

Abstract
Adaptive behaviour necessitates the formation of memories for fearful events, but also that these memories can be extinguished. Effective extinction prevents excessive and persistent reactions to perceived threat, as can occur in anxiety and ‘trauma- and stressor-related’ disorders1. However, although there is evidence that fear learning and extinction are mediated by distinct neural circuits, the nature of the interaction between these circuits remains poorly understood2,3,4,5,6. Here, through a combination of in vivo calcium imaging, functional manipulations, and slice physiology, we show that distinct inhibitory clusters of intercalated neurons (ITCs) in the mouse amygdala exert diametrically opposed roles during the acquisition and retrieval of fear extinction memory. Furthermore, we find that the ITC clusters antagonize one another through mutual synaptic inhibition and differentially access functionally distinct cortical- and midbrain-projecting amygdala output pathways. Our findings show that the balance of activity between ITC clusters represents a unique regulatory motif that orchestrates a distributed neural circuitry, which in turn regulates the switch between high- and low-fear states. These findings suggest that the ITCs have a broader role in a range of amygdala functions and associated brain states that underpins the capacity to adapt to salient environmental demands.
Similar content being viewed by others


Main
Animals are equipped with biological systems to detect and defend against environmental threats. Through associative learning, stimuli that predict threat mobilize defensive responses to mitigate harm7. When threat-associated stimuli become innocuous, these responses adapt through the process of extinction, whereby a new memory is formed that coexists in opposition to the original fear memory4,8,9. Specialized neural systems have evolved to subserve fear and extinction; when imbalanced, these systems can cause persistent reactions to threat.
ITC clusters are densely packed γ-aminobutyric acid-releasing (GABAergic) neurons that surround the basolateral amygdala (BLA), and can be distinguished from neighbouring neurons by their electrophysiological and molecular properties10,11,12,13. The medial ITC clusters, in the intermediate capsule at the BLA–central amygdala (CeA) junction, receive input from the BLA and modulate CeA activity through feedforward inhibition2,14 in a manner that is potentiated by extinction15. Although medial ITC ablation impairs extinction16, recent studies suggest that medial ITC clusters are functionally10 and anatomically17,18 heterogeneous. However, given their small size and location deep in the brain, it has proven difficult to elucidate the functions of these clusters.
ITC clusters signal an aversive stimulus
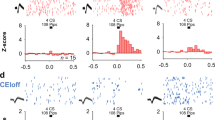
We used in vivo deep-brain imaging to monitor Ca2+ activity in individual ITC neurons in freely moving mice through a miniaturized microscope19. We separately targeted neurons in the dorsal cluster (ITCdm) and ventral cluster (ITCvm) (corresponding to previously defined medial paracapsular ITC (Imp) and main ITC nucleus (IN)10, respectively; for detailed anatomical demarcation of the clusters, see Extended Data Fig. 1, Supplementary Video 1, and Supplementary Table 1). To target ITCdm, BLA, and CeA, we injected an adeno-associated virus (AAV) encoding a Ca2+ indicator (GCaMP6f) into the amygdala (Extended Data Fig. 2a–c) and implanted a graded refractive index (GRIN) lens (Fig. 1a, b). To isolate ITCdm Ca2+ responses, we selected immunohistochemically labelled FOXP2+ neurons from GRIN lens images for analysis by registering maximum intensity projection Ca2+ images to 3D confocal images of cleared tissue (Fig. 1c–e, Supplementary Video 2; see Methods).

a, Endoscopic imaging with a miniaturized microscope in a freely moving mouse. b, AAV encoding CaMK2-GCaMP6f targeted to CeA, BLA and ITCdm. GRIN lens implanted above injection site. c, Post hoc identification of ITCdm neurons. Left, field of view (FOV)-containing horizontal sections. Right, confocal imaging of sections cleared (using the CUBIC protocol) and immunostained with anti-FOXP2 antibody. Grey lines outline GRIN lens position. d, Confocal image of tissue acquired with 5× objective (from mouse in f, g). Orange dotted line outlines area with GRIN lens implanted. Scale bar, 500 μm. DEn: Dorsal endopiriform nucleus; ITCl: lateral ITC. e, Sample shown in d, acquired with 20× objective. x–z and y–z orthogonal views visualized in an isotropic manner. Scale bar, 250 μm. See also Supplementary Video 2. n = 9 mice. f, Maximum-intensity projection image of BLA, CeA, and ITCdm neurons acquired with a miniature microscope. Dashed magenta lines indicate FOXP2-positive area outlined in e with dashed white lines (ITCdm). Image is ~600 × 600 μm. g, A ΔF/F map showing clustered ITCdm activation. See also Supplementary Video 3. h, Miniature microscope imaging in a freely moving Foxp2-Cre mouse. i, AAV encoding CAG-DIO-GCaMP6f was targeted to ITCvm. GRIN lens implanted above injection site. j, Left, averaged Ca2+ footshock (red shading) US responses in all recorded ITCdm (magenta, n = 271 neurons, 9 mice) and ITCvm neurons (green, n = 372 neurons, 6 mice). Right, averaged ΔF/F values. *P = 2.1 × 10−32, Wilcoxon rank-sum test. Centre lines, median; box limits, 25th and 75th percentiles; whiskers, range. k, Trial-averaged ΔF/F Ca2+ time-course of all recorded ITCdm (left) and ITCvm (right) neurons during US omission on the first (early) and last (late) five trials. Dashed red boxes indicate expected US delivery. l, Averaged ΔF/F responses to US omission on first (left) and last five trials (right). Mean ± s.e.m.; *P = 0.0039; NS, P = 0.98; two-sided Wilcoxon rank-sum test. Data in j–l obtained from same animals as in Fig. 2.
Implanted mice were tested in a 5-day fear conditioning and extinction paradigm (see Methods; Fig. 2a). We first limited analysis to shock-evoked Ca2+ responses during fear conditioning (day 2), comprising five pairings of an auditory conditioned stimulus (CS) with a footshock unconditioned stimulus (US). Footshocks reliably elicited strong responses in ITCdm neurons (Fig. 1f, g, j, Extended Data Fig. 2d, Supplementary Video 3), whereas simultaneously recorded neurons in CeA and BLA exhibited sparse and heterogeneous responses (Fig. 1f, g, Extended Data Fig. 2d). In vivo 2-photon Ca2+ imaging performed in head-fixed mice, which provided higher spatial resolution and simultaneous cell-type identification, confirmed that most ITCdm neurons responded to shock (84.5%, 93/110 neurons, n = 5 mice; Extended Data Fig. 3a–e). By contrast, ITCvm neurons did not show footshock responses (Fig. 1h–j, Extended Data Fig. 2e–h). The fraction of shock-responsive neurons was smaller for ITCvm than for ITCdm (P = 1.6 × 10−48, χ2 test). Activity in ITCdm, but not ITCvm, was also highly correlated, during both conditioning and a recording session in the home cage (Extended Data Fig. 2i–n). Furthermore, dual-colour in vivo fibre photometry—which simultaneously measured the activity of ITCdm and ITCvm projections in BLA—found that the activity of these two clusters was largely anti-correlated (Extended Data Fig. 3f–j).

a, Five-day fear conditioning and extinction paradigm. Hab, habituation. b, Freezing in mice with the lens targeted to the ITCdm (magenta, n = 9) or the ITCvm (green, n = 6). ITCdm: FC1–5, P = 4.7 × 10−9 (ascend); Ext1 CS1–25, P = 4.2 × 10−7 (descend); Ext2 CS1–25, P = 1.9 × 10−9 (descend); ITCvm: FC1–5, P = 1.8 × 10−7 (ascend); Ext1 CS1–25, P = 3.9 × 10−5 (descend); Ext2 CS1–25, P = 7.5 × 10−6 (descend); Jonckheere–Terpstra test for trend. Mean ± s.e.m. c, Ca2+ traces for fear and extinction neurons from ITCdm (top) and ITCvm (bottom), respectively. Five-trial averaged traces in colour and traces from individual trials in grey. d, Fractions of fear, extinction (Ext), and extinction-resistant (ExtResist) neurons in ITCdm (n = 271 neurons, 9 mice) and ITCvm (n = 372 neurons, 6 mice). Non-resp, non-responsive. P = 6.3 × 10−20, χ2 test. Fear neurons, P < 0.01; extinction neurons, P < 0.01; extinction-resistant neurons, P > 0.10. e, Averaged Ca2+ responses to CSs in ITCdm fear neurons (n = 86) and ITCvm extinction neurons (n = 67). Vertical dotted lines, CS onset and offset. Fig. 1k shows responses in entire recorded population, including task phase-classified neurons shown here, aligned and baseline-normalized to CS offset. f, Averaged ΔF/F values (solid lines) and freezing (dashed lines) from ITCdm- and ITCvm-targeted mice during each test stage. Mean ± s.e.m. g, Relationships between five-trial averaged ΔF/F values (as in f) and freezing during each test stage for ITCdm fear (left) and ITCvm extinction (middle) neurons. Data points (4 per mouse, 1 per stage) and lines (linear regression fitted) colour-coded by individual mouse (ITCdm n = 9; ITCvm n = 6). Right, correlation coefficient for fitted lines; dots coloured as in left panels. *P = 4.0 × 10−4, Wilcoxon rank-sum test. Centre lines, median; box limits, 25th and 75th percentiles; whiskers, range.
As extinction is based on the absence of an expected aversive stimulus20,21, and ITCs have been ascribed a role in extinction2,16, we investigated whether the clusters respond differentially to shock omission. We examined extinction training (day 3, see Methods; Fig. 2a), where the CS was presented without the US (25 times), and aligned Ca2+ traces to CS offset (the time at which footshocks were delivered during conditioning). During the first five extinction trials, the fraction of neurons that responded to omission of the US was greater in ITCvm (28%) than in ITCdm (4%; P = 5.8 × 10−15, χ2 test), as was the average size of the response (Fig. 1k, l, Extended Data Fig. 3k, l). On late extinction training (trials 21–25), shock-omission responses in ITCvm were markedly reduced, consistent with representing deviations from expectation.
These data show that ITCdm and ITCvm neurons represent the presence and absence of an aversive stimulus in opposing ways. Opposing functional profiles are likely to reflect differential connectivity with upstream brain regions; for example, ITCdm shock responses could be driven by multisensory and pain processing areas, including the medial geniculate nucleus–posterior intralaminar nucleus thalamic complex17,22.
ITC clusters track fear state switches
To investigate whether ITC clusters exhibit differential fear state-dependent responses to conditioned CSs, we measured Ca2+ activity in ITCdm and ITCvm neurons during CS or context habituation (day 1), CS–US fear conditioning (FC, day 2), CS-only extinction training (Ext 1, day 3; Ext 2, day 4) and extinction retrieval (Retrieval, day 5) (Fig. 2a; see Methods). CS-elicited freezing (a readout of fear state) increased over conditioning and was maintained at fear retrieval (early extinction day 3 trials) before decreasing with extinction training, and remaining low on extinction retrieval (Fig. 2b).
We functionally classified ITC neurons into three mutually exclusive categories (as for BLA neurons23). Fear neurons were selectively active during fear retrieval or early extinction; extinction neurons developed responses by late extinction; and extinction-resistant neurons showed sustained activity across extinction training (Fig. 2c, Extended Data Fig. 4a–c; see Methods). ITCdm contained a large fraction of fear neurons (75% of CS-responsive, 32% of recorded), but a small fraction of extinction neurons (15% of CS-responsive, 6% of recorded; P < 0.01) (Fig. 2d, Extended Data Fig. 4d). Conversely, extinction neurons were overrepresented in ITCvm (74% of CS-responsive, 18% of recorded), with a much smaller fraction of fear neurons (18% of CS-responsive, 5% of recorded). There was only a small fraction (less than 5%) of extinction-resistant neurons in each cluster.
Analysis of response dynamics showed that most ITCdm-fear neurons showed an onset-locked CS response during early extinction, suggesting that they rapidly detect the CS’s aversive properties. CS responses in these neurons were much reduced at extinction retrieval (Fig. 2e). Conversely, CS responses in ITCvm-extinction neurons emerged by the second extinction training session (day 4) and were maintained at extinction retrieval (day 5; Fig. 2e). This delayed increase suggests that prolonged extinction training and/or overnight extinction consolidation is required to recruit ITCvm-extinction neurons (as in BLA23).
Further indicating divergent activity in the clusters, the overall amplitude of CS responses in ITCdm-fear neurons positively correlated with freezing, but responses in ITCvm-extinction neurons anti-correlated with freezing (Fig. 2f, g). These correlations were not evident at freezing onset and offset specifically (Extended Data Fig. 5a). Notably, these relationships held when we considered all recorded (not just all classified) neurons (Extended Data Fig. 5b, c). Also, ITCvm neurons did not show CS responses during habituation (day 1), which indicates that ITCvm activity does not simply reflect a low fear state, but represents a diminished fear state produced by extinction (Extended Data Fig. 5d). By contrast, ITCdm neurons showed CS responses during habituation (day 1), which rapidly reduced across five trials, suggesting a habituation-like response to novelty (Extended Data Fig. 5e).
When we aligned responses to US omission, ITCvm-extinction neurons showed strong responses, but ITCdm-fear neurons did not (Extended Data Fig. 5f). This is consistent with the shock-omission responses observed for the entire recorded ITCvm population on test day 3 (Fig. 1k, l), which had decreased by day 5 (Extended Data Fig. 5g). Indeed, responses to US omission were weakly positively correlated with extinguished (day 4) CS responses (R = 0.36), suggesting that there is functional overlap between ITCvm neurons that signal US omission and extinction (Extended Data Fig. 5h). Hence, a prevailing fear state is represented by the relative balance of ITCdm and ITCvm neuron activity.
To assess whether interplay between ITC clusters is evident in other situations characterized by a shift in behavioural state, we used dual-cluster in vivo fibre photometry to record Ca2+ activity simultaneously in ITCdm and ITCvm as mice explored an elevated zero maze (Extended Data Fig. 6a). Transitions between quadrants corresponded to a marked shift in the balance of activity in the two clusters; ITCvm activity increased during transitions from the protected, closed, to the unprotected, open, quadrants (Extended Data Fig. 6b–e). This increase might serve to inhibit defensive behaviour and enable open quadrant exploration, analogous to the inhibition of freezing following extinction. Hence, these data suggest that shifts in ITC cluster activity correspond to transitions between defensive and explorative behavioural states across paradigms.
ITC clusters regulate fear extinction
We next assessed the causal effects of manipulating the balance of ITC cluster activity, using in vivo chemogenetics. A Cre-dependent form of the Gi-coupled κ-opioid receptor DREADD (KORD) was expressed in ITCdm or ITCvm of Foxp2-Cre mice and Cre-negative controls (Fig. 3a–f). The pharmacologically inert ligand salvinorin B (SalB) was systemically injected to selectively inactivate either cluster (for functional verification by ex vivo neuronal recordings in acute brain slices, targeting success rates, and statistics, see Extended Data Figs. 7–9, Methods, and Supplementary Table 2).

a, e, Chemogenetic manipulations of individual ITCs. b, c, ITCvm inactivation. Freezing in Cre+ and Cre− mice (injected with same AAV and ligand). Arrows and coloured shading indicate timing of SalB administration relative to full (50-trial) extinction training (b) or retrieval (c). Freezing on retrieval in b: Cre+, 37.6 ± 5.2%; Cre−, 22.5 ± 3.1%; *P = 0.0013; in c: Cre+, 45.8 ± 2.7%; Cre−, 27.3 ± 4.9%; *P = 0.0005. B, base. d, ITCvm activation. Freezing in Cre+ and Cre− mice. Freezing on retrieval: Cre+, 38.9 ± 6.1%; Cre−, 57.3 ± 4.6%; *P = 0.0079. f, ITCdm inactivation. Freezing on retrieval: Cre+, 30.9 ± 4.8%; Cre−, 55.8 ± 4.2%; *P < 0.0001. Repeated measures ANOVA, Sidak post hoc test. Mean ± s.e.m. NS, no significant differences in freezing on day 1 or day 2.
Inactivation of ITCvm during extinction training impaired extinction memory formation, as shown by higher CS-related freezing in Cre+ than in Cre− mice on (drug-free) extinction retrieval (Fig. 3a, b). In a separate experiment, inactivation of ITCvm during extinction retrieval, but not acquisition, impaired retrieval (Fig. 3a, c). Thus, ITCvm is necessary for the formation and retrieval of extinction memory. Next, we used the Gq-coupled DREADD hM3Dq and systemic injection of clozapine-N-oxide (CNO) to activate ITCvm neurons during extinction retrieval (Fig. 3a, d). Using a partial extinction training protocol (to avoid freezing floor effects; see Methods), we found that activation of ITCvm during extinction retrieval decreased CS-related freezing, relative to Cre− controls, consistent with the facilitation of retrieval. Additionally, selectively expressing hM3Dq or KORD in ITCdm and administering CNO or SalB, respectively, to activate or inactivate ITCdm during extinction retrieval produced effects opposite to these manipulations in ITCvm. Specifically, relative to Cre− controls, ITCdm activation increased CS-related freezing in Cre+ mice on retrieval (Extended Data Fig. 9a, b), whereas ITCdm inactivation decreased freezing (Fig. 3e, f). Finally, when we expressed KORD in ITCdm as well as ITCvm and injected SalB before extinction retrieval, retrieval was impaired (Extended Data Fig. 9c–f), consistent with the earlier finding that pre-retrieval lesioning of both medial clusters impairs extinction retrieval in rats16.
Collectively, these data strongly align with our imaging results, which showed that fear and extinction neurons were overrepresented (albeit not exclusively) in ITCdm and ITCvm, respectively.
ITC clusters are mutually inhibitory
We next tested whether the balance of activity between ITCdm and ITCvm was determined by mutual inhibitory interactions. We injected an AAV expressing Cre-dependent synaptophysin–GFP to target ITCdm or ITCvm in Foxp2-Cre mice (Extended Data Fig. 10a) and identified strong axonal projections and putative synaptic connections in both directions. We saw no somatic expression of GFP in projection-target areas, excluding the possibility of injection leakage or transsynaptic infection (Extended Data Fig. 10b, c). To test for functional mono-synaptic GABAergic connections, we infected ITCdm or ITCvm with an AAV that encoded Cre-dependent channelrhodopsin-2 (ChR2; Extended Data Fig. 10d, e) and photostimulated ChR2-expressing axons in acute brain slices while performing whole-cell patch-clamp recordings of neurons in the non-injected cluster (Fig. 4a).

a, Inter-cluster ex vivo electrophysiology. AAV encoding Cre-dependent ChR2–EYFP targeted to ITCvm or ITCdm neurons in Foxp2-Cre mice. Recordings were performed in one ITC cluster while photostimulating ChR2–EYFP-expressing axons from the other cluster. b, Top, example IPSC (−70 mV holding potential) evoked by photostimulating ChR2–EYFP-expressing axons. Picrotoxin (PTX) abolished IPSCs. Bottom, IPSC (−70 mV holding potential) evoked by photostimulating ChR2-expressing axons. Mono-synaptic IPSCs were isolated by subsequent application of tetrodotoxin (TTX) or TTX and 4-aminopyridine (4-AP). ACSF, artificial cerebrospinal fluid. c, PTX experiments. Magenta lines, ITCvm→ITCdm connections (n = 6 neurons, 6 mice); green lines, ITCdm→ITCvm connections (n = 5 neurons, 4 mice). *P = 0.011, paired t-test. d, TTX + 4-AP application (ITCvm→ITCdm: n = 3 neurons, 3 mice, ITCdm→ITCvm: n = 2 neurons, 2 mice). *P = 0.029, one-way ANOVA and Tukey–Kramer test. e, Long-range circuit ex vivo electrophysiology. ITCdm and ITCvm neurons were targeted with AAV encoding Cre-dependent soCoChR in Foxp2-Cre mice. Two optical fibres were placed above ITC clusters. PL- and IL-projecting BLA neurons were retrogradely labelled with CTB. f, CTB-positive BLA neuron under the microscope for slice recordings. Scale bar, 20 μm. IR, infrared; Epi, epifluorescence. g, IPSCs recorded from PL-projecting (PLp) and IL-projecting (ILp) BLA neurons (−70 mV holding potential) evoked by photostimulation of soCoChR-expressing ITC clusters. PTX blocked IPSCs. h, Mean normalized evoked IPSC (eIPSC) amplitude before and after PTX application (ITCdm→ILp, ITCvm→PLp, ITCvm→unidentified; total, n = 3 neurons, 3 mice). *P = 0.030, paired t-test. BL, baseline. i, Connection probability and mean evoked IPSC amplitudes from ITC clusters to PL- or IL-projecting and unidentified (CTB−) BLA neurons (PLp, n = 24 neurons; ILp, n = 21 neurons; unidentified, n = 38 neurons). ITCdm→PLp and ITCvm→ILp connections were not observed: datapoints superimposed at zero. j, Synaptic conductance of evoked IPSCs from connected neurons. Slopes calculated from IPSC amplitudes acquired at three holding potentials (−60, −70, −80 mV). *P = 0.0028, one-way ANOVA, Tukey-Kramer.
There were synaptic connections in all recorded ITC neurons (ITCvm→ITCdm, n = 15/15; ITCdm→ITCvm, n = 14/14), demonstrating dense reciprocal connectivity between these clusters. Optically evoked synaptic responses were blocked by the GABAA receptor blocker picrotoxin, and these responses had reversal potentials close to the equilibrium potential for chloride, confirming that they were GABAergic inhibitory postsynaptic currents (IPSCs; Fig. 4b, c, Extended Data Fig. 10f). Synaptic responses persisted when we applied the sodium channel blocker tetrodotoxin and the potassium channel antagonist 4-AP, which confirms that the connections are monosynaptic (Fig. 4b, d). We substantiated these results by using spatially focused light to directly stimulate individual clusters (Extended Data Fig. 10g–l). Thus, ITCdm and ITCvm are reciprocally and monosynaptically connected by functional inhibitory synapses.
ITC clusters control amygdala outputs
Interconnectivity between ITCdm and ITCvm could provide a mechanism for regulating shifts in cluster activity to orchestrate extinction by effects on downstream pathways. To explore this idea, we expressed a Cre-dependent, GFP-fused and soma-targeted opsin, soCoChR–EGFP, in ITCdm and ITCvm of Foxp2-Cre mice (Extended Data Fig. 11a), and selectively photostimulated each cluster in acute brain slices using two thin-fibre-coupled LEDs (Fig. 4e, Extended Data Fig. 11b–k). Stimulation of ITCvm neurons (73% of recorded)—but much less of ITCdm neurons (9% of recorded)—evoked a postsynaptic response in medial CeA (CeM), a known ITC target24 (Extended Data Fig. 12a–e). Within CeM, ventrolateral periaqueductal grey (vlPAG)-projecting neurons, a fear-promoting pathway25, preferentially received inhibitory input from ITCvm (Extended Data Fig. 12f–i).
Next, we examined ITC outputs to BLA10,17,18, focusing on infralimbic (IL)- and prelimbic (PL)-projecting neurons given prior evidence that these populations represent extinction-promoting and extinction-opposing BLA output pathways, respectively26. After selectively labelling each population with the retrograde tracer cholera toxin B (CTB) (Extended Data Fig. 12j–m; see Methods), we recorded CTB+ (and neighbouring CTB−) BLA neurons while optically stimulating ITCvm or ITCdm. This revealed GABAergic (picrotoxin-sensitive) connections from ITCvm to PL-projecting BLA neurons and from ITCdm to IL-projecting BLA neurons, but not vice versa (Fig. 4f–h). Connections from ITCdm to IL-projecting BLA neurons were also stronger than those from ITCvm to PL-projecting BLA neurons (Fig. 4i, j).
These data demonstrate a double dissociation in connectivity from specific ITC clusters to distinct amygdala output pathways. Fear-promoting BLA→PL and CeM→vlPAG pathways are more strongly inhibited by ITCvm than ITCdm, whereas ITCdm inhibits the extinction-promoting BLA→IL pathway (Extended Data Fig. 12n). mPFC-projecting BLA neurons preferentially contact IL and PL neurons that project back to BLA27, and these mPFC→BLA projections are differentially involved in fear extinction28. Thus, ITCdm and ITCvm clusters have direct, selective access to major cortex–amygdala loops that regulate fear extinction.
Discussion
Brain mechanisms that enable an appropriate balance between avoiding threat-predicting stimuli and suppressing excessive defensive behaviour after uneventful encounters are essential for survival. We have shown that the ITC clusters in the intermediate capsule boundary between BLA and CeA provide a substrate for achieving this balance during fear extinction. The relative weighting of ITC cluster activity also reflected exploration of protected and unprotected areas in the elevated zero maze. Hence, dynamic changes in the balance of ITC activity may be a general feature of behavioural state shifts during relevant changes in the environment.
The ITCdm and ITCvm clusters form a mutually inhibitory network that precisely accesses functionally distinct amygdala output pathways to orchestrate a broader circuitry that underlies a switch in defensive states. Direct mutual inhibitory connections between ITC clusters offer an efficient mechanism to subserve shifts in relative activity in response to experience with the CS, such that a weighting that favours ITCdm→ITCvm interactions during fear retrieval is dynamically transformed to ITCvm→ITCdm inhibition during extinction. This dynamic process may be modulated by as yet unidentified upstream regions that convey information to the ITCs about the sensory and valence properties of the CS and US. Although dissociable neuronal populations and subregions in other brain regions are known to exert opposing effects on extinction3,4,5, the ITCs appear to be unique in accommodating mutual inhibition as spatially separated inter-cluster connectivity.
A mutually inhibitory motif could amplify small differences in input to an all-or-none output pattern, providing a ‘winner-take-all’ mechanism that could enhance signal-to-noise to generate robust ITC circuit outputs and associated behavioural states29,30. Accordingly, ITCs could serve as a low-dimensional interpreter of sensory, cognitive and emotional information to orchestrate amygdala functions and associated brain states that underpin adaptation to salient environmental demands. ITC neurons could thereby influence a wide range of amygdala-mediated behaviours, including states associated with positive valence, which have overlapping neural representations with fear extinction31,32,33. Finally, given that ITCs are present in the human brain34, aberrant ITC function could contribute to susceptibility to various psychiatric conditions.
Methods
Mice
All animal procedures were performed in accordance with institutional guidelines and with current European Union guidelines, and were approved by the Veterinary Department of the Canton of Basel-Stadt, Switzerland, by the local government authorities for Animal Care and Use (Regierungspräsidium Tübingen, State of Baden-Württemberg, Germany), and by the NIAAA Animal Care and Use Committee. FoxP2-IRES-Cre mice (JAX#030541)35 were used for Cre-dependent expression of viral vectors. For some experiments where a Cre-dependent expression system was not required, Arc-CreER mice36 crossed with a tdTomato reporter line (Ai14) were used in addition to wild-type C57BL/6J mice. For 3D reconstruction, FoxP2-IRES-Cre mice crossed with Ai14 and D1R-Cre (JAX#37156) mice crossed with Ai14 were used. All lines were backcrossed to C57BL/6J. For behavioural experiments, only male mice (aged 1.5–3 months old at the time of virus injection) were used. Both male and female mice (2–5 months old at the time of injection, unless otherwise noted) were used for virus-based circuit tracing, ex vivo electrophysiology and immunohistochemistry. These analyses indicated no discernible differences between males and females; however, more detailed studies will be needed to examine potential sex differences in ITC circuit anatomy and function. Mice were individually housed for at least two weeks before starting behavioural paradigms. Animals were kept in a 12-h light–dark cycle with access to food and water ad libitum. All behavioural experiments were conducted during the light cycle.
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment, except during manual scoring of freezing in recorded videos.
Surgical procedures and viral vector injections
Buprenorphine (Temgesic, Indivior UK Limited; 0.1 mg/kg body weight (BW)) was injected subcutaneously 30 min before the surgery. Mice were anaesthetized using isoflurane (5% for induction, 1–2% for maintenance; Attane, Provet) in oxygen-enriched air (Oxymat 3, Weinmann) and then head-fixed on a stereotactic frame (Model 1900, Kopf Instruments). Lidocaine and ropivacain (Lidocain HCl, Bichsel, 10 mg/kg BW; Naropin, AstraZeneca, 3 mg/kg BW) were injected subcutaneously as local anaesthesia before incision to the skin. Postoperative pain medication included buprenorphine (0.01 mg/ml in the drinking water; overnight) and injections of meloxicam (Metacam, Boehringer Ingelheim; 5 mg/kg subcutaneously) for up to three days if necessary. Eyes were protected with ophthalmic ointment (Viscotears, Alcon). Rectal body temperature was maintained at 35–37 °C using a feedback-controlled heating pad (FHC) while fixed on the stereotactic frame.
Deep brain imaging
AAV2/5.CaMK2.GCaMP6f37 (600 nl, University of Pennsylvania Vector Core, UPenn, for simultaneous imaging of CeA, ITCdm, and BLA) or AAV2/9.CAG.flex.GCaMP6f (200 nl, University of Pennsylvania Vector Core, UPenn, for ITCvm) was unilaterally injected into the amygdala using a precision micropositioner (Model 2650, Kopf Instruments) and pulled volume-calibrated glass capillaries (Drummond Scientific, Cat.-No. 2-000-001, tip diameter about 15 μm) connected to a Picospritzer III microinjection system (Parker Hannifin Corporation) at the following coordinates. For ITCdm, AP −1.4 mm (from bregma), ML −3.3 mm (from bregma), DV 4.4 mm (from pia). For ITCvm, AP −1.6 mm (from bregma), ML −3.1 mm (from bregma), DV 5.0 mm (from pia). After waiting at least 10 min for diffusion of the virus, a gradient-index microendoscope (ITCdm: φ 1.0 × 9.0 mm, 1050-002179, Inscopix GRIN lens; ITCvm: φ 0.6 × 7.3 mm, 1050-002177, Inscopix GRIN lens) was implanted. The larger-diameter lens was used to record from ITCdm to increase the probability of capturing this relatively small cluster in the field of view and to provide simultaneous recordings from BLA and/or CeA neurons from the same lens. The smaller-diameter lens was used to record from ITCvm to reduce damage to the overlying CeA. For dual-colour 2-photon imaging, a cocktail of AAV2/5.CaMK2.GCaMP6f and AAV2/1.CAG.FLEX.tdTomato (University of Pennsylvania Vector Core, UPenn) (600 nl, 10–20:1 mixture ratio) was unilaterally injected, targeting ITCdm. A sterile 21-gauge needle was used to make an incision above the imaging site to avoid excessive brain pressure. The GRIN lens was subsequently lowered into the brain with a micropositioner (coordinates; for ITCdm: AP −1.4 mm (from bregma), ML 3.25 mm (from bregma), DV 4.35 mm (from pia); For ITCvm: AP −1.6 mm (from bregma), ML 3.0 mm (from bregma), DV 4.8 mm (from pia)) using a custom-built lens holder and fixed to the skull using UV light-curable glue (Henkel, Loctite 4305). The surface of the skull was sealed with Vetbond (3M). Dental acrylic (Paladur, Heraeus) was used to further seal the skull and attach a custom-made titanium head bar for fixation during the miniature microscope mounting procedure. The implanted GRIN lens was protected by rapid-curing silicone elastomers.
Chemogenetic manipulation
AAV2/8 (the genome of serotype 2 packaged in the capsid from serotype 8).hSyn.KORD.IRES.mCitrine38 (Addgene plasmid no. 65417, packaged by Virovek (Hayward, CA, USA)) or AAV2/8.hSyn.DIO.hM3D(Gq)-mCherry39 (Addgene viral prep no. 44361-AAV8) was bilaterally injected to Foxp2-Cre experimental mice (Cre+) and Cre− control mice in a volume of 0.15 μl per hemisphere for individual clusters or 0.25 μl per hemisphere for both medial clusters with a 0.5 μl syringe (Neuros model 7001 KH, Hamilton Robotics, Reno, NV, USA) connected to a UMP3 UltraMicroPump and SYS-Micro4 Controller or Nanolitre NL2010MC4 injector (World Precision Instruments, LLC, Sarasota, FL, USA) at the following coordinates; for ITCdm targeting, AP −1.4 mm, ML ±2.95 mm, DV −4.55 mm (from bregma); for ITCvm targeting, AP −1.55 mm, ML ±2.75 mm, DV −5.15 mm; for dual ITCdm and ITCvm targeting, AP −1.43 mm, ML ±2.75 mm, DV −4.75 mm.
Virus-based circuit tracings
AAV2/1.hSyn.flex.synaptophysin-EGFP (packaged by VectorBiolabs, 10–25 nl) was target-injected into either ITCdm or ITCvm (with the same coordinates as shown above) of FoxP2-IRES-Cre mice. Following viral injections, pipettes were left in place for 10 min and retracted slowly to better restrict virus infection. Three to five weeks after the injection, the animals were killed for histological analysis.
Ex vivo electrophysiology
For the KORD functionality assay (Extended Data Fig. 7), AAV2/8.hSyn.KORD.IRES.mCitrine was injected targeting both medial ITC clusters in FoxP2-IRES-Cre mice as stated above. For inter-cluster connectivity assay (Fig. 4a–d, Extended Data Fig. 10), AAV2/1.EF1a.DIO.HChR2(H134R)-EYFP (U.Penn) was target-injected using a precision stereotactic frame (Model 1900, Kopf Instruments) into either ITCdm or ITCvm of FoxP2-IRES-Cre mice as noted above. For analysis of ITC–BLA connectivity (Fig. 4e–j), FoxP2-IRES-Cre mice were unilaterally injected into the amygdala with 500 nl of AAV2/9.hSyn.flex.soCoChR-EGFP40 (Addgene no. 107712-AAV9) (coordinates: AP −1.5 mm, ML ±3.15 mm, DV 4.2–5.0 mm (from pia)) so that both ITCdm and ITCvm would be infected. Mice were allowed to recover for 5–6.5 weeks for the KORD functionality assay (Extended Data Fig. 7), and for 2–4 weeks before ex vivo electrophysiology experiments (Fig. 4, Extended Data Fig. 10). To retrogradely label PL- or IL-projecting BLA neurons, we injected 50 nl of 0.5% cholera toxin B conjugated to Alexa Fluor 555 or 647 (CTB555 or CTB647) was injected into the same hemisphere of the mPFC as the AAV injection at the following coordinates (for PL targeting: AP +1.55 mm, ML ±0.3 mm, DV −1.9 mm (from pia); for IL targeting: AP +1.75 mm, ML ±0.4 mm, DV −2.5 mm (from pia)). To label vlPAG-projecting CeM neurons, we injected 100 nl of 0.2% CTB555 in PBS into the same side of the vlPAG as the AAV injection. To avoid the subcranial midline blood sinus while targeting the vlPAG, we drilled a hole with a diameter of 0.3 mm into the skull at ±1.7 mm the from midline suture, and at the level of the lambda suture. The injection capillary was then slowly lowered at a zenith angle of 26° to the target depth of 3 mm below brain surface. CTB injections and AAV injections were performed in the same surgeries.
Fibre photometry recordings
ITCdm and ITCvm in the same hemisphere were targeted with 25 nl of AAV2/1.CAG.Flex.NES.jRGECO1a.WPRE41 (Addgene) and AAV2/1.CAG.Flex.jGCaMP7f.WPRE42, respectively, or AAV2/1.CAG.Flex.jGCaMP7f.WPRE was injected to target left ITCdm and right ITCvm. Following virus injections, either a prefabricated optical cannula comprising a bare optical fibre (Ø400 µm) and a fibre ferrule (Doric Lenses), or custom-made cannula comprising a bare optical fibre (FP400URT - 0.50 NA, Ø400 µm core multimode fiber, Thorlabs) and an in-house-designed fibre ferrule was implanted into the BLA (AP −1.4 mm, ML ±3.3 mm, DV −4.3 mm (from pia)) for unilateral recordings. For bilateral recordings, ITCdm and ITCvm clusters were directly targeted by two optical cannulas with the same coordinates as the virus injections. The surface of the skull was sealed with Vetbond (3M). Dental acrylic (Paladur, Heraeus) was used to further seal the skull and attach a custom-made titanium head bar for fixation. The ferrules were protected by rapid-curing silicone elastomers.
Immunohistochemistry
Mice were deeply anaesthetized with urethane (2 g/kg; intraperitoneally) and transcardially perfused with PBS followed by 4% paraformaldehyde (PFA) in PBS. Brains were post-fixed overnight at 4 °C and subsequently stored in PBS at 4 °C. Coronal slices (120 μm) containing the BLA were cut with a vibratome (VT1000S, Leica). Sections were washed in PBS for 10 min twice, permeabilized with permeabilization solution43 (0.2% Triton X-100, 20% DMSO, and 23 g/l glycine in PBS) for 30 min at 37 °C, and blocked in blocking solution (0.2% Triton X-100, 10% DMSO, and 6% normal donkey serum (NDS) in PBS) for 30 min at 37 °C. Slices were subsequently incubated in primary antibody solution (1:2,000 rabbit anti-FOXP2 (Abcam, ab16046), 5% DMSO, 3% NDS, 0.2% Triton X-100, 10 mg/l heparin in PBS) for 24 h at 37 °C. After washing for 10 min three times with 0.2% Triton X-100 in PBS, sections were incubated for 24 h at 37 °C with a secondary antibody solution (1:500 donkey anti-rabbit Alexa Fluor 647 (Thermo Fisher Scientific), 3% NDS, 0.2% Triton X-100, and 10 mg/l heparin in PBS. After washing for at least 30 min three times in PBS, sections were mounted on gelatin-coated glass slides and covered with anti-fade mounting medium and coverslips. Sections were scanned using a laser scanning confocal microscope (LSM700, Carl Zeiss) equipped with a 5× air objective (Plan-Apochromat 5×/0.15NA), a 10× air objective (Plan-Apochromat 10×/0.45NA), a 20× air objective (Plan-Apochromat 20×/0.8NA) or a 63× oil-immersion objective (Plan-Apochromat 63×/1.4NA).
3D reconstruction
For 3D reconstruction, thick coronal sections (3–4 mm) containing all the ITC clusters were prepared with a vibratome (VT1000S, Leica). The sections were then cleared using the CUBIC protocol44. A subset of sections was also stained with the FOXP2 antibody. The cleared tissues were imaged with a confocal microscope (Zeiss, LSM700) equipped with a 10× air objective (Zeiss, C Epiplan-Apochromat 10×/0.40NA) or a 20× air objective (Zeiss, LD Plan-NEOFLUAR, 20×/0.4NA), which have a relatively long working distance (>5 mm). The voxel size was 1–3 μm × 1–3 μm × 10 μm (x × y × z) to achieve single-cell resolution. Acquired images were imported to Imaris software (Bitplane) and individual ITC clusters were manually reconstructed plane-by-plane with the manual contour drawing function in ‘Measurement Pro’. Densely packed regions with marker positive neurons were regarded as clusters. The semi-transparent mouse brain scheme was created using Brainrender45.
Deep brain calcium imaging
Miniature microscope imaging
Two to six weeks after GRIN lens implantation, mice were head-fixed to check for sufficient expression of GCaMP6 using a miniature microscope46 (nVista HD, Inscopix). Mice were briefly anaesthetized with isoflurane to fix the microscope baseplate (1050-002192, Inscopix) to the skull using blue light curable glue (Vertise Flow, Kerr). The microscope was removed and the baseplate was capped with a baseplate cover (1050-002193, Inscopix). Mice were habituated to the brief head-fixation on a running wheel for miniature microscope mounting for at least three days before the behavioural paradigm. Imaging data were acquired using nVista HD software (Inscopix) at a frame rate of 20 Hz with an LED power of 10–60% (0.9–1.7 mW at the objective, 475 nm), analogue gain of 1, and with 1080 × 1080 pixels. For individual mice, the same imaging parameters were kept across days. Timestamps of imaging frames and behavioural stimuli were collected for alignment using the Omniplex system (Plexon).
Fear conditioning and extinction paradigm
Two different contexts were used. Context A (extinction context) consisted of a clear cylindrical chamber (diameter: 23 cm) with a smooth floor, placed in a dark-walled sound-attenuating chamber under dim light conditions. The chamber was scented and cleaned with 1% acetic acid. Context B (fear conditioning context) contained a clear square chamber (26.1 × 26.1 cm) with an electrical grid floor (Coulbourn Instruments) for footshock delivery, placed in a light-coloured sound-attenuating chamber with bright light conditions, and was scented and cleaned with 70% ethanol. A stimulus isolator (ISO-Flex, A.M.P.I.) was used for the delivery of direct current (DC) shock. Both chambers contained overhead speakers for delivery of auditory stimuli, which were generated using a System 3 RP2.1 real time processor and SA1 stereo amplifier with RPvdsEx Software (all Tucker-Davis Technologies). Cameras (Stingray, Allied Vision) for tracking animal behaviour were also placed in both chambers. Radiant Software (Plexon) was used to generate precise TTL pulses to control behavioural protocols and all the TTL signals including miniscope frame timings were recorded by Plex Control Software (Plexon) to synchronize behavioural protocols, behavioural tracking, and miniscope imaging.
On day 1 (habituation), the mice were first imaged in their homecage for 10 min, and then placed in context A and exposed to five CSs (29 pips, 200 ms, 6-kHz pure tone, repeated at 1 Hz) following a 4-min baseline period. The ITI (inter tone interval) was 30 s. On day 2 (fear conditioning), mice were first imaged in their homecage for 10 min, and then fear conditioned in context B by pairing five CSs with an unconditioned stimulus (US; 1-s footshock, 0.65 mA DC; applied 800 ms after termination of the last (29th) pip) with a variable ITI of 60–90 s (after a 2-min baseline period). Animals remained in context B for 1 min after the last CS–US pairing. On days 3 and 4 (extinction 1 and 2), fear memory was extinguished in context A. After a 4-min baseline period, animals were exposed to 25 CSs (ITI: 30 s). On day 5, extinction memory was assessed with five CS presentations (ITI: 30 s) following a 4-min baseline period.
Verification of implant sites (clearing-based, for ITCdm)
Upon completion of the behavioural paradigm, mice were lightly anaesthetized with isoflurane, head-fixed and 3D-scanned with a 2-photon microscope (Ultima, Bruker) equipped with a Ti:Sapphire laser (Insight X3, Spectra Physics) and a 16× water objective lens (0.8NA, Nikon) or a 25× water objective lens (1.05NA, Olympus) through the GRIN lens. After 2-photon microscopy, mice were transcardially perfused (as above). GRIN lenses and head bars were removed and brains were post-fixed in 4% paraformaldehyde overnight at 4 °C. Horizontal thick sections (1.5–2.0 mm) containing the imaging site were cut with a vibratome (VT1000S). Sections were cleared and stained against FOXP2 using the CUBIC protocol44 and the same combination of antibodies as above, and then imaged with a confocal microscope (Zeiss, LSM700) equipped with a 5× air objective (Zeiss, Plan-Apochromat 5×/0.15NA) or a 20× air objective (Zeiss, LD Plan-NEOFLUAR, 20×/0.4NA), which have a relatively long working distance. Using blood vessels and fibres as landmarks, a maximum intensity projection of the video acquired with a miniature microscope, a 3D stack acquired with the 2-photon microscope, and a 3D stack acquired with confocal microscopy were manually matched, and then, the area of the miniature microscope video corresponding to the FOXP2-positive area between BLA and CeA in the confocal image was assigned as ITCdm. Mice with obvious misplacement of the GRIN lens and with no FOXP2 signal were excluded from the analysis. For cases in which CeA was imaged in the periphery of the FOV, only data from ITCdm and BLA were analysed.
Verification of implant sites (slice-based, for ITCvm)
Upon completion of the behavioural paradigm, mice were transcardially perfused (as above). GRIN lenses and head bars were removed, and brains were post-fixed in 4% paraformaldehyde overnight at 4 °C. Coronal sections (120 μm) containing the imaging site were cut with a vibratome (VT1000S), immediately mounted on glass slides and coverslipped. To verify the GRIN lens position, sections were imaged with a confocal microscope as described above. Images were matched against a mouse brain atlas47. Mice with misplacement of the GRIN lens were excluded from the analysis.
Two-photon calcium imaging
In a subset of mice used for miniature microscope experiments, awake head-fixed 2-photon imaging sessions were performed through the same implanted GRIN lenses using a 2-photon microscope (Ultima Investigator, Bruker) equipped with a Ti:Sapphire femtosecond laser (InSightX3, SpectraPhysics) and a 16×/0.8NA objective (N16XLWD-PF, Nikon). GCaMP6f and tdTomato were excited at 920 nm, and signals were filtered with a 517–567-nm band-pass filter and a 573–613-nm band-pass filter, respectively. Care was taken to shield the microscope objective and the photomultipliers from stray light. Images were obtained using Prairie View software (Bruker). Square regions (approximately 800 μm × 800 μm) were imaged at 512 × 512 pixels at 30 Hz with the resonant-galvo mode. Several planes were acquired from each animal. Aversive shocks (1 s, 2.00 mA DC) were generated by a stimulus isolator (ISO-Flex, A.M.P.I.) and applied through a pair of electrodes located on the skin of the face. Shock presentations were synchronized with image acquisition by TTLs generated by the Prairie View software.
Chemogenetic manipulations and behavioural testing
Ligand injections
FoxP2-IRES-Cre mice and Cre-negative littermate controls were injected with a Cre-dependent AAV, as described above, and both groups were given salvinorin B (SalB) or clozapine-N-oxide (CNO) to control for potential behavioural effects of the compounds per se. To activate KORD, mice were subcutaneously injected with 10 mg/kg SalB (catalogue no. 11488; Cayman Chemical Company, Ann Arbor, MI, USA) 20 min before the behavioural testing. SalB was dissolved in DMSO at a 1 μl/g BW injection volume using a 50 μl Hamilton Syringe (Hamilton Company, Reno, NV, USA). To activate hM3Dq, mice were injected intraperitoneally (i.p.) with 3 mg/kg CNO (catalogue no. C0832-5MG; Sigma Aldrich) 30 min before the behavioural testing. CNO was dissolved in 10% DMSO in saline and injected at a volume of 10 μl/g BW.
Fear conditioning and extinction paradigm
Behavioural tests started 3.5 to 6 weeks after virus delivery. We did not find a systematic relationship between the period between surgery and the effects of KORD either in vivo or ex vivo. Prior to testing, each mouse was handled for 2 min per day for 6 days and habituated to subcutaneous (for KORD) or intraperitoneal (for CNO) saline injections for 3 days. Fear conditioning was conducted in a 30 × 25 × 25-cm operant chamber (Med Associates, Inc., Fairfax, VT USA) with metal walls and a metal rod floor. To provide an additional olfactory cue, the chamber was cleaned between subjects with a 79.5% water/19.5% ethanol/1% vanilla extract solution. Following a 3-min baseline period, 3 pairings (60–90 s ITI) of CS (30 s, 80-dB white noise) and US (2 s 0.6 mA, co-terminating with the CS) were presented, followed by a 120-s stimulus-free period. The Med Associates Freeze Monitor system controlled CS and US presentation. Extinction training was conducted the following day (day 2) in a 27 × 27 × 14-cm operant chamber with transparent walls and a floor covered with wood chips, cleaned between subjects with a 99% water/1% acetic acid solution and housed in a different room from training. After a 3-min baseline period, either 50 (‘full extinction’) or 10 (‘partial extinction’) CSs were presented (5-s ITI)48. Extinction memory retrieval was tested the next day (day 3) in the same context as extinction training with 5 CS presentations (5 s inter-CS interval), following a 3-min baseline period.
Post-behaviour examination of virus expression
To examine virus expression at the completion of behavioural tests, mice were terminally overdosed with pentobarbital and transcardially perfused with PBS followed by 4% PFA in PBS. Brains were post-fixed over night at 4 °C and subsequently stored in 0.1 M phosphate buffer for 1–2 days at 4 °C. Coronal sections (50 μm) were cut with a vibratome (Classic 1000 model, Vibratome, Bannockburn, IL, USA). Brain sections were incubated in 1% sodium borohydride followed by blocking solution (10% normal goat serum (Vector Laboratories) and 2% bovine serum albumin (MP Biomedicals, Santa Ana, CA, USA) in 0.05 M PBS with 0.2% Triton X-100) for 2 h at room temperature (20 °C), then incubated at 4 °C overnight in a cocktail of primary antibodies: 1) chicken anti-GFP (1:2,000 dilution, Abcam cat no. 13970) to aid visualization of KORD; 2) Living Colours DsRed polyclonal antibody (1:1,000 dilution, Clontech Labs cat no. 632496) to aid visualization of hM3Dq; and 3) rabbit anti-FOXP2 (1:2,000 dilution, Abcam cat no.16046) to visualize ITC. The next day, sections were incubated in a cocktail of secondary antibodies: Alexa 488 goat anti-chicken IgG (1:1,000 dilution, Abcam cat no.150169) and goat anti-rabbit Alexa 555 IgG (1:1,000 dilution, Abcam cat no. A21428). Sections were mounted and coverslipped with Vectashield HardSet mounting medium with DAPI (Vector Laboratories, Inc., Burlingame, CA, USA). Sections were imaged with an Olympus BX41 microscope (Olympus, Center Valley, PA, USA) and a Zeiss LSM 700 confocal microscope (Carl Zeiss Microscopy, Thornwood, NY, USA).
Images from the 139 Cre+ injected mice were inspected to determine whether virus expression was evident and restricted to the ITC cluster targeted and, if so, whether expression was present in one or both hemispheres. Targeting success rates (rounded to the nearest %) were as follows: ITCvm-targeting: 41% (21% bilateral, 20% unilateral); ITCdm-targeting: 35% (17% bilateral, 17% unilateral), dual-cluster targeting: 42% (23% bilateral, 19% unilateral). Mice with absent expression were excluded from the analysis. Data from mice with unilateral or bilateral expression were combined, as analysis of freezing behaviour on retrieval indicated generally similar results (Supplementary Table 2).
To illustrate expression patterns, virus expression in each mouse was overlayed to a corresponding coronal atlas image47 and expression within each binned 35 μm × 40 μm segment transformed to a numerical value (expression present = 1, absent = 0). Images were then aggregated across mice included in the final behavioural analysis (separately for ITCvm-targeted, ITCdm-targeted and dual-cluster targeted) to generate a population heatmap of expression indicating the fraction of animals with expression at each binned segment (0: no mice expressed; 1: all mice expressed) (Extended Data Figs. 8, 9).
Ex vivo electrophysiology
KORD functionality verification and connectivity assays between ITCs
Mice were deeply anaesthetized with 3% isoflurane in oxygen and decapitated. The brain was rapidly extracted and cooled down in ice-cold slicing ACSF containing (in mM): 124 NaCl, 2.7 KCl, 26 NaH2CO3, 1.25 NaH2PO4, 10 MgSO4, 2 CaCl2, 18 d-glucose, 4 ascorbic acid, equilibrated with carbogen (95% O2/5% CO2). Coronal brain slices (320 μm) containing the amygdala were cut in ice-cold slicing ACSF with a sapphire blade (Delaware Diamond Knives) on a vibrating microtome (Microm HM 650 V, Thermo Scientific). Slices were collected in a custom-built interface chamber with recording ACSF containing (in mM): 124 NaCl, 2.7 KCl, 26 NaH2CO3, 1.25 NaH2PO4, 1.3 MgSO4, 2 CaCl2, 18 d-glucose, 4 ascorbic acid, equilibrated with carbogen. Slices were recovered at 37 °C for 40 min and stored at room temperature. Whole-cell patch-clamp recordings were performed in a submersion chamber under an upright microscope (Olympus BX51WI), where slices were superfused with recording ACSF at 31 °C. Recordings were performed using an Axon Instruments Multiclamp 700B amplifier and a Digidata 1440A digitizer (Molecular Devices). Glass micropipettes (6–8 MΩ resistance) were pulled from borosilicate capillaries (ID 0.86 mm, OD 1.5 mm, Science Products, Germany).
For KORD functionality verification, the resting membrane potential and spikes were recorded in current-clamp mode with K-gluconate-based internal solution containing (in mM): 130 K-gluconate, 5 KCl, 4 Mg-ATP, 0.4 Na-GTP, 10 Na2-phosphocreatine, 10 HEPES, 0.6 EGTA (290–295 mOsm, pH 7.2–7.3). Signals were low-pass filtered at 10 kHz and digitized at 20 kHz. Salvinorin B (Tocris Bioscience) was prepared as 1mM stock solution in DMSO, diluted to 100 nM in ACSF on the day of recording, and perfused via the bath. Data from ITCdm and ITCvm cells were pooled for analysis.
For connectivity assays between ITC clusters, postsynaptic currents were recorded in voltage-clamp configuration with caesium-based internal solution containing (in mM): 115 Cs-methanesulfonate, 20 CsCl, 4 Mg-ATP, 0.4 Na-GTP, 10 Na2-phosphocreatine, 10 HEPES, 0.6 EGTA (290–295 mOsm, pH 7.2–7.3). Signals were low-pass filtered at 2 kHz (4-pole Bessel) and digitized at 10 kHz. Series resistance was monitored and data rejected if it changed by more than 25% over the course of an experiment. ChR2 was stimulated with 470-nm light pulses (0.5–1 ms duration) delivered by an LED (CoolLED pE) through the microscope’s submersion objective (Olympus LUMPlanFL 60×, 1.0 NA). By restricting the light path aperture, illumination was confined to a small spot within the slice (80-μm diameter). Drugs were prepared from frozen stocks, diluted in ACSF and applied via superfusion for pharmacological experiments. Picrotoxin (PTX, 100 μM, Sigma, Germany) was used to block GABAergic transmission, tetrodotoxin (TTX, 1 μM, Biotrend, Germany) was first applied alone, and subsequently together with 4-aminopyridine (4-AP, 100 μM, Tocris Bioscience) to assess monosynapticity of evoked postsynaptic currents49. In most experiments with on-cluster stimulation, 20 μM DNQX (Biotrend, Germany) was added to the recording ACSF to block fast glutamatergic transmission.
Connectivity assays from ITCs to BLA
Mice were deeply anaesthetized (ketamine 250 mg/kg and medetomidine 2.5 mg/kg bodyweight, injected intraperitoneally) and transcardially perfused with ice-cold (0–2 °C) NMDG-based solution50,51 (in mM: 2.5 KCl, 1.25 NaH2PO4 (1 H2O), 10 MgSO4 (7 H2O), 0.5 CaCl2 (2 H2O), 30 NaHCO3, 20 HEPES, 25 glucose, 93 NMDG, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 93 HCl, oxygenated with 95% O2/5% CO2, pH 7.3–4) for 3 min. The brain was rapidly removed from the skull, and coronal brain slices (300 μm) containing ITCs and BLA were prepared in ice-cold NMDG-based solution with a vibrating-blade microtome (HM650V, Microm) equipped with a sapphire blade (Delaware Diamond Knives). For recovery, slices were kept in the dark for 10 min at 33 °C in an interface chamber containing the NMDG-based solution and afterwards at room temperature (20–24 °C) in HEPES-holding solution (in mM: 20 HEPES, 92 NaCl, 2.5 KCl, 1.25 NaH2PO4 (1 H2O), 2 MgSO4 (7 H2O), 2 CaCl2 (2 H2O), 25 glucose, 30 NaHCO3, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, oxygenated with 95% O2/5% CO2, pH 7.4) for at least 1 h until recording.
Experiments were performed in a submerged chamber on an upright microscope (BX50WI, Olympus) superfused with oxygenated recording ACSF (in mM: 123 NaCl, 3 KCl, 1.25 NaH2PO4 (1 H2O), 1 MgCl2 (6 H2O), 2 CaCl2 (2 H2O), 11 glucose, 26 NaHCO3, 10 μM CNQX and 10 μM CPP) at a perfusion rate of 4 ml/min at 32 °C. EGFP+ ITC clusters and CTB+ BLA projection neurons were visualized using epifluorescence and a 5× air immersion objective (LMPlanFI 5×/0.15 NA, Olympus) or a 40× water immersion objective (LumPlanFl 40×/0.8 NA, Olympus). Patch electrodes (for BLA, 3–5 MΩ; for ITCs, 7–8 MΩ;) were pulled from borosilicate glass tubing and filled with internal solution (for voltage-clamp recordings in mM: 110 CsCl, 30 K-gluconate, 1.1 EGTA, 10 HEPES, 0.1 CaCl2, 4 Mg-ATP, 0.3 Na-GTP, 4 QX-314 chloride, pH 7.3; for current-clamp recordings in mM: 130 K-methylsulfate, 10 HEPES, 10 Na-phosphocreatine, 4 Mg-ATP, 0.3 Na-GTP, 5 KCl, 0.6 EGTA, pH 7.3). In some experiments, 0.4% biocytin was added to visualize recorded neurons post hoc. Voltage-clamp recordings from BLA neurons were acquired in whole-cell mode at a holding potential of −70 mV. soCoChR-expressing ITCdm and ITCvm were photostimulated using blue LEDs (PlexBright Blue, 465 nm, with LED-driver LD-1, Plexon) connected to optical fibres (0.39NA, FT200EMT, Thorlabs) positioned above them. Five blue light pulses of 0.6 mW (at the fibre-tip) with 10-ms duration were applied at a frequency of 0.5 Hz. Inhibitory postsynaptic currents were averaged across at least 10 light pulses. In some slices, PTX (100 μM) was administered with the recording ACSF to the last cell recorded from. In current-clamp recordings from ITCs, spikes were evoked from a holding potential of about −70 mV with the same blue light stimulation protocol as above. Data were acquired with a Multiclamp 700B amplifier, Digidata 1440A A/D converter and pClamp 10 software (all Molecular Devices) at 20 kHz and filtered at 10 kHz. Data were excluded if the access resistance exceeded 25 MΩ or 10% of the membrane resistance or changed by more than 20% during the recordings. All chemicals were purchased from Sigma-Aldrich except for CNQX, CPP and QX-314 (Tocris Bioscience).
Post hoc histological analysis for connectivity assays between ITCs
Following recordings, slices were sandwiched between two cellulose nitrate filter papers (0.45-μm pore size, Sartorius, Germany) and fixed in 4% PFA in 0.1 M phosphate buffered saline (PBS, pH 7.4) overnight. To confirm cluster-specific ChR2–EYFP expression, fixed slices were re-sectioned at 60 μm thickness with a vibrating microtome (Microm HM 650 V, Thermo Scientific). Free-floating sections were incubated in blocking solution (20% horse serum, 0.03% Triton X-100 in PBS) for 2 h at room temperature, and then overnight in PBS with 10% blocking solution and primary antibodies at a 1:1,000 dilution; in all cases against EYFP fused to channelrhodopsin2 (goat anti-GFP polyclonal), in some batches also against FOXP2 (rabbit anti-FOXP2 polyclonal). On the next day, sections were washed 3 times for 10 min in PBS, and incubated for 2 h at room temperature in PBS with 10% blocking solution and secondary antibodies at 1:1,000 dilution; in all cases with donkey-anti-goat-Alexa488, for FOXP2 staining additionally with donkey-anti-rabbit-Alexa555. Slices were then washed 3 times for 10 min in PBS, incubated for 25 min in 1:500 Neurotrace 435/455 in PBS, and washed 3 more times for 10 min in PBS. Sections were then mounted on glass slides in mounting medium (Vectashield, Vector Laboratories) and imaged with a confocal microscope (LSM 710, Zeiss).
Fibre photometry recording
A modified Doric Fibre Photometry system was used to perform the recordings. Three different excitation wavelengths were used (465 nm for Ca2+-dependent jGCaMP7f activity, 560 nm for Ca2+-dependent jRGECO1a activity, and 405 nm to record an isosbestic, Ca2+-independent, reference signal that serves to correct for photo-bleaching and movement-related artefacts52). The light intensity used for each wavelength was <100 μW at the fibre tip. Optical patch cables were extensively photo-bleached before recordings to reduce auto-fluorescence and a lock-in modulation/demodulation procedure was used to improve the signal-to-noise ratio and spectral separation of the fluorescent signals53. Data were digitized and recorded at 20 kHz.
Elevated zero-maze paradigm
The elevated zero-maze with a 55 diameter and a 5 cm width corridor was composed of two open and two closed quadrants, which are equivalent to 90° each, elevated at 50 cm above the ground. Two external walls and two internal walls with 15 cm height for the closed quadrants were made of opaque plastic and transparent plexiglass, respectively. Mice were allowed to freely explore the maze for 15 min. Mouse behaviours were video-recorded with a camera controlled with a custom written code in Bonsai54, while synchronized with photometry recording via TTL pulses. The videos were post hoc analysed by a second custom written code in Bonsai and the position of the mouse in the open versus closed quadrants determined as the central position of the body mass.
Quantification and statistical analysis
Analysis of behaviour during calcium imaging
All behavioural sessions were recorded using an overhead camera (Stingray, Allied Vision) and Cineplex software (Plexon) at 40 Hz. Mice were tracked using contour tracking, and freezing behaviour was automatically scored with the assistance of a frame-by-frame analysis of pixel change (Cineplex Editor, Plexon). The minimum duration threshold for freezing behaviour was set to 2 s. Automatically detected freezing epochs were manually inspected on the video recording to eliminate false-positive and false-negative freezing bouts (for example, during micro-grooming episodes or owing to motion artefacts caused by cable movement, respectively). Annotated freezing data were then temporally aligned with miniscope-imaged data and finally downsampled to 10 Hz.
Analysis of behaviour for chemogenetic manipulation
Behavioural sessions were video-recorded using The Med Associates Freeze Monitor system (fear conditioning) or with a GoPro HERO7 video camera (extinction training and retrieval). Based on the recorded videos, freezing was manually scored at 5-s intervals throughout testing by an experienced observer blinded to genotype. The mean numbers of freezing observations per baseline period and 5× CS blocks were converted to a percentage ((number of freezing observations/total number of observations per period) × 100) for analysis. Mice with freezing scores on any CS block more than 3 s.d. from the group mean were excluded from the analysis.
Analysis of miniature microscope calcium imaging data
Imaging frames were down-sampled to 540 × 540 pixels at 10 Hz and normalized across the whole frame by dividing each frame by a fast Fourier transform bandpass-filtered version of the frame using ImageJ (NIH)19. Motion artefact correction and PCA/ICA-based cell detection were performed with MosaicAPI (Inscopix) for MATLAB. Edges of cell masks were then smoothed by open/close functions. Raw calcium traces were obtained by averaging all the pixel values in each mask. Slow drift of the baseline signal over the course of minutes was removed using a low-cut filter (Gaussian, cutoff 2–4 min). Relative changes in calcium fluorescence F were calculated by ∆F/F0 = (F – F0)/F0 (where F0 is median fluorescence of entire trace). When a cell pair showed (1) distance between centroids <15 pixels and (2) correlation coefficient between entire time courses >0.6, one of the pair was manually eliminated to avoid double-counting. Finally, pairs of the mask and the traces of all the cells were manually inspected to exclude false-positive or false-negative cell mask allocation. For fear extinction, images from five consecutive days were concatenated before the motion correction procedure. When there was major displacement of the FOV and the same set of neurons could not be identified across days, animals were excluded.
To define responsive cells, trial-averaged Ca2+ signals were compared between the stimulus and a temporally equivalent baseline period using a Wilcoxon rank-sum test with a significance threshold of P < 0.05. The windows for averaging were 2 s (shock), 4 s (CS offset), and 29 s (CS), from stimulus onset. AUC (area under the curve) ΔF/F values were calculated using these same windows. A cell was classified as a fear neuron if it exhibited a significant tone response after fear conditioning (CS 1–5 during Ext1 (day 3), when freezing levels were highest), but no significant response after extinction (CS 21–25 during Ext2 (day 4), when freezing levels were lowest), and vice versa for extinction neurons. Finally, units were classified as extinction-resistant neurons if they displayed a significant tone response at both time points23,55,56.
Analysis of two-photon calcium imaging data
The images were analysed using custom software written in-house running on MATLAB (Mathworks). First, acquired images were motion corrected with NoRMCorre57. Artefacts caused by bi-directional scanning were corrected by shifting odd lines and even lines. Cell outlines were manually identified using ROI-manager function in ImageJ, based on motion corrected videos and maximum intensity projections. Time courses of individual cells were extracted by summing the pixel values within the cell outlines. Slow drift of the baseline signal over the course of minutes was removed using a low-cut filter (Gaussian, cutoff 2–4 min). Raw calcium signal time courses were corrected to minimize out-of-focus signal contamination: neuropil signals were subtracted from cell body signals after multiplying by a fixed contamination ratio (0.7, as previously described)58,59.
Analysis of fibre photometry recordings
Analysis was performed with custom programs written in MATLAB (Mathworks). Data with obvious motion artefacts in the isosbestic channel were discarded. De-modulated raw Ca2+ traces were down-sampled to 1 kHz and then de-trended using a low-cut filter (Gaussian, cutoff 2–4 min) to correct for slow drift of the baseline signal due to bleaching. Filtered traces were Z-scored by mean and standard error of the entire trace.
Analysis of slice whole-cell recordings
Data were analysed with custom written codes in Python 3.7 (Anaconda distribution) running the pyABF module, custom written Macros in IgorPro (Wavemetrics, USA), or custom programs written in MATLAB (Mathworks) using the abfload (Harald Hentschke/Forrest Collman) function. Connectivity was defined by comparing signal to baseline (10 trials), while statistical significance was assessed by Wilcoxon signed-rank test.
Statistical analyses and data presentation
All data are expressed as the mean ± s.e.m. unless stated otherwise. Box plots represent the median and 25th and 75th percentiles, and their whiskers represent the data range. In some of the plots, outlier values are not shown for clarity of presentation, but all data points and animals were always included in the statistical analysis. Two-sided Wilcoxon rank-sum test was used to compare two independent groups. For paired comparison, we used Wilcoxon signed-rank test or paired t-test based on distribution and n size of the data. An ANOVA was performed when more than two groups were compared, which was followed by the Tukey–Kramer post hoc method. For multiple comparisons against a baseline, Dunnett’s test was used. For comparing two distributions, the Kolmogorov–Smirnov test was applied. For trend, the Jonckheere–Terpstra test was used. For categorical data, χ2 test or Fisher’s exact test (when sample sizes and expected frequencies were small) was applied. For repeated observations of freezing scores, a repeated-measures ANOVA was used, which was followed by the Sidak post hoc method. All data analysed by ANOVA were normally distributed according to a Kolmogorov–Smirnov test on the ANOVA residuals. Throughout the study, P < 0.05 was considered statistically significant. No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those generally used in the field.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The data that support the findings of this study are available at: https://data.fmi.ch/PublicationSupplementRepo/. Source data are provided with this paper.
Code availability
Custom-written codes used to analyse data from this study are available upon reasonable request from the corresponding authors.
References
Craske, M. G. et al. Anxiety disorders. Nat. Rev. Dis. Primers 3, 17024 (2017).
Duvarci, S. & Pare, D. Amygdala microcircuits controlling learned fear. Neuron 82, 966–980 (2014).
Milad, M. R. & Quirk, G. J. Fear extinction as a model for translational neuroscience: ten years of progress. Annu. Rev. Psychol. 63, 129–151 (2012).
Orsini, C. A. & Maren, S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci. Biobehav. Rev. 36, 1773–1802 (2012).
Tovote, P., Fadok, J. P. & Lüthi, A. Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci. 16, 317–331 (2015).
Li, B. Central amygdala cells for learning and expressing aversive emotional memories. Curr. Opin. Behav. Sci. 26, 40–45 (2019).
LeDoux, J. E. Emotion circuits in the brain. Annu. Rev. Neurosci. 23, 155–184 (2000).
Bouton, M. E. Context, ambiguity, and unlearning: sources of relapse after behavioral extinction. Biol. Psychiatry 52, 976–986 (2002).
Herry, C. et al. Neuronal circuits of fear extinction. Eur. J. Neurosci. 31, 599–612 (2010).
Busti, D. et al. Different fear states engage distinct networks within the intercalated cell clusters of the amygdala. J. Neurosci. 31, 5131–5144 (2011).
Collins, D. R. & Paré, D. Spontaneous and evoked activity of intercalated amygdala neurons. Eur. J. Neurosci. 11, 3441–3448 (1999).
Millhouse, O. E. The intercalated cells of the amygdala. J. Comp. Neurol. 247, 246–271 (1986).
Waclaw, R. R., Ehrman, L. A., Pierani, A. & Campbell, K. Developmental origin of the neuronal subtypes that comprise the amygdalar fear circuit in the mouse. J. Neurosci. 30, 6944–6953 (2010).
Royer, S., Martina, M. & Paré, D. An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J. Neurosci. 19, 10575–10583 (1999).
Amano, T., Unal, C. T. & Paré, D. Synaptic correlates of fear extinction in the amygdala. Nat. Neurosci. 13, 489–494 (2010).
Likhtik, E., Popa, D., Apergis-Schoute, J., Fidacaro, G. A. & Paré, D. Amygdala intercalated neurons are required for expression of fear extinction. Nature 454, 642–645 (2008).
Asede, D., Bosch, D., Lüthi, A., Ferraguti, F. & Ehrlich, I. Sensory inputs to intercalated cells provide fear-learning modulated inhibition to the basolateral amygdala. Neuron 86, 541–554 (2015).
Mańko, M., Geracitano, R. & Capogna, M. Functional connectivity of the main intercalated nucleus of the mouse amygdala. J. Physiol. (Lond.) 589, 1911–1925 (2011).
Grewe, B. F. et al. Neural ensemble dynamics underlying a long-term associative memory. Nature 543, 670–675 (2017).
Luo, R. et al. A dopaminergic switch for fear to safety transitions. Nat. Commun. 9, 2483 (2018).
Salinas-Hernández, X. I. et al. Dopamine neurons drive fear extinction learning by signaling the omission of expected aversive outcomes. eLife 7, e38818 (2018).
Strobel, C., Marek, R., Gooch, H. M., Sullivan, R. K. P. & Sah, P. Prefrontal and auditory input to intercalated neurons of the amygdala. Cell Rep. 10, 1435–1442 (2015).
Herry, C. et al. Switching on and off fear by distinct neuronal circuits. Nature 454, 600–606 (2008).
Paré, D. & Smith, Y. GABAergic projection from the intercalated cell masses of the amygdala to the basal forebrain in cats. J. Comp. Neurol. 344, 33–49 (1994).
Tovote, P. et al. Midbrain circuits for defensive behaviour. Nature 534, 206–212 (2016).
Senn, V. et al. Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron 81, 428–437 (2014).
McGarry, L. M. & Carter, A. G. Inhibitory gating of basolateral amygdala inputs to the prefrontal cortex. J. Neurosci. 36, 9391–9406 (2016).
Arruda-Carvalho, M. & Clem, R. L. Prefrontal-amygdala fear networks come into focus. Front. Syst. Neurosci. 9, 145 (2015).
Koyama, M. & Pujala, A. Mutual inhibition of lateral inhibition: a network motif for an elementary computation in the brain. Curr. Opin. Neurobiol. 49, 69–74 (2018).
Machens, C. K., Romo, R. & Brody, C. D. Flexible control of mutual inhibition: a neural model of two-interval discrimination. Science 307, 1121–1124 (2005).
Felsenberg, J. et al. Integration of parallel opposing memories underlies memory extinction. Cell 175, 709–722.e15 (2018).
Solomon, R. L. & Corbit, J. D. An opponent-process theory of motivation. I. Temporal dynamics of affect. Psychol. Rev. 81, 119–145 (1974).
Zhang, X., Kim, J. & Tonegawa, S. Amygdala reward neurons form and store fear extinction memory. Neuron 105, 1077–1093.e7 (2020).
Braak, H. & Braak, E. Neuronal types in the basolateral amygdaloid nuclei of man. Brain Res. Bull. 11, 349–365 (1983).
Rousso, D. L. et al. Two pairs of ON and OFF retinal ganglion cells are defined by intersectional patterns of transcription factor expression. Cell Rep. 15, 1930–1944 (2016).
Guenthner, C. J., Miyamichi, K., Yang, H. H., Heller, H. C. & Luo, L. Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron 78, 773–784 (2013).
Chen, T. W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013).
Vardy, E. et al. A new DREADD facilitates the multiplexed chemogenetic interrogation of behavior. Neuron 86, 936–946 (2015).
Armbruster, B. N., Li, X., Pausch, M. H., Herlitze, S. & Roth, B. L. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl Acad. Sci. USA 104, 5163–5168 (2007).
Shemesh, O. A. et al. Temporally precise single-cell-resolution optogenetics. Nat. Neurosci. 20, 1796–1806 (2017).
Dana, H. et al. Sensitive red protein calcium indicators for imaging neural activity. eLife 5, e12727 (2016).
Dana, H. et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 16, 649–657 (2019).
Renier, N. et al. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910 (2014).
Susaki, E. A. et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157, 726–739 (2014).
Claudi, F. et al. Visualizing anatomically registered data with brainrender. eLife 10, e65751 (2020).
Ghosh, K. K. et al. Miniaturized integration of a fluorescence microscope. Nat. Methods 8, 871–878 (2011).
Franklin, K. B. J. & Paxinos, G. The Mouse Brain in Stereotaxic Coordinates (Academic, 1997).
Bukalo, O. et al. Prefrontal inputs to the amygdala instruct fear extinction memory formation. Sci. Adv. 1, e1500251 (2015).
Petreanu, L., Huber, D., Sobczyk, A. & Svoboda, K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat. Neurosci. 10, 663–668 (2007).
Tanaka, Y., Tanaka, Y., Furuta, T., Yanagawa, Y. & Kaneko, T. The effects of cutting solutions on the viability of GABAergic interneurons in cerebral cortical slices of adult mice. J. Neurosci. Methods 171, 118–125 (2008).
Ting, J. T., Daigle, T. L., Chen, Q. & Feng, G. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol. 1183, 221–242 (2014).
Lerner, T. N. et al. Intact-brain analyses reveal distinct information carried by SNc dopamine subcircuits. Cell 162, 635–647 (2015).
Gunaydin, L. A. et al. Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551 (2014).
Lopes, G. et al. Bonsai: an event-based framework for processing and controlling data streams. Front. Neuroinform. 9, 7 (2015).
An, B. et al. Amount of fear extinction changes its underlying mechanisms. eLife 6, e25224 (2017).
Baek, J. et al. Neural circuits underlying a psychotherapeutic regimen for fear disorders. Nature 566, 339–343 (2019).
Pnevmatikakis, E. A. & Giovannucci, A. NoRMCorre: an online algorithm for piecewise rigid motion correction of calcium imaging data. J. Neurosci. Methods 291, 83–94 (2017).
Hagihara, K. M., Murakami, T., Yoshida, T., Tagawa, Y. & Ohki, K. Neuronal activity is not required for the initial formation and maturation of visual selectivity. Nat. Neurosci. 18, 1780–1788 (2015).
Kerlin, A. M., Andermann, M. L., Berezovskii, V. K. & Reid, R. C. Broadly tuned response properties of diverse inhibitory neuron subtypes in mouse visual cortex. Neuron 67, 858–871 (2010).
Gründemann, J. et al. Amygdala ensembles encode behavioral states. Science 364, eaav8736 (2019).
Acknowledgements
We thank F. Ferraguti for discussions on ITC cluster nomenclature; J. Felsenberg, P. Tovote and A. Uematsu for reading and commenting on earlier versions of the manuscript; T. Mrsic-Flogel, M. Häusser and B. Roska for discussion and serving as thesis committee members for K.M.H.; E. Susaki for sharing the CUBIC protocol; Y. Tanaka for sharing the NMDG-based slice preparation protocol; L. Choo for slice physiology demonstration; the GENIE Project for making GCaMP6f, jGCaMP7f and jRGECO1a materials available; B. Roth for making KORD and DREADD materials available; K. Deisseroth for making the ChR2 material available; E. Boyden for making the soCoChR material available; all members of the Lüthi, Holmes, and Ehrlich laboratories for discussion and support, especially T. Eichlisberger for excellent animal care; and the FMI animal facility and microscopy core facility (FAIM), especially L. Gelman and J. Eglinger, and the IT department for constant support. This work was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 669582), and a Swiss National Science Foundation core grant (310030B_170268) (all to A. Lüthi); by the NIAAA Intramural Research Program (to A.H.); by the German Research Foundation DFG EH 197/3-1 (to I.E.); and by the Novartis Research Foundation.
Author information
Authors and Affiliations
Contributions
K.M.H., O.B., A. Lüthi and A.H. conceived the project. K.M.H. performed all the calcium imaging experiments, virus-based circuit mapping, and the slice experiments in Fig. 4e–j, Extended Data Figs. 11, 12 and analysed the data. O.B. performed chemogenetic manipulation experiments and analysed the data with A. Limoges, T.R., T.C., A.M. and C.W. M.Z. and A.A.-A. performed the slice experiments in Fig. 4a–d, Extended Data Figs. 7, 10 and analysed the data. I.E. conceived the slice experiments and supervised M.Z. and A.A.-A. N.K. performed fibre photometry with K.M.H. M.M. helped to establish the slice physiology setup and provided K.M.H. with technical training and advice on opsin selection. L.S.Z. and R.D.P. provided the Foxp2-Cre mouse line. K.M.H. prepared figures and drafted the manuscript. K.M.H., A. Lüthi and A.H. wrote the manuscript. All the authors commented on and finalized the manuscript and figures. A. Lüthi and A.H. supervised the entire project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Vadim Bolshakov and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 3D reconstruction of individual ITC clusters.
a, Schematic of a mouse brain volume. R, rostral; C, caudal; D, dorsal; V, ventral. b, 3D reconstructions were separately obtained via 1) anti-FOXP2 immunostaining; 2) a FoxP2-Cre × Ai14 mouse; and 3) a D1R-Cre × Ai14 mouse, in triplicate for each method. ITCap, apical ITC; ITCl, lateral ITC; ITCav, anteroventral ITC; ITCint, internal ITC; ITCpv, posteroventral ITC; M, medial; L, lateral; Scale bar, 300 μm. c, Example planes of cleared 3D tissue obtained from a wild-type mouse stained with an anti-FOXP2 antibody covering the anterior–posterior axis of ITC clusters. Bregma levels are indicated above each panel. Scale bar, 300 μm. See also Supplementary Video 1 and Supplementary Table 1.
Extended Data Fig. 2 Ca2+ imaging from ITCdm and ITCvm.
a–c, Histological validation of GCaMP6f expression. Wild-type mice (not used for in vivo imaging; n = 2) were injected with AAV-CaMK2-GCaMP6f and killed after 1 month of expression. Thin slices (120 μm) were cut and stained with an anti-FOXP2 antibody. In addition to BLA and CeA neurons, most of the FOXP2-positive ITCdm neurons expressed GCaMP6f. Blue arrow shows a putative large, FOXP2-negative ITC neuron. Scale bars, 500 μm (a), 100 μm (b), and 10 μm (c). d, Example Ca2+ traces from neurons in the CeA, ITCdm, and BLA that were simultaneously imaged with a miniature microscope during fear conditioning. Data obtained from the same mice as in Fig. 1d–g. Grey shading indicates CS presentation (30 s); red line indicates footshock US presentation (1 s). e, f, Histological validation of GCaMP6f expression in ITCvm neurons. Foxp2-Cre mice were injected with an AAV encoding Cre-dependent GCaMP6f, implanted with a GRIN lens, and killed after behavioural experiments. Thin slices (120 μm) were cut and stained with an anti-FOXP2 antibody. Scale bars, 200 μm (e), 50 μm (f). Similar results were obtained with all six mice. g, Summary of histologically confirmed GRIN lens implantation locations for ITCvm recordings. Animals with off-target implantations were excluded from analysis. h, Example Ca2+ traces from neurons in the ITCvm cluster during fear conditioning. Grey shading indicates CS presentation (30 s); red line indicates footshock US presentation (1 s). Images of GCaMP6f expression from the same mouse are shown in e, f. i, j, Correlation matrices of all simultaneously recorded neuron pairs in CeA, ITCdm, and BLA (i), or in ITCvm (j) from representative animals. The entire recording session (11 min) was used. k, Distributions of correlation coefficients from CeA–CeA, ITCdm–ITCdm, and BLA–BLA pairs. Arrowheads indicate medians of the distributions. l, Distribution of correlation coefficients from ITCvm–ITCvm pairs. Arrowhead indicates median of the distribution. m, Summary of medians of correlation coefficient distributions shown in k. Solid lines indicate individual animals in which CeA, the ITCdm cluster, and BLA were simultaneously imaged (n = 3). Dotted lines indicate animals in which only the ITCdm cluster and BLA were simultaneously imaged (n = 5). *P = 0.007, one-way ANOVA followed by Tukey–Kramer test. n, Medians of correlation coefficient distribution. The same analysis as in m was applied to data from home-cage recording sessions. ITCdm neurons also show a trend towards a higher correlation in the absence of CS or US stimulation. *P = 0.12, one-way ANOVA.
Extended Data Fig. 3 Two-photon imaging and fibre photometry.
a, Schematic showing dual-colour in vivo 2-photon imaging through the implanted GRIN lens. ITCdm neurons were labelled by co-injection of an AAV expressing Cre-dependent tdTomato in Foxp2-Cre mice. A GRIN lens was implanted above ITCdm and surrounding BLA and CeA to record Ca2+ responses to aversive skin shocks (USs). b, Mean projected FOV. Green, GCaMP6; magenta, tdTomato. Dashed lines indicate the intermediate capsule surrounding ITCdm. c, Heatplot of Ca2+ responses (ΔF/F) to US presentations showing clustered activation of ITCdm neurons. d, Regions of interest (ROIs) corresponding to ITCdm (magenta) and BLA (blue) neurons. ROI numbers correspond to traces shown in e. e, Example Ca2+ traces from ITCdm and BLA neurons. Red lines indicate US presentations (1 s). We confirmed that face skin shock used in this experiment and footshock similarly activated ITCdm neurons (data not shown). f, Schematic illustrating in vivo fibre photometry in a freely moving mouse. g, ITCdm and ITCvm clusters were targeted with AAVs encoding Cre-dependent jRGECO1a and jGCaMP7f, respectively. Recording fibres were placed in BLA to simultaneously monitor axon terminal Ca2+ dynamics of ITCdm and ITCvm axons. Isosbestic control signals were recorded in the blue channel. h, Example traces of simultaneously recorded dual-colour Ca2+ signals and a control signal during a fear conditioning session. i, Cross-correlation traces between two simultaneously recorded Ca2+ signals. Dark grey lines represent five-trial averaged traces and light grey lines represent individual trials. j, Top, minimum peak values of cross correlation. Bottom, lags of the minimum peak points (n = 2 mice). k, l, Activity heatplot of trial-averaged responses (left, first 5 trials; right, last 5 trials) to footshock US omissions of all the recorded ITCdm (k; n = 271 neurons, from 9 mice) or ITCvm neurons (l; n = 372 neurons, from 6 mice) aligned by CS offset. Cells were sorted according to their averaged ΔF/F responses in the first five trials.
Extended Data Fig. 4 Population activity of ITCdm and ITCvm neurons.
a, b, Heatplots of all recorded neurons: a, 271 ITCdm neurons from 9 mice and b, 372 ITCvm neurons from 6 mice throughout the entire 5-day fear conditioning–extinction paradigm. Neurons were sorted by their classification into fear, extinction, extinction resistant, and non-responsive neurons. c, Example Ca2+ traces from a fear neuron and an extinction neuron from ITCdm and ITCvm, respectively, for all time points indicated in a, b. Habituation, Ext1: 1–5, Ext2: 21–25, and Retrieval time points are duplicated in Fig. 2c. d, Scatter plots visualizing distributions of tone responses of all the recorded ITCdm neurons (left) and ITCvm neurons (middle and right) during Ext1: 1–5 and Ext 2: 21–25 trials. Functional cell-types (fear, extinction, and extinction-resistant neurons) are plotted with different colours; non-responsive neurons in grey (compare with Fig. 2d).
Extended Data Fig. 5 Properties of ITCdm and ITCvm neurons during fear extinction.
a, Trial-averaged ΔF/F Ca2+ responses of ITCdm fear and ITCvm extinction neurons aligned to freezing onset and offset. b, Schematics illustrating the analysis shown c. Correlation coefficients between trial-by-trial (in total, 50 CS trials) freezing levels and CS-response amplitudes of all the recorded neurons across two days of extinction. Distributions of those correlation coefficients (one value from each neuron) were first normalized in each animal, then mean ± s.e.m. values were acquired from all ITCdm and ITCvm recordings and visualized. c, The distribution of correlation coefficients for ITCdm neurons was skewed towards 1, indicating that a larger fraction showed a CS response pattern positively correlated with freezing behaviour. By contrast, ITCvm neurons exhibited the opposite tendency; a response pattern anti-correlated with freezing behaviour. *P = 1.64 × 10−40, Kolmogorov–Smirnov test. d, Trial-averaged ΔF/F Ca2+ responses of ITCvm neurons aligned to tone on habituation (day 1). e, Left, averaged Ca2+ responses to tone CS onset in ITCdm. Vertical dotted lines indicate onsets and 2-s time-window of CSs for AUC analysis. Middle, single-trial average of all the ITCdm neurons. Right, AUC quantification of single-trial responses shown in the middle panel. P = 0.027, one-way ANOVA. f, Trial-averaged ΔF/F Ca2+ responses of ITCdm fear and ITCvm extinction neurons aligned to US omission on day 3. Early, CS1–5; late, CS21–25. Dotted red boxes indicate the expected timing of US delivery. g, Trial-averaged ΔF/F Ca2+ responses of all recorded ITCvm neurons aligned to US omission on day 5. The dotted red box indicates the expected timing of US delivery. h, Relationship between CS responses during last five trials of extinction training on day 4 and US omission responses on day 3 in ITCvm extinction neurons shows a weak positive correlation.
Extended Data Fig. 6 Simultaneous photometry recordings from ITCdm and ITCvm during state transition.
a, Schematic of an elevated zero-maze apparatus. b, Top, example Z-scored Ca2+ traces of simultaneously recorded ITCdm (magenta) and ITCvm (green) neurons with the corresponding location in the maze indicated with grey (closed quadrant) and blue (open quadrant) shading. Bottom, difference between the ITCdm and ITCvm signals. c, Left, percentage of time spent in closed and open quadrants. Right, total number of transitions from closed to open quadrants (15 ± 3.9, mean ± s.e.m.) or from open to closed quadrants. n = 5 mice. Centre line, median; box limits, 25th and 75th percentiles; whiskers, range. d, Averaged activity of ITCdm or ITCvm neurons in the closed or open quadrants did not correlate with the total time spent in open quadrants. Regarding the larger variability in ITC activities in open quadrants, we note that the time an animal spent in open quadrants was, on average, much shorter than that in closed quadrants (c). As such, activity in open quadrants was to a lesser extent averaged out, resulting in higher variability. Eight sessions from n = 5 mice. e, Top, averaged Ca2+ traces during transitions between quadrants. Changes in the balance between ITCdm and ITCvm parallel a transition from a closed to an open quadrant. Bottom, difference between the ITCdm and ITCvm signals. Briefly stated, the results of this experiment make at least three points: 1) the role of the ITC clusters extends beyond signalling acute, cue-triggered fear states to conditioned fear stimuli to encompass state transitions during exploration of a potentially threatening environment; 2) the clusters exhibit markedly divergent, opposing responses to transitions between protected and unprotected environments, as they do in response to conditioned and extinguished fear cues; and 3) the pattern of results shows that increased ITCvm activity occurs when the animal moves from the protected, closed, to the unprotected, open, quadrants. Potentially, this increase in ITCvm neuron activity may serve to inhibit defensive behaviour and thereby enable exploration of the unprotected open quadrants, analogous to the inhibition of freezing behaviour following extinction. Such cross-task neuronal function is not without precedent; for example, BLA neuronal activity during exploration of the open arm in an elevated plus-maze is anti-correlated with conditioned freezing behaviour in the same mice60.
Extended Data Fig. 7 Ex vivo validation of KORD.
a, Fluorescence (mCitrine, left) and infrared (IR, right) images from a slice used for recordings in a Foxp2-Cre mouse injected with AAV-DIO-KORD-mCitrine into ITCdm and ITCvm. Inset, infrared image from a recorded ITCdm neuron. Scale bars, 200 μm. b, Representative current-clamp traces illustrating supra-threshold responses to a +60-pA current injection and continuous recording of the resting membrane potential (RMP) during application of Salvinorin B (SalB, 100 nM) from control (mCitrine−) and KORD-infected (mCitrine+) ITC neurons. Scale bars, RMP, 5 mV, 1 min; current steps, 20 mV, 200 ms. c, SalB application induced significant hyperpolarization of the membrane in mCitrine+ neurons (n = 10 neurons from 4 mice) at both time points versus baseline (12.5 min, *P = 0.003; 17.5 min, *P = 0.0001; two-sided Dunnett’s test). Changes in membrane potential were not significant in the mCitrine− control neurons (n = 5 neurons from 3 mice) at both time points versus baseline (12.5 min, P = 0.99; 17.5 min, P = 0.84; Dunnett’s test). Mean ± s.e.m. d, Comparison of membrane potential changes (ΔRMP) 17.5 min after application of SalB. Changes in KORD-infected neurons (mCitrine+) were significantly different from control (*P = 0.013, Wilcoxon rank-sum test; n = 5 neurons from 3 mice and n = 11 neurons from 5 mice for mCitrine− and mCitrine+, respectively). Mean ± s.e.m. e, Comparisons of spike rates in response to +60-pA current injections before and 17.5 min after application of SalB. KORD-infected neurons (mCitrine+, n = 11) significantly reduced spike rate (from 26.2 ± 1.69 Hz to 20.1 ± 3.30 Hz; *P = 0.016, Wilcoxon signed-rank test). By contrast, mCitrine− neurons (n = 4 neurons from 3 mice) did not show a significant reduction in spike rate (from 25.3 ± 2.89 Hz to 23.7 ± 2.27 Hz; P = 0.25, Wilcoxon signed-rank test. Mean ± s.e.m.
Extended Data Fig. 8 Histology for chemogenetic manipulations of distinct ITC clusters.
a, Examples of histological validation of KORD–mCitrine expression in ITCvm neurons across multiple anterior-to-posterior coronal sections. Following all the behavioural experiments (Fig. 3), mice were killed and slices (50 μm) cut and stained with an anti-FOXP2 antibody. Scale bar, 200 μm. b, Examples of histological validation of hM3Dq–mCherry expression in ITCvm neurons across multiple anterior-to-posterior coronal sections. Scale bar, 200 μm. c, Heatmaps illustrating virus expression aggregated across Cre+ mice in a and b. Scale bar indicates the fraction of animals exhibiting expression at each locus (0: no mice expressed; 1: all mice expressed). d, Examples of histological validation of KORD–mCitrine expression in ITCdm neurons across multiple anterior-to-posterior coronal sections. Scale bar, 200 μm. e, Examples of histological validation of hM3Dq–mCherry expression in ITCdm neurons across multiple anterior-to-posterior coronal sections. Scale bar, 200 μm. f, Heatmaps illustrating virus expression aggregated across Cre+ mice in d and e. Scale bar indicates the fraction of animals exhibiting expression at each locus (0: no mice expressed; 1: all mice expressed). Bregma levels are noted above the panels in a and d.
Extended Data Fig. 9 Additional chemogenetic manipulations of ITCdm and ITCvm clusters.
a, Chemogenetic manipulations. AAV encoding Cre-dependent hM3Dq was targeted to ITCdm neurons in Foxp2-Cre mice. b, ITCdm activation. Freezing on retrieval: Cre+, 45.7 ± 7.6%; Cre−, 28.5 ± 3.3%; *P = 0.0044. c, Schematic showing the dual-cluster chemogenetic manipulation experiment. An AAV encoding Cre-dependent KORD was targeted to both ITCvm and ITCdm neurons in Foxp2-Cre mice. d, Freezing behaviour of experimental (Cre+) and control (Cre−) mice; controls were injected with the AAV and administered the ligand. Arrows and colour-shadings indicate the timing of SalB administration before extinction retrieval. Freezing levels on extinction retrieval (day 3): Cre+, 46.4 ± 4.3%; Cre−, 27.3 ± 2.7%; *P < 0.0001, repeated-measures ANOVA followed by Sidak post hoc test. N.S., not significantly different on day 1 or day 2 freezing. Mean ± s.e.m. e, Examples of histological validation of KORD–mCitrine expression in ITCvm and ITCdm neurons across multiple anterior-to-posterior coronal sections. Following behavioural experiments, mice were killed and slices (50 μm) cut and stained with an anti-FOXP2 antibody. Scale bar, 200 μm. f, Heatmaps illustrating virus expression aggregated across Cre+ mice. Scale bar indicates the fraction of animals exhibiting expression at each locus (0: no mice expressed; 1: all mice expressed).
Extended Data Fig. 10 Reciprocal inhibitory connections between ITC clusters.
a, Inter-cluster virus-based tracing (ITCdm-targeted example shown, same strategy for ITCvm targeting). AAV encoding Cre-dependent synaptophysin–GFP targeted to ITCdm or ITCvm neurons in Foxp2-Cre mice. b, c, Confocal images from ITCdm (b) or ITCvm (c) injections. FOXP2 expression visualized using immunohistochemistry (IHC). Injections repeated in five (ITCdm), and four (ITCvm) mice. Scale bars: 500 μm (b1, c1); 200 μm (b2, 3, c2, 3). d, e, Histological validation of selective ChR2 expression in ITCdm (d1–3) and ITCvm (e1–3). Scale bars: 500 μm (d1, e1); 50 μm (d3, e3). Repeated for all mice shown in Fig. 4. f, Representative example of IPSCs recorded at different holding potentials from an ITCdm neuron upon photostimulation of ChR2-EYFP fibers from ITCvm. g, i, Schematics illustrating ex vivo slice electrophysiological experiments with direct cluster stimulation. The ITCvm (g) or ITCdm (i) cluster was targeted with an AAV encoding Cre-dependent ChR2. Recordings were performed in the other cluster while the ChR2-positive cluster or the surrounding area was selectively stimulated with aperture-restricted stimulation (see Methods). h, j, Images taken from the recording setup illustrating positions of on- and off-cluster stimulation for ITCvm while recording in ITCdm and vice versa. k, Summary of PTX application experiments (n = 3 from 3 mice). Magenta lines represent ITCvm→ITCdm connections and green lines represent ITCdm→ITCvm connections. *P = 0.005, paired t-test. l, Summary of experiments shown in g–j. On-cluster photostimulation evoked significantly larger IPSCs than off-cluster stimulation. Note that we used the highest value out of the four off-cluster stimulation sites. *P = 0.0016, paired t-test.
Extended Data Fig. 11 Selective ex vivo ITC cluster stimulation.
a, Histological validation of selective soCoChR expression. Scale bars, 50 μm. Repeated for n = 3 mice. b, Schematic illustrating ex vivo slice electrophysiological experiments. ITCdm and ITCvm neurons were targeted with an AAV encoding Cre-dependent soCoChR. Two optical fibres for photostimulation were placed above the ITC clusters. Whole-cell recordings were performed from neurons in the ITCdm, ITCvm, and ITCl clusters. To block synaptic currents, CPP (10 μM), CNQX (10 μM) and PTX (100 μM) were applied. c–e, Example responses to photostimulation of ITCdm or ITCvm clusters recorded under current-clamp configuration from ITCdm (c), ITCvm (d), and ITCl (e) neurons. f, Summary of experiments shown in b–e. All recorded ITCdm and ITCvm neurons fired action potentials selectively in response to photostimulation with the corresponding fibre. ITCl neurons did not fire action potentials upon photostimulation of ITCdm or ITCvm clusters. g, Schematic illustrating experiments wherein the position of the optical fibre was systematically moved away from the optimal location (X = 0 μm). To block synaptic currents, CPP (10 μM), CNQX (10 μM), and PTX (100 μM) were applied. These experiments further confirmed the spatial resolution of the photostimulation configuration. h, Example voltage-clamp recordings from an ITC neuron showing IPSCs evoked at different positions of the optical fibre. i, Left, quantification of peak IPSC amplitudes of the neuron shown in h. Right, normalized by the value at X = 0 μm. j, Summary of all recorded neurons (n = 3 from 3 mice). k, Summary of additionally performed current-clamp recordings (n = 3 from 3 mice).
Extended Data Fig. 12 ITC to CeM connectivity.
a, Schematic illustrating the experimental design used to assess connectivity between ITCdm or ITCvm neurons and CeM neurons. To isolate IPSCs, CPP (10 μM) and CNQX (10 μM) were applied. b, Example voltage-clamp recording from a CeM neuron showing selective IPSC elicited by stimulation of the ITCvm but not ITCdm. c, Summary of connectivity from ITCdm and ITCvm to CeM (n = 11 neurons from 4 mice). *P = 7.5 × 10−3, Fisher’s exact test. d, Evoked IPSC amplitudes recorded from CeM neurons (at −70 mV holding potential) in response to stimulation of ITCdm or ITCvm clusters. e, Synaptic conductance calculated based on IPSC amplitudes recorded at three different holding potentials (−60, −70, and −80 mV). f, Example injections of CTB555 targeted to the ventrolateral periaqueductal grey (vlPAG). Scale bar, 200 μm. g, Schematic illustrating the experimental design used to assess connectivity between ITCdm or ITCvm neurons and CeM neurons projecting to the vlPAG. vlPAG-projecting CeM neurons were retrogradely labelled with CTB injections into the vlPAG. Whole-cell recordings were performed in CTB+ CeM neurons while photostimulating incoming axons expressing ChR2–EYFP from either ITCdm or ITCvm. h, Summary of connectivity from ITCdm and ITCvm to vlPAG-projecting CeM neurons (n = 15 neurons from 4 mice and n = 10 neurons from 4 mice). *P = 0.0001, Fisher’s exact test. i, Evoked IPSC amplitudes recorded from CTB+ CeM neurons (at 0 mV holding potential) in response to stimulation of axons from ITCdm or ITCvm. j, Example injections of CTB555 and CTB647 targeted to the PL or IL, respectively. Scale bar, 500 μm. k, Example BLA histology showing PLp and ILp BLA neurons. Both subpopulations are located in the medial part of the BLA. Scale bar, 200 μm. l, Mostly non-overlapping PLp and ILp subpopulations; orange arrow indicates an example of a double-labelled neuron. Scale bar, 50 μm. m, Quantification of double-labelled neurons (n = 3 mice). Mean ± s.e.m. n, State-dependent functional changes in amygdala circuitry regulated by ITC clusters. Node size indicates activity level, connection width represents putative functional connection strength.
Supplementary information
Supplementary Tables
This file contains Supplementary Table 1, a cross-study comparison of ITC cluster nomenclature and Supplementary Table 2, statistical analyses summary for chemogenetic experiments.
Video 1
: 3D-reconstruction of individual ITC clusters. An example 3D-reconstruction of ITC clusters from a cleared FoxP2-Cre crossed with tdTomato reporter mouse brain. The same sample as shown in Extended Data Fig.1b is presented. Note, this intercross mouse line shows tdTomato signal in some CeA neurons and blood vessels in addition to ITCs, likely because of developmental transient expression of FoxP2.
Video 2
: Post-hoc identification of ITCdm in a 3D confocal image of a cleared tissue. An example confocal 3D image stack of cleared and stained tissue acquired with a 20x objective. The same sample as shown in Fig.1d, e is presented.
Video 3
: Spatially clustered robust aversive shock responses in ITCdm Example Ca2+ movies from simultaneously recorded BLA, CeA, and ITCdm regions. ITCdm shows spatially clustered and robust responses to 5 trials of aversive footshock presentation. The video was sped up 2x. The timing of shock presentation is indicated with red ‘shock’ text. The same sample as shown in Fig.1f, g is presented.
Source data
Rights and permissions
About this article

Cite this article
Hagihara, K.M., Bukalo, O., Zeller, M. et al. Intercalated amygdala clusters orchestrate a switch in fear state. Nature 594, 403–407 (2021). https://doi.org/10.1038/s41586-021-03593-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03593-1
- Springer Nature Limited
This article is cited by
-
The amygdala NT3-TrkC pathway underlies inter-individual differences in fear extinction and related synaptic plasticity
Molecular Psychiatry (2024)
-
Optogenetic stimulation of mouse Hoxb8 microglia in specific regions of the brain induces anxiety, grooming, or both
Molecular Psychiatry (2024)
-
Identifying dysfunctional cell types and circuits in animal models for psychiatric disorders with calcium imaging
Neuropsychopharmacology (2024)
-
Pharmacological stimulation of infralimbic cortex after fear conditioning facilitates subsequent fear extinction
Neuropsychopharmacology (2024)
-
A distinct cortical code for socially learned threat
Nature (2024)