the first for a eukaryotic lipid transporter within the MFS superfamily13. Compared with most known MFS transporters (the soluble substrates of which diffuse directly into the cavity), it remained unknown how lipids enter and leave MFSD2A. Our structure suggests a possible two-step mechanism: After the lipid diffuses laterally from the membrane to MFSD2A, it docks onto the lipid-binding site at the lateral entry and then moves into the central cavity. We observed lateral openings on both sides of MFSD2A, at the interface of the N- and C-domains. The TM5–TM8 opening is more likely to be the LPC entrance in an outward-facing conformation: lipid density there matches reasonably with an LPC-like molecule, the density is relatively strong and the surrounding residues are functionally important. In an inward-facing conformation, a simple scenario would be that the lateral opening on the same side also serves as the LPC entrance. This would potentially allow the LPC tail to remain in the lipid bilayer without being fully buried in the pocket during transport, helping to explain how LPCs with relatively long tails can be transported1,2. The detailed mechanisms await future investigation. Many other MFS transporters, such as GLUT3 (also in an outward-facing conformation)25, have central cavities that are better shielded from the membrane. This might reflect their different needs for substrate entry. The proposed lateral entry of lipids in MFSD2A shows parallels to other lipid transporters, such as ABC lipid transporters26, P4 ATPase27 and RND transporters28. This shared feature across structurally unrelated lipid transporter families may represent an optimal solution to overcome the high energy cost associated with moving lipids out of the membrane.
A major hurdle in developing therapeutic agents for central nervous system disorders is the limited permeability of most drug molecules across the blood–brain barrier29,30. Given the critical role of the lipid transport function of MFSD2A in suppressing transcytosis (thus ensuring the restrictive permeability of the blood–brain barrier), inhibiting MFSD2A represents an attractive strategy for drug delivery to the central nervous system. In fact, mice that lack MFSD2A activity exhibit leakage of diverse molecules, including IgG6,7,11. Our structure of MFSD2A in an outward-facing conformation provides a blueprint for structure-based discovery of MFSD2A inhibitors. In addition, biologics—such as an extracellular surface binder against MFSD2A (Extended Data Fig. 9)—have the potential to inhibit transport by impeding conformational transitions during the transport cycle.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Protein expression, purification and sample preparation
Mus musculus MFSD2A with a point mutation (Q67H) was cloned into a modified BacMam expression vector31 with an N-terminal GFP–His tag and a 3C protease cleavage site. The baculovirus was generated using sf9 insect cells (Expression Systems no. 94-001S; no further authentications or mycoplasma contamination tests were performed for this study) following a published protocol (Invitrogen LifeTechnologies). Recombinant protein was expressed in HEK293S cells (ATCC no. CRL-3022; no further authentications or mycoplasma contamination tests were performed for this study) grown in 293 Freestyle medium (LifeTechnologies) supplemented with 2% FBS. Cells were infected with baculovirus at a density of 2.5 × 106 cells per ml. After a 12-h incubation at 37 °C, 10 mM sodium butyrate was added to the culture, and the cells were moved to 30 °C for an additional 48 h before collection. All protein purification steps were carried out at 4 °C unless specifically noted otherwise. Initially, the cell pellet was resuspended in buffer containing 20 mM Tris-HCl pH 8.0 with the addition of a cocktail of protease inhibitors and DNase I, followed by a 30-min incubation. The sample was then centrifuged at 18,000g for 20 min. The crude membrane was homogenized by using either a glass Dounce tissue homogenizer or brief sonication in buffer A (20 mM Tris-HCl pH 8.0, 150 mM NaCl) supplemented with 2 mg ml−1 iodoacetamide, a cocktail of protease inhibitors and DNase I. The sample was incubated for 30 min before adding 1% lauryl maltose neopentyl glycol (LMNG) (Anatrace) and 0.1% cholesteryl hemisuccinate (CHS) (Anatrace). After an additional 2-h incubation, the sample was centrifuged at 18,000g for 45 min. The supernatant was incubated with cobalt resin for 1 h. The resin was washed with buffer A + 0.01% LMNG–0.001% CHS. The GFP–His tag was removed from MFSD2A by overnight digestion with 3C protease. The sample was further purified by gel filtration (Superdex 200 increase, GE Healthcare) in buffer A plus 0.001% (w/v) LMNG–0.00033% (w/v) glyco-diosgenin (GDN) (Anatrace)–0.00013% (w/v) CHS. The peak fraction was pooled and concentrated.
Anti-MFSD2A scFv was generated as extracellular binders for MFSD2A. scFv was isolated by phage display from chickens immunized with virus-like particles (lipoparticles)32 that contain mouse MFSD2A (Integral Molecular). The animal work was approved by Harvard University and followed the relevant ethical regulations. scFv was recombinantly expressed as a scFv-Fc fusion protein with a human Fc and purified by protein-A chromatography. The scFv fragment was generated by papain cleavage and the scFv fragment was purified by ion-exchange chromatography using a HiTrap Q HP anion exchange column (GE Healthcare). Purified mouse MFSD2A was mixed with the scFv at a 1:1 (w/w) ratio and incubated for 4 h at 4 °C. The complex was further purified by gel filtration (Superdex 200 increase, GE Healthcare) equilibrated in buffer A plus 0.001% (w/v) LMNG–0.00033% (w/v) GDN–0.00013% (w/v) CHS. The peak fraction was concentrated to 4 mg ml−1 for cryo-EM studies.
Electron microscopy sample preparation and data collection
For cryo-EM, 3 μl of the purified complex was applied to glow-discharged 300 mesh Quantifoil R2/1 holey carbon grids and blotted for 2.0 s at 96% humidity on a Leica EM GP2 before being plunge-frozen in liquid ethane cooled by liquid nitrogen. Grids were imaged on a Titan Krios operated at 300 kV using a slit width of 20 eV on a GIF-Quantum Energy Filter. Images were collected on a K3 Summit detector (Gatan) in super-resolution counting mode at a magnification of 105,000×, corresponding to a physical pixel size of 0.86 Å. Serial EM33 was used for data collection with a set of customized scripts enabling automated low-dose image acquisition. Data were collected using image shift to collect one image per hole by multiple record method (3 × 3 set of holes per stage movement).
Cryo-EM data processing
A total of 8,669 movies were collected and subjected to beam-induced motion correction using the program MotionCor234. A dose-weighted sum of all frames from each movie was used for all image processing steps. Contrast transfer function (CTF) parameters were estimated by Gctf35. Automated particle picking was first performed using cisTEM 1.0.0 Beta36 using 500 images; the picked particles were extracted with box size of 232 pixels and subjected to 2D classification in cisTEM. The good classes, representing projections in different orientations, were selected and imported to Relion3.0-Beta-237 as templates for auto picking. All the picked particles were extracted with a box size of 232 pixels with original pixel size of 0.86 Å in Relion and imported to CryoSparc (v.2.13.2)38 for further 2D classification. Rounds of 2D classifications yielded 460,956 particle images with clear features. With these particles, an initial 3D model was built by CryoSparc ab initio reconstruction without symmetry. The particles along with the initial model, which was imported as 3D template, were transferred back to Relion and subjected to 3D classification using k = 4 and tau fudge (T) value 4. The two most populated classes with good features for the MFSD2A region contained 307,951 particles after two rounds of classifications, with T = 20 in the second round. The Relion Auto-Refine of these particles resulted in a 4.2 Å map without symmetry. To improve the map quality, local 3D classification focused on the MFSD2A region was performed. The signal of multiple domains—except MFSD2A—was subtracted from the particles with a mask covering the whole complex. The modified particle set was subjected to further local 3D classification without alignment using a mask around MFSD2A and k = 4, T = 40. After classification, the class with the best features of MFSD2A was selected. The corresponding 90,577 particles were subjected to Bayesian polishing and per particle CTF refinement, then imported back to CryoSparc for final refinement. NU-Refinement of CryoSparc yielded an improved map for the whole complex with nominal resolution of 3.7 Å. By applying a mask on the transporter only, the MFSD2A domain was further refined to 3.5 Å in Relion, local resolution estimated by SAMUEL39. All refinements followed the gold-standard procedure, in which two half-datasets are refined independently. The overall resolutions were estimated based on the gold-standard Fourier shell correlation (FSC) = 0.143 criterion. The number of particles in each dataset and other details related to data processing are summarized in Extended Data Table 1.
Model building and refinement
The model was built into a 3.5 Å cryo-EM map using the Na+/melibiose symporter MelB (Protein Data Bank code 4M64) as template. Local parts were manually built in Coot40. The models were refined using Phenix real space refine41 and the geometry of the models was evaluated by Molprobity42. All the figures were prepared in PyMol (Schrödinger)43 or UCSF Chimera44.
LPC uptake assays
MFSD2A wild type and variants were cloned into pmCherry-N1 vector (ClonTech) with an mCherry fused at the C terminus. Plasmids were transfected into HEK293S cells (ATCC, CRL-3022) (seeded from the suspension culture) using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s directions. Cells grown in 12-well plates were maintained in 293 Freestyle medium (LifeTechnologies) supplemented with 10% FBS in an incubator at 37 °C and 8% CO2. The medium was replaced 24 h after transfection with 293 Freestyle medium supplemented with 10% FBS and 10 mM sodium butyrate, and cells were incubated for an additional 24 h. MFSD2A transport activity was assayed by measuring TopFluor LPC (Avanti) uptake in HEK293S cells. The cells were washed first with serum-free 293 Freestyle medium and incubated in 293 Freestyle medium supplemented with 10% FBS, 150 mM NaCl and 1 μM TopFluor LPC at 37 °C for 30 min. The cells were washed twice with ice-cold PBS, resuspended in ice-cold PBS and passed through a 35-μm cell strainer. The cells expressing MFSD2A wild type or variants in a set of experiments were gated in the same range on the basis of the mCherry level using the FL-3 channel of a BD Accuri C6 flow cytometer (Supplementary Fig. 1). The mean fluorescence of cells corresponding to TopFluor LPC level was quantified using the FL-1 channel. A one-way ANOVA test followed by Tukey’s post hoc multiple comparison test was used to calculate statistical differences using Minitab Statistical Software45 between wild-type MFSD2A and mutant transport activities. P values are indicated in the bar charts.
Conservation analysis of mouse MFSD2A structure
A multiple sequence alignment was performed with a cut-off of minimally 50% identity and maximally 90% identity to mouse MFSD2A using the UniRef database on the ConSurf web server46,47. The conservation scores were generated and coloured using a PyMOL script generated by the ConSurf web server.
System set-up for molecular dynamics simulations
We performed simulations of MFSD2A without scFv in a hydrated lipid bilayer. We performed simulations under three conditions: (1) simulations with a sodium ion initially placed in the binding pocket at a position proposed on the basis of the potential coordination environment and the cryo-EM density map, with a nonbonded interaction cut-off of 9 Å (10 independent simulations, 1.2 μs each); (2) simulations with a sodium ion initially placed in the binding pocket at a position proposed on the basis of the potential coordination environment and the cryo-EM density map, with a nonbonded interaction cut-off of 12 Å (3 independent simulations, 1.2 μs each); and (3) simulations with no sodium ions initially placed in the binding pocket, with a nonbonded interaction cut-off of 9 Å (5 independent simulations, 1.1 μs each). We observed no substantial differences in simulation behaviour between the first two conditions. For all simulation conditions, the protein structure was aligned on the Orientations of Proteins in Membranes48 entry for 6S7V49 (MFS superfamily member LtaA) using PyMOL (Schrödinger)43. Prime (Schrödinger)50 was used to model missing side chains, and to add capping groups to protein chain termini. Protonation states of all titratable residues were assigned at pH 7. Histidine residues were modelled as neutral, with a hydrogen atom bound to either the delta or epsilon nitrogen depending on which tautomeric state optimized the local hydrogen-bonding network. Dowser51 was used to add water molecules to protein cavities. Using Dabble52, the prepared protein structures were inserted into a pre-equilibrated palmitoyl-oleoyl-phosphatidylcholine bilayer, the system was solvated, and sodium and chloride ions were added to neutralize the system and to obtain a final concentration of 150 mM. Final systems comprised approximately 90,000 atoms and system dimensions were approximately 120 × 120 × 100 Å. For each simulation, initial atom velocities were assigned randomly and independently.
Molecular dynamics simulation and analysis protocols
We used the CHARMM36m force field for proteins, the CHARMM36 force field for lipids and ions, and the TIP3P model for water53,54,55. All simulations were performed using the Compute Unified Device Architecture version of particle-mesh Ewald molecular dynamics in AMBER1856 on graphics processing units.
Systems were first minimized using three rounds of minimization, each consisting of 500 cycles of steepest descent followed by 500 cycles of conjugate gradient optimization. Harmonic restraints of 10.0 and 5.0 kcal mol−1 Å−2 were applied to protein and lipids for the first and second rounds of minimization, respectively. Harmonic restraints of 1.0 kcal mol−1 Å−2 were applied to protein for the third round of minimization. Systems were then heated from 0 K to 100 K in the NVT ensemble over 12.5 ps and then from 100 K to 310 K in the NPT ensemble over 125 ps, using 10.0 kcal mol−1 Å−2 harmonic restraints applied to protein heavy atoms. Subsequently, systems were equilibrated at 310 K and 1.0 bar in the NPT ensemble, with harmonic restraints on the protein non-hydrogen atoms tapered off by 1.0 kcal mol−1 Å−2 starting at 5.0 kcal mol−1 Å−2 in a stepwise fashion every 2.0 ns for 10.0 ns, and then by 0.1 kcal mol−1 Å−2 every 2.0 ns for 20.0 ns. Production simulations were performed without restraints at 310 K and 1.0 bar in the NPT ensemble using the Monte Carlo barostat, the Langevin thermostat with a collision frequency of 1.0 ps−1, and a timestep of 4.0 fs with hydrogen mass repartitioning57. Bond lengths to hydrogen were constrained using the SHAKE algorithm58. Nonbonded interactions were cut off at either 9.0 or 12.0 Å (as described in ‘System set-up for molecular dynamics simulations’), and long-range electrostatic interactions were calculated using the particle-mesh Ewald (PME) method with an Ewald coefficient of approximately 0.31 Å, and 4th order B-splines. The PME grid size was chosen such that the width of a grid cell was approximately 1 Å. Trajectory frames were saved every 200 ps during the production simulations.
The AmberTools17 CPPTRAJ package was used to reimage trajectories59. Simulations were visualized and analysed using Visual Molecular Dynamics60 and PyMOL (Schrödinger)43. In Fig. 3c, the first 0.1 μs of the production phase of each simulation of condition (1) was discarded (as well as the equilibration phase), and trajectories were aligned on the backbone atoms of the MFSD2A cryo-EM structure Na+-binding site residues D92, T95, D96, E159, T163 and K440. The positions of Na+ ions were recorded every 10 ns for each of the 10 simulations. Each Na+ ion position was then drawn as a point superimposed on the starting MFSD2A structure. A sodium ion at a distance of 2–5 Å from the T95 side-chain oxygen forming a salt bridge with D92 and/or D96 was considered to be present at the Na1 site. A sodium ion at a distance of 5–8 Å from the T95 side-chain oxygen forming a salt bridge with D92 and/or E159 was considered to be present at the Na2 site. A salt bridge was considered to be formed if the sodium ion was within 3.5 Å of either side-chain oxygen atoms of an aspartate or glutamate residue. In Extended Data Fig. 7c, the same analysis was repeated taking into consideration only simulation no. 2 of condition (1), in which both sodium ions were bound simultaneously. In Extended Data Fig. 7a, b, the equilibration and the production phases are both plotted for each simulation. The same criteria as explained above for Fig. 3c are used to identify sodium ions present at the Na1 and Na2 sites.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
References
Nguyen, L. N. et al. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509, 503–506 (2014).
Quek, D. Q., Nguyen, L. N., Fan, H. & Silver, D. L. Structural insights into the transport mechanism of the human sodium-dependent lysophosphatidylcholine transporter MFSD2A. J. Biol. Chem. 291, 9383–9394 (2016).
Horrocks, L. A. & Yeo, Y. K. Health benefits of docosahexaenoic acid (DHA). Pharmacol. Res. 40, 211–225 (1999).
Guemez-Gamboa, A. et al. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat. Genet. 47, 809–813 (2015).
Alakbarzade, V. et al. A partially inactivating mutation in the sodium-dependent lysophosphatidylcholine transporter MFSD2A causes a non-lethal microcephaly syndrome. Nat. Genet. 47, 814–817 (2015).
Ben-Zvi, A. et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509, 507–511 (2014).
Andreone, B. J. et al. Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron 94, 581–594 (2017).
Andreone, B. J., Lacoste, B. & Gu, C. Neuronal and vascular interactions. Annu. Rev. Neurosci. 38, 25–46 (2015).
Reese, T. S. & Karnovsky, M. J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 34, 207–217 (1967).
Brightman, M. W. & Reese, T. S. Junctions between intimately apposed cell membranes in the vertebrate brain. J. Cell Biol. 40, 648–677 (1969).
Chow, B. W. & Gu, C. Gradual suppression of transcytosis governs functional blood-retinal barrier formation. Neuron 93, 1325–1333 (2017).
Yang, Y. R. et al. Mfsd2a (major facilitator superfamily domain containing 2a) attenuates intracerebral hemorrhage-induced blood-brain barrier disruption by inhibiting vesicular transcytosis. J. Am. Heart Assoc. 6, e005811 (2017).
Yan, N. Structural biology of the major facilitator superfamily transporters. Annu. Rev. Biophys. 44, 257–283 (2015).
Deng, D. et al. Crystal structure of the human glucose transporter GLUT1. Nature 510, 121–125 (2014).
Ethayathulla, A. S. et al. Structure-based mechanism for Na+/melibiose symport by MelB. Nat. Commun. 5, 3009 (2014).
Granell, M., León, X., Leblanc, G., Padrós, E. & Lórenz-Fonfría, V. A. Structural insights into the activation mechanism of melibiose permease by sodium binding. Proc. Natl Acad. Sci. USA 107, 22078–22083 (2010).
Harding, M. M. Metal-ligand geometry relevant to proteins and in proteins: sodium and potassium. Acta Crystallogr. D 58, 872–874 (2002).
Vu, T. M. et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature 550, 524–528 (2017).
Kobayashi, N. et al. MFSD2B is a sphingosine 1-phosphate transporter in erythroid cells. Sci. Rep. 8, 4969 (2018).
Scala, M. et al. Biallelic MFSD2A variants associated with congenital microcephaly, developmental delay, and recognizable neuroimaging features. Eur. J. Hum. Genet. 28, 1509–1519 (2020).
Riazuddin, S. et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol. Psychiatry 22, 1604–1614 (2017).
Hu, H. et al. Genetics of intellectual disability in consanguineous families. Mol. Psychiatry 24, 1027–1039 (2019).
Harel, T. et al. Homozygous mutation in MFSD2A, encoding a lysolipid transporter for docosahexanoic acid, is associated with microcephaly and hypomyelination. Neurogenetics 19, 227–235 (2018).
Drew, D. & Boudker, O. Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 85, 543–572 (2016).
Deng, D. et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature 526, 391–396 (2015).
Plummer, A. M., Culbertson, A. T. & Liao, M. The ABCs of sterol transport. Annu. Rev. Physiol. 83, 153–181 (2021).
Andersen, J. P. et al. P4-ATPases as phospholipid flippases-structure, function, and enigmas. Front. Physiol. 7, 275 (2016).
Nikaido, H. Structure and mechanism of RND-type multidrug efflux pumps. Adv. Enzymol. 77, 1–60 (2011).
Pardridge, W. M. The blood-brain barrier: bottleneck in brain drug development. NeuroRx 2, 3–14 (2005).
Banks, W. A. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 15, 275–292 (2016).
Goehring, A. et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protocols 9, 2574–2585 (2014).
Tucker, D. F. et al. Isolation of state-dependent monoclonal antibodies against the 12-transmembrane domain glucose transporter 4 using virus-like particles. Proc. Natl Acad. Sci. USA 115, E4990–E4999 (2018).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Grant, T., Rohou, A. & Grigorieff, N. cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383 (2018).
Scheres, S. H. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 415, 406–418 (2012).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Ru, H. et al. Molecular mechanism of V(D)J recombination from synaptic RAG1-RAG2 complex structures. Cell 163, 1138–1152 (2015).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
The PyMOL Molecular Graphics System v.2.0 (Schrödinger, 2017).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Minitab 17 Statistical Software (Minitab, 2010).
Ashkenazy, H. et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016).
Landau, M. et al. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 (2005).
Lomize, M. A., Lomize, A. L., Pogozheva, I. D. & Mosberg, H. I. OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 (2006).
Zhang, B. et al. Structure of a proton-dependent lipid transporter involved in lipoteichoic acids biosynthesis. Nat. Struct. Mol. Biol. 27, 561–569 (2020).
Jacobson, M. P., Friesner, R. A., Xiang, Z. & Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 320, 597–608 (2002).
Zhang, L. & Hermans, J. Hydrophilicity of cavities in proteins. Proteins 24, 433–438 (1996).
Betz, R. Dabble (v.2.6.3), https://doi.org/10.5281/zenodo.836914 (2017).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Klauda, J. B. et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010).
Guvench, O., Hatcher, E. R., Venable, R. M., Pastor, R. W. & Mackerell, A. D. CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 5, 2353–2370 (2009).
Case, D. A. et al. AMBER 2018 (University of California, 2018).
Hopkins, C. W., Le Grand, S., Walker, R. C. & Roitberg, A. E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874 (2015).
Ryckaert, J., Ciccotti, G. & Berendsen, H. J. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Roe, D. R. & Cheatham, T. E. IIII. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 (1996).
Vilas, J. L. et al. MonoRes: automatic and accurate estimation of local resolution for electron microscopy maps. Structure 26, 337–344 (2018).
Acknowledgements
We thank L. Montabana and D.-H. Chen for help with EM data collection. This work was made possible by support from Stanford University, the Harold and Leila Y. Mathers Charitable Foundation and NIA DP2AG052940 to L.F., a Dean’s fellowship to J.Z., the EMBO Long-Term Fellowship ALTF 544-2019 to D.A., an EMBO long-term fellowship to U.H.L., the NIH DP1 NS092473 Pioneer Award, the NIH/NINDS R35NS116820 grant, the Blavatnik Biomedical Accelerator grant and the QFASTR grant from Harvard Medical School to C.G. The research of C.G. was also supported in part by a Faculty Scholar grant from the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
C.A.P.W. and J.Z. carried out biochemical, functional and cryo-EM studies. D.A. carried out and analysed molecular dynamics simulations under the guidance of R.O.D. Y.X. assisted with functional and biochemical studies. B.A. and U.H.L. characterized the scFv. C.G. supervised the generation and characterizations of scFv. L.F. directed biochemical, functional and structural studies. C.A.P.W., J.Z. and L.F. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Diwakar Shukla and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Sequence alignment of MFSD2A and MFSD2B homologues.
Sequence alignments of MFSD2A and MFSD2B from M. musculus (Mm), Homo sapiens (Hs), Danio rerio (Dr), Xenopus laevis (Xl), Bos taurus (Bt) and Gallus gallus (Gg) are shown.
Extended Data Fig. 2 Biochemical and functional characterizations of MFSD2A.
a, Size-exclusion chromatography profile of MFSD2A. b, Size-exclusion chromatography profile of MFSD2A in complex with scFv. c, Representative SDS–PAGE gel of purified MFSD2A–scFv complex. This was carried out four times in independent experiments. d, Uptake activity of Q67H used for structural studies. Uptake activity was normalized to that of the wild type (mean ± s.e.m., n = 4 biologically independent experiments). P values from one-way ANOVA followed by Tukey’s post hoc multiple comparison test are indicated on bar chart.
Extended Data Fig. 3 Single-particle cryo-EM analysis of MFSD2A.
a, Representative cryo-EM image of MFSD2A. b, Two-dimensional class averages of MFSD2A in CryoSparc. c, The workflow of classification and refinement. d, Angle distributions of the particles for the final reconstruction. e, Local resolution of the MFSD2A map calculated by MonoRes61. f, FSC of the final reconstruction as a function of resolution. Orange, gold-standard FSC curve between two half-maps from masked MFSD2A, with indicated resolution at FSC = 0.143; blue, FSC curve between the final atomic model and the local map masked on MFSD2A only, with indicated resolution at FSC = 0.5. FSC calculation performed by SAMUEL (SAM script)39.
Extended Data Fig. 4 Representative cryo-EM density maps of MFSD2A transmembrane helices.
Electron microscopy map density for 12 transmembrane helices of MFSD2A.
Extended Data Fig. 5 Conservation analysis of mouse MFSD2A structure.
Residues are coloured from variable to conserved according to the palette below the structure.
Extended Data Fig. 6 Intracellular elements of MFSD2A.
Ribbon representation (left) and cylindrical representation (right) of MFSD2A viewed from the intracellular side. N- and C-domains are coloured in cyan and green, respectively. IL, intracellular linker (orange). The helix after the last transmembrane helix is also coloured in orange.
Extended Data Fig. 7 Sodium-binding sites in molecular dynamics simulations.
a, Shaded regions (black, blue and green) indicate points in time during each simulation when a sodium ion was present at the Na1 site—in particular, points at which a sodium ion at a distance of 2–5 Å from the T95 side-chain oxygen formed a salt bridge with D92 and/or D96. b, Shaded regions indicate points in time during each simulation when a sodium ion was present at the Na2 site—in particular, points at which a sodium ion at a distance of 5–8 Å from the T95 side-chain oxygen formed a salt bridge with D92 and/or E159. Both a and b show data for simulations under three conditions. In the first two conditions (black and blue), a sodium ion is initially placed in the binding pocket at a position suggested by the potential coordination environment and the cryo-EM density, whereas in the third (black), no sodium ions are initially placed in the binding pocket. The first and third conditions (black and green) used a 9 Å nonbonded interaction cut-off, whereas the second (blue) used a 12 Å cut-off. Plots include equilibration as well as production phases of each simulation. c, Sodium positions from simulation no. 2 of the first condition (highlighted by red box), in which sodium ions bind simultaneously at the Na1 and Na2 sites. Positions of sodium bound at the Na1 site are shown as purple spheres, and positions of sodium bound at the Na2 site are shown as orange spheres (Methods). In this simulation, a Na+ ion was initially placed at a position proposed on the basis of the potential coordination environment and the cryo-EM density map, shown as a yellow circle. d, Sodium-binding sites in a representative frame from the same simulation. Sodium-coordinating residues are shown as sticks. Sodium bound at the Na1 site is shown as a purple sphere and sodium bound at the Na2 site is shown as an orange sphere. Oxygen atoms of water molecules are shown as red spheres.
Extended Data Fig. 8 Structural mapping of disease-causing mutations.
a, Close-up view of S170. S170 and R190 (sticks) are within hydrogen-bond distance. b, Zoomed-in view of S343, near the helical bend of TM8 that gives rise to lateral opening. c, Uptake activities of mouse MFSD2A variants with equivalent point mutations to human microcephaly-associated mutations. Uptake activities are normalized to that of the wild type (mean ± s.e.m., n = 6 biologically independent experiments). P values from one-way ANOVA followed by Tukey’s post hoc multiple comparison test are indicated on bar chart.
Extended Data Fig. 9 Structure of MFSD2A in complex with scFv.
a, The cryo-EM map of the MFSD2A–scFv complex. b, The ribbon representations of the MFSD2A–scFv complex. A model scFv was docked into the density.
Supplementary information
Supplementary Figure 1
This file contains an example of the gating strategy for flow cytometry in uptake assays.
Rights and permissions
About this article

Cite this article
Wood, C.A.P., Zhang, J., Aydin, D. et al. Structure and mechanism of blood–brain-barrier lipid transporter MFSD2A. Nature 596, 444–448 (2021). https://doi.org/10.1038/s41586-021-03782-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03782-y
- Springer Nature Limited
This article is cited by
-
Structures of the Mycobacterium tuberculosis efflux pump EfpA reveal the mechanisms of transport and inhibition
Nature Communications (2024)
-
LDL receptor-related protein 5 selectively transports unesterified polyunsaturated fatty acids to intracellular compartments
Nature Communications (2024)
-
Structural basis for triacylglyceride extraction from mycobacterial inner membrane by MFS transporter Rv1410
Nature Communications (2023)
-
Single-cell sequencing reveals that endothelial cells, EndMT cells and mural cells contribute to the pathogenesis of cavernous malformations
Experimental & Molecular Medicine (2023)
-
Lipid flipping in the omega-3 fatty-acid transporter
Nature Communications (2023)