
Abstract
The major facilitator superfamily domain-containing protein 2A (MFSD2A) is a constituent of the blood-brain barrier and functions to transport lysophosphatidylcholines (LPCs) into the central nervous system. LPCs such as that derived from docosahexanoic acid (DHA) are indispensable to neurogenesis and maintenance of neurons, yet cannot be synthesized within the brain and are dependent on MFSD2A for brain uptake. Recent studies have implicated MFSD2A mutations in lethal and non-lethal microcephaly syndromes, with the severity correlating to the residual activity of the transporter. We describe two siblings with shared parental ancestry, in whom we identified a homozygous missense mutation (c.1205C > A; p.Pro402His) in MFSD2A. Both affected individuals had microcephaly, hypotonia, appendicular spasticity, dystonia, strabismus, and global developmental delay. Neuroimaging revealed paucity of white matter with enlarged lateral ventricles. Plasma lysophosphatidylcholine (LPC) levels were elevated, reflecting reduced brain transport. Cell-based studies of the p.Pro402His mutant protein indicated complete loss of activity of the transporter despite the non-lethal, attenuated phenotype. The aggregate data of MFSD2A-associated genotypes and phenotypes suggest that additional factors, such as nutritional supplementation or modifying genetic factors, may modulate the severity of disease and call for consideration of treatment options for affected individuals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The blood-brain barrier (BBB), localized at the level of tight junctions between adjacent brain endothelial cells, regulates delivery of metabolites and essential nutrients into the brain while limiting entry of toxic plasma and cell constituents. Substrate-specific transport proteins are responsible for the transport of specific compounds across the BBB. Examples include the SLC2A1-encoded glucose transporter 1 (GLUT1) that ensures sufficient supply of glucose as the main energy substrate of the brain. The gradient of amino acids across the BBB is strictly controlled by amino acid transporters, such as the large neutral transporter LAT1, encoded by SLC7A5, and additional amino acid-specific transporters. Lactate and other monocarboxylates are dependent upon transporters of the SLC16 family. ATP-binding cassette (ABC) transporters limit penetration of drugs into the brain [1,2,3]. Recently, the major facilitator superfamily domain-containing protein 2A (MFSD2A, MIM 614397) was identified as a sodium-dependent transporter of docosahexanoic acid (DHA), an omega-3 long chain polyunsaturated fatty acid (LC-PUFA) highly enriched in the brain, across the BBB. DHA and other fatty acids are transported by MFSD2A in the form of lysophosphatidylcholine (LPC) [4]. The abundance of LPCs in the blood and efficient conversion to phosphatidylcholines (PCs) in cellular membranes suggest a key role for LPCs in cellular homeostasis and growth, with proposed roles in neural progenitor proliferation and neurite outgrowth [5].
In vitro and in vivo experiments in embryonic stem (ES) cells and rodent models have implicated DHA in differentiation of neural stem cells into neurons [6, 7], as well as in proliferation of cells undergoing differentiation into neural cells [7]. Moreover, DHA regulates membrane fluidity and thus supports signal transduction and neurotransmission. Dietary deficiency of omega-3 fatty acid during development is associated with decreased DHA in brain membrane phospholipids, inhibition or delay of neurogenesis, reduced learning ability and memory consolidation, altered activity of membrane receptors and proteins, and altered dopaminergic and serotonergic signaling in the fetal brain [8, 9].
MFSD2A is highly expressed in endothelium constituting the BBB [10] and encodes a 12-pass transmembrane protein which transports LPCs via a rocker-switch model [11]. In addition to its role in promoting specific transport of LPCs, MFSD2A lipid transport activity has been implicated in maintenance of BBB integrity by inhibiting transport of most molecules by transcellular vesicular transport (transcytosis) [10, 12]. Mfsd2a-knockout mice show microcephaly, cognitive defects and severe anxiety, markedly reduced levels of DHA in the brain, and neuronal cell loss in the hippocampus and cerebellum, overall reminiscent of omega-3 fatty acid deficiency [4]. Recently, inactivating and partially inactivating MFSD2A mutations were identified in lethal and non-lethal microcephaly syndromes, respectively [5, 13]. Plasma concentrations of LPCs containing mono- and polyunsaturated fatty acyl chains were significantly increased in affected individuals, suggesting reduced brain uptake of LPCs in the pathogenesis [5, 13]. Here, we report two siblings with a non-lethal microcephaly syndrome despite a homozygous inactivating mutation in MFSD2A, suggesting that additional factors may be involved in modulating the clinical expression. Thus, prediction of phenotypic severity cannot be based on the residual activity of the transporter alone.
Case reports
The subjects of this study were two siblings, a 5-year 8-month-old male (individual II-1 in Fig. 1a), and a 4-year 2-month old female (II-2 in Fig. 1a). These were the only children to non-consanguineous healthy parents of Jewish Moroccan ancestry. Individual II-1 was delivered after an uneventful pregnancy at 40 weeks of gestation with a birthweight of 2.8 kg (12th percentile) and a head circumference of 31 cm (− 2.1 SD). He was brought to clinical attention at 2 months of age due to hypotonia, feeding difficulties, stridor, and esotropic strabismus. Within the next few months, obvious global developmental delay became apparent, without regression. At the time of his last exam, he spoke few words and indicated wants by gestures. He could understand simple commands such as pointing to colors or shapes and was weaned from diapers during daytime. Motor function was impaired; he was able to reach for objects with difficulty and ambulated with a walker and had stable sitting from 2 years of age.

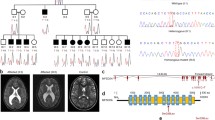
Pedigree and molecular analysis. a Pedigree indicating two affected children (solid fill) and genotypes of family members. b Sanger sequencing of heterozygous parent (upper panel) and affected individual (lower panel) showing the c.1205C > A variant. c The Pro402 residue and neighboring amino acids are highly conserved throughout evolution. d Predicted consequences of P402H mutation on MFSD2A structure. The left panel depicts 3D structural models of MFSD2A WT and P402H mutant, highlighting specific structural changes. The common parts of both models are shown in green (partially transparent), the largely unstructured helix X in the WT model is shown in cyan (partially transparent), and the helix X in the P402H mutant is shown in yellow. The P402 sidechain in the WT models is highlighted in cyan. The H402 sidechain in the mutant model is highlighted in yellow, forming hydrogen bond with Y431 sidechain in helix XI. In the right panel, LPC-oleate is shown docked to both the MFSD2A WT (docking pose in cyan, partially transparent) and P402H mutant models. The main difference between the two binding orientations is in the hydrophobic hydrocarbon tail
On examination at 5.5 years, the boy was communicative and interested in his surroundings and had microcephaly (head circumference 46.5 cm, < 1st percentile, − 3.5 SD) and a tented upper lip. Truncal hypotonia, appendicular spasticity with dystonia, and esotropic strabismus were prominent. Physical examination including funduscopic examination was otherwise unremarkable. Routine laboratory investigations as well as metabolic work-up including plasma amino acids, acylcarnitines, lactate and ammonia and urine organic acids were all within the normal range. Brain magnetic resonance imaging (MRI) at 4 years of age disclosed paucity of white matter with attendant lateral ventricular widening (Fig. 2a).

Neuroimaging studies of the affected individuals. a T1 and T2 weighted images (left and right, respectively) of individual II-1 at age 4 years showing paucity of white matter with lateral ventricular widening. b T1 and T2 weighted images (left and right, respectively) of individual II-2 at age 21 months showing paucity of white matter with lateral ventricular widening
His younger sister, individual II-2, followed a similar course with unremarkable perinatal course and congenital microcephaly (30.5 cm, − 2.9 SD, at birth; 41 cm, − 3.6 SD, at 14 months; and 45 cm, − 3.0 SD, at 4 years of age), global developmental delay with a mean developmental quotient of 0.49, hypotonia with appendicular spasticity, esotropic strabismus, and dystonia. Physical exam at 4 years of age showed microcephaly, esotropia, wide nasal bridge with epicanthal folds, tented upper lip, and a dimpled chin. Brain MRI performed at age 21 months was similar to that of the older brother, showing paucity of white matter with attendant lateral ventricular widening (Fig. 2b).
In view of the uneventful perinatal course, congenital microcephaly, parental common ancestry, and the similarity between the two siblings, further genetic molecular studies were pursued.
Materials and methods
Exome analysis
Exonic sequences were enriched in the DNA sample of individual II-2, using SureSelect Human All Exon 50 Mb Kit (Agilent Technologies, Santa Clara, CA). Sequences were determined by HiSeq2500 (Illumina, San Diego, CA) as 100-bp paired-end runs. Data analysis including read alignment and variant calling was performed by DNAnexus software (Palo Alto, CA) using the default parameters with the human genome assembly hg19 (GRCh37) as reference. Parental consent was given for DNA studies. The study was performed with the approval of the ethical committees of Hadassah Medical Center and the Israeli Ministry of Health.
Transport assays
Transport assays using HEK293 cells were performed as previously described [4]. Briefly, plasmids encoding wild-type, Asp97Ala, and Pro402His MFSD2A were transfected into HEK293 cells. Uptake assays were performed after 24 h of transfection with increasing concentrations of LPC-[14C]DHA. Experiments were repeated twice in triplicate in 12-well plates. Uptake activity is expressed in dpm per well. Radiolabeled LPC-[14C]DHA was purchased from ARC. Non-radiolabeled LPC-DHA was synthesized as described previously [14].
Lipid extraction from plasma samples
Briefly, plasma phospholipids were extracted using a butanol/methanol-based protocol. Plasma samples (5 μl) were re-suspended in 50 μl of butanol/methanol (1/1, v/v) containing a mixture of internal standards (Avanti Polar Lipids), vortexed for 30 s, and sonicated for 30 min on ice. Samples were centrifuged at 14,000 rpm for 10 min at 4 °C to pellet insoluble. The supernatants were transferred to MS vial and injected (2 μl) as is into a liquid chromatography-tandem mass spectrometry (LC-MS/MS) instrument. Each sample was extracted in triplicate. Technical QC samples and blank extract were prepared together with the samples to ensure data quality.
Lipidomics LC-MS/MS analysis
Samples were randomized for injection into a liquid chromatography-tandem mass spectrometry (LC-MS/MS) instrument (1290 Liquid Chromatography System, and 6460 QqQ, Agilent Technologies). Quality controls and blanks were injected after every six sample injections to monitor stability of the instrument response and carryover. The chromatographic column was a Kinetex HILIC (150 × 2.1 mm, 2.6 μM, 100 Å; Phenomenex). Gradient elution was undertaken with solvents A (50% acetonitrile/50% 25 mM ammonium formate buffer, pH 4.6) and B (95% acetonitrile/5% 25 mM ammonium formate buffer, pH 4.6), with a gradient range from 75 to 25% solvent B for 6 min, 90 to 10% solvent B for 1 min, 0.1 to 99.9% solvent B for 0.1 min, followed by 0.1 to 99.9% solvent B for 3 min (total run time of 10.1 min). Phospholipids (see Supplemental Table 1) were quantified using multiple reaction monitoring with precursor to headgroup transitions. MS parameters were: gas temperature of 300 °C, gas flow of 5 l/min, sheath gas flow of 11 l/min, and capillary voltage of 3500 V. Quantification data were extracted using MassHunter Quantitative Analysis (QQQ) software, and data were manually curated to ensure correct peak integration. Areas under the curve for the extracted ion chromatogram peaks for each multiple reaction monitoring transition and lipid species were normalized to internal standards. Isotope correction was then done on all relevant lipid species.
Immunoblotting and immunofluorescence microscopy
Immunoblotting and immunofluorescence microscopy for MFSD2A were carried out as described previously using a rabbit polyclonal antibody against MFSD2A in HEK293 cells transfected overnight with constructs expressing WT and mutant human MFSD2A in a pcDNA3.1 vector [11].
3D structural modeling of P402H
Starting from the published 3D model of MFSD2A WT in the outward occluded state [11], P402 in the WT model was mutated to histidine by sidechain prediction using SCWRL [15]. Then, the neighboring region of H402, from residue 379 to residue 407, was explored in conformational space by loop modeling method implemented in MODELER [16]. The resulted 2500 models were evaluated by the DOPE (discrete optimized protein energy) score [17] and the best ranked model was selected. Finally, known substrates of Mfsd2a including LPC-oleate and molecules not to be transported were docked into the selected 3D model of MFSD2A P402H with DOCK version 3.6 [18].
Results
Exome sequencing identifies a homozygous missense variant in MFSD2A
Exome analysis of the proband yielded 42.8 million mapped reads with a mean coverage of 75×. Following alignment to the reference genome [hg19] and variant calling, we performed a series of filtering steps. These included removing variants, which were called less than 8×, were off-target, on the X chromosome, synonymous, had MAF > 0.5% at ExAC (Exome Aggregation Consortium, Cambridge, MA, URL: http://exac.broadinstitute.org) or MAF > 1% at the Hadassah in-house database. A total of 358 variants remained, and only one of them was homozygous. This was chr1.hg19:g.40433585 C > A, NM_032793.4: c.1205C > A; p.Pro402His in the major facilitator superfamily domain containing 2A (MFSD2A) gene. Using Sanger sequencing, we confirmed homozygosity in both affected individuals and heterozygosity in the parents (Fig. 1a, b). The variant was carried by only one of the ~ 125,000 individuals whose exome analyses were deposited at gnomAD (URL:http://gnomad.broadinstitute.org/, accessed Oct. 2016) and was not present in our in-house database (~ 2650 exome analyses).
The Pro402His variant is predicted to alter structure and transport function
MFSD2A consists of 14 exons encoding the 543 amino acid MFSD2A protein, the sodium-dependent lysophosphatidylcholine (LPC) transporter which is the major transporter for uptake of omega-3 fatty acid docosahexaenoic acid (DHA) into the brain [4]. Since the Pro402 residue is highly conserved throughout evolution (Fig. 1c), we set to determine the effect of the mutation on the function of the MFSD2A protein. To examine the predicted consequences of the P402H mutation on the structure of MFSD2A, we generated 3D structural models of human MFSD2A P402H mutant based on our predicted 3D model of human MFSD2A WT [11]. Mutation of P402 to His resulted in a higher alpha helix content in the neighboring region (helix X) with respect to that in the WT model. The H402 sidechain forms a hydrogen bond with the Y431 sidechain in helix XI (Fig. 1d). These predicted changes in the mutant model may lead to multiple consequences in the protein structure and dynamics, including increased structural rigidity and reduced mobility due to enhanced helix packing.
To investigate how the mutant model may affect transport, we docked to this mutant model known LPC substrates of MFSD2A as well as molecules that are not transported. In comparison to the WT model, the mutant model could not distinguish the substrates from the non-substrates. The docking predicted that the binding geometry of the known substrate LPC-oleate in the mutant model mainly differed from that in the WT model with respect to the orientation of the LPC hydrophobic fatty acid tail (Fig. 1d). Taken together, the P402H mutant would be predicted to be inactivating.
The Pro402His variant disturbs LPC transport and results in increased plasma LPCs
To test the hypothesis that P402H is an inactivating mutation, we expressed wild-type (WT), D97A (sodium binding mutant that is completely inactive as a negative control), and P402H (proband mutant) human MFSD2A in HEK293 cells; of note, these cells do not endogenously express MFSD2A and are thus essentially null. Expression of P402H in HEK293 cells resulted in plasma membrane localization and expression similar to WT MFSD2A (Fig. 3a, b). The cells were then treated with increasing concentrations of [14C]-LPC-DHA. The amount of the [14C]-LPC-DHA transported by the P402H mutant cells was significantly reduced compared to that transported by the WT cells. Moreover, the transport curve of the P402H mutant cells was comparable to that of the D97A mutant cells, indicating complete inactivity of the transporter (Fig. 3c).

Functional analysis of the MFSD2A variant. a Western blot analysis of WT and P402H expressed in HEK293 cells relative to non-transfected control cells (Mock). Duplicate transfected wells of cells are shown for each construct; beta-actin served as a loading control. MFSD2A shows a typical complex banding pattern due to glycosylation. b Immunofluorscence localization of WT and P402H expressed in HEK293 cells indicated that P402H is expressed on the plasma membrane similar to WT MFSD2A. c Transport assay of LPC-[14C]DHA in WT, P402H, and D97A mutants in HEK293 cells. Transport of LPC-[14C]DHA in cells expressing P402H is similar to D97A mutant, indicating complete inactivity
We also performed a targeted plasma lipidomic analysis including quantification of LPCs in plasma in the affected children, parents, and a control, hypothesizing that inactivation of MFSD2A would result not only in LPC depletion within brain tissue but also in a reciprocal increase of plasma LPC levels as previously observed in humans and mice with inactivating MFSD2A mutations [5, 13]. Consistent with this hypothesis, we found that the levels of plasma LPCs were increased in the affected individuals as compared to the control and the mother (Fig. 4; Supplemental Table 2); the significance of the increase of LPC level correlated with the chain length of the LPC and with its degree of unsaturation, consistent with higher affinity of unsaturated fatty acyl chains for MFSD2A compared to the saturated acyl chains [4]. Surprisingly, the father exhibited similarly elevated plasma LPCs as the affected children; repeat plasma sampling confirmed initial findings. This unexpected result may be due to MFSD2A haploinsufficiency coupled with other unknown factors that influence plasma LPC metabolism.

Targeted lipidomic analysis of plasma lysophosphatidylcholine (LPC). Heatmaps show levels of saturated, mono- or polyunsaturated fatty acid species in LPCs in blood plasma. a Low abundant LPC species and b high abundant LPC species in plasma. Fatty acid identity is designated as number of carbons/number of double bonds, e.g., 20:4 denotes a lysophospholipid with 20 carbons and 4 double bonds. Scale bar represents normalized, isotope-corrected LPC levels
Discussion
Recently, two consortia reported MFSD2A mutations in association with clinical findings that may be classified as a genetic leukoencephalopathy [19]. Alakbarzade et al. [2015] reported multiple affected siblings from a Pakistani family who suffered from microcephaly, spastic quadriparesis, and intellectual disability with absent speech. Neuroimaging disclosed paucity of white matter volume, particularly posteriorly around the lateral ventricles [13]. Guemez-Gamboa et al. [2015] studied two affected families, from Libya and Egypt, with microcephaly, developmental delay, intellectual disability, spastic quadriparesis, seizures, and ultimately death within the first few years of life. Brain imaging showed gross hydrocephalus ex evacuo, effacement of the cortical surface, and cerebellar and brainstem hypoplasia and/or atrophy [5]. All three MFSD2A mutations identified were localized to the transmembrane domains of the protein. Cell-based assays showed that the mutant proteins were stably expressed and localized to the plasma membrane, comparable to wild-type protein. However, transport activity of the mutants was reduced in vitro and plasma levels of LPC were increased in affected individuals. The p.Ser339Leu mutant of the Pakistani family led to partial inactivity of the transporter, whereas the p.Thr159Met and p.Ser166Leu alterations in the Libyan and Egyptian families nearly abolished transport function [5, 13]. The hydroxyl group on Thr-159 is essential for sodium binding, and Ser-166 was found to be crucial for normal transport [11]. Accordingly, the more severe phenotype of the latter families was attributed to greater severity of the mutations. Surprisingly, the p.Pro402His mutant identified in this study renders the transporter essentially inactive and leads to increased plasma LPC levels in affected individuals; however, it leads to a non-lethal phenotype with neuroimaging similar to that of the Pakistani family. Additionally, plasma LPC levels in the father (heterozygous for the mutation) were also increased, likely reflecting the fact that LPC levels are regulated by other factors beyond MFSD2A. Consequently, dietary influences or other genetic factors may act to modify the clinical presentation by compensating for reductions in brain DHA levels, and outcome cannot be predicted by the effect of the mutant on transport function alone.
MFSD2A belongs to the major facilitator superfamily (MFS), one of the largest families of transporters with over 70 functionally diverse subfamilies. Ligands range from simple and complex sugars and amino acids to drugs and organic anions [14, 20]. Several MFS transporters have been associated with disease phenotypes, including SLC2A1, MFSD8, and FLVCR2. Deficiency of glucose transporter 1 (GLUT1), otherwise known as solute carrier family 2, member 1 (SLC2A1) [MIM 606777], is associated with epileptic encephalopathy and other neurological phenotypes resulting from decreased glucose concentration in the central nervous system [21]. GLUT1 deficiency is treatable by a ketogenic diet which provides an alternative source of energy to the brain [22]. Mutations in the major facilitator superfamily domain-containing protein 8 (MFSD8) underlie neuronal ceroid lipofuscinosis [MIM 610951], an autosomal recessive neurodegenerative lysosomal storage disorder presenting with developmental regression, seizures, mental and motor regression, speech impairment, ataxia, visual failure, and myoclonus [23]. Finally, feline leukemia virus subgroup C receptor 2 (FLVCR2), a MFS transporter hypothesized to be involved in regulation of growth, calcium exchange, and homeostasis, is mutated in proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (PVHH; MIM 225790), a severe prenatal lethal disorder characterized by hydranencephaly and fetal akinesia deformation sequence with muscular neurogenic atrophy [24].
Transport defects may be treatable by providing supra-physiologic doses of substrates. Mutations in SLC6A8, encoding the creatine transporter, cause X-linked cerebral creatine deficiency syndrome [MIM 300352], manifesting with intellectual disability, developmental delay, epilepsy, and autistic behavior. Magnetic resonance spectroscopy (MRS) in such cases reveals significant reduction of the creatine signal [25, 26]. Supplementation with creatine or with its precursors L-arginine and L-glycine may lead to clinical improvement in individuals with residual protein function, provided that treatment is initiated early [27]. Mutations in SLC1A4, which encodes the ASCT1 transporter responsible for shuttling serine and other neutral amino acids into neuronal cells, are associated with progressive microcephaly, spastic tetraplegia, hypomyelination, and variable seizures [28,29,30]. Similarly, L-serine supplementation has been proposed as a therapy for serine biosynthesis and transport defects [31]. Accordingly, the role of DHA or omega-3 fatty acid dietary supplementation should be assessed in individuals with MFSD2A mutations, especially in cases with residual transport function of the protein.
In conclusion, we report a non-lethal presentation of syndromic microcephaly associated with an inactivating mutation in MFSD2A. This case illustrates the complexity of genotype-phenotype correlations, calls for additional clinical and molecular studies, and suggests the contribution of genetic or nutritional modifying factors to the disease.
Accession numbers
The variant was submitted to ClinVar; the accession number for the DNA variant data will be available prior to publication.
References
Zhao Z, Zlokovic BV (2014) Blood-brain barrier: a dual life of MFSD2A? Neuron 82:728–730
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201
Nałęcz KA (2016) Solute carriers in the blood-brain barier: safety in abundance. Neurochem Res 42:795–809
Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, Zhang X, Wenk MR, Goh EL, Silver DL (2014) Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509:503–506
Guemez-Gamboa, A., Nguyen, L.N., Yang, H., Zaki, M.S., Kara, M., Ben-Omran, T., Akizu, N., Rosti, R.O., Rosti, B., Scott, E., et al. (2015). Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat Genet 47, 809–813
Kawakita E, Hashimoto M, Shido O (2006) Docosahexaenoic acid promotes neurogenesis in vitro and in vivo. Neuroscience 139:991–997
He C, Qu X, Cui L, Wang J, Kang JX (2009) Improved spatial learning performance of fat-1 mice is associated with enhanced neurogenesis and neuritogenesis by docosahexaenoic acid. Proc Natl Acad Sci U S A 106:11370–11375
Coti Bertrand P, O'Kusky JR, Innis SM (2006) Maternal dietary (n-3) fatty acid deficiency alters neurogenesis in the embryonic rat brain. J Nutr 136:1570–1575
Gharami K, Das M, Das S (2015) Essential role of docosahexaenoic acid towards development of a smarter brain. Neurochem Int 89:51–62
Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, Gu C (2014) Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509:507–511
Quek DQ, Nguyen LN, Fan H, Silver DL (2016) Structural insights into the transport mechanism of the human sodium-dependent lysophosphatidylcholine transporter MFSD2A. J Biol Chem 291:9383–9394
Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, Bullock K, Deik AA, Ginty DD, Clish CB, Gu C (2017) Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron 94:581–594.e585
Alakbarzade, V., Hameed, A., Quek, D.Q., Chioza, B.A., Baple, E.L., Cazenave-Gassiot, A., Nguyen, L.N., Wenk, M.R., Ahmad, A.Q., Sreekantan-Nair, A., et al. (2015). A partially inactivating mutation in the sodium-dependent lysophosphatidylcholine transporter MFSD2A causes a non-lethal microcephaly syndrome. Nat Genet 47, 814–817
Berger JH, Charron MJ, Silver DL (2012) Major facilitator superfamily domain-containing protein 2a (MFSD2A) has roles in body growth, motor function, and lipid metabolism. PLoS One 7:e50629
Krivov GG, Shapovalov MV, Dunbrack RL (2009) Improved prediction of protein side-chain conformations with SCWRL4. Proteins 77:778–795
Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815
Shen MY, Sali A (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci 15:2507–2524
Mysinger MM, Shoichet BK (2010) Rapid context-dependent ligand desolvation in molecular docking. J Chem Inf Model 50:1561–1573
Vanderver A, Prust M, Tonduti D, Mochel F, Hussey HM, Helman G, Garbern J, Eichler F, Labauge P, Aubourg P et al (2015) Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab 114:494–500
Reddy VS, Shlykov MA, Castillo R, Sun EI, Saier MH (2012) The major facilitator superfamily (MFS) revisited. FEBS J 279:2022–2035
Seidner G, Alvarez MG, Yeh JI, O'Driscoll KR, Klepper J, Stump TS, Wang D, Spinner NB, Birnbaum MJ, De Vivo DC (1998) GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet 18:188–191
Klepper J, Voit T (2002) Facilitated glucose transporter protein type 1 (GLUT1) deficiency syndrome: impaired glucose transport into brain-- a review. Eur J Pediatr 161:295–304
Siintola E, Topcu M, Aula N, Lohi H, Minassian BA, Paterson AD, Liu XQ, Wilson C, Lahtinen U, Anttonen AK et al (2007) The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am J Hum Genet 81:136–146
Meyer E, Ricketts C, Morgan NV, Morris MR, Pasha S, Tee LJ, Rahman F, Bazin A, Bessières B, Déchelotte P et al (2010) Mutations in FLVCR2 are associated with proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (fowler syndrome). Am J Hum Genet 86:471–478
Salomons GS, van Dooren SJ, Verhoeven NM, Cecil KM, Ball WS, Degrauw TJ, Jakobs C (2001) X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am J Hum Genet 68:1497–1500
van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S, Abulhoul L, Grünewald S, Anselm I, Azzouz H, Bratkovic D, de Brouwer A, Hamel B et al (2013) Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J Med Genet 50:463–472
Dunbar M, Jaggumantri S, Sargent M, Stockler-Ipsiroglu S, van Karnebeek CD (2014) Treatment of X-linked creatine transporter (SLC6A8) deficiency: systematic review of the literature and three new cases. Mol Genet Metab 112:259–274
Srour M, Hamdan FF, Gan-Or Z, Labuda D, Nassif C, Oskoui M, Gana-Weisz M, Orr-Urtreger A, Rouleau GA, Michaud JL (2015) A homozygous mutation in SLC1A4 in siblings with severe intellectual disability and microcephaly. Clin Genet 88:e1–e4
Damseh N, Simonin A, Jalas C, Picoraro JA, Shaag A, Cho MT, Yaacov B, Neidich J, Al-Ashhab M, Juusola J et al (2015) Mutations in SLC1A4, encoding the brain serine transporter, are associated with developmental delay, microcephaly and hypomyelination. J Med Genet 52:541–547
Heimer G, Marek-Yagel D, Eyal E, Barel O, Oz Levi D, Hoffmann C, Ruzzo EK, Ganelin-Cohen E, Lancet D, Pras E et al (2015) SLC1A4 mutations cause a novel disorder of intellectual disability, progressive microcephaly, spasticity and thin corpus callosum. Clin Genet 88:327–335
El-Hattab AW (2016) Serine biosynthesis and transport defects. Mol Genet Metab 118:153–159
Acknowledgements
The authors wish to thank the family for their participation in this study.
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
The work was supported in part by National Research Foundation grants, Singapore NRF2016NRF-NRFI001-15 (to D.L.S.), NRFI2015-05 (to M.R.W.); by the Biomedical Research Council of A*STAR (to H. F.); and by a BMRC-SERC joint grant (BMRC-SERC 112 148 0006, to M.R.W.) from the Agency for Science, Technology and Research, Singapore.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Harel, T., Quek, D.Q.Y., Wong, B.H. et al. Homozygous mutation in MFSD2A, encoding a lysolipid transporter for docosahexanoic acid, is associated with microcephaly and hypomyelination. Neurogenetics 19, 227–235 (2018). https://doi.org/10.1007/s10048-018-0556-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-018-0556-6