
Abstract
Objective
Emerging studies have revealed that macrophages possess different dependences on the uptake, synthesis, and metabolism of serine for their activation and functionalization, necessitating our insight into how serine availability and utilization impact macrophage activation and inflammatory responses.
Methods
This article summarizes the reports published domestically and internationally about the serine uptake, synthesis, and metabolic flux by the macrophages polarizing with distinct stimuli and under different pathologic conditions, and particularly analyzes how altered serine metabolism rewires the metabolic behaviors of polarizing macrophages and their genetic and epigenetic reprogramming.
Results
Macrophages dynamically change serine metabolism to orchestrate their anabolism, redox balance, mitochondrial function, epigenetics, and post-translation modification, and thus match the distinct needs for both classical and alternative activation.
Conclusion
Serine metabolism coordinates multiple metabolic pathways to tailor macrophage polarization and their responses to different pathogenic attacks and thus holds the potential as therapeutic target for types of acute and chronic inflammatory diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As the key executor of innate immunity, macrophages can be tissue residential or derived from monocytes, and exist as M0, M1, and M2 phenotypes in the body. Upon the crosstalk of PAMPs or DAMPs with their PPRs, quiescent M0 cells are primed to start the gene reprogramming-based polarization and differentiate into proinflammatory M1 type or anti-inflammatory M2 type to fulfill pathogen clearance and damage healing, respectively. Certain activated macrophages even obtain immunity memory after retraction. In vivo, macrophages often adopt mixed phenotypes dependent on the stimulation and differentiation progress [1].
Accompanying with the priming of M0 macrophages is their metabolic remodeling that coordinates their polarization and effector functioning. Comparing to M0 cells running a low level of oxidative phosphorylation (OXPHOS) [2], activated macrophages conduct exuberant bioenergic and biosynthetic metabolisms, like glycolysis, glutaminolysis, OXPHOS, lipogenesis, etc [3]. Metabolic activity greatly differs among macrophage subsets and decides their destiny and immunological exertion [4]. Generally, M1 type has a highly mobilized glycolysis and interrupted tri-carboxylic acid (TCA) cycle, accumulating itaconate and succinate, and producing NO and cytokines; the reparative and proliferative M2 subset exploits glucose for TCA and lipogenesis, burns free fatty acids, and generates arginine; and memorizing macrophages conduct a moderate level of OXPHOS and lipid metabolism [2, 3, 5]. Notably, metabolism rewiring impacts the epigenetics of macrophages crucial for their polarization, as represented by the decision of macrophage fate by the α-ketoglutarate (α-KG)-dependent dioxygenases (like TETs and JMJD3), and the acetyl-CoA-producers ACLY and ASSC [6]. Overall, metabolic reprogramming plays critical roles in macrophage polarization, inflammatory responses, and trained innate immunity [4]; its deregulation destroys macrophage subtype equilibrium and elicits various diseases like sepsis and chronic immunological disorders.
Mounting studies have indicated that macrophages differently and dynamically metabolize serine for their activation and functioning. Cell acquires serine mainly through the uptake from extracellular environment via alanine–serine–cysteine–threonine transporters (ASCT) 1 and 2, and the de novo serine biosynthesis (SSP) where the glycolytic intermediate 3-phosphoglycerate is converted into L-serine under the orderly catalyzation of PHGDH (phosphoglycerate dehydrogenase), PSAT1 (phosphoserine aminotransferase 1), and PSPH (phosphoserine phosphatase) with the concomitant reduction of NAD+ to NADH and α-KG production (Fig. 1). Well-fed cell freely ingests seine to maintain intracellular serine while the SSP becomes vigorous and vital upon serine deprivation or over-consumption [7]. L-serine and its derivatives are structural elements for protein synthesis, the ceramide and membrane phospholipids and sphingolipid [7, 8], and the D-serine, a physiological co-agonist of the N-methyl D-aspartate type of glutamate receptor [9, 10]. The cleavage of serine to glycine and one-carbon (1C) unit by SHMT starts the 1C metabolism for the anabolism of vital cellular components like purine, thymidylate, cysteine and methionine, and the production of formate, ATP, glutathione, S-adenosyl methionine (SAM), etc [11, 12]. Serine metabolism also impacts NAD(P)H yield and mitochondrial fitness and maintains redox balance via promoting pyruvate entry to TCA, OXPHOS, FAO, and SGOC [13,14,15], and more significantly, tunes the levels of metabolites (like SAM, acetyl-CoA, and α-KG) and the (de)methylation of histones (and non-histone proteins) [11, 16,17,18]. With these metabolic contributions (Fig. 1), serine metabolism actively modulates cell growth, survival, and differentiation, and is supportive for proliferative cells like tumor cells, activated T cells, or multipotent cells, tumor initiation, development and metastasis, and immune responses in the body [12, 19, 20].

A schematic presentation of serine metabolism. Cell takes serine from microenvironment or synthesize serine via converting glycolytic intermediate 3-phosphoglycerate. Serine participates in the bio-syntheses of proteins, cell membrane components phosphatidylserine and sphingosine, D-serine, etc. Importantly, serine metabolism conjugates together with one-carbon metabolism that includes the folate cycle and the methionine cycle, and thereby mediates the de novo production of nuclear acid precursors purine and pyrimidine, redox maintaining agents NADPH and glutathione, and methylation donor S-adenosylmethionine, playing a vital role in cell proliferation, survival, differentiation, and epigenetics. SSP de novo serine biosynthesis, PPP pentose phosphate pathway, ASCT1/2 Alanine–serine–cysteine–threonine transporter 1/2, G-6-P glucose 6-phosphate, R-5-P Ribose-5-phosphate, 3-PG 3-Phosphoglycerate, 3-PPyr 3-Phosphohydroxypyruvate, 3P-Serine 3-Phosphoserine, PHGDH 3-Phosphoglycerate dehydrogenase, PSAT1 Phosphoserine aminotransferase 1, PSPH Phosphoserine phosphatase, NAD+ Nicotinamide adenine dicriboglycine (oxidized), NADH Nicotinamide adenine dicriboglycine(reduced), SHMT1 Serine hydroxymethyltransferase 1, SHMT2 Serine hydroxymethyltransferase 2, α-KG α-ketoglutarate; Glu, glutamate; PEP Phosphoenolpyruvate, TCA Tricarboxylic acid, OAA Oxaloacetic acid, Ac-CoA Acetyl-CoA, ACLY ATP citrate lyase, PDH Pyruvate dehydrogenase, ETCs, Electron transport chain, THF tetrahydrofolate, NADP+ Nicotinamide adenine dinucleotide phosphate (oxidized), NADPH Nicotinamide adenine dinucleotide phosphate(reduced), SAM S-adenosylmethionine, Met Methionine, SAH S-adenosyl-l-homocysteine, HCY Homocysteine
Here, we summarize the involvement of serine metabolism in macrophage activation and innate immunity and emphasize how it coordinates multiple metabolic pathways to tailor macrophage polarization and their responses to different pathogenic attacks, aiming to display the connection of macrophages’ serine metabolism with acute and chronic diseases such as cancer and its potential as therapeutic targets.
Mobilization of serine metabolism during macrophage polarization
It is well known that metabolic rewiring orchestrates the activation of quiescent naïve macrophage to M1 or M2 phenotype. Complying with its metabolic contribution, serine metabolism (namely, serine uptake and the SSP) is also significantly mobilized in a dynamic manner during this process (Table 1).
Altered serine metabolism during macrophage polarization
Transcriptomics, metabolomics, and isotopic tracing study have confirmed the mobilization of serine metabolism during macrophage activation. As early as 4 h or 6 h after LPS stimulation, murine peritoneal macrophages (periMΦs) started to increase their uptake of exogenous serine [21, 22] and glycine to a much smaller extent [21]. Differently, the expression of PHGDH and PSAT1 was unchanged in the bone marrow-derived macrophages (BMDMs) primed for 4 h [21]; however, after two more hours, both were drastically upregulated together with Asct1 and 2 and their positive regulators (e.g., Atf4), and this increase gradually peaked at 24 h and declined at 48 h post-stimulation [22,23,24]. Accordingly, the periMΦs stimulated by LPS for 6 h actively consumed glucose for de novo synthesis of serine and glycine besides promoting serine uptake [22]. Importantly, similarly increased expression of ATF4, PSPH, and ASCT1 existed in peripheral blood mononuclear cells of the patients with rotavirus infection or rheumatoid arthritis, highlighting the tight connection of inflammation with macrophage demand for serine [23]. Oppositely, RNA virus infection downregulated the genes encoding serine transporters (Asct1 and 2) and SSP-related enzymes (Phgdh, Psat1, and Psph) in periMΦs 5–10 h post-transfection and decreased intracellular serine and glycine at 6 h, indicating the reduced SSP and serine uptake as a hallmark of virus-infected cells [25]. In contrast, the long exposure (e.g., 24 h) to LPS or LPS + IFN-γ also repressed the levels of Phgdh (or Psat1 and Psph) in the BMDMs and the PHGDH activity 24 h post-stimulation [26, 27]. Overall, naïve macrophages likely enhance the uptake of exogenous serine initially and then turn to upregulate the SSP pathway at a later timepoint, to synergize the increasing demand for serine during their early response phase to LPS stimulation.
On the other side, the alternatively activated macrophages (M2) by IL-4 and/or IL-13 have been shown to also highly express SSP enzymes and accrue intracellular serine. In BMDMs and periMΦs, the Phgdh moderately upregulated 8 h after IL-4 treatment was more strongly induced along with Psat1 and Psph after another 16 h [26, 27]; and the activity of PHGDH enzyme was validly stimulated by IL-4 but not by LPS [26], suggesting that de novo serine biosynthesis may coordinate with M2 polarization specifically. Indeed, in IL-4-polarized periMΦs, IL-4 plus IL-13-primed BMDMs and immunosuppressive tumor-associated macrophages (TAMs) in tumor microenvironment (TME), the PERK/ATF4 axis was activated and the PSAT1 and serine biosynthesis got upregulate, both of which were proved to be critical for immunosuppressive function [27,28,29]. Moreover, the higher levels of Phgdh and Psat1 existed in M2- versus M1-polarized BMDMs, the Phgdh transcripts decreased along the LPS stimulation (0–16 h) but reclimbed to a peak at 8 h post-simulation with IL-4, and a better PHGDH catalytic activity was displayed in IL-4 primed BMDMs regardless of the presence of exogenous serine/glycine [26], all marking the significance of the SSP for M2 polarization.
Serine metabolism-related metabolic rewiring during macrophage polarization
Isotopic tracing demonstrates that polarized macrophages mobilize serine metabolism to meet their anabolic and catabolic needs in multiple ways (Table 1). As early as 4–6 h after LPS stimulation, periMΦs upregulated mitochondrial SHMT2 and its catalyzation of either exogenously ingested or biosynthesized serine to glycine and 1C unit, which were employed for the biosynthesis of purines and thymidylate via 1C metabolism, GSH synthesis, and SAM generation via contributing ATP [21, 22]. Upon the SSP activation at the late stage of LPS-stimulated polarization, macrophages shunted more glucose to the SSP and drove the synthesized and exogenously taken serine to cooperate with methionine for the PPP, methionine cycle, and 1C/folate cycle, urging the production of purine, ATP and SAM and thus the high ratio of SAM/SAH, which is responsible for the histone methylation required for M1 phenotypic reprogramming [22, 25]. Intriguing is that though integrating 13C-serine/glycine into GSH and accumulating GSSG at 4 h post-priming with LPS [21], the periMΦs stopped more incorporation of serine carbons into GSH at 6 h; instead, GSH became the most decreased metabolite and the GSH/GSSG ratio greatly declined [22]. Besides, serine depletion unaltered GSH level in the thioglycolate-elicited periMΦs (TGPMs) treated with LPS + INF-γ for 15 h [30]. This on/off adjustment of GSH/GSSG ratio and the underlying reason are especially meaningful considering the role of generate ROS in macrophage polarization. Mentionable is that viral infection reduced the SPP of periMΦs and their glucose consumption, serine makeup and SAM pool and finally activated TBK1-mediated IRF responses [25]. Despite still taking exogenous serine [31], M2 macrophages were much stronger in SSP expression and catabolic ability than M1 phenotype, and their serine biosynthesis was often tightly linked to SGOC metabolism and affected the steady levels of serine/glycine, α-KG, SAM, nucleotides [27, 31].
Aside from offering serine for above-mentioned metabolic utilizations, the amplified SSP has been suggested as a nexus among central carbon metabolism in tumor cells, and its inhibition would disrupt mass balance and compromise the support for the biosynthesis of purine and pyrimidine [32]. Serine metabolism similarly casts wide influences over macrophages’ cellular metabolism, especially glycolysis, OXPHOS and lipid metabolism, the major metabolic hallmarks distinctly involved in their polarization (Table 2). As for M1 macrophages primed with LPS (or plus IFN-γ), depriving nutrimental serine/glycine inhibited glycolysis (e.g., the declined glycolytic enzyme genes, basal ECAR, and glycolytic capacity) and markedly lessened the intracellular pyruvate, lactate and acetyl-CoA [21, 23, 30]; however, the ECAR remained unchanged if specifically deleting Phghd from the periMΦs primed with IFN-γ for 12 h [31], despite a lower ECAR appearing for the BMDMs deficient in Perk and thus defective of the SSP after 24-h treatment of LPS + IFN-γ [27], suggesting that the nutrient supply of serine rather than its biosynthesis is positively associated with glycolysis tightly during M1 polarization. Interestingly, serine starvation promoted glucose uptake in TGPMs stimulated with LPS + IFN-γ for 15 h [30], probably as a feedback response to the amino acid deficiency-evoked bioenergic peril. Meanwhile, both mitochondrial mass and the expression of genes related to mitochondrial electron transport chain (ETC) complexes seemed unaltered when priming periMΦs or BMDMs with LPS (or plus IFN-γ) for 4 h, 8 h, or 24 h [21, 23, 27], and the OXPHOS (e.g., OCR, ATP, and NADH/NAD+) was insignificantly impacted in either M1 macrophages differentiated from TGPMs devoid of serine/glycine [30] or BMDMs defective in the SSP and primed with LPS plus IFN-γ for a day [27], supporting that the OXPHOS is not under the control of serine metabolism in M1 macrophages. Moreover, both lipogenic gene (Scd1) and intermediate metabolite malonyl-CoA were diminished in periMΦs stimulated with LPS for 6 h and BMDMs primed with LPS plus IFN-γ for 24 h [23, 27]. Noticeably, three above OXPHOS indexes in serine-starved BMDMs fell at 6 h but not 4 h post-LPS stimulation [23]. Apparently, M1 polarization more likely relies on serine uptake to maintain vigorous glycolysis and leave OXPHOS unimpacted.
Surprisingly, naïve macrophages enhance the expression of SSP enzymes and PHGDH activity when polarized toward M2 phenotype, not to yield more serine but mainly to ensure their mitochondrial fitness to match the increased requirements for OXPHOS, α-KG production and FAO and thus for proliferation [26, 27, 31]. Applying the inhibitors against PERK or PHGDH or deleting Perk or Psat1 in IL-4 primed BMDMs downregulated the genes key for lipid metabolism and mitochondrial respiration and resulted in a deviated glucose oxidation (lower ECAR, OCR, lipid uptake/lipolysis and ATP yield) and a compromised mitochondrial calcium flux [27, 31]. Specifically, ATF4 overexpression could drive more transcription of SSP genes to rescue the basal OCR and ECAR repressed by Psat1 deletion [27], highlighting the essential contribution of the SSP to a metabolic network tailoring M2 polarization. Noticeably, serine biosynthesis regulates mitochondrial fitness of M2 macrophages seemingly not via altering mitochondrial mass and ETC [27]. Mentionable is that in contrast to Psat1 knockout, the myeloid deletion of PHGDH and SG deprivation insignificantly altered the ECAR and OCR in IL-4 primed BMDMs [31], hinting that PSAT1-supported α-KG production may already sufficiently maintains the mitochondrial fitness of M2 macrophages.
Modulation of macrophage polarization by serine metabolism
In line with the role in polarization-associated metabolic reprogramming as above discussed, mounting evidences have demonstrated that the uptake and biosynthesis of serine actively modulate macrophage polarization and their effector function in vitro and in vivo (Table 2).
M1 polarization
Either serine starvation or SSP disruption alters the expression of proinflammatory cytokines and iNOS in M1-polarized macrophages and thus affects other immune cells in vitro and in vivo, but the detailed responses vary with polarization timings and test models. The deprivation of serine/glycine or the application of serine transporter blockers dramatically diminished the mRNAs of Il-1β in periMΦs or BMDMs primed for 4 h with LPS [21] and TGPMs primed for 15 h with LPS + IFN-γ [30], but elevated the mRNAs and proteins of Il-1β in BMDMs primed for 12 h with IFN-γ or for 24 h with LPS [26, 31], showing that intracellular serine could be supportive or inhibitive for IL-1β secretion. Likewise, the rapid upregulation of IL-10 (one cytokine inducing IL-6) during M1 polarization was suppressed if restricting exogenous serine [23], while more IL-10 proteins were expressed by BMDMs with serine deficiency and one-day LPS priming [26]. As for TNF-α, though being unaffected by nutrimental serine in some M1 polarization experiments [21, 26], its mRNA/protein expression were upregulated along with Il-6 and iNOS when depriving serine and glycine in culture media or restricting intracellular import of serine [23, 31]. Notably, serine starvation or supplementation would respectively up- or downregulate Tnf-α in TGPMs primed for 15 h with LPS plus IFN-γ [30]. Obviously, serine availability influences M1 effector function quite context-dependently.
The SSP blockade negatively or positively affects M1 polarization too. PHGDH inhibitors (NCTs or CBR5884) were seen to greatly lower the mRNA expression of Il-1α and β and Tnf-α in LPS-stimulated periMΦs, the protein level of pro-IL-1β in human peripheral monocyte-derived macrophages stimulated with LPS for 6 h [22], and the secretion of IL-1β in the supernatant of cultured synovial fluid cells from gouty patients treated with LPS for 12 h, and of IL-10 to a smaller extent in BMDMs primed with LPS for 24 h [26]. Oppositely, both the IL-1β secretion by BMDMs stimulated with LPS for 24 h and the mRNA and protein levels of NOS2, IL-1β, and IL-6 in BMDMs treated with IFN-γ for 12 h were increased by PHGDH inhibitors [26], [31]. Besides, PH-755-003N blocked the SSP in vitro and in vivo but failed to affect Il-1β and Il-10 in both periMΦs and BMDMs stimulated for 4 h with LPS [21]. Genetic manipulation also suggests the role of the SPP to be either negligible, promotive, or suppressive for M1 polarization and effector functioning. Six-hour LPS priming markedly lowered the mRNAs of Il-1β and the proteins of pro-IL-1β (but not TNF-α) in Phgdh-/-iBMDMs [22], while deleting myeloid Psat1 did not change either the percentage of CD206+CD301+ cells and iNOS level in the BMDMs primed with LPS + IFN-γ or the proliferation of their co-cultured CD8+ cells [27]. The upregulation of Nos2 (and iNOS protein), IL-1β and IL-6 by INF-γ in BMDMs was even enhanced by Phgdh silencing or knockout, and only the enzymatically active PHGDH could eliminate this impact31, implying that the activated SSP may impede M1 functioning. So, how serine metabolism affects M1 polarization remains unsure. Such uncertainties were clearly reflected by the distinct responses to serine deficiency made by the BMDMs where M1 features were markedly enhanced by CBR5884 pretreatment or myeloid specific Phgdh deletion. Serine/glycine shortage eliminated the enhancement of Il-1β by CBR5884 [26] but strengthened the upregulation of NOS2 (mRNA and protein), Il-6 and Il-1β by Phgdh deletion [31], indicating that exogenous serine can either sustain IL-1β elevation or counter against M1 polarization.
Despite the results from cell models somewhat contradicting from each other, in vivo studies suggest that serine metabolism tends to be more supportive than suppressive for M1 polarization. Serum IL-1β and TNF-α were reduced in either the mice treated with PHGDH inhibitors and challenged with LPS for 1.5 h or 6 h or the animals fed with serine-free diet for 4 wk and treated with LPS for 15 h [21, 22, 30]. Their organic levels and inflammasome activation were also largely reduced in the mice offered with serine-free diet and M1 condition, and such reductions were even causally related to the attenuation of LPS-caused pneumorrhagia [30]. However, the tumor xenografts grew more slowly and presented more F4/80+ cells, iNOS, and immune-positive signals for IL-1β, iNOS, and IL-6 in Phgdh−/− mice than in wild-type animals, while restricting serine for two weeks could subside tumor growth too [31], hinting the in vivo promotion of proinflammatory features by limiting serine metabolism.
M2 polarization
In comparison to M1 polarization, more evidences support the participation of serine metabolism (mainly the SSP) in M2 polarization; and either serine supply restriction or SSP disruption has been shown to drive M1 activation but undermines M2 polarization. Basing on their inverse data-driven modeling and multiomics analysis for Tsc2/mTORC1, Wilson et al. have first proposed that PHGDH acts as a mTORC1-dependent metabolic checkpoint for macrophage polarization and proliferation [26]. The expression and activity PHGDH were enhanced during IL-4-primed M2 polarization and it was the PHGDH-mediated SSP but not exogenous serine that warranted the expression of the M2 signature genes (Arg1, Retnla, and Chil3) and the secretion of the anti-inflammatory cytokine IL-10 [26]. Afterward, the application of PHGDH inhibitors or the knockdown/knockout of myeloid Phgdh or Psat1 were seen to decrease CD301+CD206+ cells derived from IL-4 stimulated BMDMs [26] and attenuate the proliferation and expression of Arg1 (and ARG1 proteins as well), Retnla, Chil3, and Mgl1 in polarized macrophages [26, 27, 31]. The absence of the SSP in M2 macrophages even markedly retarded the proliferation of their co-cultured tumor cells and yielded good antitumoral immune effects in the xenograft-bearing mice (namely, a reduced tumor size and weight); and these tumor tissues all had a downregulation of ARG proteins, a smaller absolute number of TAMs, a lower frequency of CD206+ TAMs, and more TILs and IFNγ+CD8+ T cells [27, 31]. Importantly, either ablating myeloid Phgdh or PHGDH inhibitors reduced ARG+ macrophages and eosinophile recruitment in chitin-treated mice, and exacerbated liver fibrosis and steatosis in CCl4-induced animals [31], strongly suggesting that an intact SSP is indispensable for immunosuppressive macrophages.
Relatively, different answers exist to the contribution of serine uptake to M2 polarization. IL-4-stimulation of M2 marker genes like Arg1, RetlnA, Chil3 and Igf1 in BMDMs was once reported to be insignificantly responsive to the depletion of serine and glycine [26]; and oppositely, either serine deprivation or serine transport blockade was seen to decrease the mRNA expression of Arg1, RetlnA and Mgl1 and the protein level of ARG1 in IL-4 primed BMDMs, regardless of the SSP activity [31]. In vivo, two-week feeding with serine/glycine-free chow slowed down the growth of tumor xenografts in mice and reduced eosinophil recruitment and macrophage Arg1 in chitin-treated animals, and applying CBR5884 to serine-starved animals further synergistically suppressed M2 polarization [31], convincing the pivotal role of serine metabolism in realizing M2 function in vivo.
Action mechanism of serine metabolism in macrophage polarization
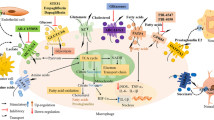
Currently, whether and how metabolic activities including serine metabolism intervene in the canonical signaling cascades and epigenetic reprogramming decisive for macrophage activation remains obscure. Among various action mechanisms proposed for serine metabolism in macrophage polarization, mainly appreciated are its contributions to mTORC signaling, ROS production, and the modification of histones, non-histones, and DNAs (Fig. 2).

The contribution of serine metabolism to macrophage polarization. The contributions of serine metabolism to mTORC signaling, ROS production, posttranslational modification, and epigenetics are major action mechanisms that have been well appreciated for the control of macrophage polarization. Importantly, serine metabolism holds a great potential as a critical epigenetic modulator. Via tuning the availability for methyl-group, acetyl-CoA, lactate, serine metabolism has been demonstrated to be able to affect the histone/DNA modification (methylation/methylation and acetylation). M1/M2: Red represents positively modulate M polarization; Blue represents negatively modulate M polarization. 5-hmC 5-hydroxymethylcytosine, TET2 the ten eleven translocation (Tet) methylcytosine dioxygenase family member 2, JMJD3 Jumonji domain-containing protein-3, FAO fatty acid oxidation, OXPHOS oxidative phosphorylation, Mt fitness mitochondrial fitness, PHD Prolyl Hydroxylase, IKKβ inhibitor kappa B kinaseβ, NF-κB Nuclear factor-kappa B, H3K27me3 trimethylated histone H3 at lysine 27, H3K4me3 trimethylated histone H3 at lysine 4, H3K36me3 trimethylated histone H3 at lysine 36, S6K1 ribosomal protein S6 kinase, ATF4 Activating transcription factor 4, Ac-CoA acetyl-CoA
Metabolic contribution
Via its direct or indirect dedications, serine metabolism serves as a nexus of multiple metabolic activities especially involved glucose, amino acids, lipids, nucleotides, etc., and contributes to cell bioenergy, biomass, and redox homeostasis.
Akt/mTORC pathway
MTOR pathway is a central knob not only reacting nutrient and metabolic status of cells but also integrating growth signals in guiding cell cycle progression, cell growth and differentiation [33]. Unsurprisingly, it plays essential regulatory roles in the differentiation and function of immune cells [33,34,35]. Via tuning the levels of amino acids (e.g., serine, glycine, branched-chain amino acids, etc.), nucleotides, and lipids, serine metabolism yields the signals acting through the Akt/mTORC1/ATF4 axis [36], [37, 38].
Compared to naïve macrophages, M1 populations own a more activated mTOR signaling [39]. In an early study, serine deprivation was shown to inhibit the mTORC signaling, and thus to subside IL-1β production and inflammasome activation and to reprogram the transcriptomic and metabolic profile in the M1-primed TGPMs [30]. After the priming for 1 or 6 h, the starved TGPMs significantly reduced the phosphorylation levels of mTORC1 and S6K1 and the mRNA expression of ATF4. MTORC1 inhibitor rapamycin could block the inhibitory effect of serine deprivation on NF-κB activation and the production of IL-1β and TNF-α and excite caspase 1 and cell death, while the mTOR activators (MHY1485 or leucine) rescued the IL-1β yield in macrophages cultured with serine-depleted medium [30]. A recent study revealed that PHGDH inhibition could shunt more glucose carbons to BCAAs, and the resultantly accumulated leucine and isoleucine then activated mTORC1 and reduced cell death in osteosarcoma [36]. However, whether SSP disruption activates mTORC1 similarly in macrophages remains to be unveiled.
As for M2 polarized macrophages, the mTORC1 signaling sustains their active SSP and probably the production of α-KG [26, 34]. In the CSF1-derived BMDMs from Tsc2 fl/flLyz2-Cre mice the levels of both SSP enzymes and PHGDH activity got escalated, which was obviously due to the persistently active signaling of myeloid mTORC1 and could be reduced by rapamycin [26]. The SSP inhibition evidently suppressed their IL-4-mediated M2 polarization and effector function but conversely enhanced their M1 polarization [23]. M2 polarization was noticed to become defective due to the downregulation of Akt signaling as a feedback response to the constitutive activation of mTORC1 [40], as reflected by the decreased uptake and consumption of glucose for glycolysis and oxidative metabolism and the suppressed induction of a subset M2 genes responsible for the signal inputs additional from the Jak–Stat pathway including Arg1, Retnla and Mgl2 [41]. Besides, the canonical TGF-β1/Smad3 signaling cooperated with the activated mTORC1/4E-BP1 to drive the ATF4-dependent SSP and glycine yield for TGF-β1–induced collagen biosynthesis[42], showing the mediation of ATF4/SSP axis in the healing signaling of TGF-β and mTORC. Notably, IL-4 stimulates the activating phosphorylation of Akt, mTORC1 and ACLY, the cytosolic production of acetyl-CoA, and ultimately the acetylation of proteins including histones for M2 phenotype. For instance, mTORC1 itself could upregulate the ACLY enzyme protein levels and stimulate its activating phosphorylation in M2 macrophages [41]; and these effects were impaired in BMDMs deficient in Raptor while the elevated ACLY levels stood out in Tsc1−/−BMDMs [40]. Obviously, it would be valuable to dig more details of the impacts of serine metabolism on TCA cycle, lipogenesis, FAO, glutaminolysis, and acetyl-CoA synthesis, all of which affect the level of acetyl-CoA during macrophage polarization.
Redox balancing
Redox balance weighs much in macrophage physiology. M1 and M2 macrophages are, respectively, characterized as oxidative and reductive macrophages according to their intracellular glutathione content. Besides cytotoxic effects, ROS exert a fundamental regulation of M1 polarization as signal molecules via driving MAPK/JNK/p38 and IKK/IκBα to activate NF-κB, and also implicate in the early stage of M2 differentiation probably via the redox-dependent STAT6 translocation or even in the acquirement of immunosuppressive TAMs and their PD-L1 expression [43,44,45,46]. NADPH-dependent oxidases and mitochondrial respiration complex are two major ROS producers in macrophages, and serine metabolism appears to modulate their activities and thereby affect M1 activation.
As discussed above, the serine-converted glycine directly participates in the transient de novo GSH synthesis in macrophages during the first few hours of their priming as a non-transcriptional response to increased ROS [21]. Serine starvation lowered the levels of GSH and GSH/GSSG ratio in LPS-primed murine periMΦs and the supplementation of cell-permeable GSH ethyl ester rescued their inhibited transcription of Il-1β. Recently, the mTORC1/ATF4 axis that was suppressed by serine deficiency but induced by the SSP was reported to promote GSH synthesis through activating the SLC7A11-mediated cystine uptake in MEFs [37], hinting another contribution of serine to GSH yield. In addition, despite staying away from the early GSH induction [21], the SSP may influence central carbon metabolism and thus the PPP, NADPH production and NADPH/NADP+ ratio in macrophages. Both the PPP and the SSP were identically activated by LPS [22, 32], even though GSH was identified as the metabolite most reduced by LPS and the incorporation of 13C-serine carbons to GSH remained unchanged [22]. Interestingly, consuming SAM to methylate phosphatidylethanolamine facilitated SAM turnover for the syntheses of cysteine and glutathione via transsulfuration [47], and the NAPDH from the cytosolic serine catabolism was required to generate acetyl-CoA for hepatic lipogenesis [48], unveiling more potential mechanisms for the LPS-promoted SGOC metabolism to regulate GSH synthesis and redox homeostasis in macrophages.
Serine metabolism also affects mitochondrial fitness via regulating the mitochondrial translation and the intactness of ETCs and avoid mtROS overproduction [49]. And PHGDH loss was seen to impair heme synthesis and mitochondrial respiration and cause lethal oxidative stress in endothelial cells [50]. However, serine deprivation did not significantly impact the ETCs of LPS-primed macrophages but impeded their glucose oxidation and mitochondrial import of pyruvate, leading to impaired OXPHOS and reduced NADH and NADH/NAD+ ratio [23] and contributing to mtROS induction and aberrant cytokine expression (e.g., suppressed IL-10 and elevated IL-6) [23]. Mentionable is that the NAD(H) pool reduced by mitochondrially interrupted copper signaling could pose the human monocyte-derived macrophages treated with LPS and IFN-γ for 24 h in the metabolic and epigenetic states against M1 activation, and reduced the inflammation in mouse models of bacterial and viral infections [51], showing that the serine may sustain NADH and NADH/NAD+ ratio and finally proinflammatory response.
Epigenetic modulation
The modulation of epigenetics by cellular metabolic reprogramming has been the most extensively studied. Serine metabolism holds a great potential as a critical epigenetic modulator via knobbing the metabolism of glucose, amino acids, lipids, and nuclear acids. Indeed, serine metabolism really affects the modification of (non) histones and DNA regulatory for macrophage activation through tuning the availability of methyl-group, acetyl-CoA, lactate, etc.
Methylation of histones and DNAs
The trimethylation of histone H3 at lysine 4, 27, and 36 (H3K4me3, H3K27me3, and H3K36me3) substantially defines chromatin states and gene expression [52]. M1 polarization is known to link to H3K4me3 and H3K36me3, two histone marks often associated with an active chromatin, while the suppressive H3K27me3 is more implicated in M2 polarization. Owing to elevating SAM/SAH ratio and histone methylation or promoting α-KG supply and dioxygenase-catalyzed demethylation, serine metabolism can act as an epigenetic check point for macrophage polarization.
As discussed previously, the SGOC metabolism-mediated production of glycine and formate contributes to the de novo generation of purine and ATP and then the synthesis of SAM (the universal substrate for all methylation reactions) and SAH [22]. As expected, serine depletion due to PHGDH inhibition, Phgdh knockout, or SG deprivation significantly reduced intracellular SAM in activated macrophages and serum SAM in mice [31], and the damaged macrophage polarization could only be rescued by supplementing formate or SAM, showing the tight connection between serine metabolism and SAM level in vitro and in vivo. After six-hour priming the cooperation of the SSP with the PPP was triggered to elevate SAM/SAH ratio in BMDMs and directly increased the H3K36me3 over the gene regulatory regions of Il-1βs (+ 1126 and + 3627), Il-1α and Il-18 except Tnfα, and the Il-1 transcription; and all these effects could be significantly abolished by the inhibitors against PHGDH, SHMT2, G2PD, and MAT2A, by the deficiency of nutrimental serine and methionine, or by the combinational deprivation of serine, glycine and methionine from culture media [22]. In addition, H3K27me3 was also regulated by the SGOC metabolism in the mouse primary periMΦs infected with RNA viruses for 5–10 h [22]. The SGOC enzymes were prominently downregulated in these cells, and SSP blockade via Phgdh knockout/knockdown or PHGDH inhibitor or nutrimental depletion of serine and glycine all dramatically reduced H3K27me3 but not H3K4me3 and H3K9me3 and the occupancy of H3K27me3 over the regulatory region of the gene encoding the lysosomal ATPase subunit (ATP6v0D2) (-3221, -1795, and -918) was specifically diminished in a SAM/serine-dependent manner, resulting in a higher V-ATPase expression and activity. The correspondingly enhanced lysosomal degradation of YAP conversely promoted the TBK1-IRF3 axis and augmented the IFN-β-mediated antiviral innate immunity. PHGDH knockout or restriction of exogenous serine and glycine also downregulated the H3K27me3 at the upstream of Igf1 gene (− 3000, − 2000, − 1000), which could be restored SAM or serine or overexpressed PHGDH. The resultantly elevated IGF1 then activated the p38-dependent JAK–STAT1 axis to promote M (IFN-γ) polarization and suppress STAT6-mediated M (IL-4) activation in vitro and in vivo [31]. Generally, both serine uptake and the SSP adjust the SAM/SAH ratio and histone methylation and enable a communication between ‘‘metabolic state’’ and ‘‘chromatin state’’ for macrophage activation.
Serine metabolism modulates the level of α-KG and the demethylation of histones and DNAs for macrophage polarization too. Indeed, the PSAT1 activity significantly contributes to the intracellular α-KG in stem cells [18], [53], TSC2−/− cells [54], and polarizing macrophages [27, 55]. Aside from elevating the α-KG/glutamate ratio to prompt FAO and OXPHOS and benefit mitochondrial fitness and cell proliferation, the escalated α-KG production and α-KG/succinate ratio strengthen the α-KG-dependent demethylases such as JMJD3 or TETs and M2 polarization [55, 56]. It is mentionable that compared to LPS IL-4 triggered a higher α-KG/succinate ratio and stronger dioxygenase activity in BMDMs, and the α-KG-supported JMJD3 (the H3K27 demethylase) deeply implicates in the macrophage activation toward to M2 but not M1 phenotype [55]. In IL-4-stimulated BMDMs deficient in PSAT1 or PERK, the level of α-KG and the histone demethylation declined significantly and the methylation marks on H3K27 became greater, especially at the loci of M2 genes containing Irf4, Pparg and Mgl2; supplementing dm-KG restored those suppressed M2 genes but the JMJD3 inhibitor GSK-J4 did blocked this rescue [27]. Consistently, the heightened α-KG/succinate ratio was not connected to the enhanced transcription of other M2 marker genes (Arg1, Yml, Tetnla, and Mrcl) in IL-4-stimulated Jmjd3−/− BMDMs, while their escalation of oxygen consumption was lost even in the presence of glutamine or DM-α-KG [55]. All these studies highly suggest that that the PSAT1/SSP-based availability of α-KG is indispensable for the JMJD3-mediated epigenetic reprogramming supportive for M2 polarization. Notably, IFN-β was found to restrict the α-KG/succinate ratio and the sustained activation of JMJD3-IRF4 pathway and antagonize the GM-CSF-induced CCL-17 secretion and inflammatory pain in vivo [56], revealing a possible role of the SSP-supplied α-KG in M1 polarization and inflammatory response.
The climb of intracellular α-KG also promotes the conversion of 5-methylcytosine into 5-hydroxymethylcytosine (5-hmC) and DNA demethylation by TETs and generally triggers gene activation in cells including activated macrophages [57,58,59,60]. In lung interstitial macrophages, endothelial respondin3 mobilized the LGR4/β-catenin signaling to enhance TCA cycle via glutaminolysis and the resultantly driven α-KG production specifically induced the reprogrammed DNA hydroxymethylation for anti-inflammatory transition. Rspondin3 increased the H3K4me3 on the promoters of anti-inflammatory genes (Mrc1, Arg1, Chil3, and Retnla) too, but the enhanced α-KG production increased the activity of TET rather than JMJDs [61]. Though whether serine metabolism plays a role in DNA hydroxymethylation in macrophages via affecting the α-KG-dependent DNA demethylation remains to be investigated, PHGDH was disclosed to be supportive of the yield of D-2-hydroxyglutarate in PHGDH-high cancer cells [17], an oncometabolite structurally alike to α-KG and thus serving as a competitive inhibitor against α-KG-reliant demethylation of histones and DNA [62]. D-2-hydroxyglutarate was enriched by macrophages during the later stage of LPS-stimulated inflammatory reaction, and negatively regulated inflammatory signaling in vitro and modulated local and systemic inflammatory responses in vivo [24], implying that the SSP may tune the regulatory effects of α-KG on macrophage epigenetics in a similar way. Meanwhile, the upregulated serine metabolism and SAM generation were seen to be implicated in the elevation of DNA methylation that was particularly enriched at retrotransposon elements and associated with their transcriptional silencing [63]; and the addition of methionine and SAM to LPS-stimulated RAW 264.7 cells intensified the methylation of promoters, especially the CpG sites over Il-6, Tnf-α, and Ifn-β, and attenuated their induction [64], indicating that serine metabolism may modulate DNA methylation for M1 polarization via raising SAM level.
Acetylation of histone
Histone acetylation/deacetylation is a major epigenetic regulation of gene expression [65]. Cell uses acetyl-CoA to indicate metabolic state and do distinctive acetylation of proteins [4, 65]. Overall, a high level of nucleocytosolic acetyl-CoA is a signature of “growth” or “fed” state and employed for lipid synthesis and histone acetylation; in “survival” or “fasted” state, cell preferentially directs acetyl-CoA into mitochondria to assure mitochondrial-dependent activities. Acetyl-CoA fluctuations within subcellular compartments enable the substrate-level regulation of acetylation modifications and lay profound influences on numerous life processes.
The Akt/mTORC/ACLY axis has been determined to mediate TLR and metabolic signaling in promoting acetyl-CoA production and histone acetylation (H3 and H4) and tuning M1 versus M2 polarization, and the glycolytic intermediate pyruvate, FAO, lactate, and acetate are the factors known to affect the level of acetyl-CoA and its supported histone acetylation [35, 41, 66,67,68]. As discussed above, one-day serine starvation markedly reduced intracellular pyruvate, lactate, acetyl-CoA, and malonyl-CoA (a substrate for lipogenesis) in LPS-stimulated periMΦs [23], and the serine biosynthetic pathway did serve as a major contributor of α-KG production in M2-polarized macrophages [26], both suggesting that it is highly possible for serine metabolism to adjust the levels of acetyl-CoA and histone acetylation. In line with this hypothesis, serine starvation was recently reported to reduce acetyl-CoA levels via limiting the glucose flux to glycolysis and TCA cycle and thus to induce histone hypoacetylation (including H3K27 acetylation), decreasing the expression of estrogen receptor (ER) α and its transcriptional targets like progesterone receptor (PR) and the sensitivity to antiestrogens, and eventually triggering a transition of ER-positive breast cancer cells to ER/PR-negative state [69]. Obviously, it would be very interesting to investigate whether serine starvation induces such a global loss of histone acetylation in macrophages and controls their polarization.
Lactylation of histone
Despite the elevated lactate secretion is critical for M1 polarization or is transiently activated at the early stage of M2 polarization, it was until 2019 that the secreted lactate was found to be exploited for the lactyl-CoA yield and histone lactylation at lysine residues (Kla) to promote M2 macrophage differentiation and the expression of homeostatic genes for wound healing [70]. A temporal dynamics of histone lactylation different from acetylation appeared in bacteria-exposed M1 macrophages, which was particularly increased in the late phase of M1 macrophage polarization and induced wound healing-involved homeostatic genes [70]. Later, the TLR signaling adapter BCAP (B-cell adapter for PI3K) was shown to regulate the inflammatory to reparatory macrophage transition via the induction of aerobic glycolysis and lactate production and thus histone lactylation rather than via the activation of NF-κB or MAPK [71], and the dysregulated glycolysis and lactate export in the monocytes at the early stage of myocardial infarction (MI) were found to promote their histone lactylation (like H3K18la) and downstream reparative gene expression post-MI [72], both marking the significance of histone lactylation in immunosuppression and inflammation. Even though no direct evidence is available for serine metabolism to affect the histone lactylation of macrophages, either serine deprivation or SSP blockade could alter the bifurcation of glucose carbons and the pyruvate import into the TCA cycle and FAO, and hence influenced the level of lactate [23]. Really, PHGDH downregulation reduced glucose consumption, LDH activity and lactate production in sertoli cells [73], and the SSP-supported astrocyte-specific phosphorylation demonstrated the shift between lactate and serine [10]. It is attractive to clarify if serine metabolism conduces to the histone lactylation crucial for macrophage polarization.
Posttranslational modification of non-histones
Serine metabolism and the related metabolites also participate in the post-translation modification of non-histones and the involved biochemical events critical for macrophage polarization [74, 75]. For instance, the prolyl hydroxylation of IKKβ at P191 would suppress its phosphorylation so the upregulated SSP and α-KG yield may enhance PDH activity and thus inhibit NF-κB nuclear translocation [55]. In addition, lactate could induce HMGB1 acetylation and lactylation via stimulating the suppression of deacetylase SIRT1 and the nuclear recruitment of acetylases p300/CBP in RAW 264.7 cells and drive the secretion of HMGB1-contained exosomes to increase endothelium permeability [76], and the enhanced mevalonate pathway urged GTPase Rac1 activation via enhancing Rac1 geranylgeranylation at its C-terminal cysteine residue and promoted profibrotic polarization of macrophages and the fibrotic repair in idiopathic pulmonary fibrosis [77]. Interestingly, HDAC3 could deacetylate HADHA (an FAO enzyme, mitochondrial trifunctional enzyme subunit a) at K303 and repress its activity in macrophage mitochondria, thus restricting the FAO to optimize NLRP3 inflammasome activation [78]. Obviously, whether and how much serine metabolism and relevant metabolites contribute to these potential PTM in macrophages deserve further inspection.
Altered serine metabolism in inflammatory diseases
Improper and sustained activation/polarization of macrophages leads to tissue damage and immune dysfunction and thus to the occurrence of illnesses, e.g., various infectious diseases, chronic inflammation, auto-inflammatory disease, and cancers as well. Owing to its influences over macrophage polarization, the alteration in the serine metabolism of macrophages not only contributes to inflammatory responses to pathogen invasion and to chronic and autoimmune inflammation, but also substantially implicates in tumor growth and progression. Both serine restriction and SSP inhibition have been applied to tackle a series of pathological inflammatory disease in model animals, including lethal sepsis, viral infections, liver fibrosis, and cancer.
Infectious diseases
M1 phenotype macrophages are the key effector of the host response against intracellular pathogens like bacteria and virus. Their exerted potent cytotoxic function supports the elimination of infected cells, but also incurs the injury to normal cells and tissues and their death if overstimulated and prolonged without a control. On one side, in line with the dependence of M1 polarization and Th1 cytokine production on serine supply, a rapid consumption of serine happened upon Pasteurella multocida infection in murine lungs. Supplying sufficient serine could support the secretion of proinflammatory cytokines (L-1β, IL-17, IFN-γ, and TNF-α in the lungs and serum) and nicely restrict bacterial load and thus macrophage- and neutrophil-mediated inflammatory response and animal death [79]. On the other side, in several animal models of lethal sepsis, both the uptake of exogenous serine and the SSP got obviously enhanced by microbial stimuli like LPS, which was tightly correlated with the elevated M1 phenotype and the secretion of proinflammatory cytokines (e.g., IL-1β and TNF-α). Accordingly, the serine-free feeding for four weeks effectively lowered the LPS-caused proinflammatory cytokines in multiple organs of mice and lessened their lung hemorrhage and inflammatory diseases [80], while the pre-administration of the SSP inhibitor PH-755-003N nicely protected the mice from LPS-induced endotoxemia and improved their survival [81]. These results suggest that serine metabolism could be a target for controlling acute inflammation and avoiding tissue injury. Exogenous serine was also shown to be indispensable for the induction of IL-10 in macrophages by LPS, which is critical for the suppression of sustained inflammation [82].
Noticeably, bacterial infection may reprogram the serine metabolism of macrophages to facilitate their own intracellular replication and virulence. As reported by Jiang et al., the macrophages infected by Salmonella Typhimurium had a decreased use of glucose carbon for serine biosynthesis but the production of glycolytic intermediates that were supportive for bacterial replication turned escalated; and targeting this metabolic reprogramming via enhancing host SSP could effectively impair the replication and virulence of Salmonella typhimurium and save infected animals [83]. Interestingly, it was shown that during infection of Salmonella typhimurium, the macrophage-specific ATP6V0D2 and its mediated autophagosome–lysosomal fusion were downregulated, which could prompt inflammasome activation to better eliminate bacterial infection [84]. Considering the inhibition of ATP6V0D2 by the SSP via promoting histone methylation [85], propelling the serine biosynthesis of macrophages is helpful for their combat against Salmonella typhimurium.
Serine metabolism is also implicated in the antiviral responses of macrophages. Gene expression of transcription factor and enzymes for serine biosynthesis and neutral amino acid transporters in peripheral blood mononuclear cells was stronger in ten patients infected with rotavirus when compared to 8 age-matched healthy controls [82]. However, infection of RNA viruses Sendai virus (SEV) and vesicular stomatitis virus (VSV) were linked to the downregulation of enzymes of SGOC metabolism in macrophages, and PHGDH inhibition and SG starvation could subside the histone methylation-mediated ATP6V0D2 downregulation and then enhanced YAP lysosomal degradation and IFN-β production in infected macrophages, hence yielding a series of antiviral innate immunity like the lower viral titers in liver, lung and spleen, restricted lung damage, and declined survival rate seen in the VSV-infected Phgdh−/− mice [85].
Chronic inflammation and autoimmune diseases
Delayed transition of macrophages from M1 to M2 polarization or the inclination of M1/M2 balance to M1 phenotype result in attenuated wound healing and thus tissue injury, two events leading to chronic inflammation and autoimmune diseases [86, 87]. High level of serine uptake was seen to exist in the macrophages from synovial fluids of patients with rheumatoid arthritis [82], and the inhibition of the SSP, 1C metabolism and SAM production suppressed IL-1β production in freshly isolated synovial fluid cells from gouty patients [88], indicating the potential of serine metabolism inhibition in treating these diseases. On the other hand, since serine metabolism is indispensable for the proliferation and anti-inflammatory activity of M2 macrophages, suppressing serine metabolism could shift M2 macrophages toward to M1 phenotype and thus contribute to the aggravation of tissue injury and chronic inflammation symptoms. Indeed, the CCl4-induced liver fibrosis was exacerbated in the mice fed with the serine and glycine-free diet [89], and more eosinophils recruitment appeared in the peritonitis mice induced by intraperitoneally injected chitin with nutrimental restriction or myeloid PHGDH deficiency [89].
Cancers
A variety of factors released by cancer cells such as monocyte chemotactic factors would recruit monocytes/macrophages into the TME and turn them into TAMs. These macrophages often adopt a M2-related profile, secret angiogenic and immunosuppressive factors (e.g., the PD-L1 to suppress cytotoxic T cells and the IL-10 to induce regulatory T lymphocytes), often promote rather than counteract tumor growth and progression [90]. As mentioned above, IL-4 primed macrophages depend upon active serine metabolism to gain and sustain M2 phenotype and anti-inflammatory function; and as for TAMs, their immunosuppressive roles have also been shown to be modulated by the altered intracellular serine, via either the PERK-ATF4-PSAT1 or the IGF1-p38 axes [89, 91]. Consistently, the SSP inhibitor NCT503 was confirmed to greatly reduce TAM numbers and immunosuppressive activity but induce the expansion of TILs and IFN-γ+ T cells in B16-F10 melanoma model animals and ultimately prolonged animal survival time effectively [91]. Likewise, the volume and weight of the tumors developed from the inoculated murine Lewis lung carcinoma (LLC) cells increased with a much slower speed in PHGDH-KO mice where more IL-6 and IL-1β and M1-like macrophages existed. In control diet-fed mice with restricted M1 macrophage phenotype, the depletion of TAMs suppressed the growth of LLC tumor; whereas in the serine-restricted diet-fed mice dominated with M1-like phenotype, the removal of TAMs drove tumor growth [89]. Obviously, targeting L-serine metabolism in TAMs should be seriously taken account into cancer immunotherapy.
Conclusion and perspectives
Collectively, combining its direct and indirect metabolic contributions, serine metabolism serves as a knob of glycolysis, the PPP, the TCA/OXPHOS, the synthesis and oxidation of fatty acids, the metabolism of amino acids and the nucleotide metabolism, and thus is responsible for the genetic and epigenetic reprogramming critical for macrophage polarization and effector functions. However, the ambiguities exist and wait to be clarified; e.g., the discrepancies in the roles of exogenous and endogenously synthesized serine in M1 or M2 polarization, in the activity and action kinetics of serine metabolism in different activation stages, and in how various metabolic signals deriving from serine metabolism are well orchestrated to shape the immunometabolism of macrophages and guide their phenotypic adaptation. More research is also required to determine the involvement of serine metabolism in certain action mechanisms like the lactylation and acetylation of histone or non-histone targets. Clearly, how serine availability adjusts macrophage activation and inflammatory responses in vivo (particularly under diseased states) is of high significancy and deserves a more comprehensive investigation.
Currently available evidences from model animal studies have demonstrated that serine metabolism actively participates via different mechanisms in bacterial and viral infection, liver fibrosis, gout, rheumatoid arthritis, and cancers. It would be worth to explore the more contribution of serine metabolism in the pathology and treatment of many other macrophage-mediated inflammatory diseases. In fact, serine metabolism-linked metabolic activities and action mechanisms in macrophages have been discovered to mediate the occurrence and progression of multiple inflammatory diseases. For instance, human sarcoidosis patients hold mTORC1 activation, macrophage proliferation as hallmarks that correlated with clinical disease progression [92], the mTORC1-mediated downregulation of Akt signaling and the resultant defective M2 polarization impacted type 2 immunity, inflammation, and allergy [40].
Due to the distinct demands for serine and serine-metabolism-related inter-metabolites and the different responses to the alternating serine availability, macrophage subsets manifest an exquisite susceptiveness to inhibitors against serine metabolism, providing therapeutic opportunities for selectively tailoring immune responses in cancers [93]. Types of cancer cells addict to serine and serine metabolism for their onset, expansion, metastasis and drug resistance [94,95,96]; and limiting the entry of L-serine into tumor cells and their mitochondria have produced promising anticancer effects in animal trials [97]. Such curative approaches may restrict the accessibility of serine to the macrophages that are already inferior and immunologically inert in the immunosuppressive and metabolically challenging TME to further dampen their immuno-clearance capability, and may also conversely elicit the immunosuppression mediated by myeloid derived suppressor cells and TAMs to paralyze antitumor immunity in TME. Notedly, silencing PHGDH in cancer cells with high PHGDH levels was found to condition macrophages for polarization toward the M1 phenotype [91], while cancer cells overproduce and release serine and glycine into the immunity-defective TME to induce IL-1β liberation and further shift TAMs into the PD-L1-expressing suppressive type [98]. Anyhow, future anticancer therapy against L-serine metabolism should seriously take account of their side-by impacts on TME, especially on immune cell subsets differently responding to serine metabolism [99].
Notably, two recent studies on mitochondrial fitness and unfolded protein response have suggested that de novo serine biosynthesis takes part in the longevity associated with mitochondrial protein homeostasis[100, 101]. Considering the influence of immune-metabolism over immune-paralyzing and trained immunity and the metabolic coordination of serine metabolism in rewiring multiple metabolic pathways in macrophages, it would be very valuable to give prominence to the connection of serine metabolism with immune-paralyzing and trained immunity.
Data availability
The data that support the findings of this study are openly available in public and free resources.
References
Smith TD, Tse MJ, Read EL, et al. Regulation of macrophage polarization and plasticity by complex activation signals. Integr Biol. 2016;8:946–55.
Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. 2017;38:395–406.
Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. 2021;81:1201–8.
Jung J, Zeng H, Horng T. Metabolism as a guiding force for immunity. Nat Cell Biol. 2019;21:85–93.
Galli G, Saleh M. Immunometabolism of macrophages in bacterial infections. Front Cell Infect Microbiol. 2020;10(3):607–50.
Soto-Heredero G, Gomez de Heras MM, et al. Glycolysis—a key player in the inflammatory response. FEBS J. 2020;287:3350–69.
Gao X, Lee K, Reid MA, et al. Serine availability influences mitochondrial dynamics and function through lipid metabolism. Cell Rep. 2018;22:3507–20.
Muthusamy T, et al. Serine restriction alters sphingolipid diversity to constrain tumour growth. Nature. 2020;586:790–5.
Neame S, Safory H, Radzishevsky I, Touitou A. The NMDA receptor activation by D-serine and glycine is controlled by an astrocytic Phgdh-dependent serine shuttle. P Natl Acad Sci USA. 2019;116:20736–42.
I Fernandez Moncada, U Fundazuri, G Lavanco. A lactate-dependent shift of glycolysis mediates synaptic and cognitive processes. bioRxiv. 2023;12:143–58.
Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16:650–62.
Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab. 2017;25:27–42.
Reid MA, Allen AE, Liu S, Liberti MV, Liu P, Liu X, et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat Commun. 2018;9(1):5442. https://doi.org/10.1038/s41467-018-07868-6.
Mehrmohamadi M, Liu X, Shestov AA, et al. Characterization of the usage of the serine metabolic network in human cancer. Cell Rep. 2014;9:1507–19.
Murphy JP, Giacomantonio MA, Paulo JA, et al. The NAD(+) salvage pathway supports PHGDH-driven serine biosynthesis. Cell Rep. 2018;24:2381–91.
Newman AC, Maddocks ODK. Serine and functional metabolites in cancer. Trends Cell Biol. 2017;27:645–57.
Fan J, et al. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem Biol. 2015;10:510–6.
Baksh SC, et al. Extracellular serine controls epidermal stem cell fate and tumour initiation. Nat Cell Biol. 2020;22:779–90.
Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between metabolism and cancer biology. Cell. 2017;168:657–69.
Mayers JR, Vander Heiden MG. Nature and nurture: what determines tumor metabolic phenotypes? Cancer Res. 2017;77:3131–4.
Rodriguez AE, Ducker GS, Billingham LK, et al. Serine metabolism supports macrophage IL-1beta production. Cell Metab. 2019;29:1003–11.
Yu W, Wang Z, Zhang K, et al. One-carbon metabolism supports s-adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol Cell. 2019;75:1147–60.
Kurita K, Ohta H, Shirakawa I, Tanaka M, Kitaura Y, et al. Macrophages rely on extracellular serine to suppress aberrant cytokine production. Sci Rep. 2021;11:11137–45.
de Goede KE, Harber KJ, Gorki FS, et al. d-2-Hydroxyglutarate is an anti-inflammatory immunometabolite that accumulates in macrophages after TLR4 activation. Biochim Biophys Acta Mol Basis Dis. 2022;1868:166427–38.
Shen L, Hu P, Zhang Y, Ji Z, Shan X, et al. Serine metabolism antagonizes antiviral innate immunity by preventing ATP6V0d2-mediated YAP lysosomal degradation. Cell Metab. 2021;33:971–87.
Wilson JL, Nägele T, Linke M, et al. Inverse data-driven modeling and multiomics analysis reveals phgdh as a metabolic checkpoint of macrophage polarization and proliferation. Cell Rep. 2020;30:1542–64.
Raines LN, Zhao H, Wang Y, et al. PERK is a critical metabolic hub for immunosuppressive function in macrophages. Nat Immunol. 2022;23:123–31.
Pratap UP, Vadlamudi RK. PERK promotes immunosuppressive M2 macrophage phenotype by metabolic reprogramming and epigenetic modifications through the PERK-ATF4-PSAT1 axis. Immunometabolism. 2022;4:2346–56.
Willenborg S, Sanin DE, Jais A, et al. Mitochondrial metabolism coordinates stage-specific repair processes in macrophages during wound healing. Cell Metab. 2021;33:2398–414.
Chen S, Xia Y, He F, et al. Serine supports IL-1beta production in macrophages through mTOR signaling. Front Immunol. 2020;11:1866–73.
Shan X, Hu P, Ni L, et al. Serine metabolism orchestrates macrophage polarization by regulating the IGF1-p38 axis. Cell Mol Immunol. 2022;19:1263–78.
Mafi S, Mansoori B, Taeb S, Sadeghi H, Abbasi R, Cho WC, et al. mTOR-Mediated Regulation of Immune Responses in Cancer and Tumor Microenvironment. Front Immunol. 2021;12:774103. https://doi.org/10.3389/fimmu.2021.774103.
Linke M, Pham HT, Katholnig K, Schnoller T, Miller A, Demel F, et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol. 2017;18(3):293–302. https://doi.org/10.1038/ni.3655.
Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity. 2016;45(4):817–30. https://doi.org/10.1016/j.immuni.2016.09.016.
Rathore R, Caldwell KE, Schutt C, Brashears CB, Prudner BC, Ehrhardt WR, et al. Metabolic compensation activates pro-survival mTORC1 signaling upon 3-phosphoglycerate dehydrogenase inhibition in osteosarcoma. Cell Rep. 2021;34(4):108678. https://doi.org/10.1016/j.celrep.2020.108678.
Margaret E, Torrence MRM, Hosios AM, Alexander J. The mTORC1-mediated activation of ATF4 promotes protein and glutathione synthesis downstream of growth signals. Elife. 2021;10:e63326. https://doi.org/10.7554/eLife.63326.
Tait-Mulder J, Hodge K, Sumpton D, Zanivan S, Vazquez A. The conversion of formate into purines stimulates mTORC1 leading to CAD-dependent activation of pyrimidine synthesis. Cancer Metab. 2020;8:20. https://doi.org/10.1186/s40170-020-00228-3.
Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25(7):771–84. https://doi.org/10.1038/cr.2015.68.
Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun. 2013;4:2834. https://doi.org/10.1038/ncomms3834.
Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife. 2016;5:348–59. https://doi.org/10.7554/eLife.11612.
Brintha Selvarajah IA, Platé M. mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-β1–induced collagen biosynthesis. Sci Signal. 2019(34):231–42. https://doi.org/10.1126/scisignal.aav3048.
Tan HY, Wang N, Li S, Hong M, Wang X, Feng Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid Med Cell Longev. 2016;2016:2795090. https://doi.org/10.1155/2016/2795090.
Roux C, Jafari SM, Shinde R, Duncan G, Cescon DW, Silvester J, et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc Natl Acad Sci U S A. 2019;116(10):4326–35. https://doi.org/10.1073/pnas.1819473116.
Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu ZG. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013;23(7):898–914. https://doi.org/10.1038/cr.2013.75.
Formentini L, Santacatterina F, Nunez de Arenas C, Stamatakis K, Lopez-Martinez D, Logan A, et al. Mitochondrial ROS production protects the intestine from inflammation through functional M2 macrophage polarization. Cell Rep. 2017;19(6):1202–13. https://doi.org/10.1016/j.celrep.2017.04.036.
Ye C, Sutter BM, Wang Y, Kuang Z, Tu BP. A metabolic function for phospholipid and histone methylation. Mol Cell. 2017;66(2):180–93 e8. https://doi.org/10.1016/j.molcel.2017.02.026.
Zhang Z, TeSlaa T, Xu X, Zeng X, Yang L, Xing G, et al. Serine catabolism generates liver NADPH and supports hepatic lipogenesis. Nat Metab. 2021;3(12):1608–20. https://doi.org/10.1038/s42255-021-00487-4.
Morscher RJ, Ducker GS, Li SH, Mayer JA, Gitai Z, Sperl W, et al. Mitochondrial translation requires folate-dependent tRNA methylation. Nature. 2018;554(7690):128–32. https://doi.org/10.1038/nature25460.
Vandekeere S, Dubois C, Kalucka J, Sullivan MR, Garcia-Caballero M, Goveia J, et al. Serine synthesis via PHGDH is essential for heme production in endothelial cells. Cell Metab. 2018;28(4):573–87 e13. https://doi.org/10.1016/j.cmet.2018.06.009.
Solier S, Muller S, Caneque T, Versini A, Mansart A, Sindikubwabo F, et al. A druggable copper-signalling pathway that drives inflammation. Nature. 2023;617(7960):386–94. https://doi.org/10.1038/s41586-023-06017-4.
Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015;22(5):861–73. https://doi.org/10.1016/j.cmet.2015.08.024.
Hwang IY, Kwak S, Lee S, Kim H, Lee SE, Kim JH, et al. Psat1-Dependent Fluctuations in alpha-Ketoglutarate Affect the Timing of ESC Differentiation. Cell Metab. 2016;24(3):494–501. https://doi.org/10.1016/j.cmet.2016.06.014.
Wang J, Filippakis H, Hougard T, Du H, Ye C, Liu HJ, et al. Interleukin-6 mediates PSAT1 expression and serine metabolism in TSC2-deficient cells. Proc Natl Acad Sci U S A. 2021;118(39):3451–65. https://doi.org/10.1073/pnas.2101268118.
Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18(9):985–94. https://doi.org/10.1038/ni.3796.
Ming-Chin Lee K, Achuthan AA, De Souza DP, Lupancu TJ, Binger KJ, Lee MKS, et al. Type I interferon antagonism of the JMJD3-IRF4 pathway modulates macrophage activation and polarization. Cell Rep. 2022;39(3):110719. https://doi.org/10.1016/j.celrep.2022.110719.
Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345(6204):1251086. https://doi.org/10.1126/science.1251086.
Li J, Li L, Sun X, Deng T, Huang G, Li X, et al. Role of Tet2 in Regulating Adaptive and Innate Immunity. Front Cell Dev Biol. 2021;9:665897. https://doi.org/10.3389/fcell.2021.665897.
Sun F, Abreu-Rodriguez I, Ye S, Gay S, Distler O, Neidhart M, et al. TET1 is an important transcriptional activator of TNFalpha expression in macrophages. PLoS One. 2019;14(6):e0218551. https://doi.org/10.1371/journal.pone.0218551.
Liu N, Zhang J, Yan M, Chen L, Wu J, Tao Q, et al. Supplementation with alpha-ketoglutarate improved the efficacy of anti-PD1 melanoma treatment through epigenetic modulation of PD-L1. Cell Death Dis. 2023;14(2):170. https://doi.org/10.1038/s41419-023-05692-5.
Saeed S, Quintin J, Hindrik H, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086.
Zhou B, Magana L, Hong Z, Huang LS, et al. The angiocrine Rspondin3 instructs interstitial macrophage transition via metabolic-epigenetic reprogramming and resolves inflammatory injury. Nat Immunol. 2020;21:1430–43.
Xu W, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30.
Kottakis F, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539:390–5.
Ji J, et al. Methionine attenuates lipopolysaccharide-induced inflammatory responses via DNA methylation in macrophages. ACS Omega. 2019;4:2331–6.
Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2021;13:877–919.
Langston PK, Nambu A, Jung J, Shibata M, Aksoylar HI, Lei J, et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat Immunol. 2019;20(9):1186–95. https://doi.org/10.1038/s41590-019-0453-7.
Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe CC, Hess T, et al. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via atp-citrate lyase. Immunity. 2019;51(6):997–1011 e7. https://doi.org/10.1016/j.immuni.2019.11.009.
Li AM, Li Y, He B, Jiang H, Ramirez Y, Zhou M-N, et al. Serine starvation silences estrogen receptor signaling through histone hypoacetylation. Biorxiv. 2021. https://doi.org/10.1101/2021.09.05.459037.
Noe JT, Rendon BE, Geller AE, Conroy LR, Morrissey SM, Young LEA, et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci Adv. 2021;7(46):234–45. https://doi.org/10.1126/sciadv.abi8602.
Zhang D, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–80.
Irizarry-Caro RA, McDaniel MM, Overcast GR, Jain VG, Troutman TD, Pasare C. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc Natl Acad Sci U S A. 2020;117:30628–38.
Wang N, et al. Histone Lactylation Boosts Reparative Gene Activation Post-Myocardial Infarction. Circ Res. 2022;131:893–908.
Guo WB, et al. Down regulating PHGDH affects the lactate production of sertoli cells in varicocele. Reprod Biol Endocrinol. 2020;18:70.
Diskin C, Ryan TAJ, O’Neill LAJ. Modification of proteins by metabolites in immunity. Immunity. 2021;54:19–31.
Liu Y, Vandekeere A, Xu M, Fendt SM, Altea-Manzano P. Metabolite-derived protein modifications modulating oncogenic signaling. Front Oncol. 2022;12:988–96.
Yang K, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022;29:133–46.
Larson-Casey JL, et al. Increased flux through the mevalonate pathway mediates fibrotic repair without injury. J Clin Invest. 2019;129:4962–78.
Chi Z, et al. Histone deacetylase 3 couples mitochondria to drive IL-1beta-dependent inflammation by configuring fatty acid oxidation. Mol Cell. 2020;80(43–58): e47.
Wu Q, Chen X, Li J, Sun S. Serine and metabolism regulation: a novel mechanism in antitumor immunity and senescence. Aging Dis. 2020;11(6):1640–53. https://doi.org/10.14336/AD.2020.0314.
Zhang X, Ji L, Li MO. Control of tumor-associated macrophage responses by nutrient acquisition and metabolism. Immunity. 2023;56(1):14–31. https://doi.org/10.1016/j.immuni.2022.12.003.
Sullivan MR, Mattaini KR, Dennstedt EA, Nguyen AA, Sivanand S, Reilly MF, et al. Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab. 2019;29(6):1410–21 e4. https://doi.org/10.1016/j.cmet.2019.02.015.
Rinaldi G, Pranzini E, Van Elsen J, Broekaert D, Funk CM, Planque M, et al. In vivo evidence for serine biosynthesis-defined sensitivity of lung metastasis, but not of primary breast tumors, to mTORC1 inhibition. Mol Cell. 2021;81(2):386–97 e7. https://doi.org/10.1016/j.molcel.2020.11.027.
Wei L, Lee D, Law CT, Zhang MS, Shen J, Chin DW, et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat Commun. 2019;10(1):4681. https://doi.org/10.1038/s41467-019-12606-7.
Pranzini E, Pardella E, Muccillo L, Leo A, Nesi I, Santi A, et al. SHMT2-mediated mitochondrial serine metabolism drives 5-FU resistance by fueling nucleotide biosynthesis. Cell Rep. 2022;40(7):111233. https://doi.org/10.1016/j.celrep.2022.111233.
Tajan M, Hennequart M, Cheung EC, Zani F, Hock AK, Legrave N, et al. Serine synthesis pathway inhibition cooperates with dietary serine and glycine limitation for cancer therapy. Nat Commun. 2021;12(1):366. https://doi.org/10.1038/s41467-020-20223-y.
Su S, Zhao J, Xing Y, Zhang X, Liu J, Ouyang Q, et al. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell. 2018;175(2):442–57 e23. https://doi.org/10.1016/j.cell.2018.09.007.
Buque A, Galluzzi L, Montrose DC. Targeting Serine in Cancer: Is Two Better Than One? Trends Cancer. 2021;7(8):668–70. https://doi.org/10.1016/j.trecan.2021.06.004.
Lionaki E, Gkikas I, Daskalaki I, Ioannidi MK, Klapa MI, Tavernarakis N. Mitochondrial protein import determines lifespan through metabolic reprogramming and de novo serine biosynthesis. Nat Commun. 2022;13(1):651. https://doi.org/10.1038/s41467-022-28272-1.
Lima TI, Laurila PP, Wohlwend M, Morel JD, Goeminne LJE, Li H, et al. Inhibiting de novo ceramide synthesis restores mitochondrial and protein homeostasis in muscle aging. Sci Transl Med. 2023;15(696):547–56. https://doi.org/10.1126/scitranslmed.ade6509.
Kadomoto S, Izumi K, Mizokami A. Macrophage polarity and disease control. Int J Mol Sci. 2021;23:144.
Ma B, Nie X, Liu L, Li M (2023) GSK2656157, a PERK inhibitor, alleviates pyroptosis of macrophages induced by mycobacterium bacillus calmette–guerin infection. Int J Mol Sci. 2023;24(22):16239. https://doi.org/10.3390/ijms242216239.
Linke M, et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol. 2017;18:293–302.
Qi W, Chen X, Li J, Sun S. Serine and metabolism regulation: a novel mechanism in antitumor immunity and senescence. Aging Dis. 2020;11:1640–53.
Sullivan MRM, Katherine R, et al. Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab. 2019;29:1410–21.
Rinaldi G, EricaVan E, et al. In vivo evidence for serine biosynthesis-defined sensitivity of lung metastasis, but not of primary breast tumors, to mTORC1 inhibition. Mol Cell. 2020;5:134–46.
Wei L, Derek Law C-T, et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nature Commun. 2019;10:569–89.
Tajan MH, Cheung EM, et al. Serine synthesis pathway inhibition cooperates with dietary serine and glycine limitation for cancer therapy. Nat Commun. 2021;12:366–80.
Su S, et al. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell. 2018;175(442–457): e423.
Buque AG, Montrose LDC. Targeting serine in cancer: is two better than one? Trends Cancer. 2021;7:668–70.
Lionaki EG, Daskalaki I, et al. Mitochondrial protein import determines lifespan through metabolic reprogramming and de novo serine biosynthesis. Nat Commun. 2022;13:651–70.
Tanes I, Lima P-PL. Martin Wohlwend Inhibiting de novo ceramide synthesis restores mitochondrial and protein homeostasis in muscle aging. Sci Transl Med. 2023;15:547–56.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81371675).
Author information
Authors and Affiliations
Contributions
Yuping Chen conceptualised the manuscript. Yuping Chen and Xinqiong Huang wrote the manuscript. Xue Yang and Li Xiang designed and prepared the figures and tables. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Responsible Editor: L Li.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, X., Yang, X., Xiang, L. et al. Serine metabolism in macrophage polarization. Inflamm. Res. 73, 83–98 (2024). https://doi.org/10.1007/s00011-023-01815-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-023-01815-y