

Abstract
Neonatal cholestasis is a group of rare disorders of impaired bile flow characterized by conjugated hyperbilirubinaemia in the newborn and young infant. Neonatal cholestasis is never physiological but rather is a sign of hepatobiliary and/or metabolic disorders, some of which might be fatal if not identified and treated rapidly. A step-wise timely evaluation is essential to quickly identify those causes amenable to treatment and to offer accurate prognosis. The aetiology of neonatal cholestasis now includes an expanding group of molecularly defined entities with overlapping clinical presentations. In the past two decades, our understanding of the molecular basis of many of these cholestatic diseases has improved markedly. Simultaneous next-generation sequencing for multiple genes and whole-exome or whole-genome sequencing now enable rapid and affordable molecular diagnosis for many of these disorders that cannot be directly diagnosed from standard blood tests or liver biopsy. Unfortunately, despite these advances, the aetiology and optimal therapeutic approach of the most common of these disorders, biliary atresia, remain unclear. The goals of this Review are to discuss the aetiologies, algorithms for evaluation and current and emerging therapeutic options for neonatal cholestasis.
Key points
-
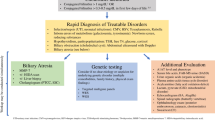
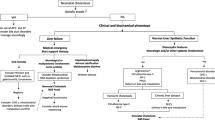
Early recognition and expedited evaluation of an infant with cholestasis are of utmost importance, as neonatal cholestasis is never physiological and often requires immediate treatment or intervention.
-
Cost-effective methods to reliably screen for biliary atresia in the first month of life are needed to improve age at diagnosis and Kasai hepatoportoenterostomy for infants with biliary atresia.
-
New genetic causes of neonatal cholestasis are being discovered at a rapid rate owing to the advent of next-generation gene-sequencing technologies and sophisticated bioinformatics.
-
Use of genetic testing might enable us to rapidly identify genetic causes of cholestasis without the need for invasive procedures and might lead to new precision treatments.
-
Multiple sites exist within the hepatobiliary tree where bile formation or flow can be impaired, resulting in neonatal cholestasis; these sites are potential targets for new pharmacological therapies.





Similar content being viewed by others

References
Kelly, D. A. & Stanton, A. Jaundice in babies: implications for community screening for biliary atresia. BMJ 310, 1172–1173 (1995).
Bartlett, M. & Gourley, G. in Liver Disease in Children 4th edn (eds Suchy, F. J., Sokol, R. J. & Balistreri, W. F.) 177–198 (Cambridge Univ. Press, 2014).
Fawaz, R. et al. Guideline for the evaluation of cholestatic jaundice in infants: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J. Pediatr. Gastroenterol. Nutr. 64, 154–168 (2017).
Feldman, A. & Suchy, F. J. in Liver Disease in Children 4th edn (eds Suchy, F. J., Sokol, R. J. & Balistreri, W. F.) 101–110 (Cambridge Univ. Press, 2014).
Harpavat, S., Finegold, M. J. & Karpen, S. J. Patients with biliary atresia have elevated direct/conjugated bilirubin levels shortly after birth. Pediatrics 128, e1428–e1433 (2011).
Harpavat, S., Garcia-Prats, J. A. & Shneider, B. L. Newborn bilirubin screening for biliary atresia. N. Engl. J. Med. 375, 605–606 (2016).
Harpavat, S. et al. Newborn direct or conjugated bilirubin measurements as a potential screen for biliary atresia. J. Pediatr. Gastroenterol. Nutr. 62, 799–803 (2016).
Chardot, C. et al. Improving outcomes of biliary atresia: French national series 1986–2009. J. Hepatol. 58, 1209–1217 (2013).
Serinet, M. O. et al. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics 123, 1280–1286 (2009).
Schreiber, R. A. et al. Biliary atresia: the Canadian experience. J. Pediatr. 151, 659–665 (2007).
Hussein, M., Howard, E. R., Mieli-Vergani, G. & Mowat, A. P. Jaundice at 14 days of age: exclude biliary atresia. Arch. Dis. Child 66, 1177–1179 (1991).
Mieli-Vergani, G., Howard, E. R., Portman, B. & Mowat, A. P. Late referral for biliary atresia — missed opportunities for effective surgery. Lancet 1, 421–423 (1989).
Lee, W. S. Pre-admission consultation and late referral in infants with neonatal cholestasis. J. Paediatr. Child Health 44, 57–61 (2008).
Sokol, R. J. et al. Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology 46, 566–581 (2007).
Lien, T. H. et al. Effects of the infant stool color card screening program on 5-year outcome of biliary atresia in Taiwan. Hepatology 53, 202–208 (2011).
Hopkins, P. C., Yazigi, N. & Nylund, C. M. Incidence of biliary atresia and timing of hepatoportoenterostomy in the United States. J. Pediatr. 187, 253–257 (2017).
Herbst, S. M. et al. Taking the next step forward - diagnosing inherited infantile cholestatic disorders with next generation sequencing. Mol. Cell. Probes 29, 291–298 (2015).
Wang, N. L. et al. A specially designed multi-gene panel facilitates genetic diagnosis in children with intrahepatic cholestasis: simultaneous test of known large insertions/deletions. PLOS ONE 11, e0164058 (2016).
Matte, U. et al. Analysis of gene mutations in children with cholestasis of undefined etiology. J. Pediatr. Gastroenterol. Nutr. 51, 488–493 (2010).
Togawa, T. et al. Molecular genetic dissection and neonatal/infantile intrahepatic cholestasis using targeted next-generation sequencing. J. Pediatr. 171, 171–177 (2016).
Karpen, S. et al. Use of a comprehensive 66 gene panel to diagnose the causes of cholestasis in >700 individuals [abstract 1213]. Hepatology 66 (Suppl. 1), 655A (2017).
Suchy, F. J. Neonatal cholestasis. Pediatr. Rev. 25, 388–396 (2004).
Balistreri, W. F. & Bezerra, J. A. Whatever happened to “neonatal hepatitis”. Clin. Liver Dis. 10, 27–53 (2006).
Yerushalmi, B. et al. Niemann-pick disease type C in neonatal cholestasis at a North American center. J. Pediatr. Gastroenterol. Nutr. 35, 44–50 (2002).
Lu, Y. B., Peng, F., Li, M. X., Kobayashi, K. & Saheki, T. [Progresses and perspectives in the study on citrin deficiency]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 23, 655–658 (2006).
Satrom, K. & Gourley, G. Cholestasis in preterm infants. Clin. Perinatol. 43, 355–373 (2016).
Hoang, V. et al. Percutaneously inserted central catheter for total parenteral nutrition in neonates: complications rates related to upper versus lower extremity insertion. Pediatrics 121, e1152–e1159 (2008).
Hsieh, M. H. et al. Parenteral nutrition-associated cholestasis in premature babies: risk factors and predictors. Pediatr. Neonatol. 50, 202–207 (2009).
Lee, W. S. & Sokol, R. J. Intestinal microbiota, lipids, and the pathogenesis of intestinal failure-associated liver disease. J. Pediatr. 167, 519–526 (2015).
Balistreri, W. F. et al. Intrahepatic cholestasis: summary of an American Association for the Study of Liver Diseases single-topic conference. Hepatology 42, 222–235 (2005).
Gonzales, E. et al. Liver diseases related to MDR3 (ABCB4) gene deficiency. Front. Biosci. (Landmark Ed) 14, (4242–4256 (2009).
Liu, L. Y., Wang, X. H., Lu, Y., Zhu, Q. R. & Wang, J. S. Association of variants of ABCB11 with transient neonatal cholestasis. Pediatr. Int. 55, 138–144 (2013).
Goldschmidt, M. L. et al. Increased frequency of double and triple heterozygous gene variants in children with intrahepatic cholestasis. Hepatol. Res. 46, 306–311 (2016).
Boyer, J. L. Bile formation and secretion. Compr. Physiol. 3, 1035–1078 (2013).
Wagner, M. & Trauner, M. Recent advances in understanding and managing cholestasis. F1000Res 5, 705 (2016).
Lee, W. S. & Sokol, R. J. Mitochondrial hepatopathies: advances in genetics, therapeutic approaches, and outcomes. J. Pediatr. 163, 942–948 (2013).
Herzog, D., Chessex, P., Martin, S. & Alvarez, F. Transient cholestasis in newborn infants with perinatal asphyxia. Can. J. Gastroenterol. 17, 179–182 (2003).
Stieger, B. Recent insights into the function and regulation of the bile salt export pump (ABCB11). Curr. Opin. Lipidol. 20, 176–181 (2009).
Feldman, A. & Sokol, R. J. Alpha-1-antitrypsin deficiency: an important cause of pediatric liver disease. Lung Health Prof. Mag. 4, 8–11 (2013).
Koo, K. A. et al. Biliatresone, a reactive natural toxin from Dysphania glomulifera and D. littoralis: discovery of the toxic moiety 1,2-diaryl-2-propenone. Chem. Res. Toxicol. 28, 1519–1521 (2015).
Waisbourd-Zinman, O. et al. The toxin biliatresone causes mouse extrahepatic cholangiocyte damage and fibrosis through decreased glutathione and SOX17. Hepatology 64, 880–893 (2016).
El Kasmi, K. C. et al. Phytosterols promote liver injury and Kupffer cell activation in parenteral nutrition-associated liver disease. Sci. Transl Med. 5, 206ra137 (2013).
Berauer, J. P. et al. Identification of PKD1L1 gene variants in children with the biliary atresia splenic malformation syndrome. Hepatology https://doi.org/10.1002/hep.30515 (2019).
Feranchak, A. P. & Sokol, R. J. Cholangiocyte biology and cystic fibrosis liver disease. Semin. Liver Dis. 21, 471–488 (2001).
Chinsky, J. M. et al. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet. Med. 19, 1380 (2017).
Demir, H. et al. Serum alpha-fetoprotein levels in neonatal cholestasis. Turk. J. Pediatr. 55, 152–157 (2013).
Sundaram, S. S., Bove, K. E., Lovell, M. A. & Sokol, R. J. Mechanisms of disease: inborn errors of bile acid synthesis. Nat. Clin. Pract. Gastroenterol. Hepatol. 5, 456–468 (2008).
Mittal, V. et al. Role of abdominal sonography in the preoperative diagnosis of extrahepatic biliary atresia in infants younger than 90 days. AJR Am. J. Roentgenol. 196, W438–W445 (2011).
Kianifar, H. R. et al. Accuracy of hepatobiliary scintigraphy for differentiation of neonatal hepatitis from biliary atresia: systematic review and meta-analysis of the literature. Pediatr. Radiol. 43, 905–919 (2013).
Yang, J. G., Ma, D. Q., Peng, Y., Song, L. & Li, C. L. Comparison of different diagnostic methods for differentiating biliary atresia from idiopathic neonatal hepatitis. Clin. Imaging 33, 439–446 (2009).
Liu, B. et al. Three-dimensional magnetic resonance cholangiopancreatography for the diagnosis of biliary atresia in infants and neonates. PLOS ONE 9, e88268 (2014).
Meyers, R. L. et al. Percutaneous cholecysto-cholangiography in the diagnosis of obstructive jaundice in infants. J. Pediatr. Surg. 39, 16–18 (2004).
Jensen, M. K. et al. HIDA, percutaneous transhepatic cholecysto-cholangiography and liver biopsy in infants with persistent jaundice: can a combination of PTCC and liver biopsy reduce unnecessary laparotomy? Pediatr. Radiol. 42, 32–39 (2012).
Shanmugam, N. P. et al. Selective use of endoscopic retrograde cholangiopancreatography in the diagnosis of biliary atresia in infants younger than 100 days. J. Pediatr. Gastroenterol. Nutr. 49, 435–441 (2009).
Zerbini, M. C. et al. Liver biopsy in neonatal cholestasis: a review on statistical grounds. Mod. Pathol. 10, 793–799 (1997).
Russo, P. et al. Design and validation of the biliary atresia research consortium histologic assessment system for cholestasis in infancy. Clin. Gastroenterol. Hepatol. 9, 357–362 (2011).
Russo, P. et al. Key histopathologic features of liver biopsies that distinguish biliary atresia from other causes of infantile cholestasis and their correlation with outcome: a multicenter study. Am. J. Surg. Pathol. 40, 1601–1615 (2016).
Haafiz, A. B. Liver fibrosis in biliary atresia. Expert Rev. Gastroenterol. Hepatol. 4, 335–343 (2010).
Hanquinet, S. et al. Contribution of acoustic radiation force impulse (ARFI) elastography to the ultrasound diagnosis of biliary atresia. Pediatr. Radiol. 45, 1489–1495 (2015).
Leschied, J. R. et al. Shear wave elastography helps differentiate biliary atresia from other neonatal/infantile liver diseases. Pediatr. Radiol. 45, 366–375 (2015).
Wu, J. F. et al. Transient elastography is useful in diagnosing biliary atresia and predicting prognosis after hepatoportoenterostomy. Hepatology 68, 616–624 (2018).
Metzker, M. L. Sequencing technologies - the next generation. Nat. Rev. Genet. 11, 31–46 (2010).
Trauner, M., Fuchs, C. D., Halilbasic, E. & Paumgartner, G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology 65, 1393–1404 (2017).
Shneider, B. L. et al. Portal hypertension in children and young adults with biliary atresia. J. Pediatr. Gastroenterol. Nutr. 55, 567–573 (2012).
Feldman, A. G. & Sokol, R. J. Neonatal cholestasis. Neoreviews https://doi.org/10.1542/neo.14-2-e63 (2013).
Yazigi, N. A. Long term outcomes after pediatric liver transplantation. Pediatr. Gastroenterol. Hepatol. Nutr. 16, 207–218 (2013).
Martin, S. R., Atkison, P., Anand, R., Lindblad, A. S. & Group, S. R. Studies of pediatric liver transplantation 2002: patient and graft survival and rejection in pediatric recipients of a first liver transplant in the United States and Canada. Pediatr. Transplant. 8, 273–283 (2004).
Heubi, J. E., Bove, K. E. & Setchell, K. D. R. Oral cholic acid is efficacious and well tolerated in patients with bile acid synthesis and Zellweger spectrum disorders. J. Pediatr. Gastroenterol. Nutr. 66, e57–e59 (2018).
Santra, S. & Baumann, U. Experience of nitisinone for the pharmacological treatment of hereditary tyrosinaemia type 1. Expert Opin. Pharmacother. 9, 1229–1236 (2008).
Demirbas, D., Brucker, W. J. & Berry, G. T. Inborn errors of metabolism with hepatopathy: metabolism defects of galactose, fructose, and tyrosine. Pediatr. Clin. North Am. 65, 337–352 (2018).
Wang, K. S. et al. Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasis. Hepatology 65, 1645–1654 (2017).
Lane, E. & Murray, K. F. Neonatal cholestasis. Pediatr. Clin. North Am. 64, 621–639 (2017).
Sundaram, S. S., Mack, C. L., Feldman, A. G. & Sokol, R. J. Biliary atresia: indications and timing of liver transplantation and optimization of pretransplant care. Liver Transpl. 23, 96–109 (2017).
Feranchak, A. P., Suchy, F. J. & Sokol, R. J. in Liver Disease in Children 4th edn (eds Suchy, F. J., Sokol, R. J. & Balistreri, W. F.) 111–140 (Cambridge Univ. Press, 2014).
Sullivan, J. S., Sundaram, S. S., Pan, Z. & Sokol, R. J. Parenteral nutrition supplementation in biliary atresia patients listed for liver transplantation. Liver Transpl. 18, 120–128 (2012).
Utterson, E. C. et al. Biliary atresia: clinical profiles, risk factors, and outcomes of 755 patients listed for liver transplantation. J. Pediatr. 147, 180–185 (2005).
DeRusso, P. A. et al. Growth failure and outcomes in infants with biliary atresia: a report from the Biliary Atresia Research Consortium. Hepatology 46, 1632–1638 (2007).
Campbell, A. L. & Herold, B. C. Immunization of pediatric solid-organ transplantation candidates: immunizations in transplant candidates. Pediatr. Transplant. 9, 652–661 (2005).
Centers for Disease Control and Prevention. Recommended child and adolescent immunization schedule for ages 18 years or younger. CDC https://www.cdc.gov/vaccines/schedules/downloads/child/0-18yrs-child-combined-schedule.pdf (2019).
Feldman, A. G., Feudtner, C. & Kempe, A. Reducing the underimmunization of transplant recipients. JAMA Pediatr. 172, 111–112 (2017).
Feldman, A. G., Sundaram, S. S., Beaty, B. L. & Kempe, A. Hospitalizations for respiratory syncytial virus and vaccine-preventable infections in the first 2 years after pediatric liver transplant. J. Pediatr. 182, 232–238 (2017).
Alonso, E. M. et al. Factors predicting health-related quality of life in pediatric liver transplant recipients in the functional outcomes group. Pediatr. Transplant. 17, 605–611 (2013).
de Vries, W. et al. Overall quality of life in adult biliary atresia survivors with or without liver transplantation: results from a national cohort. Eur. J. Pediatr. Surg. 26, 349–356 (2016).
Cai, S. Y. et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2, e90780 (2017).
Lindor, K. D. et al. Ursodeoxycholic acid in the treatment of primary biliary cirrhosis. Gastroenterology 106, 1284–1290 (1994).
Heathcote, E. J. et al. The Canadian multicenter double-blind randomized controlled trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology 19, 1149–1156 (1994).
Combes, B. et al. Prolonged follow-up of patients in the U. S. multicenter trial of ursodeoxycholic acid for primary biliary cirrhosis. Am. J. Gastroenterol. 99, 264–268 (2004).
Poupon, R. E., Balkau, B., Eschwege, E. & Poupon, R. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N. Engl. J. Med. 324, 1548–1554 (1991).
Pares, A. et al. Long-term effects of ursodeoxycholic acid in primary biliary cirrhosis: results of a double-blind controlled multicentric trial. UDCA-Cooperative Group from the Spanish Association for the Study of the Liver. J. Hepatol. 32, 561–566 (2000).
Corpechot, C. et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology 48, 871–877 (2008).
Paumgartner, G. & Pusl, T. Medical treatment of cholestatic liver disease. Clin. Liver Dis. 12, 53–80 (2008).
Zollner, G. et al. Expression of bile acid synthesis and detoxification enzymes and the alternative bile acid efflux pump MRP4 in patients with primary biliary cirrhosis. Liver Int. 27, 920–929 (2007).
Marschall, H. U. et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology 129, 476–485 (2005).
Lindor, K. D. et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 50, 808–814 (2009).
Halilbasic, E. et al. Side chain structure determines unique physiologic and therapeutic properties of norursodeoxycholic acid in Mdr2−/− mice. Hepatology 49, 1972–1981 (2009).
Hohenester, S. et al. A biliary HCO3- umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology 55, 173–183 (2012).
Fickert, P. et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 130, 465–481 (2006).
Moustafa, T. et al. Alterations in lipid metabolism mediate inflammation, fibrosis, and proliferation in a mouse model of chronic cholestatic liver injury. Gastroenterology 142, 140–151 (2012).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: the diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 67, 145–172 (2017).
Ali, A. H., Carey, E. J. & Lindor, K. D. Recent advances in the development of farnesoid X receptor agonists. Ann. Transl Med. 3, 5 (2015).
Kast, H. R. et al. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 277, 2908–2915 (2002).
Beuers, U., Trauner, M., Jansen, P. & Poupon, R. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J. Hepatol. 62, S25–37 (2015).
Huang, L. et al. Farnesoid X receptor activates transcription of the phospholipid pump MDR3. J. Biol. Chem. 278, 51085–51090 (2003).
Moschetta, A., Bookout, A. L. & Mangelsdorf, D. J. Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nat. Med. 10, 1352–1358 (2004).
Halilbasic, E., Baghdasaryan, A. & Trauner, M. Nuclear receptors as drug targets in cholestatic liver diseases. Clin. Liver Dis. 17, 161–189 (2013).
Tran, M., Liu, Y., Huang, W. & Wang, L. Nuclear receptors and liver disease: summary of the 2017 basic research symposium. Hepatol. Commun. 2, 765–777 (2018).
Shneider, B. L. et al. Placebo-controlled randomized trial of an intestinal bile salt transport inhibitor for pruritus in alagille syndrome. Hepatol. Commun. 2, 1184–1198 (2018).
Baghdasaryan, A. et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J. Hepatol. 64, 674–681 (2016).
Miethke, A. G. et al. Pharmacological inhibition of apical sodium-dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout mice. Hepatology 63, 512–523 (2016).
Ding, L., Yang, L., Wang, Z. & Huang, W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 5, 135–144 (2015).
Thompson, R. J. et al. Phase 2 open label efficacy and safety study of the apical sodium-depnedent bile acid transporter inhibitor maralixibat in children with progressive familial intrahepatic cholestasis: 48-week interim efficacy analysis. Hepatology 66, 57A (2017).
Shneider, B. L. et al. Results of ITCH, a multi-center randomized double-blind placebo-controlled trial of maralixibat, an ileal Apical Sodium-dependent Bile Acid Transporter Inhibitor (ASBTi), for pruritus in Alagille Syndrome (ALGS). Hepatology 66, 84A (2017).
Slijepcevic, D. et al. Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology 66, 1631–1643 (2017).
Zema, M. J. Colesevelam hydrochloride: evidence for its use in the treatment of hypercholesterolemia and type 2 diabetes mellitus with insights into mechanism of action. Core Evid. 7, 61–75 (2012).
Cortez, L. & Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 8, 28938 (2014).
Hayashi, H. & Sugiyama, Y. 4-Phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatology 45, 1506–1516 (2007).
Hasegawa, Y. et al. Intractable itch relieved by 4-phenylbutyrate therapy in patients with progressive familial intrahepatic cholestasis type 1. Orphanet J. Rare Dis. 9, 89 (2014).
van der Velden, L. M. et al. Folding defects in P-type ATP 8B1 associated with hereditary cholestasis are ameliorated by 4-phenylbutyrate. Hepatology 51, 286–296 (2010).
Gonzales, E. et al. Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatology 62, 558–566 (2015).
Verkade, H. J. et al. Biliary atresia and other cholestatic childhood diseases: advances and future challenges. J. Hepatol. 65, 631–642 (2016).
Bezerra, J. A. et al. Use of corticosteroids after hepatoportoenterostomy for bile drainage in infants with biliary atresia: the START randomized clinical trial. JAMA 311, 1750–1759 (2014).
Sokol, R. J. et al. Intravenous immunoglobulin (IVIG) following portoenterostomy in infants with biliary atresia: a phase 1/2A trial [abstract LB-8]. Hepatology 64 (Suppl.), 1123A (2016).
Friedman, S. L. et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 67, 1754–1767 (2018).
Wang, K. S. Newborn screening for biliary atresia. Pediatrics 136, e1663–e1669 (2015).
Tseng, J. J., Lai, M. S., Lin, M. C. & Fu, Y. C. Stool color card screening for biliary atresia. Pediatrics 128, e1209–e1215 (2011).
Hsiao, C. H. et al. Universal screening for biliary atresia using an infant stool color card in Taiwan. Hepatology 47, 1233–1240 (2008).
Mogul, D., Zhou, M., Intihar, P., Schwarz, K. & Frick, K. Cost-effective analysis of screening for biliary atresia with the stool color card. J. Pediatr. Gastroenterol. Nutr. 60, 91–98 (2015).
Woolfson, J. P. et al. Province-wide biliary atresia home screening program in British Columbia: evaluation of first 2 years. J. Pediatr. Gastroenterol. Nutr. 66, 845–849 (2018).
Franciscovich, A. et al. PoopMD, a mobile health application, accurately identifies infant acholic stools. PLOS ONE 10, e0132270 (2015).
Lertudomphonwanit, C. et al. Large-scale proteomics identifies MMP-7 as a sentinel of epithelial injury and of biliary atresia. Sci. Transl Med. 9, eaan8462 (2017).
Acknowledgements
R.J.S. is supported in part by US National Institutes of Health (NIH) grants U01 DK062453 and UL1 TR002535. A.G.F. is supported by an NIH and National Center for Advancing Translational Sciences Clinical and Translational Science Award (KL2 TR002534) and a Children’s Hospital Colorado Research Scholar Award.
Author information
Authors and Affiliations
Contributions
Both authors contributed equally to the preparation of this manuscript.
Corresponding author
Ethics declarations
Competing interests
R.J.S. has consulted with Albireo, Alexion, Retrophin and Shire. A.G.F. declares no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Gilbert syndrome
-
A benign condition caused by a decrease in the activity of UGT1A1 that leads to intermittent elevations in serum unconjugated or indirect bilirubin levels.
- Crigler–Najjar syndrome
-
An autosomal recessive disorder caused by mutations in UGT1A1 that lead to markedly elevated serum unconjugated or indirect bilirubin levels.
- Galactosaemia
-
An autosomal recessive disorder caused by mutations in GALT that result in an inability to metabolize galactose normally, affecting the liver, brain and lens of the eye.
- Choledochal cyst
-
Congenital dilatation of the biliary system.
- Biliary atresia
-
A progressive sclerosing inflammatory process of the extrahepatic and intrahepatic bile ducts in infants under 3 months of age that leads to fibrosis and obliteration of the biliary tree.
- Hepatoportoenterostomy (HPE) or Kasai procedure
-
A surgical procedure for treatment of biliary atresia in which a Roux-en-Y loop of jejunum is connected to the porta hepatitis, enabling bile to flow from the liver to the intestines.
- Alagille syndrome
-
An autosomal dominant disorder in which a mutation in JAG1 or NOTCH2 results in phenotypic abnormalities including interlobular bile duct paucity.
- Tyrosinaemia type 1
-
An autosomal recessive disorder caused by a defect in the enzyme fumarylacetoacetate hydrolase resulting in accumulation of toxic intermediates including succinylacetone in tissues and organs, leading to hepatocellular damage, hepatocellular carcinoma and neurotoxicity.
- Biliary atresia splenic malformation syndrome
-
A phenotype of biliary atresia in which patients have a combination of laterality (left–right differentiation) defects, including asplenia or polysplenia, midline liver, preduodenal portal vein, interruption of the inferior vena cava, intestinal malrotation, situs inversus or cardiac malformations.
- Posterior embryotoxon
-
A prominent Schwalbe’s line, in which the corneal endothelium and the uveal trabecular meshwork join, that is seen commonly in patients with Alagille syndrome.
- Hepatobiliary scintigraphy
-
A diagnostic imaging technique that evaluates hepatocellular function and patency of the biliary system by following a radiolabeled tracer into the liver and out through the biliary system into the small intestine.
- Dubin–Johnson syndrome
-
A benign autosomal recessive disorder caused by mutations in ABCC2 that result in isolated elevated serum conjugated or direct bilirubin levels and a dark coloured liver.
- Niemann–Pick disease type C
-
A lysosomal storage disease associated with mutations in NPC1 and NPC2, which results in cholesterol and lipid accumulation in the lysosomes of hepatocytes, causing cholestasis, and affects the spleen and brain.
Rights and permissions
About this article

Cite this article
Feldman, A.G., Sokol, R.J. Neonatal cholestasis: emerging molecular diagnostics and potential novel therapeutics. Nat Rev Gastroenterol Hepatol 16, 346–360 (2019). https://doi.org/10.1038/s41575-019-0132-z
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41575-019-0132-z
- Springer Nature Limited
This article is cited by
-
Biliary atresia
Nature Reviews Disease Primers (2024)
-
Diagnostic yield and novel candidate genes by next generation sequencing in 166 children with intrahepatic cholestasis
Hepatology International (2024)
-
The value of blood and urine metabolomics in differential diagnosis of cholestasis in infants
Egyptian Liver Journal (2023)
-
Alterations in gut microbiota and metabolite profiles in patients with infantile cholestasis
BMC Microbiology (2023)
-
Determination of Optimal Vitamin D Dosage in Children with Cholestasis
BMC Pediatrics (2023)