
Abstract
Targeting MDM2-p53 interaction has emerged as a promising antitumor therapeutic strategy. Several MDM2-p53 inhibitors have advanced into clinical trials, but results are not favorable. The lack of appropriate biomarkers for selecting patients has been assumed as the critical reason for this failure. We previously identified ZER6 isoform p52-ZER6 as an oncogene upregulated in tumor tissues. In this study we investigated whether p52-ZER6 acted as a blocker of MDM2-p53 binding inhibitors, and whether p52-ZER6 could be used as a biomarker of MDM2-p53 binding inhibitors. In p53 wild-type colorectal carcinoma HCT116, hepatocarcinoma HepG2 and breast cancer MCF-7 cells, overexpression of p52-ZER6 enhanced MDM2-p53 binding and promoted p53 ubiquitination/proteasomal degradation. Furthermore, overexpression of p52-ZER6 in the tumor cells dose-dependently reduced their sensitivity to both nutlin and non-nutlin class MDM2-p53 binding inhibitors. We showed that p52-ZER6 restored tumor cell viability, which was suppressed by nutlin-3, through restoring their proliferation potential while suppressing their apoptotic rate, suggesting that MDM2-p53 binding inhibitors might not be effective for patients with high p52-ZER6 levels. We found that nutlin-3 treatment or p52-ZER6 knockdown alone promoted the accumulation of p53 protein in the tumor cells, and their combinatorial treatment significantly increased the accumulation of p53 protein. In HCT116 cell xenograft nude mouse model, administration of shp52-ZER6 combined with an MDM2-p53 binding inhibitor nutlin-3 exerted synergistic antitumor response. In conclusion, this study reveals that p52-ZER6 might be a potential biomarker for determining patients appropriate for MDM2-p53 binding inhibition-based antitumor therapy, and demonstrates the potential of combinatorial therapy using MDM2-p53 binding inhibitors and p52-ZER6 inhibition.
Similar content being viewed by others


Introduction
Since its discovery more than 40 years ago, tumor suppressor p53, whose expression is commonly downregulated in tumor patients, has been well known as one of the most important tumor suppressors. It plays a pivotal role in maintaining genome integrity by triggering various cellular events such as cell cycle arrest, cellular senescence, DNA repair, and cell death, thereby conferring protection to cells against malignant transformation [1, 2]. While p53 mutations are observed in ~50% of the tumor patients [2], p53 downregulation attributed to the aberrant expression of its negative regulators, particularly mouse double minute 2 (MDM2), is commonly reported in patients with wild-type p53 [3]. This abnormality has been assumed to be the main reason underlying uncontrolled tumor cell proliferation and antiapoptotic potential [4]. More recent studies have revealed that p53 is also a critical regulator of other hallmarks of cancer, including tumor angiogenesis, metabolism, and stemness [5,6,7,8], further emphasizing its role as a tumor suppressor.
Considering that p53 is crucial for stimulating apoptosis upon exposure to unrepairable DNA damage, triggering the accumulation of p53 protein has emerged as an attractive antitumor therapeutic strategy. Inhibiting the binding between p53 and MDM2, an E3 ligase that enhances the ubiquitination and degradation of p53 protein, has attracted attention as a strategy for inducing p53 protein accumulation in tumor patients with wild-type p53 [9]. Several small compounds have been developed for this purpose [10, 11]. A few such inhibitors, belonging to the nutlin family, including RG7388 (idasanutlin), RG7112, and RO8994, as well as from the non-nutlin family, such as HDM201 (siremadlin), have advanced into clinical trials [12,13,14,15,16,17]. The nutlin family, comprising the first potent non-genotoxic small molecule specific inhibitors of MDM2-p53 binding, is a group of cis-imidazoline compounds that mimic MDM2 binding on the pocket of p53 protein, thereby promoting the accumulation of p53 protein and mediating antitumor therapeutic effects [18]. Meanwhile, HDM201 ((4S)-5-(5-chloro-1-methyl-2-oxopyridin-3-yl)-4-(4-chlorophenyl)-2-(2,4-dimethoxypyrimidin-5-yl)-3-propan-2-yl-4H-pyrrolo[3,4-d] imidazol-6-one) is an MDM2-p53 binding inhibitor containing a pyrrolidonoimidazole scaffold [19].
Despite the promising pre-clinical results, clinical trials examining MDM2-p53 binding inhibitors have only shown evidence of a modest therapeutic response [20]. The lack of appropriate biomarkers for predicting the antitumor efficacy of compounds targeting the binding of MDM2 and p53 and, subsequently, for selecting patients appropriate for these treatments has been assumed to be a critical reason underlying this failure [21]. Hence, there is an urgent need to identify an appropriate biomarker for improving the applicability of MDM2-p53 binding inhibitors and enhancing the antitumor efficacy of these inhibitors by adopting a combinatory strategy with therapeutic approaches targeting the biomarker [22].
p52-ZER6 is an isoform of zinc-finger estrogen receptor interaction clone 6 (ZER6), a zinc-finger protein that contains six C2H2-type zinc fingers [23, 24]. Through high-throughput screening of factors regulating p21 transcriptional activity using a small hairpin RNA (shRNA) expression vector library covering 2,065 genes, we previously identified ZER6 isoform p52-ZER6 as an oncogene which was upregulated in tumor tissues [25]. p52-ZER6, but not its splicing isoform p71-ZER6, reduces the protein levels of p53 by inducing degradation, leading to enhanced cell cycle progression and tumor cell proliferation [25].
Considering that the mechanism of action of p52-ZER6 is contrary to that of the inhibitors of MDM2-p53 binding in regulating p53 levels, in the present study, we aimed to examine whether p52-ZER6 acts as a blocker of these inhibitors. As nutlin-3, belonging to the third generation of the nutlin family, exerts its antitumor effect by inhibiting the MDM2-p53 interaction and promoting p53 protein accumulation, we hypothesized that p52-ZER6 would attenuate its antitumor effect and could be used as a biomarker of MDM2-p53 binding inhibitors. Furthermore, we aimed to analyze the effect of combining the suppressor of p52-ZER6 and the inhibitors of MDM2-p53 binding on cell cycle progression, tumor cell survival, proliferation, and apoptotic rate. Finally, we aimed to examine the potential of combining an MDM2-p53 binding inhibitor with p52-ZER6 inhibition as a novel antitumor therapeutic strategy.
Materials and methods
Vectors construction
shRNA expression vectors were constructed as described previously [26]. Target sites (shp52-1: GAGAGGACGTAGTCTTGCT; shp52-2: GACTGAGAGGCAGCGAGAA) were predicted using the algorithm as previously reported. Oligonucleotides bringing the hairpin structure, as well as overhanging and terminator sequences were synthesized, annealed and inserted into the BspMI sites of a pcPUR+U6i cassette vector containing a U6 promoter [26].
p52-ZER6, p53, MDM2, and ubiquitin overexpression vectors (p52-ZER6, pcp53, pcMDM2, and pcUbi), were constructed as described previously. Briefly, the corresponding coding sequences were obtained by reverse-transcribing total RNA extracted from HCT116 cells using the PrimeScript Reagent Kit with gDNA Eraser (Takara Bio, Dalian, China) and amplifying the corresponding regions using Takara Prime STAR Max DNA Polymerase (Takara Bio). The amplicons were cloned into pcDNA3.1(+) (Invitrogen Life Technologies, Carlsbad, CA), respectively [27]. For FLAG-tagged p53 (pcFLAG-p53) and FLAG-tagged p52-ZER6 overexpression vectors, corresponding fragments were amplified from pcp53 and pcp52-ZER6, respectively, and inserted into the BamHI and Not I sites of pcFLAG [25].
Cell lines, cell culture, and reagent
Wild-type HCT116, HepG2, and MCF-7 cell lines were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). HCT116 cell lines were cultured in McCoy’s 5A medium (Gibco, Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Biological Industries, Beit Haemek, Israel), while HepG2 and MCF-7 cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% FBS. All cell lines were verified using short-tandem repeat profiling method, and have been routinely tested and found negative for mycoplasma contamination using Mycoplasma Detection Kit QuickTest (Biotool, Houston, TX). Transfection was performed using Lipofectamine 2000 (Invitrogen Life Technologies) according to the manufacturer’s instruction. Nutlin-3 (Medchem Express, Monmouth Junction, NJ; Mw: 581.49; purity ≥98.90%) as well as RG7388 and HDM201 (ApexBio, Shanghai, China; Mw: 616.48 and 555.41, respectively; purities ≥99%) were dissolved in DMSO.
For gene silencing experiments, cells were seeded in 6-well plate and transfected with 2 µg of the indicated shRNA expression vector. Twenty four hours later, cells were subjected to puromycin selection (final concentration: 1.2 μg/mL) for 36 h to eliminate untransfected cells.
Animal experiment
For the in vivo tumor study, BALB/c-nu/nu mice (male; body weight, 18–22 g; 6 weeks old) were purchased from Chongqing Medical University (Chongqing, China). Animal study was approved by the Institutional Ethics Committee of Chongqing Medical University (Permit No. SYXK-2018-003), and carried out at the Chongqing Medical University. All animal experiments conformed to the approved guidelines of the Animal Care and Use Committee of Chongqing Medical University. All efforts were made to minimize suffering.
For the xenograft experiment, BALB/c-nu/nu mice were first injected subcutaneously with 5 × 106 of HCT116WT cells. Tumors were allowed to grow for 12 days until their volume reached 100–150 mm3. Mice were then randomly separated into 4 groups (n = 7), and treated with shCon or shp52-ZER6 (5 μg, intratumor injection, once every 2 days) together with vehicle or nutlin-3 (40 mg/kg body weight) diluted in vehicle (intraperitoneal injection, once a day). 1: 1: 1: 7 solution of DMSO: Tween 80: propanediol: phosphate buffered saline was used as vehicle. Prior to injection, vectors were mixed with transfection reagent Entransfer-in vivo (Engreen, Beijing, China). Tumor size (V) was evaluated by a caliper every 2 days before treatment, or every day after treatment, and calculated using the following equation: V = a × b2/2, where a and b are the major and minor axes of the tumor, respectively. The investigator was blinded for the group allocation and during the assessment.
Calculation of growth delay and enhancement factor was performed as described previously [28,29,30]. Doubling time was calculated using Doubling Time Calculator (http://www.radclass.mudr.org/content/doubling-time-calculation-growth-rate-lesion-or-mass) [31]. Absolute growth delay for each treated group was obtained by subtracting the doubling time of the tumor in the treated group from that of the control group. Normalized growth delay was calculated using the following equation: Normalized growth delay (for a) = absolute growth delay (a and b combinatorial treatment) − absolute growth delay (b); and vice versa. Enhancement factor was calculated using the following equation: Enhancement factor (for a) = normalized growth delay (for a)/absolute growth delay (a).
Western blotting and quantitative real time-PCR (qRT-PCR) analysis
Detailed methods for Western blotting and qRT-PCR analysis are described in the Supplementary Materials and Methods. The sequences of the primers and antibodies used are shown in the Supplementary Tables S1 and S2, respectively.
Statistical analysis
All quantification results are presented as mean ± SD (n = 3; unless further indicated). Statistical analysis was performed using Student’s t test. For xenograft experiments, statistical analysis was performed using one‐way ANOVA. A value of P < 0.05 was considered statistically significant.
Results
p52-ZER6 attenuates the p53/p21 signaling pathway in various tumor cells
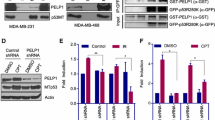
To explore the role of p52-ZER6 in p53 signaling in tumor cells, p52-ZER6 was overexpressed in p53 wild-type colorectal carcinoma (CRC), hepatocarcinoma (HCC), and breast cancer cell lines (HCT116, HepG2, and MCF-7, respectively; Supplementary Fig. 1), and its effects on p53 protein accumulation were examined. p52-ZER6 overexpression robustly reduced p53 protein accumulation in these cell lines along with the mRNA and protein expression levels of its target gene p21 (Fig. 1a, b). Knockdown of p52-ZER6 (Supplementary Fig. 2a, b) robustly enhanced the accumulation of p53 protein and the expression of p21 (Fig. 1c, d). These results indicate that p52-ZER6 negatively regulates p53 protein expression; however, as shown in Supplementary Fig. 3, p52-ZER6 does not affect p53 mRNA expression. Our previous study showed that p52-ZER6 could enhance p53 ubiquitination/proteasomal degradation [25]. Indeed, immunoprecipitation results markedly showed that p52-ZER6 strengthened the binding between p53 and its E3 ligase, MDM2, in the abovementioned tumor cells (Fig. 1e). Concomitantly, knockdown of p52-ZER6 evidently weakened the binding between MDM2 and p53 proteins in HCT116 cells as well as in HepG2 and MCF-7 cells (Fig. 1f). Together, these results indicate that p52-ZER6 regulates p53 protein expression in various tumor cells by strengthening the integrity of the MDM2-p53 complex.

a p53 and p21 protein expression levels in HCT116 colorectal carcinoma (CRC), HepG2 hepatocarcinoma (HCC), and MCF-7 breast cancer cells overexpressing p52-ZER6, as determined using Western blotting. b p21 mRNA expression level in p52-ZER6-overexpressed HCT116, HepG2, and MCF-7 cells, as analyzed using quantitative reverse-transcription PCR (qRT-PCR). c p53 and p21 protein expression levels in p52-ZER6-knocked down HCT116, HepG2, and MCF-7 cells, as determined using Western blotting. d p21 mRNA expression level of in p52-ZER6-knocked down HCT116, HepG2, and MCF-7 cells, as analyzed using qRT-PCR. Binding capacity of MDM2 to p53 protein in p52-ZER6-overexpressed (e) or p52-ZER6-knocked down (f) HCT116, HepG2, and MCF-7 cells, as determined using immunoprecipitation assay. Cell lysates were immunoprecipitated against anti-p53 antibody. The presence of MDM2 was detected using Western blotting. Cells transfected with pcCon or shCon were used as controls. β-actin was used for qRT-PCR normalization and as Western blotting loading control. Quantitative data are presented as relative to control and expressed as mean ± SD (n = 3). pcCon: pcDNA3.1(+); **P < 0.01.
p52-ZER6 is a novel blocker of the MDM2-p53 binding inhibitors
To explore whether p52-ZER6 can act antagonistically with MDM2-p53 binding inhibitors, we investigated the effect of various concentrations of nutlin-3 on tumor cells overexpressing p52-ZER6. The effect of nutlin-3 on tumor cell viability was significantly attenuated at all concentrations in HCT116 cells overexpressing p52-ZER6 (Fig. 2a), indicating that p52-ZER6 suppressed the sensitivity of HCT116 cells to nutlin-3. Similarly, the sensitivities of HepG2 (Fig. 2b) and MCF-7 (Fig. 2c) cells to nutlin-3 also decreased significantly in p52-ZER6-overexpressing cells. Accordingly, the IC50 value of nutlin-3 increased ~3.9-fold in p52-ZER6-overexpressed HCT116 cells (Fig. 2d), while those of HepG2 and MCF-7 cells increased ~3.5- and 3.2-fold, respectively (Fig. 2e, f). Furthermore, p52-ZER6 overexpression inhibited the increase in the accumulation of p53 and p21 proteins induced by various concentrations of nutlin-3 in HCT116, HepG2, and MCF-7 cells (Fig. 2g–i). These results clearly showed that p52-ZER6 suppresses the sensitivity of various tumor cells to nutlin-3 by impeding the nutlin-3-induced p53 accumulation.

Viabilities of p52-ZER6-overexpressed HCT116 (a), HepG2 (b), and MCF-7 (c) cells treated with indicated doses of nutlin-3. IC50 values of nutlin-3 in HCT116 (d), HepG2 (e), and MCF-7 (f) cells overexpressing p52-ZER6 based on the results of cell viability assays in a–c. p53 and p21 protein accumulations in HCT116 (g), HepG2 (h), and MCF-7 (i) cells overexpressing p52-ZER6 and treated with nutlin-3, as determined using Western blotting. j p53 and p21 protein accumulations in HCT116 cells transfected with indicated amounts of p52-ZER6 overexpression vector and treated with nutlin-3, as determined using Western blotting. k Viabilities of HCT116 cells transfected with indicated amounts of p52-ZER6 and treated with nutlin-3. l IC50 values of nutlin-3 in HCT116 cells transfected with indicated amounts of p52-ZER6 overexpression vector and treated with nutlin-3, based on the results of cell viability assay in k. m Combination index (CI) of nutlin-3 and p52-ZER6 overexpression in HCT116 cells, as calculated by CompuSyn analysis based on the viabilities of HCT116 cells in a. Cells transfected with pcCon and treated with DMSO were used as control. β-actin was used as Western blotting loading control. For nutlin-3 treatment, cells were treated with 5 μM nutlin-3 (final concentration), unless further indicated. Quantitative data are presented as relative to control and expressed as mean ± SD (n = 3). pcCon: pcDNA3.1(+); **P < 0.01.
To further confirm the effect of p52-ZER6 on nutlin-3-induced p53 protein accumulation, we examined the p52-ZER6 dose-dependent inhibition of nutlin-3-induced p53 accumulation. The accumulation of p53 and p21 proteins in HCT116 cells decreased with an increase in p52-ZER6 expression (Fig. 2j). Concomitantly, at every concentration of nutlin-3 tested, cell viability was positively correlated with the levels of p52-ZER6 (Fig. 2k). Furthermore, the IC50 values of nutlin-3 increased in a p52-ZER6-dependent manner (Fig. 2l). These results indicate that the sensitivity of tumor cells to nutlin-3 is negatively correlated with the levels of p52-ZER6, most plausibly due to the decrease in the accumulation of p53. To determine whether p52-ZER6 acts as a blocker of nutlin-3, we calculated the CI values of nutlin-3 in p52-ZER6-overexpressing HCT116 cells. For every dose of nutlin-3 tested, the CI values were between 2.42 and 3.19 (Fig. 2m). These results indicate the existence of a marked antagonism between nutlin-3 and p52-ZER6 [32, 33].
To examine the possibility of the interaction of nutlin-3 with p52-ZER6, we first predicted the structure of the p52-ZER6 protein using the Swiss model (https://swissmodel.expasy.org/). Docking simulation using AutodockTools revealed that nutlin-3 might bind to p52-ZER6 at its Lys412 and Ser417 (Supplementary Fig. S4a). We further assessed the possibility of interactions between the MDM2, p53, and p52-ZER6 proteins and the binding pocket of nutlin-3 using PyMOL 1.8 X. The structures of the p53 and MDM2 proteins were obtained from the PDB database (http://www.rcsb.org/pdb/home/home.do; ID: P04637 and Q00987, respectively). As shown in Supplementary Fig. S4b, p52-ZER6 binds to MDM2 and p53 near the binding pocket of nutlin-3. Hence, these results demonstrate the possibility that p52-ZER6 might reduce the accessibility of nutlin-3 to its binding pocket on MDM2. Together, these results show that p52-ZER6 acts as a blocker of the nutlin family and is negatively correlated with the sensitivity of tumor cells to the members of the nutlin family by inhibiting nutlin-induced p53 protein accumulation.
We further investigated whether p52-ZER6 can act as a blocker of other MDM2-p53 binding inhibition-based antitumor compounds. To this end, we used RG7388 (idasanutlin, a second-generation member of nutlin-3) and HDM201 (siremadlin, a non-nutlin class MDM2-p53 binding inhibitor), which are MDM2-p53 binding inhibitors currently under clinical trials [10]. p52-ZER6 overexpression markedly suppressed the RG7388 and HDM201-induced accumulation of p53 and p21 proteins in HCT116 cells (Fig. 3a, b). Concomitantly, p52-ZER6 overexpression increased the viability of HCT116 cells at every concentration of RG7388 and HDM201 tested, resulting in ~12-fold and 8-fold increase in their IC50 values, respectively (Fig. 3c, d). Similar tendencies were observed in HepG2 and MCF-7 cells (Supplementary Fig. 5a–d). Furthermore, transfection of the p52-ZER6 overexpression vector at different amounts revealed that HCT116 cell sensitivity towards RG7388 and HDM201 decreased in a p52-ZER6-dependent manner (Fig. 3e, f). Furthermore, as revealed by the CI values, p52-ZER6 showed strong, or even very strong, antagonism against these compounds (Fig. 3g, h). These results clearly indicated that p52-ZER6 could impede the antitumor effects of both nutlin and non-nutlin classes of MDM2-p53 binding inhibitors.

p53 and p21 protein accumulations in p52-ZER6-overexpressed HCT116 cells treated with 0.5 μM (final concentration) of RG7388 (a) or HDM201 (b), as determined using Western blotting. Viabilities of p52-ZER6 overexpressed HCT116 cells treated with indicated doses of RG7388 (c) or HDM201 (d). Viabilities of HCT116 cells transfected with indicated amounts of p52-ZER6 overexpression vectors and treated with indicated doses of RG7388 (e) or HDM201 (f). Combination indexes between RG7388 (g) or HDM201 (h) and p52-ZER6 overexpression in HCT116 cells as analyzed with CompuSyn based on the results of cell viability assay in c and d, respectively. β-actin was used as Western blotting loading control. Cell transfected with pcCon or shCon and treated with DMSO were used as control. Quantitative results are presented as relative to control and expressed as means ± SD (n = 3). **P < 0.01.
p52-ZER6 suppresses the antitumor effects of nutlin-3
We next investigated the effect of p52-ZER6 on the antitumor potential of nutlin-3. As shown in Fig. 1, p52-ZER6 significantly attenuated the inhibitory effect of nutlin-3 on tumor cell viability while suppressing the activation of the p53/p21 pathway. The decrease in tumor cell viability could indeed be the result of reduced cell proliferation, increased apoptosis, or a synergistic effect of both. Meanwhile, the p53/p21 pathway is the main negative regulator of cell cycle progression, which could subsequently lead to the inhibition of cell proliferation and induction of apoptosis [18]. Treatment with nutlin-3 induced cell cycle arrest in the G0/G1 phase in HCT116 cells; however, this effect was significantly suppressed in HCT116 cells overexpressing p52-ZER6 (Fig. 4a). Concomitantly, p52-ZER6 also restored the suppressive effect of nutlin-3 on the levels of cyclin D1 and cyclin E, which are G1 cyclins, suggesting that p52-ZER6 disrupted the nutlin-3-induced cell cycle arrest (Fig. 4b).

a Percentage of cells at each cell cycle phase of p52-ZER6-overexpressed HCT116 cells treated with nutlin-3. Cells were stained with propidium iodide (PI), and the percentages were determined by flow cytometry. b Protein expression levels of cyclin D1 and cyclin E in p52-ZER6-overexpressed HCT116 cells treated with nutlin-3, as determined using Western blotting. c Number of proliferative p52-ZER6-overexpressed HCT116 cells treated with nutlin-3, as analyzed using EdU incorporation assay. Representative images (scale bars: 200 μm; left) and the percentage of EdU-positive cells to Hoechst-positive cells (right; n = 6) were shown. d Apoptotic rate of p52-ZER6-overexpressed HCT116 cells treated with nutlin-3, as examined using Annexin V/PI staining and flow cytometry. Representative images (left) and the percentage of apoptotic cells (right) were shown. e Protein expression levels of cleaved caspases 3 and 7 in p52-ZER6-overexpressed HCT116 cells treated with nutlin-3, as examined using Western blotting. f Relative activity of executioner caspases in p52-ZER6-overexpressed HCT116 cells treated with nutlin-3, as examined using caspase 3/7 activity assay. g Colony formation potential of p52-ZER6-overexpressed HCT116 cells treated with nutlin-3. Representative images (left) and the quantification results (right, n = 6) were shown. Cells transfected with pcCon and treated with DMSO were used as control. For nutlin-3 treatment, cells were treated with 5 μM nutlin-3 (final concentration). β-actin was used as Western blotting loading control. Quantitative data are presented as relative to control and expressed as mean ± SD (n = 3; unless further indicated). **P < 0.01.
Disruption of cell cycle arrest might affect cell proliferation and apoptosis. Indeed, the results of EdU incorporation assay showed that while nutlin-3 significantly reduced the percentage of EdU-positive cells, p52-ZER6 overexpression restored it (Fig. 4c), indicating that p52-ZER6 impeded the suppressive effect of nutlin-3 on tumor cell proliferation. Furthermore, p52-ZER6 overexpression also robustly attenuated the effect of nutlin-3 on HCT116 cell apoptosis (Fig. 4d). Together with the fact that p52-ZER6 overexpression suppressed the nutlin-3-induced cleavage and activities of caspases 3 and 7 (Fig. 4e, f), these results clearly showed that p52-ZER6 significantly suppressed the effect of nutlin-3 on the induction of tumor cell apoptosis. Collectively, these data indicated that p52-ZER6 restores tumor cell viability, which is suppressed by nutlin-3, by restoring the proliferation potential and suppressing the apoptotic rate of these cells.
We next investigated the effect of p52-ZER6 and nutlin-3 on the colony formation potential of tumor cells. Overexpression of p52-ZER6 robustly attenuated the effect of nutlin-3 in suppressing colony formation potential of HCT116 cells (Fig. 4g). Moreover, the effect of p52-ZER6 on the suppression of tumor cell proliferation and colony formation potential of nutlin-3 was dose-dependent, further confirming the antagonistic effect of p52-ZER6 on nutlin-3 (Supplementary Fig. 6a, b). Collectively, these results show that p52-ZER6 acts as a blocker of nutlin-3, impeding its antitumor potential.
p52-ZER6 inhibition increases the accumulation of p53 protein synergistically with both nutlin and non-nutlin classes of MDM2-p53 binding inhibitors
Considering that p52-ZER6 acts as a blocker of nutlin-3, we next investigated whether p52-ZER6 inhibition can augment the effect of nutlin-3 on increasing p53 protein accumulation in tumor cells. As shown in Fig. 5a, while treatment with nutlin-3 alone suppressed p53 ubiquitination, p52-ZER6 silencing further suppressed it. Concomitantly, while nutlin-3 treatment or p52-ZER6 knockdown alone promoted the accumulation of p53 protein in these cells, their combinatory treatment significantly increased the accumulation of p53 protein (Fig. 5b–d).

a p53 ubiquitination level in p52-ZER6-silenced and nutlin-3 treated HCT116 cells were analyzed using anti-ubiquitin immunoblotting of cell lysates immunoprecipitated with anti-p53 antibody. Cells were treated with MG132 to inhibit proteasomal degradation. For inputs of immunoprecipitation assay, 40 μg of corresponding samples was loaded. Ubi ubiquitin, IP immunoprecipitation, IB immunoblotting. Cells transfected with shCon was used as controls. p53 protein level in p52-ZER6-knocked down HCT116 (b), HepG2 (c), and MCF-7 (d) cells treated with 5 μM nutlin-3 (final concentration), as determined by Western blotting. e Viabilities of p52-ZER6-knocked down HCT116 cells treated with indicated doses of nutlin-3. f IC50 values of nutlin-3 in p52-ZER6-knocked down HCT116 cells based on the results of cell viability assay in e. g CI between nutlin-3 and p52-ZER6 knockdown in HCT116 cells as calculated by CompuSyn analysis based on the results of cell viability assay in e. β-actin was used as Western blotting loading control. Cell transfected with shCon or transfected with shCon and treated with DMSO were used as control. Quantitative data are presented as relative to control and expressed as mean ± SD (n = 3). **P < 0.01.
Next, we investigated the viability of p52-ZER6-knocked down HCT116 cells treated with nutlin-3. At every dose of nutlin-3, the viability of p52-ZER6-knocked down HCT116 cells was significantly lower than that of the corresponding control cells (Fig. 5e). Correspondingly, the IC50 values of nutlin-3 in the cells with p52-ZER6-knocked down were reduced to ~0.3-fold compared to those observed in the control (Fig. 5f). The results observed with the HepG2 and MCF-7 cells further confirmed this tendency (Supplementary Fig. 7a–d), as their IC50 values were also reduced to ~0.3-fold. These results indicated that inhibition of p52-ZER6 could enhance the effect of nutlin-3 on the viability of various tumor cells. Furthermore, the results showed that the combination treatment of p52-ZER6 knockdown and 5 μM to 20 μM nutlin-3 had a synergistic effect (Fig. 5g).
To examine whether p52-ZER6 inhibition can exert a synergistic effect in combination with the RG7388 and HDM201, we examined the IC50 values of RG7388 and HDM201 in HCT116, HepG2, and MCF-7 cells with p52-ZER6 knockdown. At every concentration tested, tumor cell sensitivity towards these drugs increased significantly in cells with p52-ZER6 knockdown compared to that in controls, leading to a robust reduction in their IC50 values (Supplementary Fig. 8a–f). Furthermore, calculation of CI values revealed synergistic or strong synergistic effects between p52-ZER6 knockdown with RG7388 as well as with HDM201 treatment (Supplementary Fig. 8g, h). Concomitantly, p52-ZER6 knockdown markedly increased the RG7388- and HDM201-induced accumulation of p53 and p21 proteins in HCT116 cells (Supplementary Fig. 8i, j). These results demonstrated a potent synergistic effect upon the suppression of p52-ZER6 and treatment with both nutlin and non-nutlin classes of MDM2-p53 binding inhibitors.
Altogether, these results clearly suggest that the combinatory treatment of p52-ZER6 inhibition with both nutlin and non-nutlin classes of MDM2-p53 binding inhibitors exerts a strong synergistic effect, most plausibly by synergistically promoting p53 protein accumulation.
p52-ZER6 silencing augments the antitumor potential of nutlin-3
Next, we explored whether p52-ZER6 silencing could augment the antitumor potential of nutlin-3. While knockdown of p52-ZER6 or the conduction of nutlin-3 treatment alone induced a G0/G1 cell cycle arrest, a combination of both led to nearly 90% G0/G1 phase arrest (Fig. 6a). These results were further confirmed by their synergistic effects exerted on the G1 cyclins (Fig. 6b). Furthermore, while knockdown of p52-ZER6 and the conduction of nutlin-3 treatment suppressed the proliferation of HCT116 cells, the percentage of EdU-positive cells in nutlin-3-treated tumor cells with p52-ZER6 knockdown was significantly lower (Fig. 6c). Moreover, compared to p52-ZER6-knockdown or nutlin-3 treatment alone, treatment of cells with nutlin-3 in combination with p52-ZER6-knockdown led to a significant increase in apoptotic cells to more than 40%. This combinatory treatment also induced the cleavage of executor caspases and their activities (Fig. 6d–f). Similar results were obtained in the colony formation assay (Fig. 6g). Together, these results demonstrated a potent synergistic effect observed upon suppressing of p52-ZER6 and treatment with nutlin-3, thereby enhancing their combined antitumor potential.

a Percentage of p52-ZER6-knocked down HCT116 cells treated with nutlin-3 in each cell cycle phase. Cells were stained with PI, and the percentages were determined by flow cytometry. b Protein expression levels of cyclin D1 and cyclin E in p52-ZER6-knocked down HCT116 cells treated with nutlin-3, as determined by Western blotting. c Number of proliferative p52-ZER6-knocked down HCT116 cells treated with nutlin-3, as analyzed using EdU incorporation assay. Representative images (left; scale bars: 200 μm) and the percentage of EdU-positive cells to Hoechst-positive cells (right; n = 6) were shown. d Apoptotic rate of p52-ZER6-knocked down HCT116 cells treated with nutlin-3, as examined using Annexin V/PI staining and flow cytometry. Representative images (left) and the percentage of apoptotic cells (right) were shown. e Protein expression levels of cleaved caspase 3 and cleaved caspase 7 in p52-ZER6-knocked down HCT116 cells treated with nutlin-3, as examined using Western blotting. f Relative activity of executioner caspases in p52-ZER6-knocked down HCT116 cells treated with nutlin-3, as examined using caspase 3/7 activity assay. g Colony formation potential of p52-ZER6-knocked down HCT116 cells treated with nutlin-3. Representative images (left) and the quantification results (right, n = 6) were shown. Cells transfected with shCon and treated with DMSO were used as control. For nutlin-3 treatment, cells were treated with 5 μM nutlin-3 (final concentration). β-actin was used as Western blotting loading control. Quantitative data are presented as relative to control and expressed as means ± SD (n = 3; unless further indicated). *P < 0.05, **P < 0.01.
p52-ZER6 silencing synergistically enhances the antitumor therapeutic effect of nutlin-3 in vivo
To further confirm the synergistic effects of p52-ZER6 silencing and nutlin-3 treatment in vivo, we performed xenograft experiments by transplanting HCT116 cells subcutaneously into BALB/c-nu/nu mice. The xenografted tumors were subjected to treatments with nutlin-3 or shp52-ZER6 expression vector either separately or in combination beginning from day 12 after subcutaneous injection, when the tumor volumes reached 100–150 mm3. As evidenced by the tumor volumes (Fig. 7a, b), while subjection to treatment with nutlin-3 or p52-ZER6 knockdown alone inhibited tumor progression; the combinatory treatment conspicuously enhanced this inhibitory effect. Within 3 weeks, the volumes of tumors in the control group increased ~11-fold and those in the single treatment groups with either nutlin-3 or p52-ZER6 silencing increased ~5-fold. Meanwhile, the increase in tumor volume with the combinatory treatment was suppressed to less than 2.5-fold (Fig. 7a, b). These results were further confirmed by the morphological appearance and weights of tumors (Fig. 7c, d). Combinatory treatments involving p52-ZER6 knockdown and nutlin-3 administration increased the doubling time of the xenografted tumors from 8 days (treatment by nutlin-3 alone) or 9 days (treatment by p52-ZER6 knockdown alone) to 16 days. Accordingly, the synergistic effect of p52-ZER6 knockdown and nutlin-3 treatment enhanced the efficacy of treatment with p52-ZER6 knockdown or nutlin-3 alone with an enhancement factor of 2.7 or 3.5, respectively (Fig. 7e).

Volume of xenografted tumors formed by HCT116 cells in BALB/c-nu/nu mice following indicated treatment at indicated time points (n = 7/group; a), and the fold-change of tumor volumes at day 32 compared to those at the starting point of the treatment (day 12; b). Morphological appearance (c) and tumor weight (d) at day 32 are shown. e Tumor growth delay and enhancement factor of combinatorial treatment of nutlin-3 and p52-ZER6 knockdown. (A) Absolute growth delay was calculated by subtracting the doubling time of the tumor in the treated group from that of the shCon group; (B) normalized growth delay for shp52-ZER6 was calculated by subtracting the absolute growth delay of the shp52+nutlin-3 group from that of the shCon+nutlin group; (C) normalized growth delay for nutlin-3 was calculated by subtracting the absolute growth delay of the shp52+nutlin-3 group from that of the shp52 group; (D) enhancement factor for shp52-ZER6 was calculated by dividing the normalized growth delay for shp52-ZER6 (B) with the absolute growth delay of the shp52 group; (E) enhancement factor for nutlin-3 was calculated by dividing the normalized growth delay for nutlin-3 (C) with the absolute growth delay of the shCon+nutlin group. f p52-ZER6, p53, and p21 protein expression levels in xenografted tumors treated with indicated treatments, as determined using Western blotting. β-actin was used as a loading control. g Immunohistochemistry staining against p53, p21, and PCNA, as well as TUNEL assay in tissue sections of xenografted tumors treated with indicated treatments. Scale bars: 50 μm. Quantitative data are presented as mean ± SD. **P < 0.01.
We next investigated the effect of p52-ZER6 knockdown and nutlin-3 in inducing p53 accumulation in vivo. The combination of p52-ZER6 knockdown and nutlin-3 treatment could robustly induce the accumulation of p53 and p21 proteins compared to single treatment (Fig. 7f, g). Immunohistochemistry results further confirmed these findings, as p53 and p21 expression levels were upregulated in xenografted tumor lesions subjected to treatment with nutlin-3 or those subjected to treatment involving the suppression of p52-ZER6. These effects were further enhanced in xenografted tumor lesions subjected to the combinatory therapy (Fig. 7g). Furthermore, the results of proliferating cell nuclear antigen (PCNA) staining and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay revealed that combinatory therapy further enhanced the suppressive effects of nutlin-3 and p52-ZER6 silencing on tumor cell proliferation while promoting nutlin-3- and p52-ZER6-induced apoptosis. These results clearly showed that p52-ZER6 inhibition could synergistically enhance the antitumor effect of nutlin-3 in vivo.
Overall, in this study, we demonstrated that p52-ZER6 acts as a novel blocker of both nutlin and non-nutlin classes of MDM2-p53 binding inhibitors, disrupting the effect of these inhibitors to promote p53 protein accumulation by enhancing the binding between MDM2 and p53 proteins. These findings suggest that p52-ZER6 is a potential marker for determining the efficacy of this class of drug. Furthermore, we showed the potential applicability of combinatory therapy using a p52-ZER6 inhibitor and an MDM2-p53 binding inhibitor as a novel antitumor therapeutic strategy (Fig. 8).

Schematic diagram showing ZER6 isoform p52-ZER6 as an antitumor therapeutic response determiner and blocker of MDM2-p53 binding inhibitors.
Discussion
Inhibition of the MDM2/p53 pathway, which aims to induce p53 protein accumulation, has garnered interest as a promising antitumor strategy [2, 34]. While many MDM2-p53 binding inhibitors have entered the clinical trial phase, a considerable proportion has not shown satisfactory results compared to those in their pre-clinical trials. Several nutlin analogs have been evaluated in phase I and phase II clinical trials for their efficacy against solid tumors, acute myeloid leukemia, hematological cancers, and melanoma [15, 35,36,37,38,39]. However, even for the compound that has entered the most advanced phase of the trials, RG7388 (idasanutlin), the overall response in the phase II clinical trial for polycythemia vera and essential thrombocythemia is reportedly 58%. For the non-nutlin family inhibitor, HDM201, the overall response in the phase I clinical trial for acute myeloid leukemia was 21%. These discrepancies, together with their adverse effects, are the main hurdles for the clinical application of these drugs.
One of the most critical reasons underlying the discrepancy between the results observed in pre-clinical trials and clinical trials was the lack of a marker for ascertaining the selection criteria, as most trials only excluded patients with mutant p53 and focused on the examination of the primary endpoints of safety, tolerability, and efficiency. MDM4 and MYCN are involved in the development of resistance to nutlin-3 and might function as markers for antitumor therapeutic strategies involving the use of nutlin family-based drugs; however, these two factors inactivate the p53 signaling pathway by regulating MDM2 expression and do not directly affect its binding with p53, the direct target of this class of drug [40]. Herein, our results clearly demonstrate antagonism between p52-ZER6 expression levels and tumor cell sensitivity to MDM2-p53 binding inhibitors, including nutlin-3, RG7388, and HDM201. Considering that both p52-ZER6 and MDM2-p53 binding inhibitors target the MDM2-p53 binding and exert antagonistic effects on MDM2/p53 complex integrity and p53 protein accumulation, the level of p52-ZER6 reflects the therapeutic antitumor response of these inhibitors. This indicates the possibility of considering the p52-ZER6 level as a criterion for determining patients who would benefit from the use of MDM2-p53 binding inhibitors.
As p52-ZER6 exerts an antagonistic effect with MDM2-p53 binding inhibitors, most plausibly by blocking their accessibility to their binding pocket in the MDM2 protein, aiming for a reduction in the p52-ZER6 levels might be a potential strategy for increasing tumor cell sensitivity to these inhibitors. Our results demonstrated that p52-ZER6 suppression significantly increased tumor cell sensitivity towards MDM2-p53 binding inhibitors, as it reduced the IC50 values of these inhibitors in CRC, HCC, and breast cancer cells. These synergistic effects resulted in enhanced cell cycle arrest and apoptosis as well as a significant inhibition of tumor cell proliferation. Our in vivo experiment further confirmed the potential applicability of combinatory antitumor therapy using an shRNA expression vector targeting p52-ZER6 and an MDM2-p53 binding inhibitor, as this combination significantly suppressed the progress of xenografted tumors compared to treatment with only shp52-ZER6 or nutlin-3. Hence, our results demonstrate the effects of a novel, potential antitumor combinatory therapy with a p52-ZER6 blocker and an MDM2-p53 binding inhibitor. Moreover, this combinatory strategy might increase the sensitivity of patients with high p52-ZER6 expression towards MDM2-p53 binding inhibitors. Together, p52-ZER6 could be used to determine the suitability of patients for MDM2-p53 binding inhibitor-based therapy and to ascertain the necessity of subjection to a combinatory therapy using a p52-ZER6 blocker and MDM2-p53 binding inhibitors.
Adverse effects are another hurdle faced by MDM2-p53 binding inhibitors [41, 42]. Combinatory therapy, which may help decrease the required dose of drugs, may also mitigate treatment-associated toxicities [43]. Considering that inhibition of p52-ZER6 could sensitize tumor cells towards MDM2-p53 binding inhibitors, as indicated by a significant decrease in their IC50 values, and although a further detailed investigation is necessary, combinatory therapy using a p52-ZER6 blocker and MDM2-p53 binding inhibitors may help reduce the dose of inhibitors needed to achieve similar antitumor therapeutic effects, and thus reduce the adverse effects.
In conclusion, we identified p52-ZER6 as a determinant of tumor cell sensitivity to MDM2-p53 binding inhibitors, potentially exerting its antagonistic effect by blocking the accessibility of MDM2-p53 binding inhibitors to its binding pocket. Furthermore, combining p52-ZER6 inhibition and MDM2-p53 binding inhibitor treatment clearly augment the antitumor therapeutic effect. Hence, our study provides a potential biomarker to determine patient suitability for MDM2-p53 binding inhibition-based antitumor therapy and prevent unnecessary treatment with this class of drugs in inappropriate patients. We also provide a potential antitumor therapeutic strategy through the combination of a p52-ZER6 inhibitor and MDM2-p53 binding inhibitor, which might, subsequently, enable the development of a precise, tailored therapeutic antitumor strategy for patients harboring wild-type p53.
References
Kaiser AM, Attardi LD. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018;25:93–103.
Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83.
Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453–9.
Bieging KT, Attardi LD. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012;22:97–106.
Ni T, Li XY, Lu N, An T, Liu ZP, Fu R, et al. Snail1-dependent p53 repression regulates expansion and activity of tumour-initiating cells in breast cancer. Nat Cell Biol. 2016;18:1221–32.
Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST, et al. Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol Cell. 2017;68:281–92.e5.
Yamakuchi M, Lotterman CD, Bao C, Hruban RH, Karim B, Mendell JT, et al. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci USA. 2010;107:6334–9.
Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metab. 2013;18:617–33.
Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res. 2008;14:5318–24.
Konopleva M, Martinelli G, Daver N, Papayannidis C, Wei A, Higgins B, et al. MDM2 inhibition: an important step forward in cancer therapy. Leukemia. 2020;34:2858–74.
Sanz G, Singh M, Peuget S, Selivanova G. Inhibition of p53 inhibitors: progress, challenges and perspectives. J Mol Cell Biol. 2019;11:586–99.
Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56:5979–83.
Montesinos P, Beckermann BM, Catalani O, Esteve J, Gamel K, Konopleva MY, et al. MIRROS: a randomized, placebo-controlled, phase III trial of cytarabine±idasanutlin in relapsed or refractory acute myeloid leukemia. Future Oncol. 2020;16:807–15.
Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett. 2013;4:466–9.
Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res. 2016;22:868–76.
Zhang Z, Ding Q, Liu JJ, Zhang J, Jiang N, Chu XJ, et al. Discovery of potent and selective spiroindolinone MDM2 inhibitor, RO8994, for cancer therapy. Bioorg Med Chem. 2014;22:4001–9.
Guerreiro N, Jullion A, Ferretti S, Fabre C, Meille C. Translational modeling of anticancer efficacy to predict clinical outcomes in a first-in-human phase 1 study of MDM2 inhibitor HDM201. AAPS J. 2021;23:28.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8.
Liao G, Yang D, Ma L, Li W, Hu L, Zeng L, et al. The development of piperidinones as potent MDM2-P53 protein-protein interaction inhibitors for cancer therapy. Eur J Med Chem. 2018;159:1–9.
Shen H, Moran DM, Maki CG. Transient nutlin-3a treatment promotes endoreduplication and the generation of therapy-resistant tetraploid cells. Cancer Res. 2008;68:8260–8.
Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical overview of MDM2/X-targeted therapies. Front Oncol. 2016;6:7.
Duffy MJ, Synnott NC, O’Grady S, Crown J. Targeting p53 for the treatment of cancer. Semin Cancer Biol. 2020;79:58–67.
Conroy AT, Sharma M, Holtz AE, Wu C, Sun Z, Weigel RJ. A novel zinc finger transcription factor with two isoforms that are differentially repressed by estrogen receptor-alpha. J Biol Chem. 2002;277:9326–34.
Stabach PR, Thiyagarajan MM, Weigel RJ. Expression of ZER6 in ERalpha-positive breast cancer. J Surg Res. 2005;126:86–91. discussion 1-2.
Huang C, Wu S, Li W, Herkilini A, Miyagishi M, Zhao H, et al. Zinc-finger protein p52-ZER6 accelerates colorectal cancer cell proliferation and tumour progression through promoting p53 ubiquitination. Ebiomedicine. 2019;48:248–63.
Miyagishi M, Taira K. Strategies for generation of an siRNA expression library directed against the human genome. Oligonucleotides. 2003;13:325–33.
Huang C, Wu S, Ji H, Yan X, Xie Y, Murai S, et al. Identification of XBP1-u as a novel regulator of the MDM2/p53 axis using an shRNA library. Sci Adv. 2017;3:e1701383.
Iwasa T, Okamoto I, Suzuki M, Nakahara T, Yamanaka K, Hatashita E, et al. Radiosensitizing effect of YM155, a novel small-molecule survivin suppressant, in non-small cell lung cancer cell lines. Clin Cancer Res. 2008;14:6496–504.
He T, Guo J, Song H, Zhu H, Di X, Min H, et al. Nutlin-3, an antagonist of MDM2, enhances the radiosensitivity of esophageal squamous cancer with wild-type p53. Pathol Oncol Res. 2018;24:75–81.
Park JK, Chung YM, Kim BG, Yoo YA, Yang BS, Kim JS, et al. N’-(phenyl-pyridin-2-yl-methylene)-hydrazine carbodithioic acid methyl ester enhances radiation-induced cell death by targeting Bcl-2 against human lung carcinoma cells. Mol Cancer Ther. 2004;3:403–7.
Mehrara E, Forssell-Aronsson E, Ahlman H, Bernhardt P. Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res. 2007;67:3970–5.
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6.
Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81.
Vilgelm AE, Cobb P, Malikayil K, Flaherty D, Johnson CA, Raman D, et al. MDM2 antagonists counteract drug-induced DNA damage. Ebiomedicine. 2017;24:43–55.
Patnaik A, Tolcher A, Beeram M, Nemunaitis J, Weiss GJ, Bhalla K, et al. Clinical pharmacology characterization of RG7112, an MDM2 antagonist, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2015;76:587–95.
Zhong H, Chen G, Jukofsky L, Geho D, Han SW, Birzele F, et al. MDM2 antagonist clinical response association with a gene expression signature in acute myeloid leukaemia. Br J Haematol. 2015;171:432–5.
Abdul Razak AR, Miller WH Jr., Uy GL, Blotner S, Young AM, Higgins B, et al. A phase 1 study of the MDM2 antagonist RO6839921, a pegylated prodrug of idasanutlin, in patients with advanced solid tumors. Invest N Drugs. 2020;38:1156–65.
Yee K, Papayannidis C, Vey N, Dickinson MJ, Kelly KR, Assouline S, et al. Murine double minute 2 inhibition alone or with cytarabine in acute myeloid leukemia: Results from an idasanutlin phase 1/1b study small star, filled. Leuk Res. 2021;100:106489.
Mascarenhas J, Lu M, Kosiorek H, Virtgaym E, Xia L, Sandy L, et al. Oral idasanutlin in patients with polycythemia vera. Blood. 2019;134:525–33.
Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2-p53 pathway revisited. J Biomed Res. 2013;27:254–71.
Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol. 2017;10:133.
Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–36.
Chen SH, Lahav G. Two is better than one; toward a rational design of combinatorial therapy. Curr Opin Struct Biol. 2016;41:145–50.
Acknowledgements
We thank Professor Xia Zhang (Institute of Pathology, Southwest Hospital, Third Military Medical University) for his helpful comments in preparing this manuscript. This study was supported by grants from the National Natural Science Foundation of China (Nos. 31871367, 81872273, 32070715, 82173029).
Author information
Authors and Affiliations
Contributions
VK and SRW conceived the project, designed the experiments, analyzed and interpreted the experimental results, and wrote the manuscript. WFL, LA and CH performed most of the experiments. WFL and LA performed the statistical analysis and assisted with the design of the study and drafting of the manuscript. CH constructed the plasmids and performed the immunoprecipitations. YT and LQ performed part of cell viability assay. MM designed shRNA target sites and analyzed part of the data. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Patents related to the results of this study had been filed with Chinese patent applications (No. 201910626899.0 and 201910626900.X). The authors declare no competing interests.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Wf., Alfason, L., Huang, C. et al. p52-ZER6: a determinant of tumor cell sensitivity to MDM2-p53 binding inhibitors. Acta Pharmacol Sin 44, 647–660 (2023). https://doi.org/10.1038/s41401-022-00973-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00973-9
- Springer Nature Singapore Pte Ltd.