
Abstract
Serine metabolism is reportedly involved in immune cell functions, but whether and how serine metabolism regulates macrophage polarization remain largely unknown. Here, we show that suppressing serine metabolism, either by inhibiting the activity of the key enzyme phosphoglycerate dehydrogenase in the serine biosynthesis pathway or by exogenous serine and glycine restriction, robustly enhances the polarization of interferon-γ-activated macrophages (M(IFN-γ)) but suppresses that of interleukin-4-activated macrophages (M(IL-4)) both in vitro and in vivo. Mechanistically, serine metabolism deficiency increases the expression of IGF1 by reducing the promoter abundance of S-adenosyl methionine-dependent histone H3 lysine 27 trimethylation. IGF1 then activates the p38-dependent JAK–STAT1 axis to promote M(IFN-γ) polarization and suppress STAT6-mediated M(IL-4) activation. This study reveals a new mechanism by which serine metabolism orchestrates macrophage polarization and suggests the manipulation of serine metabolism as a therapeutic strategy for macrophage-mediated immune diseases.
Similar content being viewed by others


Introduction
Macrophages polarized to different activation states perform diverse functions in processes including host defense, inflammation, and tumor development [1,2,3]. M1 macrophages, classically activated by interferon γ (IFN-γ) and/or bacterial lipopolysaccharide (LPS) (also termed M(IFN-γ) or M(LPS + IFN-γ)), express inducible nitric oxide synthase (iNOS) and proinflammatory cytokines such as interleukin (IL)-6, IL-1β, and tumor necrosis factor α. M2 macrophages, alternatively activated by IL-4 (i.e., M(IL-4)) or IL-13, produce arginase 1 (ARG1), resistin-like alpha (Retnla), macrophage galactose-type lectin 1, and anti-inflammatory cytokines such as IL-10. Accordingly, M1 macrophages perform bactericidal, proinflammatory, and antitumor functions, while M2 macrophages perform worm clearance, tissue repair, and anti-inflammatory and protumor functions [1,2,3]. In vivo, macrophages are polarized into distinct phenotypes in response to complex microenvironmental stimuli. Tumor-associated macrophages (TAMs) are considered to fall within the spectrum of M2 activation because of their anti-inflammatory, immunosuppressive, and tissue remodeling effects [2]. Revealing and explaining the mechanisms regulating the phenotype of TAMs may provide insights for reprogramming TAMs into antitumor effector cells for therapy.
An accumulating body of literature supports the idea that the activation of macrophages is closely related to changes in cell metabolism [4]. During macrophage activation, various metabolic pathways are reprogrammed to adapt to changes in cell function, thus promoting specialized immunophenotypes [5, 6]. Altered metabolism of certain amino acids is one of the earliest occurring and most important characteristics that define macrophage subsets [7]. L-arginine is converted to NO by iNOS in M1 macrophages but metabolized by ARG1 in M2 macrophages [7,8,9]. Furthermore, glutamine has been shown to orchestrate macrophage polarization through α-ketoglutarate production [10]. However, whether the metabolism of other amino acids plays a role in fine-tuning macrophage polarization is largely unknown.
Serine can be either synthesized de novo or taken up from the microenvironment. In the serine biosynthesis pathway, phosphoglycerate dehydrogenase (PHGDH) catalyzes the first rate-limiting step in the conversion of glycolysis-derived 3-phosphoglyceric acid. Serine supports LPS-stimulated production of IL-1β by feeding one-carbon metabolism or enhancing mechanistic target of rapamycin kinase (mTOR) signaling [11,12,13]. Another study revealed that PHGDH inhibition promotes the M(LPS + IFN-γ) state and that this effect is dependent on the presence of exogenous serine and glycine [14]. In addition, PHGDH activity can enhance M(IL-4) macrophage polarization independent of exogenous serine and glycine [14]. However, the roles and mechanism of PHGDH and serine in macrophage polarization, especially polarization induced by the major endogenous macrophage-activating factor IFN-γ [15], are largely unknown.
Signaling by insulin-like growth factor 1 (IGF1), a hepatogenic endocrine hormone, plays an important role in regulating growth, metabolism, and lifespan [16, 17]. However, the role of IGF1 signaling in macrophage polarization is inconsistent in different experimental models [18,19,20,21,22].
Here, we aimed to elucidate how serine metabolism affects macrophage polarization through the IGF1–mitogen-activated protein kinase 14 (p38) axis. First, blocking the activity of PHGDH, a key enzyme in the serine biosynthesis pathway, by genetic ablation or by treatment with the inhibitor CBR-5884 robustly enhanced M(IFN-γ) but suppressed M(IL-4) polarization both in vitro and in vivo. Moreover, restricting exogenous serine and glycine yielded similar effects. Mechanistically, inhibiting both endogenous and exogenous serine metabolism promoted the expression of IGF1 via a reduction in S-adenosyl methionine (SAM)-mediated histone H3 lysine 27 trimethylation (H3K27me3). Subsequently, IGF1 activated the p38-dependent Janus kinase (JAK)–signal transducer and activator of transcription 1 (STAT1) axis, thus promoting M(IFN-γ) polarization and inhibiting polarization toward STAT6-mediated M(IL-4) phenotype. In summary, our findings clarify a previously unknown mechanism by which serine metabolism plays a key role in orchestrating macrophage polarization.
Materials and methods
Mice
Phgdhfl/fl mice were generated by Cyagen Biosciences (Guangzhou, China) using CRISPR‒Cas9 gene editing. IGF1Rfl/fl mice were provided by Dr. Yingjie Wu (Dalian Medical University, China), and Lyz2-Cre mice were provided by Dr. Xiaoyue Tan (Nankai University, China). Phgdhfl/fl and IGF1Rfl/fl mice were separately crossed with Lyz2-Cre mice to generate mice with myeloid cell-specific PHGDH knockout and IGF1R knockout, respectively, and their littermates. Age- and sex-matched 6–12-week-old mice were used to compose all experimental groups. Murine genotypes were determined by PCR analysis of tail-snip DNA using the following primers: Phgdh lox forward, 5ʹ-GCGTTTACAGCCATCTTCCTTCC-3ʹ; Phgdh lox reverse, 5ʹ-ATCCTCTAAGCTGCATCCCTTATC-3ʹ; IGF1R lox forward, 5ʹ-CTTCCCAGCTTGCTACTCTAGG-3ʹ; IGF1R lox reverse, 5ʹ-CAGGCTTGCAATGAGACATGGG-3ʹ; Lyz2-cre forward, 5ʹ-CCCAGAAATGCCAGATTACG-3ʹ; and Lyz2-cre reverse, 5ʹ-CTTGGGCTGCCAGAATTTCTC-3ʹ. For the serine and glycine deprivation experiments, mice were fed a control diet (#M10006, BioPike) or serine- and glycine-free chow (#M19053001, BioPike) for 2 weeks before the experiments. All mice were on a C57BL/6 background and were maintained on a 12 h light/12 h dark cycle at a controlled temperature (22 °C ± 2 °C) under specific-pathogen-free conditions in the animal facility of Tianjin Medical University. Mice were fed a standard chow diet with water available ad libitum unless otherwise specified. All animal experiments were approved by the Ethics Committee of Tianjin Medical University.
Cell lines and primary cell culture
RAW264.7 cells (ATCC, TIB-71), HCC70 (ATCC, CRL-2315) and T47D (ATCC, HTB-133) were cultured at 37 °C and 5% CO2 in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin‒streptomycin (PS). MCF-7 (ATCC, HTB-22) and murine Lewis lung carcinoma (LLC) cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% FBS and 1% PS. MDA-MB-231 (ATCC, HTB-26) and MDA-MB-468 (ATCC, HTB-132) cells were cultured at 37 °C without CO2 in Leibovitz’s L-15 medium supplemented with 10% FBS and 1% PS. The cell lines were routinely subjected to a full quality analysis for authentication, including examination of their morphological and specific properties, microorganism detection, short tandem repeat profiling, isoenzymology, and PCR species identification [23]. The PLKO.1-shCtrl/shPhgdh, VSV-G, and psPAX2 plasmids were transfected into 293T cells for lentiviral production, and RAW264.7 cells were then infected with the lentivirus to establish a stable shRNA-mediated PHGDH knockout RAW264.7 cell line. After 48 h of infection, stably transduced cells were screened by incubation with medium containing 8 μg/ml puromycin for 1 week. For isolation of mouse peritoneal macrophages (PMs), each mouse was injected intraperitoneally with 1 ml of Brewer thioglycollate medium (3:100; #225650, BD). On the 4th day, the mice were sacrificed, and 8 ml of PBS supplemented with 3% FBS precooled to 4 °C was injected into the abdominal cavity. The peritoneal fluid was slowly extracted and centrifuged. Red blood cells were lysed with red blood cell lysis buffer (#R1010, Solarbio) and resuspended in RPMI-1640 medium containing 10% FBS. Bone marrow-derived macrophages (BMDMs) were isolated from the tibias and femurs of mice and cultured in RPMI-1640 medium containing 10% FBS and 30% L929 supernatant at 37 °C and 5% CO2 for 7 days. For serine and glycine deprivation experiments, PMs and BMDMs were cultured overnight in assay medium: MEM (#11090-081, Thermo Fisher Scientific) supplemented with dialyzed FBS (1:10; #26400-044, Gibco), D-glucose (17 mM; #G7021, Sigma-Aldrich), PS (1:100; #15140-122, Gibco), serine (400 μM; #S4311, Sigma-Aldrich) and glycine (400 μM; #G5417, Sigma-Aldrich). Cells that were subjected to serine and glycine starvation were cultured in medium without serine or glycine. For the macrophage polarization assay, macrophages were treated with IFN-γ (100 ng/ml; #50709-MNAH, Sino Biological), IFN-γ + LPS (100 ng/ml; #L4516, Sigma-Aldrich), or IL-4 (20 ng/ml; #404-ML, R&D Systems).
Xenograft tumor model and macrophage removal
LLC (1 × 106) cells were injected subcutaneously into PHGDH-KO-Mφ and PHGDH-WT-Mφ mice (Fig. 3a, b) and into IGF1R-KO-Mφ and IGF1R-WT-Mφ mice (Fig. S10j, k) (n = 6–10 mice in each group). Mice were matched for age, sex and body weight. In the experiment shown in Fig. S4c, d, C57BL/6 mice were fed an SG-deficient diet or standard chow for 2 weeks prior to subcutaneous injection of LLC cells. Beginning on the 2nd day after injection, mice in the macrophage removal group (Mφ−) were intraperitoneally injected with clodronate liposomes (0.1 ml/10 g, #F70101C-N-2, FormuMax) every 3 days to clear macrophages. Mice in the macrophage presentation group (Mφ + ) mice were given the same amount of PBS liposomes as mice in the control group. Mice were sacrificed 20–27 days after subcutaneous injection of LLC cells for tumor harvesting. The width (W) and length (L) of the tumors were measured, and the volume (V) of each tumor was calculated using the equation V = (W2 × L/2). Tumors were preserved for subsequent staining.
Chitin model
Chitin (#C9752, Sigma-Aldrich) was washed twice with PBS and then sonicated on ice. The suspension was filtered through a 100 μm filter and diluted in 30 ml of PBS. Each mouse was intraperitoneally injected with ~3 μg of chitin. Two days after chitin injection, the peritoneal cells were collected for flow cytometry and qPCR.
CCl4-induced liver fibrosis model
Liver fibrosis was induced by thrice-weekly intraperitoneal injection of CCl4 (1.0 ml/kg; #C27710, Acmec Biochemical) diluted 1:3 in corn oil for 4 weeks (twelve injections in total). For histological analysis, livers were dissected and fixed with 4% paraformaldehyde solution overnight prior to staining with hematoxylin–eosin for routine examination or with Sirius red (#G1471, Solarbio) for visualization of fibrous deposits. Fibrotic areas were quantified with ImageJ software (NIH). OCT-embedded liver sections were stained with Oil red O (#G1261, Solarbio) to visualize lipids or subjected to immunofluorescence staining. Serum alanine aminotransferase (ALT) was measured using a commercial kit (#C009-2-1, Nanjing Jiancheng Bioengineering Institute). Liver RNA was extracted to evaluate the expression of fibrosis-related cytokine genes by qPCR.
RNA extraction, RT‒PCR, and RNA-seq analysis
Total RNA was extracted with TRIzol reagent (#15596018, Invitrogen) and then reverse-transcribed with a reverse transcription system (#24408, Bimake). Quantitative RT‒PCR analysis was then performed with SYBR Green PCR Mix (#B21203, Bimake) and the designed primers in an ABI 7300 Detection System. β-Actin was used as the housekeeping gene. The qPCR primers used were as follows: Phgdhforward, 5ʹ-CCTCATTGTCCGGTCTGCTAC-3ʹ and reverse, 5ʹ-CATCTTTCATCGAAGCTGTTGC-3ʹ; Nos2 forward, 5ʹ-GAAACGCTTCACTTCCAATG-3ʹ and reverse, 5ʹ-AATCCACAACTCGCTCCAA-3ʹ; Il-6 forward, 5ʹ-TTGCCTTCTTGGGACTGAT-3ʹ and reverse, 5ʹ-TTGCCATTGCACAACTCTT-3ʹ; Il-1β forward, 5ʹ-GAAATGCCACCTTTTGACAGTG-3ʹ and reverse, 5ʹ-TGGATGCTCTCATCAGGACAG-3ʹ; Arg1 forward, 5ʹ-CCACAGTCTGGCAGTTGGAAG-3ʹ and reverse, 5ʹ-GGTTGTCAGGGGAGTGTTGATG-3ʹ; Mgl1 forward, 5ʹ-CAGAATCGCTTAGCCAATGTGG-3ʹ and reverse, 5ʹ-TCCCAGTCCGTGTCCGAAC-3ʹ; Mgl2 forward, 5ʹ-TTCAAGAATTGGAGGCCACT-3ʹ and reverse, 5ʹ-CAGACATCGTCATTCCAACG-3ʹ; Retnla forward, 5ʹ-CCAATCCAGCTAACTATCCCTCC-3ʹ and reverse, 5ʹ-CACTTGGTGGTTTGCTACG-3ʹ; Tnf-α forward, 5ʹ-CCTCTTCTCATTCCTGCTTG-3ʹ and reverse, 5ʹ-GTCACCCCGTCCACATCTT-3ʹ; Igf1 forward, 5ʹ-CTGGACCAGAGACCCTTTGC-3ʹ and reverse, 5ʹ-GGACGGGGACTTCTGAGTCTT-3ʹ; Rgs1 (regulator of G-protein signaling 1) forward, 5ʹ-TCTGGGATGAAATCGGCCAAG-3ʹ and reverse, 5ʹ-GCATCTGAATGCACAAATGCTT-3ʹ; Atp6v0d2 (ATPase H+ transporting V0 subunit D2) forward, 5ʹ-CAGAGCTGTACTTCAATGTGGAC-3ʹ and reverse, 5ʹ-AGGTCTCACACTGCACTAGGT-3ʹ; S1pr1 (sphingosine-1-phosphate receptor 1) forward, 5ʹ-ATGGTGTCCACTAGCATCCC-3ʹ and reverse, 5ʹ-CGATGTTCAACTTGCCTGTGTAG-3ʹ; Tgm2 (transglutaminase 2) forward, 5ʹ-GACAATGTGGAGGAGGGATCT-3ʹ and reverse, 5ʹ-CTCTAGGCTGAGACGGTACAG-3ʹ; Slc6a8 (solute carrier family 6 member 8) forward, 5ʹ-GCAGGGTGTGCATATCTCCAA-3ʹ and reverse, 5ʹ-TACCCCCACTCACATCAGTCA-3ʹ; C77080 (expressed sequence C77080) forward, 5ʹ-CCACAGGGATTCCGAACCC-3ʹ and reverse, 5ʹ-CAGAGCACTCGCAGGACTC-3ʹ; Oit3 (oncoprotein induced transcript 3) forward, 5ʹ-CACCTGCGGTCCTAGATCCT-3ʹ and reverse, 5ʹ-GCAAAGAGGTTGATTCTGGGA-3ʹ; Gpnmb (glycoprotein nonmetastatic melanoma protein B) forward, 5ʹ-GCTGGTCTTCGGATGAAAATGA-3ʹ and reverse, 5ʹ-CCACAAAGGTGATATTGGAACCC-3ʹ; Ezh2 forward, 5ʹ-AGTGACTTGGATTTTCCAGCAC-3ʹ and reverse, 5ʹ-AATTCTGTTGTAAGGGCGACC-3ʹ; and Jmjd3 forward, 5ʹ- TGAAGAACGTCAAGTCCATTGTG-3ʹ and reverse, 5ʹ-TCCCGCTGTACCTGACAGT-3ʹ. For RNA-seq analysis, WT and KO BMDMs, BMDMs treated with or without serum starvation, and RAW264.7 cells transfected with siControl or siPHGDH were collected for extraction of total RNA. RNA-seq was performed using the MGISEQ-2000 platform, and the transcript data were profiled and compared according to standard protocols (Beijing Huada Gene Research Institute). All sequencing data have been submitted to the National Center for Biotechnology Information Gene Expression Omnibus repository under GEO accession numbers GSE165684 and GSE196840.
Dual-luciferase reporter assays
Igf1-Luc was transfected into RAW264.7 cells, and Renilla luciferase under the control of the thymidine kinase promoter was used as an internal control. Cells were treated as indicated in the figures and were then collected with passive lysis buffer, and luciferase activity was analyzed using a dual luciferase assay kit (#E1910, Promega).
Flow cytometry
Red blood cells in samples were lysed with red blood cell lysis buffer. Cells were then filtered to prepare a single-cell suspension and stained with specific surface antibodies in staining buffer (containing 2% BSA) (as shown below). Cells were washed three times, incubated with antibodies on ice, and then subjected to FACS using a FACSVerse flow cytometer (BD). The antibodies used were anti-CD45 PE (#103105, Biolegend), anti-CD170 FITC (#155504, Biolegend), anti-F4/80 PE-Cy5 (#123112, Biolegend), anti-CD11b APC (#101211, Biolegend), anti-IFNγ-Rα (#12753, Santa Cruz), anti-IFNγ-Rβ (#12752, Santa Cruz), and Alexa Fluor 488 goat anti-hamster (Armenian) IgG (#405508, Biolegend). Data were analyzed with FlowJo (Tree Star).
Western blot analysis
Cells were lysed using radioimmunoprecipitation assay lysis buffer supplemented with protease inhibitor cocktail. Equal amounts of protein were separated by SDS‒PAGE for western blotting to analyze the expression of the target proteins with specific antibodies. Visualization was performed with an ECL chemiluminescence kit (WBKLS0500, Millipore). Antibodies specific for the following proteins were used: PHGDH (1:1000; ab240744, Abcam); iNOS (1:1000; #13120S, CST); ARG1 (1:1000; #93668S, CST); β-actin (1:1000; #sc-47778, Santa Cruz); Tubulin (1:1000; #ab7792, Abcam); FLAG (1:1000; #F1804, Sigma); p-JAK1 (Y1034/1035) (1:1000; #74129S, CST); JAK1 (1:1000, #3344S, CST); p-JAK2 (Y1008) (1:1000; #8082S, CST); JAK2 (1:1000, #3230S, CST); p-STAT1 (Y701) (1:1000; #8826S, CST); STAT1 (1:1000; #14994S, CST); p-p38 (T180/Y182)(1:1000; #4511T, CST); p38 (1:1000; #8690T, CST); p-STAT6 (Y641) (1:1000; #56554S, CST); STAT6 (1:1000; #5397S, CST); p-ERK (T202/Y204) (1:1000; #4370T, CST); ERK(1:1000; #4695T, CST); p-JNK (T183/Y185) (1:1000; #4668T, CST); JNK (1:1000; #9252T, CST); p-AKT (S473) (1:1000; #9271S, CST); AKT (1:1000; # D153569-0025, Sangon Biotech); p-mTOR (S2448) (1:1000; #5536T, CST); mTOR (1:1000; #2983T, CST); p-S6K(S371) (1:1000; #9208T, CST); S6K (1:1000; #AF0258, Beyotime); H3K4me3 (1:1000; #ab8580, Abcam); H3K9me3 (1:1000; #ab8898, Abcam); H3 (1:1000; #ab201456, Abcam); and EZH2 (1:1000; #5246T, CST).
Measurement of serine and SAM concentrations and PHGDH activity
The IGF-1 protein content in conditioned medium was measured with a mouse IGF-1 ELISA Kit (#EK0378, Boster). The SAM and serine concentrations in the samples were measured with a SAM ELISA kit (#CEG414Ge, Cloud-Clone) and a DL-Serine Assay Kit (#K743, BioVision), respectively. The PHGDH activity in cells was measured with a PHGDH Activity Assay Kit (Colorimetric) (#ab273328, Abcam). The assays were performed in accordance with the reagent manufacturers’ protocols.
Plasmid transfection, RNA interference, and reagents
The PHGDH expression plasmids were constructed by standard molecular biology techniques, and the generated sequences were inserted into the pLenti-3xFlag/pcDNA3.1-HA vector. A point mutation (PHGDH-V425M) was generated by site-directed mutagenesis using a plasmid encoding the wild-type PHGDH protein as the template. The transcription reporter gene Igf1-Luc was constructed by standard molecular biology techniques under the control of the Igf1 promoter and was inserted into a vector containing the open reading frame encoding firefly luciferase. RAW264.7 cells and primary macrophages were transfected with the corresponding plasmids and siRNA using Lipofectamine 2000 (#11668, Invitrogen) or Lipofectamine RNAiMAX (#3778, Invitrogen). The siRNA sequences were as follows: siPhgdh, 5ʹ-GGGAGCGGGAGATCGAGAA-3ʹ; siRgs1, 5ʹ-CCAAGTCCAAAGACATACTTT-3ʹ; siAtp6v0d2, 5ʹ-GCAGCTATATGATAGACAATA-3ʹ; siS1pr1, 5ʹ-CGTCTGGAAACGTCAATTCTT-3ʹ; siTgm2,5ʹ-CCTGACAGAGTCAAACCTCAT-3ʹ; siSlc6a8, 5ʹ-CGTGTACTTCACTGCTACATT-3ʹ; siC77080, 5ʹ-GGCTGAACTTCGGAGCATTTC-3ʹ; siOit3, 5ʹ-CCCACTGCAAAGATGGATGAA-3ʹ; siGpnmb, 5ʹ-GATGTGTATGTGATAACAGAT-3ʹ; siIgf1, 5ʹ-CAGGCATTGTGGATGAGTGTT-3ʹ; siIgf1r-1, 5ʹ-CCAACGAGCAAGTTCTTCGTT-3ʹ; siIgf1r-2, 5ʹ-CAATGGTAACTTGAGTTACTA-3ʹ; and siEzh2, 5ʹ-GCACAAGTCATCCCGTTAAAG-3ʹ. The reagents used were as follows: CBR-5884 (#HY-100012, MCE); L-phenylglycine (#237647, Sigma-Aldrich); SB203580 (#HY-10256, MCE); fludarabine (#HY-B0069, MCE); BMS-345541 hydrochloride (#HY-10518, MCE); AS1517499 (#HY-100614, MCE); and ruxolitinib (#HY-50856, MCE). IFN-β (100 ng/ml; #50708-MCCH, Sino Biological); sodium formate (1 mM; #247596, Sigma-Aldrich); glutathione ethyl ester (1 mM; #G1404, Sigma-Aldrich); and SAM (0.2 mM; #A506555, Sigma-Aldrich).
Chromatin immunoprecipitation (ChIP) assay
For the H3K27me3 ChIP assay, BMDMs were crosslinked with 1% formaldehyde, and the reaction was then stopped with 125 mM glycine. After ultrasonication, the cell lysates were incubated with an anti-H3K27me3 antibody (#ab6002, Abcam) for immunoprecipitation. qPCR was used to detect the promoter sequences of the target genes in the recovered DNA immune complexes and the input DNA. Transcript levels were normalized to the corresponding DNA input control, and the experimental steps were carried out using a Simple Chip Plus Sonication Chromatin IP Kit (#56383, CST). The primers used were as follows: Igf1-3000 (IP: H3K27me3) forward, 5ʹ-TCCTCTCTTTAGCACATTGTG-3ʹ and reverse, 5ʹ-GAGTTGGAGGAAAGGGGCTCT-3ʹ; Igf1-2000 (IP: H3K27me3) forward, 5ʹ-CTGGAATCTACAAAGAAAATG-3ʹ and reverse, 5ʹ-CCATTAGCTGTGCTGTGGCCA-3ʹ; Igf1-1000 (IP: H3K27me3) forward, 5ʹ-AATGATAGCCTGTTAGTGGAA-3ʹ and reverse, 5ʹ-GGGAAGGAGAGAATGCTATGT-3ʹ; and Igf1-200 (IP: H3K27me3) forward, 5ʹ-TCTGAAAGACCACTGAGAAAT-3ʹ and reverse, 5ʹ-GCTGGCTAGCAATACTCTCTC-3ʹ.
Immunofluorescence staining
Primary antibodies specific for F4/80 (1:200; #ab16911, Abcam), CD11b (1:200; #ab133357, Abcam), Arg1 (1:1000; #93668S, CST), IL-6 (1:200; # AF-406-NA, R&D Systems), IL-1β (1:200; #9722, Abcam), and iNOS (1:200; #13120S, CST) were used for immunofluorescence staining of 5-μm-thick frozen sections. Alexa Fluor 488- or Alexa Fluor 594-conjugated secondary antibodies were then used to detect the primary antibodies before imaging of the stained sections with a fluorescence microscope. Images were analyzed using ImageJ software. For semiquantitative histological analysis, ten microscopic fields were randomly selected, and the percentage of positive cells was counted.
Immunohistochemistry
Tumors were harvested and fixed with 4% paraformaldehyde for 24–48 h. After being dehydrated in an ethanol gradient and embedded in paraffin, the tumors were sliced into 5 μm sections. The tumor sections were then immunostained by sequential incubation with primary and secondary antibodies. Nuclei were stained with DAPI, and imaging analysis was carried out under a fluorescence microscope. Semiquantitative histological analysis was performed using ten randomly selected microscopic fields, and the percentage of positive cells was counted. Antibodies specific for the following proteins were used: CD11b (1:200; #ab133357, Abcam), CD4 (1:200; #14-9766-82, Invitrogen), CD8a (1:200; #14-0808-82, Invitrogen), Foxp3 (1:200; # 14-5773-80, Invitrogen), F4/80 (1:200; #ab16911, Abcam), iNOS (1:200; #13120S, CST), and ARG1 (1:1000; #93668S, CST).
Mitochondrial and glycolysis stress tests
PMs were seeded in 24-well Seahorse XFe-24 assay plates (#10421, Agilent) and treated with IFN-γ or IL-4. The cells were then washed and cultured in XF RPMI medium (pH 7.4) for 1 h. By mitochondrial and glycolysis stress tests, the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), respectively, were automatically calculated and recorded using Seahorse XFe-24 software (Agilent). For OCR measurement, ATP production was determined using 2.0 μM oligomycin. The maximal respiration was measured with 2.0 μM FCCP. The cells were then treated with 1.0 μM antimycin A/rotenone to measure nonmitochondrial respiration. Measurements were taken after the addition of each mitochondrial inhibitor and before the addition of the next inhibitor. For ECAR measurement, the basal glycolytic capacity was determined using 10 μM glucose. The maximal glycolytic capacity was measured with 1.5 μM oligomycin. The cells were then treated with 50 μM 2-deoxy-D-glucose to confirm that the extracellular acidification in the experiment had originated via the glycolytic pathway. Measurements were taken after the addition of each glycolysis inhibitor and before the addition of the next inhibitor.
Statistical analyses
The numbers of samples and levels of significance are indicated in the figure legends. The significance levels were determined by two-tailed Student’s t test. Fluorescence imaging analysis was performed in a blinded manner. All quantitative data are presented as the mean ± SEM of at least three independent experiments. Statistical analyses were performed using GraphPad Prism software (GraphPad Software). A p value of <0.05 was considered significant (NS, not significant (p ≥ 0.05); *p < 0.05; **p < 0.01; ***p < 0.001).
Results
PHGDH inhibits M(IFN-γ) but promotes M(IL-4) polarization through its enzymatic activity
We first explored whether PHGDH, the rate-limiting enzyme in the serine biosynthesis pathway, plays a role in M1 macrophage polarization. Inhibition of PHGDH by treatment with the specific inhibitor CBR-5884 [24] or a small interfering RNA (siRNA) significantly upregulated the mRNA expression of M1-specific marker genes, including Nos2, Il-6, and Il-1β, and markedly enhanced iNOS protein expression in M(LPS + IFN-γ) RAW264.7 cells and M(IFN-γ) BMDMs (Fig. 1a, b and Fig. S1a–c). Conversely, PHGDH inhibition suppressed the expression of M2-specific marker genes and reduced ARG1 protein expression (Fig. 1c, d and Fig. S1d). Next, we generated Phgdhfl/flLyz2-Cre+ mice (KO-Mφ), which, unlike their littermate Phgdhfl/flLyz2-Cre− (WT-Mφ) mice, have myeloid cell-specific Phgdh deletion [25], as confirmed in BMDMs and primary PMs (Fig. S1e, f). Macrophage differentiation and numbers were not influenced by Phgdh ablation, as we previously reported [25]. Consistent with this finding, we observed similar upregulation of M1 markers and suppression of M2 markers in Phgdh-ablated primary macrophages (Fig. 1e–h and Fig. S1e, f).

PHGDH inhibits M(IFN-γ) but promotes M(IL-4) polarization through its enzymatic activity. Wild-type bone marrow-derived macrophages (BMDMs) were pretreated with the PHGDH inhibitor CBR-5884 (15 μM) for 12 h and were then stimulated with IFN-γ (100 ng/ml) for 12 h (a, b) or with IL-4 (20 ng/ml) for 24 h (c, d), followed by qRT‒PCR or western blot analysis of M1 (a, b) and M2 (c, d) marker expression. Phgdhfl/flLyz2-Cre- (PHGDH-WT) and Phgdhfl/flLyz2-Cre+ (PHGDH-KO) mouse BMDMs were stimulated with IFN-γ for 12 h or the indicated times, followed by qRT‒PCR (e) or western blot (f) analysis of M1 marker expression. PHGDH-WT and PHGDH-KO BMDMs were stimulated with IL-4 for 24 h or the indicated times, followed by qRT‒PCR (g) or western blot (h) analysis of M2 marker expression. RAW264.7 cells with siRNA-mediated PHGDH silencing or stable shRNA-mediated PHGDH knockdown were transfected with an RNA interference (RNAi)-resistant PHGDH ectopic expression plasmid or a plasmid expressing a catalytically dead PHGDH mutant (V425M) and were then either stimulated with IFN-γ for 12 h to evaluate M1 marker expression by qRT‒PCR (i) or western blot analysis (j) or stimulated with IL-4 for 24 h to detect M2 marker expression by qRT‒PCR (k) or western blot analysis (l). siCtrl siControl, shCtrl shControl, EV empty vector. The data are from three independent experiments with biological duplicates in each and are shown as the mean ± SEM values (n = 3) (a, c, e, g, i, k) or are representative of three independent experiments (b, d, f, h, j, l). *p < 0.05, **p < 0.01, ***p < 0.001
Conversely, iNOS expression was reduced in PHGDH-overexpressing RAW264.7 cells under M(LPS + IFN-γ) polarization conditions (Fig. S1g). Furthermore, PHGDH overexpression significantly reduced the enhanced expression of M1 markers in RAW264.7 cells with siRNA-mediated PHGDH silencing and RAW264.7 cells with stable shRNA-mediated PHGDH knockdown (Fig. 1i, j and Fig. S1h). However, cells overexpressing the enzymatically inactive PHGDH V425M mutant had levels of M1 markers comparable to those in PHGDH-deficient RAW264.7 cells, suggesting that the enzymatic activity of PHGDH is critical for its suppression of macrophage M1 polarization (Fig. 1i, j and Fig. S1i). Moreover, PHGDH enzymatic activity is critical for the promotion of macrophage M2 polarization (Fig. 1k, l). Collectively, these findings indicate that PHGDH regulates macrophage polarization through its enzymatic activity.
Serine metabolism modulates macrophage polarization
We next sought to determine whether exogenous serine also regulates macrophage polarization. Inhibition of alanine-serine-cysteine-threonine transporters by the specific inhibitor L-phenylglycine [26] decreased the level of intracellular serine and led to elevated expression of IFN-γ-induced M1 markers but reduced expression of IL-4-induced M2 markers (Fig. 2a–d and Fig. S2a). Furthermore, depletion of serine (-S) or glycine (-G) alone in the medium did not enhance M1 marker expression, whereas their combined depletion (-SG) resulted in increased M1 polarization (Fig. 2e, f). In addition, the expression of M2 markers was significantly reduced in SG-starved BMDMs (Fig. 2g, h). Furthermore, M1 polarization markers were significantly upregulated in PHGDH-KO cells with exogenous SG depletion compared to SG-treated PHGDH-KO cells (Fig. 2i, j and Fig. S2b, c), indicating the strong combined inhibition of M1 polarization by suppression of the de novo serine biosynthesis pathway and depletion of exogenous serine. Moreover, a synergistic effect of PHGDH deficiency and exogenous serine deprivation on the regulation of M2 polarization was observed (Fig. 2k, l). Taking these results together, we thus concluded that serine metabolism modulates macrophage polarization.

Serine metabolism modulates macrophage polarization in vitro. Wild-type BMDMs were pretreated with the ASCT-specific inhibitor L-phenylglycine (50 μM) for 12 h and were then stimulated with IFN-γ (100 ng/ml) for 12 h (a, b) or with IL-4 (20 ng/ml) for 24 h (c, d), followed by qRT‒PCR or western blot analysis of M1 (a, b) and M2 (c, d) marker expression. e WT BMDMs were starved of serine and/or glycine for 12 h and were then stimulated with IFN-γ for 12 h, followed by qPCR analysis of M1 marker expression. f WT BMDMs were cultured with the indicated concentration of serine in the absence of glycine for 12 h and were then stimulated with IFN-γ for 12 h, followed by western blot analysis of iNOS. BMDMs were starved of serine and glycine (SG) and were then stimulated with IL-4 for 24 h or the indicated times, followed by qPCR (g) or western blot (h) analysis of M2 marker expression. PHGDH-WT and PHGDH-KO BMDMs were starved of SG for 12 h and were then stimulated with IFN-γ for 12 h (i, j) or IL-4 for 24 h (k, l), followed by qPCR or western blot analysis of M1 (i, j) or M2 (k, l) marker expression. The data are from three independent experiments with biological duplicates in each and are shown as the mean ± SEM values (n = 3) (a, c, e, g, i, k) or are representative of three independent experiments (b, d, f, h, j, l). *p < 0.05, **p < 0.01, ***p < 0.001
Serine metabolism regulates macrophage polarization in vivo
To further investigate the role of myeloid PHGDH in macrophage polarization in vivo, we transplanted murine LLC cells into PHGDH-WT-Mφ and PHGDH-KO-Mφ mice and found that myeloid PHGDH deficiency robustly inhibited tumor growth (Fig. 3a, b). Next, we examined immune cell infiltration in tumors by immunohistochemical staining of tumor sections. In sections from PHGDH-KO-Mφ mice, the numbers of CD11b-positive cells and CD4-positive cells almost doubled and the population of CD8a-positive cells increased three-fold compared with those in sections from PHGDH-WT-Mφ mice (Fig. S3a). The number of regulatory T cells decreased by ~50%, as shown by forkhead box P3 (FoxP3) staining (Fig. S3a). We then investigated macrophage infiltration and polarization in tumor sections. Our results showed that the population of F4/80-positive macrophages increased nearly two-fold (Fig. S3b). The expression of iNOS was significantly upregulated, while that of ARG1 was downregulated (Fig. S3b). Consistent with these findings, immunofluorescence staining showed that macrophages that expressed IL-1β, iNOS, or IL-6 were significantly more abundant than those that did not (Fig. 3c), indicating that PHGDH deficiency promotes M(IFN-γ) but suppresses M(IL-4) macrophage polarization in vivo. Next, we investigated the effect of serine starvation on macrophage polarization in vivo. C57BL/6 mice were maintained on standard or serine/glycine-free chow for 2 weeks and were then inoculated with LLC cells. Consistent with a previous report [27], serine-restricted diet-fed mice, along with significantly lower concentrations of serum serine and the SG-derived metabolite SAM, showed reduced tumor growth (Fig. S4a–d). Furthermore, in vivo macrophage depletion with clodronate liposomes [28,29,30,31], which efficiently depletes both M1 and M2 TAMs, inhibited LLC tumor growth in control diet-fed mice but accelerated tumor growth in serine-restricted diet-fed mice (Fig. S4c–e). These results strongly suggest that serine restriction reprograms M2 TAMs to the M1 phenotype, an antitumoral phenotype, in LLC tumors.

Serine metabolism regulates macrophage polarization in vivo. a–c PHGDH-WT and PHGDH-KO mice were subcutaneously injected with 2 × 106 LLC cells per mouse (n = 6). Tumor volume was calculated every 2 days beginning 4 days after cell inoculation (a). Tumor xenografts and tumor weights on the 21st day are shown (b). Immunofluorescence staining was performed with the indicated antibodies on tumor sections from PHGDH-WT and PHGDH-KO mice. Representative images are shown (c). Scale bars, 100 μm. Semiquantitative histological analysis was performed (10 fields of 5 biological replicates for each group). d–g Eosinophil populations in the peritoneal cavities of PHGDH-WT and PHGDH-KO mice (n = 4) were analyzed by flow cytometry 2 days post-chitin administration. The percentages of eosinophils among CD45+ cells (d, e) and the total numbers of eosinophils (f) are shown. The expression of Arg1 mRNA in isolated peritoneal macrophages was measured by qPCR (g). h–k Eosinophil populations in the peritoneal cavities of wild-type mice treated with or without CBR-5884 and/or SG starvation (n = 4) were analyzed as described above. Shown are the percentages of eosinophils among CD45+ cells (h, i) and the total numbers of eosinophils (j). The expression of Arg1 mRNA in isolated peritoneal macrophages was measured by qPCR (k). The data are shown as the mean ± SEM values (n = 6 in a and the right panel of b; n = 10 in the right panel of c; n = 4 in e–g and i–k) or are representative of four mice (d, h). *p < 0.05, **p < 0.01, ***p < 0.001
As ARG1-expressing macrophages have a critical function in promoting the resolution of fibrosis [32, 33], we further tested the role of myeloid PHGDH in regulating the M(IL-4) phenotype in the CCl4-induced liver fibrosis model. We found that mice with myeloid PHGDH deficiency showed more severe liver fibrosis, as demonstrated by increased serum levels of alanine aminotransferase (ALT) (Fig. S5a) and exacerbated hepatic steatosis (Fig. S5b), as well as expanded fibrotic areas (Fig. S5c, d). Furthermore, we found that myeloid PHGDH deficiency did not affect Th2 cytokine expression but significantly reduced the number of ARG1-positive macrophages in the liver, indicating that PHGDH acts as a positive regulator of the M(IL-4) phenotype (Fig. S5e, f).
Another important function of M(IL-4)-polarized macrophages is to recruit eosinophils through ARG1, a process that can be modeled by chitin treatment [32]. Myeloid PHGDH deficiency induced by either genetic ablation or chemical inhibitor treatment reduced the recruitment of eosinophils but did not change the percentage of macrophages and also reduced Arg1 expression in macrophages (Fig. 3d–g and Fig. S5g), indicating that PHGDH deficiency impaired M(IL-4) functions in vivo. Next, we investigated the effect of serine starvation on the M(IL-4) phenotype in vivo. C57BL/6 mice were maintained on standard or serine/glycine-free chow for 2 weeks and then treated with chitin. Serine-restricted diet-fed mice, along with a significantly lower concentration of serum serine, showed reduced recruitment of eosinophils and reduced Arg1 expression in macrophages (Fig. 3h–k). Moreover, the synergistic effect of PHGDH inhibition and serine deprivation on the suppression of M2 polarization was also observed in vivo (Fig. 3h–k), further implying a vital role for serine metabolism in promoting the function of M(IL-4) macrophages in vivo.
Serine metabolism modulates macrophage polarization through the p38–JAK–STAT1 signaling axis
Next, we explored how serine metabolism modulates macrophage polarization. STAT1 activation reportedly plays a critical role in M1 macrophage polarization, whereas M2 macrophage activation requires STAT6 activity [1]. Both PHGDH-KO primary BMDMs and siPHGDH-transfected RAW264.7 cells showed enhanced activation of the STAT1-upstream kinases JAK1 and JAK2 upon IFN-γ treatment (Fig. 4a and Fig. S6a); SG starvation also induced a similar effect (Fig. 4b). Furthermore, PHGDH deficiency enhanced the activation of STAT1 and JAK1 upon interferon-β (IFN-β) treatment (Fig. S6b), suggesting that JAK1–STAT1 signaling is generally inhibited by PHGDH. Conversely, PHGDH overexpression resulted in reduced activation of JAK–STAT1 signaling induced by IFN-γ (Fig. 4c). Moreover, treatment with specific inhibitors either for STAT1 (fludarabine) or for JAK1/2 (ruxolitinib) significantly reduced the enhanced phosphorylation of STAT1 and expression of iNOS in PHGDH-deficient RAW264.7 cells (Fig. 4d), suggesting that canonical JAK–STAT1 signaling is responsible for the PHGDH deficiency-induced upregulation of M(IFN-γ) marker genes. Of note, the expression of the IFN-γ receptor subunits IFN-γR1 and IFN-γR2 was not upregulated in PHGDH-deficient cells, either at the basal level or by IFN-γ treatment (Fig. S6c, d).

Serine metabolism modulates macrophage polarization through the p38–JAK–STAT1 signaling axis. a–i Western blot analyses were performed with the indicated antibodies. PHGDH-WT and PHGDH-KO BMDMs were treated with IFN-γ for the indicated times (a). WT BMDMs were starved of SG for 12 h and were then treated with IFN-γ for 30 min (b). RAW264.7 cells were transfected with the PHGDH ectopic expression plasmid and were then stimulated with IFN-γ for the indicated times (c). RAW264.7 cells were transfected with siControl or siPHGDH, treated with JAK (ruxolitinib), STAT1 (fludarabine), or p38 (SB203580) inhibitors for 6 h, and were then stimulated with IFN-γ for 12 h (d). PHGDH-WT and PHGDH-KO BMDMs were pretreated with the p38 inhibitor SB203580 for 6 h and were then stimulated with IFN-γ for the indicated times (e). WT BMDMs were starved of SG for 12 h, treated with the p38 inhibitor SB203580 for 6 h, and then stimulated with IFN-γ for 30 min (f). WT BMDMs were pretreated with the PHGDH inhibitor CBR-5884 for 6 h and were then stimulated with IL-4 for the indicated times (g). PHGDH-WT and PHGDH-KO BMDMs were stimulated with IL-4 for the indicated times (h). WT BMDMs were starved of SG for 12 h and were then stimulated with IL-4 for the indicated times (i). PHGDH-WT and PHGDH-KO BMDMs were pretreated with the STAT6 inhibitor AS1517499 for 6 h and were then stimulated with IL-4 for 24 h, followed by analysis of M2 markers by qRT‒PCR (j) or western blotting (k). l WT BMDMs were starved of SG for 12 h, treated with AS1517499 for 6 h, and then stimulated with IL-4 for 24 h, followed by analysis of ARG1 by western blotting. PHGDH-WT and PHGDH-KO BMDMs were pretreated with the STAT1 inhibitor fludarabine for 6 h and were then stimulated with IL-4 for 24 h, followed by analysis of M2 markers by qRT‒PCR (m) or western blotting (n). o WT BMDMs were starved of SG for 12 h, treated with fludarabine for 6 h, and then stimulated with IL-4 for 24 h, followed by analysis of ARG1 by western blotting. The data are from three independent experiments with biological duplicates in each and are shown as the mean ± SEM values (n = 3) (j, m) or are representative of three independent experiments (a–i and k, l, n, o); NS not significant (p ≥ 0.05); *p < 0.05; **p < 0.01, ***p < 0.001
We then investigated whether other signaling pathways are altered in the setting of PHGDH deficiency. We found that PHGDH deficiency induced enhanced activation of p38 but had minimal influence on the regulation of other MAP kinases or metabolic signaling pathways (Fig. S6e). Furthermore, we found that the p38-specific inhibitor SB203580 significantly reduced IFN-γ−STAT1 activation and M1 marker expression in the setting of PHGDH deficiency (Fig. 4d, e and Fig. S6f). Moreover, the enhanced IFN-γ−JAK–STAT1 signaling induced by SG deprivation was also dependent on p38 activity (Fig. 4f and Fig. S6g).
Next, we explored the mechanisms by which PHGDH promotes M(IL-4) polarization. We found that either PHGDH deficiency or SG deprivation significantly reduced the phosphorylation of STAT6 but not that of other metabolic signaling molecules (Fig. 4g–i and Fig. S7a–c). Furthermore, the STAT6-specific inhibitor AS1517499 significantly disrupted the phenotype (Fig. 4j–l). Moreover, the STAT1 inhibitor fludarabine reversed the suppression of the M(IL-4) phenotype (Fig. 4m–o). Taken together, these data indicate that serine metabolism modulates macrophage polarization through the p38–JAK–STAT1 signaling axis.
SAM derived from serine metabolism modulates macrophage polarization
Energy metabolism remodeling has been reported to support macrophage polarization [4,5,6]. By using a Seahorse extracellular flux analyzer to measure the ECAR and OCR, we found that PHGDH deficiency or SG starvation did not significantly alter energy metabolism in IFN-γ- or IL-4-treated PMs, consistent with the minimal regulation of metabolic signaling pathways (Figs. S8a–e, S6e and S7b).
We then sought to identify the SG-derived metabolites that might be responsible for modulating macrophage polarization. Formate, glutathione (GSH), and SAM have been separately documented to play important roles in serine-modulated cellular functions such as cancer and T-cell proliferation and cytokine secretion by virus-infected or LPS-treated macrophages [11, 12, 25, 34,35,36]. Interestingly, we found that the addition of SAM but not formate or GSH rescued the alteration in macrophage polarization induced by SG deprivation (Fig. 5a, b). As expected, SG deprivation and PHGDH knockout significantly reduced the cellular SAM level compared with that in control macrophages (Fig. S8f, g). Consistent with this finding, the serum concentration of SAM in mice fed a serine-restricted diet was significantly lower than that in mice in the control group (Fig. S4b), indicating that the concentration of SAM is closely associated with serine metabolism in vitro and in vivo.

SAM derived from serine metabolism modulates macrophage polarization. qPCR analysis of M1 (a) or M2 markers (b) in WT BMDMs starved of SG for 12 h; supplemented with the indicated concentration of formate, GSH, or SAM; and then treated with IFN-γ for 12 h (a) or IL-4 for 24 h (b). qPCR analysis of M1 and M2 markers in PHGDH-WT and PHGDH-KO BMDMs supplemented with or without 400 μM serine (c, d) or 400 μM SAM (e, f) for 12 h and then treated with IFN-γ for 12 h or IL-4 for 24 h. g–j Western blot analysis with the indicated antibodies. PHGDH-WT and PHGDH-KO BMDMs supplemented with or without 400 μM SAM for 12 h were treated with IFN-γ for 12 h (g) or IL-4 for 24 h (i). WT BMDMs starved of SG for 12 h and supplemented with 400 μM SAM were treated with IFN-γ for 12 h (h) or IL-4 for 24 h (j). The data are from three independent experiments with biological duplicates in each and are shown as the mean ± SEM values (n = 3) (a–f) or are representative of three independent experiments (g–j). NS not significant (p ≥ 0.05); *p < 0.05; **p < 0.01; ***p < 0.001
We further found that the addition of exogenous serine and SAM significantly suppressed M(IFN-γ) but supported M(IL-4) polarization (Fig. 5c–f). In addition, SAM treatment restored the levels of phenotypic markers in PHGDH-KO BMDMs to the levels seen in WT BMDMs (Fig. 5e, f), whereas exogenous serine exhibited only a partial rescue effect (Fig. 5c, d). This may be because serine treatment only modestly increased the SAM level in PHGDH-KO BMDMs (Fig. S8g), in agreement with our previous report that PHGDH deficiency reduces the efficiency of one-carbon flux, even in the presence of exogenous serine [25]. SAM treatment also restored IFN-γ-induced p38–JAK–STAT1 signaling and IL4–STAT6 signaling in PHGDH-deficient or SG-deprived BMDMs to the levels seen in WT BMDMs (Fig. 5g–j). Taken together, these data indicate that SAM derived from serine metabolism is crucial for regulating macrophage polarization.
Serine metabolism reduces IGF1 expression by increasing the promoter abundance of SAM-dependent H3K27me3
To further investigate the mechanism by which serine metabolism orchestrates macrophage polarization, we used a high-throughput RNA sequencing (RNA-seq) to assess gene expression in PHGDH-deficient BMDMs and RAW264.7 macrophages (these two RNA-seq datasets were used in [25]) and in SG-depleted BMDMs (Fig. S9a). Nine genes that were identified as upregulated in all three RNA-seq datasets were selected for further analysis of macrophage polarization (Fig. S9a). Knocking down IGF1 alone by siRNA in BMDMs reduced M(IFN-γ) and enhanced M(IL-4) macrophage polarization (Fig. S9b–d).
Next, we sought to determine how the IGF1 expression level correlates with serine metabolism. Igf1 mRNA expression was upregulated in PHGDH-deficient RAW264.7 cells, BMDMs, and PMs compared with the corresponding control cells (Fig. 6a and Fig. S9e, f). Furthermore, the IGF1 level was elevated in the supernatant of PHGDH-KO cells (Fig. 6b). Conversely, PHGDH overexpression significantly reduced IGF1 expression in RAW264.7 cells (Fig. 6c, d). SG starvation also enhanced IGF1 expression in BMDMs (Fig. 6e, f). We then transfected cells with a promoter–reporter construct of Igf1 and found that stable knockdown of PHGDH significantly increased promoter activity and that serine starvation further enhanced this effect in RAW264.7 cells (Fig. 6g); collectively, these results suggest that Igf1 expression is affected by serine metabolism at the transcriptional level.

Serine metabolism reduces IGF1 expression by increasing the promoter abundance of SAM-dependent H3K27me3. qPCR analysis of Igf1 mRNA expression (a) and ELISA (b) of IGF1 in the supernatants of PHGDH-WT and PHGDH-KO BMDMs. qPCR analysis of Igf1 mRNA expression (c) and ELISA (d) of IGF1 in the supernatants of RAW264.7 cells transfected with empty vector or the PHGDH overexpression plasmid. qPCR analysis of Igf1 mRNA expression (e) or ELISA (f) of IGF1 in the supernatants of BMDMs starved of SG. g Analysis of Igf1-Luc activity in PHGDH-WT and PHGDH-KO BMDMs starved of SG. h qPCR analysis of Igf1 mRNA expression in PHGDH-WT and PHGDH-KO BMDMs supplemented with or without 400 μM SAM. i Analysis of Igf1-Luc activity in RAW264.7 cells transfected with siControl or siPHGDH and supplemented with or without 400 μM SAM. j, k Western blot analysis of histone modification markers in PHGDH-WT and PHGDH-KO BMDMs supplemented with or without 400 μM SAM for 12 h. l ChIP‒qPCR analysis of H3K27me3 enrichment at the indicated positions in the Igf1 promoter in PHGDH-WT and PHGDH-KO BMDMs. PHGDH-WT and PHGDH-KO BMDMs were supplemented with or without 400 μM serine or 400 μM SAM for 12 h and were then treated with or without IFN-γ for 12 h (m–o) or IL-4 for 24 h (p, q), followed by ChIP‒qPCR analysis of H3K27me3 enrichment in the promoter of the Igf1 gene. r, s qPCR analysis of Jmjd3 and Ezh2 mRNA expression in PHGDH-WT and PHGDH-KO BMDMs. t Western blot analysis of EZH2 expression in PHGDH-WT and PHGDH-KO BMDMs. BMDMs with siRNA-mediated EZH2 silencing were transfected with empty vector or the PHGDH expression plasmid. qPCR analysis of Igf1 mRNA expression (u), western blot analysis performed with the indicated antibodies (v), and ChIP‒qPCR analysis of H3K27me3 enrichment in the promoter of the Igf1 gene (w). The data are from three independent experiments with biological duplicates in each and are shown as the mean ± SEM values (n = 3) (a–i, l–s, u, w) or are representative of three independent experiments (j, k, t, v). NS not significant (p ≥ 0.05); *p < 0.05; **p < 0.01; ***p < 0.001
PHGDH and serine metabolism are involved in histone methylation, which is critical for regulating chromatin status and gene transcription, through the major methyl donor SAM [12, 25, 37,38,39,40]. Therefore, we hypothesized that PHGDH and serine metabolism decrease the expression of Igf1 in a SAM-dependent manner. Our results showed that SAM treatment significantly reduced the increases in both Igf1 promoter activity and Igf1 mRNA expression induced either by PHGDH deficiency (Fig. 6h, i) or by SG starvation (Fig. S9g, h). Therefore, we sought to determine whether serine deprivation changes the histone modification status of the Igf1 promoter. Consistent with our previous findings, the level of the major repressive histone methylation mark H3K27me3 (trimethylation of histone H3 at lysine 27) but not the levels of H3K9me3 and H3K4me3 (trimethylation of histone H3 at lysine 9 and lysine 4, respectively) [25, 40, 41] was substantially decreased in PHGDH-deficient macrophages and SG-starved macrophages (Fig. 6j and Fig. S9i). Moreover, we found that the decrease in the H3K27me3 level caused by knockout of the Phgdh gene was rescued by exogenous supplementation with SAM (Fig. 6k). Thus, we next performed chromatin immunoprecipitation followed by qPCR (ChIP‒qPCR) to investigate how the H3K27me3 chromatin occupancy in the nine genes with upregulated transcription in all three datasets (listed in Fig. S9a) changed under serine deficiency. Our results showed that H3K27me3 occupancy in the promoter of the Igf1 gene was significantly decreased in both PHGDH-deficient cells and SG-starved cells (Fig. 6l and Fig. S9j, k). A similar reduction in the H3K27me3 abundance was found in the promoters of another four upregulated genes in cells deprived of serine (Fig. S9j). Furthermore, we found that exogenous supplementation with serine and SAM enhanced H3K27me3 enrichment at the Igf1 gene promoter in PHGDH-deficient macrophages during macrophage polarization (Fig. 6m–q). Consistent with our previous report [25], the rescue effect of serine was weaker than that of SAM, especially in KO BMDMs, indicating a role for the PHGDH-mediated serine biosynthesis pathway in coordinating efficient one-carbon flux (Figs. 5c–f and 6m–q). Together, these results indicate that serine metabolism can inhibit Igf1 transcription by increasing the promoter abundance of SAM-mediated H3K27me3. Given that enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) is a key H3K27 methyltransferase and regulates Igf1 expression through epigenetic regulation [42,43,44], we sought to determine whether serine metabolism inhibits Igf1 expression through suppression of EZH2-dependent methylation of H3K27 in the promoter of Igf1. Interestingly, we found that the expression of EZH2 but not that of the H3K27 demethylase jumonji domain containing 3 (JMJD3) was significantly decreased in PHGDH-deficient cells and SG-starved cells (Fig. 6r–t and Fig. S9l–n), consistent with a previous report [45]. EZH2 knockdown significantly increased the mRNA level of Igf1, which was decreased by PHGDH overexpression (Fig. 6u). Further experiments showed that EZH2 knockdown reduced the PHGDH overexpression-induced increases in the total H3K27me3 level and the H3K27me3 abundance in the promoter of the Igf1 gene (Fig. 6v, w and Fig. S9o, p). These data suggest that serine metabolism deficiency increases the expression of Igf1 by decreasing the levels of SAM and EZH2 and subsequently decreasing the level of H3K27me3 in the Igf1 promoter region.
Serine metabolism deficiency modulates macrophage polarization and JAK–STAT1 signaling via IGF1-dependent p38 activation
To explore whether Igf1 signaling regulates macrophage polarization, we generated IGF1Rfl/flLyz2-Cre+ (IGF1R-KO-Mφ) mice, which undergo deletion of loxP-flanked IGF1R alleles (IGF1Rfl/fl) specifically in myeloid cells. IGF1R-KO-Mφ mice displayed normal PM differentiation and numbers compared with the corresponding IGF1Rfl/flLyz2-Cre− (IGF1R-WT-Mφ) mice (Fig. S10a–c). Macrophage-specific deletion of IGF1R was confirmed in primary PMs from IGF1Rfl/flLyz2-Cre+ mice (Fig. S10d). IGF1R KO reduced M(IFN-γ) phenotype polarization and activation of the p38–JAK–STAT1 signaling axis but supported M(IL-4) phenotype polarization and enhanced STAT6 phosphorylation (Fig. 7a–d). Similar results were obtained by inhibition of IGF1 signaling in BMDMs with siIGF1R or siIGF1 (Fig. S10e–i). To address whether IGF1 signaling can protect against tumor progression in mice, IGF1R-KO-Mφ and IGF1R-WT-Mφ mice were challenged with inoculation of LLC cells for xenograft formation. IGF1R-KO-Mφ mice showed larger tumor sizes (Fig. S10j, k) and reduced M1 macrophage polarization but enhanced M2 polarization in tumor sections (Fig. S10l). These data indicate that IGF1 signaling supports M1 phenotype polarization but inhibits M2 macrophage activation.

Serine metabolism deficiency orchestrates macrophage polarization and JAK–STAT1 signaling via IGF1-dependent p38 activation. Igf1fl/flLyz2-Cre− (IGF1R-WT) and Igf1Rfl/flLyz2-Cre+ mouse (IGF1R-KO) BMDMs were stimulated with IFN-γ for 12 h or IL-4 for 24 h, followed by qRT‒PCR analysis of M1 (a) and M2 (b) marker expression. IGF1R-WT and IGF1R-KO BMDMs were treated with IFN-γ (c) or IL-4 (d) for the indicated times, followed by western blot analysis with the indicated antibodies. PHGDH-WT and PHGDH-KO BMDMs were transfected with siControl or siIGF1R and were then stimulated either with IFN-γ for 12 h (e, f) or the indicated times (i) or with IL-4 for 24 h (g, h) or the indicated times (j), followed by qRT‒PCR or western blot analysis. WT BMDMs transfected with siControl or siIGF1R were starved of SG, supplemented with or without 400 μM SAM, and then stimulated either with IFN-γ for 12 h (k) or 30 min (m) or with IL-4 for 24 h (l) or 30 min (n), followed by qRT‒PCR or western blot analysis. The data are from three independent experiments with biological duplicates in each and are shown as the mean ± SEM values (n = 3) (a, b, e, g, k, l) or are representative of three independent experiments (c, d, f, h, i, j, m, n). *p < 0.05, **p < 0.01, ***p < 0.001
Moreover, the enhanced transcription of IFN-γ-induced M1 markers and reduced transcription of IL-4-induced M2 markers in siPHGDH-transfected BMDMs (Fig. S11a, b) and PHGDH-KO BMDMs (Fig. 7e–h) were reversed by inhibition of IGF1 signaling with siIGF1 or siIGF1R, respectively. The elevated IFN-γ-induced phosphorylation of p38–STAT1 and decreased IL-4-induced phosphorylation of STAT6 in PHGDH-KO BMDMs were also restored by siIGF1R transfection (Fig. 7i, j). Similar reversals in M1 and M2 marker transcription were also observed in IGF1R-knockdown SG-starved BMDMs (Fig. 7k, l). Supplementation with SAM restored the elevated p38–JAK–STAT1 signaling and reduced IL-4–STAT6 signaling in SG-starved WT BMDMs, and this restoration was further enhanced in IGF1-knockdown cells (Fig. 7m, n). Collectively, these results indicate that the SAM–IGF1 axis mediates serine deficiency-induced p38 activation. Taken together, these data indicate that serine metabolism deficiency modulates macrophage polarization and JAK–STAT1 signaling via IGF1-dependent p38 activation (Fig. 8).

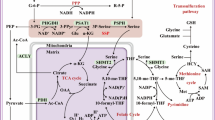
Serine metabolism regulates the SAM-IGF1-p38 axis to orchestrate macrophage polarization. Suppressing the serine biosynthesis pathway either by inhibition of PHGDH activity or by exogenous serine and glycine restriction, robustly enhances M(IFN-γ) polarization but suppresses M(IL-4) polarization both in vitro and in vivo. Serine metabolism deficiency increases the expression of IGF1 by reducing the abundance of SAM-dependent H3K27me3 in the promoter of the Igf1 gene. IGF1 then activates the p38-dependent JAK–STAT1 axis to promote M(IFN-γ) polarization and suppress STAT6-mediated M(IL-4) activation
PHGDH has been reported to be overexpressed in various types of tumors. We next sought to determine whether tumor PHGDH can affect macrophage polarization through secreted IGF1. By utilizing several human breast tumor cell lines, we found that the level of intracellular PHGDH was negatively correlated with the level of secreted IGF1 in the culture supernatant (Fig. S11c). Furthermore, PHGDH knockdown in HCC70 cells, with a high level of PHGDH expression, enhanced the mRNA level and secretion of IGF1. In contrast, PHGDH overexpression in T47D cells, with barely detectable PHGDH expression, reduced the mRNA level and secretion of IGF1 (Fig. S11d). Furthermore, the increase in M(IL-4) and decrease in M(IFN-γ) polarization induced by the HCC70 cell supernatant were reversed by chemical inhibition of PHGDH in HCC70 cells, and these effects were reinduced by the addition of an IGF1 neutralizing antibody (Fig. S11e, f). Similar anti-IGF1 treatment reversed the alterations in macrophage polarization phenotype induced by the supernatant of T47D cells (Fig. S11g, h), indicating the crucial role of secreted IGF1 in macrophage polarization. Taken together, the above data show that tumor PHGDH can modulate macrophage polarization through secreted IGF1.
Discussion
The importance of the essential amino acids L-arginine and glutamine and their derived metabolites in regulating classical and alternative macrophage activation states has been well documented [7,8,9,10]. Intriguingly, our study shows that serine, a nonessential amino acid, can regulate macrophage-mediated immune responses by fine-tuning the balance of macrophage polarization states. Specifically, inhibiting serine metabolism by pharmacological inhibition or genetic ablation of the key de novo serine biosynthesis enzyme PHGDH or by exogenous serine restriction markedly promoted M(IFN-γ) but inhibited M(IL-4) polarization both in vitro and in vivo. Moreover, PHGDH deficiency and serine deprivation had a synergistic effect on the regulation of macrophage polarization. The addition of exogenous serine further influenced the macrophage polarization state, indicating that intracellular serine in macrophages is not saturated and can be bidirectionally manipulated to modulate serine-related activities. It would be interesting to investigate whether and how serine concentrations in different tissue microenvironments contribute to distinct resident macrophage phenotypes.
Our mechanistic study also revealed a modulatory role for SAM derived via the serine synthesis pathway (SSP) or from exogenous serine in macrophage polarization. The addition of exogenous serine partially reversed the reprogramming of macrophage polarization phenotypes in PHGDH-deficient cells, indicating that the blockade of one-carbon flux caused by SSP inhibition was only partial, consistent with our previous findings [25]. This conclusion was also supported by the synergistic effect of PHGDH deficiency and serine starvation on macrophage polarization (Fig. 2i–l and Fig. S2c).
Our further experiments showed that serine metabolism deficiency reduced the abundance of SAM-mediated H3K27me3 at the promoter of Igf1 and thus promoted Igf1 expression. The contradictory reported effects of IGF1 on macrophage polarization may be attributable to differences in the disease models and tissue microenvironments utilized [18, 19, 21, 22].
The IGF1–p38 axis further supported STAT1-dependent M(IFN-γ) polarization but inhibited STAT6-mediated M(IL-4) activation, consistent with the reported enhancing function of stress-induced p38 in STAT1-dependent transcription [46, 47]. Notably, we found that p38 is indispensable for the activation of the STAT1 upstream kinase JAK. However, we could not exclude the possibility that p38 directly influences STAT transcriptional activity, as reported in previous studies [46,47,48].
TAMs are highly plastic and are important regulators of the tumor microenvironment [2, 49, 50]. Because serine metabolism promotes tumor proliferation by participating in many important processes, the antitumor effects of PHGDH inhibitor treatment and/or dietary SG limitation have been extensively studied in animal models [27, 51,52,53,54,55,56]. Interestingly, our experiments showed that the PHGDH levels in cancer cells were negatively correlated with the secreted IGF1 levels in the corresponding supernatants. Knocking down PHGDH in cancer cells with high PHGDH levels conditioned macrophages for polarization toward the M1 phenotype by elevating IGF1 secretion (Fig. S11e). Therefore, our study shows that manipulating serine metabolism might target TAM plasticity and antitumor immunity not only by directly influencing macrophage effector functions but also by interfering with the communication between tumor cells and TAMs.
Overall, this work reveals the important roles of PHGDH and serine metabolism in orchestrating macrophage polarization. Our work not only identifies the serine–SAM–IGF1–p38–STAT1 axis as a new “metabo-epi-immune” mechanism but also extends our understanding of IGF1 as another critical cytokine secreted from cancer cells that affects the antitumor immune function of TAMs.
References
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Investig. 2012;122:787–95.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20.
Langston PK, Shibata M, Horng T. Metabolism supports macrophage activation. Front Immunol. 2017;8:61.
Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. 2017;38:395–406.
Stienstra R, Netea-Maier RT, Riksen NP, Joosten LAB, Netea MG. Specific and complex reprogramming of cellular metabolism in myeloid cells during innate immune responses. Cell Metab. 2017;26:142–56.
Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. 1998;160:5347–54.
Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. 2014;5:532.
Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–54.
Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18:985–94.
Rodriguez AE, Ducker GS, Billingham LK, Martinez CA, Mainolfi N, Suri V, et al. Serine metabolism supports macrophage IL-1beta production. Cell Metab. 2019;29:1003–11.e4.
Yu W, Wang Z, Zhang K, Chi Z, Xu T, Jiang D, et al. One-carbon metabolism supports S-adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol Cell. 2019;75:1147–60.e5.
Chen S, Xia Y, He F, Fu J, Xin Z, Deng B, et al. Serine supports IL-1beta production in macrophages through mTOR signaling. Front Immunol. 2020;11:1866.
Wilson JL, Nagele T, Linke M, Demel F, Fritsch SD, Mayr HK, et al. Inverse data-driven modeling and multiomics analysis reveals Phgdh as a metabolic checkpoint of macrophage polarization and proliferation. Cell Rep. 2020;30:1542–52.e7.
Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18:545–58.
Hakuno F, Takahashi SI. IGF1 receptor signaling pathways. J Mol Endocrinol. 2018;61:T69–86.
Kineman RD, Del Rio-Moreno M, Sarmento-Cabral A. 40 years of IGF1: understanding the tissue-specific roles of IGF1/IGF1R in regulating metabolism using the Cre/loxP system. J Mol Endocrinol. 2018;61:T187–98.
Higashi Y, Sukhanov S, Shai SY, Danchuk S, Tang R, Snarski P, et al. Insulin-like growth factor-1 receptor deficiency in macrophages accelerates atherosclerosis and induces an unstable plaque phenotype in apolipoprotein E-deficient mice. Circulation. 2016;133:2263–78.
Spadaro O, Camell CD, Bosurgi L, Nguyen KY, Youm YH, Rothlin CV, et al. IGF1 shapes macrophage activation in response to immunometabolic challenge. Cell Rep. 2017;19:225–34.
Barrett JP, Minogue AM, Falvey A, Lynch MA. Involvement of IGF-1 and Akt in M1/M2 activation state in bone marrow-derived macrophages. Exp Cell Res. 2015;335:258–68.
Ieronymaki E, Theodorakis EM, Lyroni K, Vergadi E, Lagoudaki E, Al-Qahtani A, et al. Insulin resistance in macrophages alters their metabolism and promotes an M2-like phenotype. J Immunol. 2019;202:1786–97.
Youssif C, Cubillos-Rojas M, Comalada M, Llonch E, Perna C, Djouder N, et al. Myeloid p38alpha signaling promotes intestinal IGF-1 production and inflammation-associated tumorigenesis. EMBO Mol Med. 2018;10:e8403.
American Type Culture Collection Standards Development Organization Workgroup ASN. Cell line misidentification: the beginning of the end. Nat Rev Cancer. 2010;10:441–8.
Mullarky E, Lucki NC, Beheshti Zavareh R, Anglin JL, Gomes AP, Nicolay BN, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci USA. 2016;113:1778–83.
Shen L, Hu P, Zhang Y, Ji Z, Shan X, Ni L, et al. Serine metabolism antagonizes antiviral innate immunity by preventing ATP6V0d2-mediated YAP lysosomal degradation. Cell Metab. 2021;33:971–87.e6.
Foster AC, Rangel-Diaz N, Staubli U, Yang JY, Penjwini M, Viswanath V, et al. Phenylglycine analogs are inhibitors of the neutral amino acid transporters ASCT1 and ASCT2 and enhance NMDA receptor-mediated LTP in rat visual cortex slices. Neuropharmacology. 2017;126:70–83.
Muthusamy T, Cordes T, Handzlik MK, You L, Lim EW, Gengatharan J, et al. Serine restriction alters sphingolipid diversity to constrain tumour growth. Nature. 2020;586:790–5.
Xia L, Huang W, Tian D, Zhang L, Qi X, Chen Z, et al. Forkhead box Q1 promotes hepatocellular carcinoma metastasis by transactivating ZEB2 and VersicanV1 expression. Hepatology. 2014;59:958–73.
D’Errico G, Alonso-Nocelo M, Vallespinos M, Hermann PC, Alcala S, Garcia CP, et al. Tumor-associated macrophage-secreted 14-3-3zeta signals via AXL to promote pancreatic cancer chemoresistance. Oncogene. 2019;38:5469–85.
Wang Y, Sun Q, Ye Y, Sun X, Xie S, Zhan Y, et al. FGF-2 signaling in nasopharyngeal carcinoma modulates pericyte-macrophage crosstalk and metastasis. JCI Insight. 2022;7:e157874.
Tan HY, Wang N, Man K, Tsao SW, Che CM, Feng Y. Autophagy-induced RelB/p52 activation mediates tumour-associated macrophage repolarisation and suppression of hepatocellular carcinoma by natural compound baicalin. Cell Death Dis. 2015;6:e1942.
Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009;5:e1000371.
Ji L, Zhao X, Zhang B, Kang L, Song W, Zhao B, et al. Slc6a8-mediated creatine uptake and accumulation reprogram macrophage polarization via regulating cytokine responses. Immunity. 2019;51:272–84.e7.
Ducker GS, Ghergurovich JM, Mainolfi N, Suri V, Jeong SK, Hsin-Jung Li S, et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2017;114:11404–9.
Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 2017;25:345–57.
Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 2016;24:104–17.
Gao X, Locasale JW. Serine metabolism links tumor suppression to the epigenetic landscape. Cell Metab. 2016;24:777–9.
Li S, Swanson SK, Gogol M, Florens L, Washburn MP, Workman JL, et al. Serine and SAM responsive complex SESAME regulates histone modification crosstalk by sensing cellular metabolism. Mol Cell. 2015;60:408–21.
Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 2015;22:861–73.
Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–69.
Iwabata H, Yoshida M, Komatsu Y. Proteomic analysis of organ-specific post-translational lysine-acetylation and -methylation in mice by use of anti-acetyllysine and -methyllysine mouse monoclonal antibodies. Proteomics. 2005;5:4653–64.
Giambra V, Gusscott S, Gracias D, Song R, Lam SH, Panelli P, et al. Epigenetic restoration of fetal-like IGF1 signaling inhibits leukemia stem cell activity. Cell Stem Cell. 2018;23:714–26.e7.
Galvis LA, Holik AZ, Short KM, Pasquet J, Lun AT, Blewitt ME, et al. Repression of Igf1 expression by Ezh2 prevents basal cell differentiation in the developing lung. Development. 2015;142:1458–69.
Wang Y, Hou N, Cheng X, Zhang J, Tan X, Zhang C, et al. Ezh2 acts as a tumor suppressor in Kras-driven lung adenocarcinoma. Int J Biol Sci. 2017;13:652–9.
Bialopiotrowicz E, Noyszewska-Kania M, Kachamakova-Trojanowska N, Loboda A, Cybulska M, Grochowska A, et al. Serine biosynthesis pathway supports MYC-miR-494-EZH2 feed-forward circuit necessary to maintain metabolic and epigenetic reprogramming of Burkitt lymphoma cells. Cancers. 2020;12:580.
Kovarik P, Stoiber D, Eyers PA, Menghini R, Neininger A, Gaestel M, et al. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proc Natl Acad Sci USA. 1999;96:13956–61.
Ramsauer K, Sadzak I, Porras A, Pilz A, Nebreda AR, Decker T, et al. p38 MAPK enhances STAT1-dependent transcription independently of Ser-727 phosphorylation. Proc Natl Acad Sci USA. 2002;99:12859–64.
Goh KC, Haque SJ, Williams BR. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J. 1999;18:5601–8.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–96.
Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30:36–50.
Tajan M, Hennequart M, Cheung EC, Zani F, Hock AK, Legrave N, et al. Serine synthesis pathway inhibition cooperates with dietary serine and glycine limitation for cancer therapy. Nat Commun. 2021;12:366.
Yoshino H, Nohata N, Miyamoto K, Yonemori M, Sakaguchi T, Sugita S, et al. PHGDH as a key enzyme for serine biosynthesis in HIF2alpha-targeting therapy for renal cell carcinoma. Cancer Res. 2017;77:6321–9.
Wang Q, Liberti MV, Liu P, Deng X, Liu Y, Locasale JW, et al. Rational design of selective allosteric inhibitors of PHGDH and serine synthesis with anti-tumor activity. Cell Chem Biol. 2017;24:55–65.
Ngo B, Kim E, Osorio-Vasquez V, Doll S, Bustraan S, Liang RJ, et al. Limited environmental serine and glycine confer brain metastasis sensitivity to PHGDH inhibition. Cancer Discov. 2020;10:1352–73.
Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJ, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12:452–8.
Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–83.
Acknowledgements
We thank Dr. Xiaoyue Tan (Nankai University, China) for kindly providing the Lyz2-Cre mice and several cell lines. This research was supported by grants from the Tianjin Municipal Natural Science Foundation of China (20JCYBJC00220, QY) and from the National Natural Science Foundation of China: 81672710 (QY), 81872239 (QY), 82073051 (TW), 81874055 (TW), and 81902900 (LS).
Author information
Authors and Affiliations
Contributions
QY conceived the project and designed the experiments. QY, YW, and ZY wrote and edited the manuscript. TW, LW, and BC helped with editing the manuscript. XS, PH, LN, LS, YZ, ZJ, and YC conducted the experiments. MG, HW, LR, and KY contributed the IGF1Rfl/fl mice.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shan, X., Hu, P., Ni, L. et al. Serine metabolism orchestrates macrophage polarization by regulating the IGF1–p38 axis. Cell Mol Immunol 19, 1263–1278 (2022). https://doi.org/10.1038/s41423-022-00925-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-022-00925-7
- Springer Nature Limited
Keywords
This article is cited by
-
Regulation of macrophage polarization by targeted metabolic reprogramming for the treatment of lupus nephritis
Molecular Medicine (2024)
-
The significant role of amino acid metabolic reprogramming in cancer
Cell Communication and Signaling (2024)
-
The roles and molecular mechanisms of non-coding RNA in cancer metabolic reprogramming
Cancer Cell International (2024)
-
Trophoblast-derived miR-410-5p induces M2 macrophage polarization and mediates immunotolerance at the fetal-maternal interface by targeting the STAT1 signaling pathway
Journal of Translational Medicine (2024)
-
Metabolic reprogramming and immune evasion: the interplay in the tumor microenvironment
Biomarker Research (2024)