
Summary
Purpose MDM2 is a negative regulator of the tumor suppressor p53. RO6839921 is an inactive pegylated prodrug of idasanutlin, an MDM2 antagonist, developed for intravenous administration. On cleavage by plasma esterases, the active principle (AP = idasanutlin) is released. This phase 1 study investigated the safety, pharmacokinetics, and pharmacodynamics of RO6839921 in patients with advanced solid tumors (NCT02098967). Methods Patients were evaluated on a 5-day dosing schedule every 28 days. Dose escalation used the Bayesian new continual reassessment model. Accelerated dose titration was permitted until grade ≥2 drug-related AEs were observed. The target DLT rate to define the MTD was 16–25%. p53 activation was assessed by measuring macrophage inhibitory cytokine-1 (MIC-1). Results Forty-one patients received 14–120 mg AP; 39 were DLT evaluable. The MTD was 110-mg AP (8% DLT rate), whereas 120-mg AP had a 44% DLT rate. DLTs were neutropenia, thrombocytopenia, and stridor. The most common treatment-related AEs (≥30%) were nausea, fatigue, vomiting, and thrombocytopenia. Pharmacokinetic analyses indicated rapid conversion of prodrug to AP and an approximately linear and dose-proportional dose-exposure relationship, with a 2-fold increase in exposure between Days 1 and 5 of AP. MIC-1 increases were exposure dependent. Stable disease was observed in 14 patients (34%). Conclusions RO6839921 showed reduced pharmacokinetic exposure variability and a safety profile comparable with that of oral idasanutlin. Although this study indicated that RO6839921 could be administered to patients, the results did not provide sufficient differentiation or improvement in the biologic or safety profile compared with oral idasanutlin to support continued development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Murine double minute 2 (MDM2) is a master negative regulator of the tumor suppressor protein p53 (gene TP53), which is frequently inactivated in human cancer [1]. MDM2 overexpression may drive tumorigenesis by down-regulating p53 [1]. Tumors with functional p53 and intact p53 downstream signaling, which account for approximately half of all solid tumors and most hematologic malignancies, are candidates for therapy with MDM2 antagonists [1, 2].
MDM2 antagonists block p53-MDM2 binding, stabilize p53, and activate p53 signaling to induce cell-cycle arrest and apoptosis [2]. This release of MDM2 inhibition restores p53 function in cancers overproducing MDM2 and/or with wild-type p53 signaling. Idasanutlin is an oral MDM2 antagonist of the nutlin family of compounds that is currently in development for solid and hematologic malignancies in several phase 1–3 studies [2,3,4,5].
RO6839921 is an inactive pegylated prodrug of idasanutlin (active principle = AP) [5,6,7]. On cleavage of the pegylated tail by esterases in the blood, the AP (idasanutlin) is released. The AP is a potent and selective inhibitor of p53-MDM2 binding to activate the p53 pathway and induce cell-cycle arrest and/or apoptosis. Intravenous (IV) RO6839921 has shown anti-tumor activity at nontoxic doses against established osteosarcoma and acute myeloid leukemia (AML) xenograft models in immunocompromised mice [6]. These preclinical pharmacological results supported further evaluation of RO6839921 in clinical studies.
RO6839921 was developed with the aim to further improve exposure variability and pharmacokinetic properties, reduce gastrointestinal toxicity in absence of prophylaxis, and potentially improve efficacy compared with oral idasanutlin. This phase 1 study investigated the safety, tolerability, pharmacokinetics, and pharmacodynamics of the MDM2 antagonist RO6839921 following IV administration in patients with advanced malignancies.
Methods
Study design
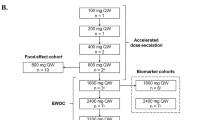
This phase 1 study was an open-label, first-in-human, multicenter, dose-escalation study of RO6839921 in patients with solid tumors and in patients with AML. Results for the AML cohort will be reported independently. On cleavage of the prodrug by plasma esterases, the AP idasanutlin is released. Therefore, doses are reported in milligrams of AP. A dose-escalation trial was planned with an initial pharmacokinetic and safety cohort (Cohort 0; 14 mg) to confirm prodrug cleavage followed by the solid tumor and AML cohorts. The 2 arms were escalated independently.
The escalation scheme for patients with solid tumors included single-evaluable patient dose escalation. On evidence of grade ≥2 study treatment–related adverse events (AEs) (excluding treatable nausea, vomiting, constipation, diarrhea, fatigue/asthenia, and alopecia; thrombocytopenia required confirmation with a second complete blood count within 48 h), cohorts were then expanded to 3 evaluable patients for determination of dose increments using the new continual reassessment method [8]. Subsequent doses tested per the new continual reassessment method were 80 mg, 95 mg, 110 mg, and 120 mg.
RO6839921 was administered as an IV infusion over approximately 1 h once daily for 5 days followed by 23 days of rest. Treatment of patients continued until disease progression, unacceptable toxicity, withdrawal of consent, or investigator discretion.
The primary objectives of the study were to determine the maximum tolerated dose (MTD), determine the recommended phase 2 dose, and characterize the dose-limiting toxicities (DLTs) and the overall safety profile of the escalated dose levels. The secondary objectives were to determine the pharmacokinetic parameters of RO6839921 and the AP; to assess the pharmacodynamic effects of the compound in blood, bone marrow, and tumor; and to assess any clinical responses.
The study was conducted in accordance with the principles set forth by the International Conference on Harmonisation good clinical practice guidelines. All patients provided written informed consent. The study was registered under ClinicalTrials.gov (NCT02098967).
Patients
Patients were aged ≥18 years with solid tumors who had measurable disease per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1), including patients with stable central nervous system metastasis, with a life expectancy of ≥12 weeks. Patients were required to have an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 to 1 and adequate hematologic, hepatic, and renal function. No limitations on the amount or type of prior anti-tumor therapy were stipulated. Patients were excluded if they had any severe and/or uncontrolled medical conditions or other conditions that could affect their participation, bone marrow disorders or delayed recovery from prior chemoradiotherapy (cytopenias are on-target toxicities of MDM2 antagonists), or known coagulopathies, known platelet disorders, or history of non–drug-induced thrombocytopenia.
Safety assessments
Patients receiving ≥1 dose of RO6839921 were considered safety evaluable. The safety outcome measures were incidence and severity of AEs regardless of attribution, incidence of laboratory abnormalities, electrocardiograms, and vital signs. The AE severity grading scale for the NCI CTCAE (version 4.03) was used to assess AE severity.
Patients who received ≥80% of study medication and completed the 28-day cycle or who had a DLT event but did not meet the dosing or duration requirements were considered DLT-evaluable patients. DLTs were assessed during the first treatment cycle (28 days) except for cytopenias, which were followed to nadir (or onset of grade 4 toxicity). The following events were considered DLTs:grade ≥3 febrile neutropenia; grade 3 hematologic toxicity requiring transfusion or any grade ≥4 hematologic toxicity;grade ≥ 3 nonhematologic toxicity not clearly resulting from the underlying malignancy, except alopecia; grade 3 fatigue/asthenia, anorexia, constipation, nausea, or vomiting, despite adequate supportive care measures (>24 h of treatment); total parenteral nutrition; or hospitalization. The targeted MTD was the dose at which the DLT rate was 16% to 25% [8]. Descriptive statistics were used to summarize all safety data.
Pharmacokinetic and pharmacodynamic assessments
Patients were required to provide blood samples for pharmacokinetic/pharmacodynamic assessment and archival tumor tissue for biomarker testing (if available). Plasma pharmacokinetic assessments were conducted for all patients during the first cycle of treatment on the first and fifth days of dosing, immediately before dosing, and at multiple postdose time points for measurements of RO6839921 and AP concentrations using a validated liquid-chromatography tandem mass spectrometry method. The following pharmacokinetic parameters were estimated using standard noncompartmental methods: the area under the plasma concentration-time curve from 0 to infinity, the area under the concentration-time curve from time 0 to 24 h; the maximum observed plasma concentration, the time to reach maximum plasma concentration, and the terminal half-life computed as (ln 2)/kel, where kel is the apparent elimination rate computed as the magnitude of the slope from the log-linear regression of the apparent terminal elimination phase of the plasma concentration-time curve.
Pharmacodynamic samples were collected before and after administration of RO6839921 to measure macrophage inhibitory cytokine 1 (MIC-1) protein in serum using an enzyme-linked immunosorbent assay. Data were reported as percentage of change from baseline. Baseline p53 next-generation sequencing was used to assess TP53 status from archival tumor tissue (Almac, Craigavon, United Kingdom).
Efficacy analysis
Tumor responses were assessed by radiology at baseline and after every 2 cycles of treatment (approximately every 8 weeks ±7 days) according to RECIST v1.1. The clinical benefit rate was calculated by determining the number of patients who achieve a complete response (CR) or partial response (PR) or stable disease divided by the total number of patients.
Results
Patients
This study was conducted at 6 sites in the United States and Canada between April 2014 and May 2018. In the dose-escalation phase and expanded MTD cohort, 41 patients were treated at 5 doses: 14 mg AP (n = 5), 80 mg AP (n = 7), 95 mg AP (n = 6), 110 mg AP (MTD, n = 14), and 120 mg AP (n = 9). All patients discontinued the study; the majority (29 patients; 71%) discontinued due to disease progression (Table 1). Six patients (15%) discontinued due to AEs of grade 4 stridor (in a patient with supraglottic squamous cell carcinoma), grade 2 dyspepsia, nausea, and leukopenia, which were considered treatment-related by the investigator. Additional reasons included withdrawal of consent (10%), refusal of further treatment after first dose in cycle 3 but did return for safety follow-up (2%), and study termination (2%).
Most patients were male (73%) and white (85%) (Table 2). The median age was 56 (range, 26–83) years, and 27% were aged ≥65 years. The most common tumor type was sarcoma (39%), followed by lung (15%), head and neck (12%), and salivary gland (5%). Singlet tumor types (22%) are footnoted under Table 1. Patients had an ECOG PS of either 0 (56%) or 1 (44%). Most patients had received ≥2 lines of systemic anti-cancer therapy (78%), prior cancer-related surgery (73%), and prior radiotherapy (56%).
DLT and MTD determination
A total of 39 patients were evaluable for DLTs per protocol. Of the 2 patients ineligible for DLT evaluation, 1 had rapid central nervous system progression undetected at screening after 2 doses in cycle 1 at 14 mg AP and 1 withdrew consent on study Day 22 at 110 mg AP.
Five (13%) patients experienced DLTs; 4 of 9 patients (44%) in the 120-mg AP cohort experienced 5 DLTs, and 1 of 13 patients (8%) in the 110-mg AP cohort experienced 1 DLT. One patient (3%) in the 120-mg cohort experienced 2 DLTs (neutropenia and thrombocytopenia). All other DLTs–—decreased neutrophils, thrombocytopenia, and stridor (serious)—were experienced by 1 patient (3%) each. All DLTs were grade 4 and resolved with study discontinuation, except for the patient who had solitary thrombocytopenia. Because these 2 doses were within 10% of each other, it was decided that further evaluation of intermediate doses was not warranted, and the MTD was determined to be 110 mg AP (8% DLT rate).
Safety
All 41 patients received ≥1 dose of RO6839921 and were considered safety evaluable. Almost all patients (40 of 41 patients; 98%) experienced ≥1 AE. The most common (≥20%) all-cause AEs of any grade were nausea (76%), fatigue (59%), vomiting (49%), thrombocytopenia (39%), diarrhea (34%), decreased appetite (34%), constipation (29%), hypotension (24%), and abdominal pain (24%). Twenty-three (56%) patients experienced all-cause grade ≥ 3 AEs, most commonly (≥10%) neutropenia (15%) and thrombocytopenia (10%). Thirty-seven (90%) patients experienced treatment-related AEs of any grade, most frequently (≥20%) nausea (73%), fatigue (54%), vomiting (44%), thrombocytopenia (39%), decreased appetite (29%) and diarrhea (24%; Table 3). Fifteen patients (37%) had grade ≥ 3 treatment-related AEs, most commonly neutropenia (15%) and thrombocytopenia (10%).
Six patients (15%) reported 7 serious AEs (SAEs) (Table 4). Urinary tract staphylococcal infection and hypersensitivity vasculitis were reported in 1 patient, and the remaining 5 SAEs were reported in 1 patient each. Four patients (9.8%) experienced 1 treatment-related SAE each: pancytopenia (120 mg AP), stridor (120 mg AP), abdominal pain (95 mg AP), and nausea (110 mg AP).
Six patients (15%) reported 8 AEs that resulted in withdrawal of study treatment. All doses of AP >14 mg resulted in ≥1 patient who experienced AEs leading to withdrawal from treatment, including 3 patients (21%) in the 110-mg AP cohort. One patient in the 80-mg AP cohort experienced 3 AEs of dyspepsia, nausea, and influenza-like illness. All other AEs that resulted in discontinuation—including atrial flutter, spinal compression fracture, stridor, deep vein thrombosis, and leukopenia—were reported by 1 patient each.
All patients with AEs leading to dose modification were reported at the higher AP doses (95 mg, 110 mg, and 120 mg) in 8 patients (20%). AEs leading to dose modification were thrombocytopenia (4 patients; 10%), neutropenia (2 patients, 5%), and leukopenia, decreased platelet count, abdominal pain, and vomiting (1 patient each; 2%).
No deaths due to study drug were reported. Two patients died of progressive disease within 30 days of the last infusion. Patients received a median of 10 (range, 1–146) doses, leading to a median duration of exposure of 32 (range, 0–962) days.
Pharmacokinetics
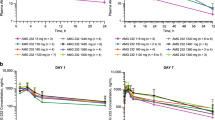
Mean plasma prodrug concentration-time profiles following administration of RO6839921 AP are presented in Fig. 1. The pharmacokinetic conversion of the prodrug RO6839921 to AP in patients started during the 1-h IV infusion and was rapid, yielding peak AP at ≈3.5 h postinfusion. There was a lack of accumulation (mean Day 5/Day 1 ratio ≈1) of prodrug RO6839921. These characteristics have been confirmed at high doses (Tables 5 and 6). The dose-exposure relationship of the AP in maximum observed plasma concentration and area under the concentration-time curve from time 0 to 24 h on Day 5 is approximately linear and dose proportional (Fig. 2).

Mean (SD) plasma concentrations of AP (idasanutlin, top) and prodrug (RO6839921, bottom) concentration-time profiles following RO6839921 intravenous administration of prodrug on Day 5. AP active principle

AP dose-exposure relationship on Day 5 of Cmax (a) and AUC0–24 (b). AP active principle, AUC0–24 area under the concentration-time curve from 0 to 24 h, Cmax maximum observed plasma concentration
Pharmacodynamics
Serum levels were assessed for pharmacodynamic effects. The pharmacodynamic association of change in MIC-1 levels—a secreted protein that is strongly induced by activated p53—with RO6839921 AP exposure was similar to that previously established for oral idasanutlin (Fig. 3) [5].

Pharmacodynamic association of MIC-1 levels with steady-state AP exposure. AP active principle, AUC0–24 area under curve at 24 h, FCBL fold-change from baseline, MIC-1 macrophage inhibitory cytokine 1
Next-generation sequencing was used to assess TP53 status from archival tumor tissue. In the 19 patients who had samples available for sequencing, 5 (26%) had ≥1 mutation. There was 1 patient who had >1 mutation detected. Because no objective responses were reported, an analysis of correlation with response was not performed.
Efficacy
None of the patients achieved objective responses (CR or PR). Of 41 patients, 17 (41%) had stable disease and 21 (51%) had progressive disease as best response (Table 7). The clinical benefit rate was 41% (median, 4 cycles; range, 1–15 cycles). Three patients had no on-treatment response assessment results available. Thirty-three of the 41 patients had target lesion assessments beyond baseline (Fig. 4). No apparent relationship was observed between dose (or pharmacokinetic exposure) and best overall response. Patients with stable disease remained on study for a median of 143 (range, 22–995) days (Fig. 5).

Best overall percentage change in the sum of target lesions. AP active principle

Duration of response for patients who had stable disease. AP active principle, PD progressive disease, SD stable disease
Four of 5 patients with liposarcoma had stable disease as best response. One patient receiving 95 mg AP had sustained stable disease of 396 days. Another patient receiving 95 mg AP had a dose reduction to 80 mg at cycle 16 for better tolerability and had sustained stable disease of ≥946 days (34 cycles) when transferred to compassionate use oral idasanutlin. Of these 4 patients with liposarcoma, 2 were confirmed to have wild-type p53. Unfortunately, p53 results were not available for the 2 patients with sustained stable disease and the 1 patient with liposarcoma who lacked disease control.
Discussion
RO6839921 is a potent pegylated prodrug and selective new-generation antagonist of the p53-MDM2 interaction for IV administration. Once RO6839921 is metabolized to idasanutlin, it selectively inhibits p53 binding to MDM2 with high affinity, resulting in stabilization and accumulation of p53 and activation of the pathway [6]. Clinical studies with the oral idasanutlin have indicated that p53 may be activated by this novel therapeutic strategy, which releases p53 from MDM2 inhibition. In particular, idasanutlin and its predecessor molecule RG7112 have shown sustained stable disease, especially in patients with sarcoma and AML [5, 9, 10]. RO6839921 was developed to decrease variability in exposure seen with the oral compounds and allow expansion into indications, in which patients cannot swallow (such as pediatrics) or absorb the oral compound. It was designed with the intention to combine the potency of idasanutlin but to potentially improve on pharmacokinetic properties. In this phase 1 study in patients with advanced solid tumors, RO6839921 demonstrated modestly improved pharmacokinetic properties with a safety, pharmacokinetic, and pharmacodynamic profile similar to that of oral idasanutlin.
RO6839921 was generally well tolerated. In patients with solid tumors, the MTD was 110 mg/d for 5 days in a 28-day cycle. Neutropenia, thrombocytopenia, and stridor were DLTs. Predominant study drug–related AEs (≥20%) included thrombocytopenia, nausea, vomiting, fatigue, neutropenia, diarrhea, and decreased appetite.
This study demonstrated that a soluble form of an MDM2 antagonist in the form of the prodrug to idasanutlin could be administered IV to patients with solid tumors. The rapid cleavage of the prodrug to the AP (idasanutlin) accounts for the similar AE profile to that of oral idasanutlin in patients with solid tumors. Analysis of the pharmacokinetics data showed that this prodrug provides an improved interpatient variability compared with oral idasanutlin (coefficient of variation 27% vs 46%) [7]. In this study, variability ranged from 14.7% to 32.7% compared with 50% variability often observed with oral idasanutlin [7].
Evidence of p53 activation, as measured by the pharmacodynamics marker MIC-1, was observed [5, 11]. The MIC-1–fold change from baseline was associated with exposure and was observed at doses below the MTD, similar to that previously established for oral idasanutlin [5].
No objective responses of CR or PR were reported. Seventeen of 41 (41.5%) patients achieved stable disease as best response. Two patients (both with liposarcoma) had stable disease ongoing for >12 months at data cutoff. Of 20 patients with neoadjuvant liposarcoma treated with the predecessor MDM2 antagonist molecule RG7112, 1 patient had a confirmed PR and the predominant response was stable disease in 14 patients [9]. In this phase 1 study, 4 of 5 patients with liposarcoma had stable disease as a best response; 2 of the patients had sustained stable disease of 396 and > 946 days, respectively.
Overall, this study showed that a soluble form of an MDM2 antagonist (RO6839921) as the prodrug to oral idasanutlin could be administered IV to patients with solid tumors. The rapid pharmacokinetics of the prodrug to oral idasanutlin accounts for the similar AE profile. In patients with solid tumors, a moderate improvement in variability was observed for the prodrug compared with oral idasanutlin. However, these data did not demonstrate sufficient safety improvement or differentiation—compared with oral idasanutlin—to support further development of the IV prodrug RO6839921. Pediatric oral formulations of idasanutlin are in development for clinical testing. A phase 3 study of oral idasanutlin with cytarabine vs cytarabine plus placebo (MIRROS) is currently recruiting patients with relapsed or refractory AML (NCT02545283) [12].
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Qualified researchers may request access to individual patient level data through the ClinicalStudyDataRequest platform (www.clinicalstudydatarequest.com). Further details on Roche’s criteria for eligible studies are available here (https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
References
Vassilev LT (2007) MDM2 inhibitors for cancer therapy. Trends Mol Med 13(1):23–31
Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, Chu XJ, Bartkovitz D, Podlaski F, Janson C, Tovar C, Filipovic ZM, Higgins B, Glenn K, Packman K, Vassilev LT, Graves B (2013) Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem 56(14):5979–5983
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303(5659):844–848
Yee K, Martinelli G, Vey N, Dickinson MJ, Seiter K, Assouline S, Drummond M, Yoon S-S, Kasner M, Lee J-H, Kelly KR, Blotner S, Higgins B, Middleton S, Nichols G, Chen G, Zhong H, Pierceall WE, Zhi J, Chen L-C (2014) Phase 1/1b study of RG7388, a potent MDM2 antagonist, in acute myelogenous leukemia (AML) patients (Pts) [abstract]. Blood 124(21):116
Siu LL, Italiano AI, Miller WH, Blay J-Y, Gietema JA, Bang Y-J, Mileshkin LR, Hirte HW, Reckner M, Higgins B, Jukofsky L, Blotner S, Zhi J, Middleton S, Nichols GL, Chen LC (2014) Phase 1 dose escalation, food effect, and biomarker study of RG7388, a more potent second-generation MDM2 antagonist, in patients (pts) with solid tumors [abstract]. J Clin Oncol 32(15 suppl):abstract 2535
Higgins B, Tovar C, Glen K, Railkar A, Filipovic Z, Qureshi F, Vu B, Ehrlich G, Fishlock D, Chen L-C, Middleton S, Nichols G, Packman K, Vassilev L (2014) Preclinical activity of MDM2 antagonist RO6839921, a pegylated prodrug for intravenous administration [abstract]. Mol Cancer Ther 14(12 suppl 2):abstract 156
Razak A, Gore L, Britten CD, Miller WH, Uy GL, Nichols G, Middleton S, Blotner S, Zhi J, Jukofsky L, Pierceall W, Higgins B, Chen LC (2016) A phase I study of the MDM2 antagonist RO6839921, a pegylated prodrug of idasanutlin, for intravenous (IV) administration in patients with advanced solid tumors. Eur J Cancer 69:S21–S22
O'Quigley J, Pepe M, Fisher L (1990) Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics 46(1):33–48
Yang H, Filipovic Z, Brown D, Breit SN, Vassilev LT (2003) Macrophage inhibitory cytokine-1: a novel biomarker for p53 pathway activation. Mol Cancer Ther 2(10):1023–1029
Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, Geho D, Middleton SA, Vassilev LT, Nichols GL, Bui BN (2012) Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol 13(11):1133–1140
Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, Bowen D, Martinelli G, Drummond MW, Vyas P, Kirschbaum M, Iyer SP, Ruvolo V, González GM, Huang X, Chen G, Graves B, Blotner S, Bridge P, Jukofsky L, Middleton S, Reckner M, Rueger R, Zhi J, Nichols G, Kojima K (2016) Results of the phase i trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res 22(4):868–876
Montesinos P, Esteve J, Konopleva M, Martinelli G, Ottmann O, Rodriguez-Veiga R, Röllig C, Wei A, Vey N, Chiu I, Monnet A, Ott MG, Fenaux P (2019) MIRROS: an ongoing randomized phase 3 trial of idasanutlin + ARA-C in patients with relapsed or refractory acute myeloid leukemia [abstract]. J Clin Oncol 37(15 suppl):abstract TPS7063
Acknowledgements
We thank the participants of the study. Special thanks to Steven Middleton, Gwen Nichols, William Pierceall, Jianguo Zhi, Lori Jukofsky, Patricia Delora, David Stephen Ward, and Ene Kelly and in the memory of Zoran Filipovic.
Funding
This study was funded by F. Hoffmann-La Roche Ltd. The sponsor was responsible for the clinical operations oversight, data management, medical monitoring, drug supply, statistical analysis, drug safety process, medical writing, and journal article processing charges. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Author information
Authors and Affiliations
Contributions
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical Writing, Editorial, and Other Assistance.
Support for third-party writing assistance for this manuscript, furnished by writer Christine Gould, PhD, CMPP, of Health Interactions, Inc., was provided by F. Hoffmann-La Roche Ltd. and Genentech, Inc.
Corresponding author
Ethics declarations
Conflict of interest
All authors report support of the parent study and funding of editorial support from F. Hoffmann-La Roche. SB, A-MY, BH, and L-CC are employees of Roche. ARR has received research funding from Genentech/Roche. WHM has served as a consultant for Amgen, BMS, GSK, Merck, Novartis and Roche; has received research funding to his institution from Amgen, AstraZeneca, BMS, MedImmune, Merck, MethylGene, Novartis, Pfizer and Roche; and has received honoraria from BMS, GSK, Merck, Novartis and Roche. LG has served as a consultant for Amgen, Genentech/Roche and Novartis; has stock with Amgen, Anchiano, Blueprint Medicines, Celgene, Clovis, Mirati and Sanofi; and has a spouse employed with Anchiano.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abdul Razak, A.R., Miller, W.H., Uy, G.L. et al. A phase 1 study of the MDM2 antagonist RO6839921, a pegylated prodrug of idasanutlin, in patients with advanced solid tumors. Invest New Drugs 38, 1156–1165 (2020). https://doi.org/10.1007/s10637-019-00869-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-019-00869-2