
Abstract
Dysregulation of NLRP3 inflammasome results in uncontrolled inflammation, which participates in various chronic diseases. TWIK2 potassium channel mediates potassium efflux that has been reported to be an essential upstream mechanism for ATP-induced NLRP3 inflammasome activation. Thus, TWIK2 potassium channel could be a potential drug target for NLRP3-related inflammatory diseases. In the present study we investigated the effects of known K2P channel modulators on TWIK2 channel expressed in a heterologous system. In order to increase plasma membrane expression and thus TWIK2 currents, a mutant channel with three mutations (TWIK2I289A/L290A/Y308A) in the C-terminus was expressed in COS-7 cells. TWIK2 currents were assessed using whole-cell voltage-clamp recording. Among 6 known K2P channel modulators tested (DCPIB, quinine, fluoxetine, ML365, ML335, and TKDC), ML365 was the most potent TWIK2 channel blocker with an IC50 value of 4.07 ± 1.5 μM. Furthermore, ML365 selectively inhibited TWIK2 without affecting TWIK1 or THIK1 channels. We showed that ML365 (1, 5 μM) concentration-dependently inhibited ATP-induced NLRP3 inflammasome activation in LPS-primed murine BMDMs, whereas it did not affect nigericin-induced NLRP3, or non-canonical, AIM2 and NLRC4 inflammasomes activation. Knockdown of TWIK2 significantly impaired the inhibitory effect of ML365 on ATP-induced NLRP3 inflammasome activation. Moreover, we demonstrated that pre-administration of ML365 (1, 10, 25 mg/kg, ip) dose-dependently ameliorated LPS-induced endotoxic shock in mice. In a preliminary pharmacokinetic study conducted in rats, ML365 showed good absolute oral bioavailability with F value of 22.49%. In conclusion, ML365 provides a structural reference for future design of selective TWIK2 channel inhibitors in treating related inflammatory diseases.
Similar content being viewed by others

Introduction
Inflammasomes are cytosolic multiprotein complexes and core components of innate immunity [1, 2]. The NLR family pyrin domain containing 3 (NLRP3) inflammasome is composed by the sensor protein NLRP3, the adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), and the effector protein caspase-1 [3,4,5]. Assembly of the NLRP3 inflammasome triggers caspase-1 activation to result in the secretion of IL-1β and IL-18, as well as pyroptosis [6, 7]. NLRP3 inflammasome responds to a surprisingly diverse set of stimuli, including extracellular ATP, pore-forming toxins (nigericin and maitotoxin, etc.), crystalline, particulate matter (such as uric acid crystals, silica, asbestos, and alum), and misfolded proteins related to neurodegenerative diseases [1, 3]. The dysregulation of NLRP3 inflammasome has been reported to participate in the development of diverse autoimmune, metabolic and neurodegenerative diseases, including gout, atherosclerosis, Alzheimer’s disease, type II diabetes, and others. Thus, NLRP3 inflammasome has been suggested to be a potential drug target for the treatment of related inflammatory diseases [8,9,10,11].
Potassium (K+) efflux has been reported to be an essential upstream mechanism for NLRP3 inflammasome activation with most NLRP3 agonists [12,13,14]. Various NLRP3 agonists induce potassium efflux by mechanistically distinct pathways [14]. Extracellular ATP, which increases massively to several tens or hundreds μmol/L within stressed or dying cells, activates NLRP3 inflammasome by ligating purinergic P2X7 receptor [15, 16]. Recently, TWIK2 channel, which belongs to TWIK subfamily of two-pore domain potassium (K2P) channels, has been reported to cooperate with P2X7 receptor to mediate potassium efflux and be fundamental for ATP-induced NLRP3 inflammasome. Deletion of TWIK2 prevented ATP-induced NLRP3 activation in vitro and sepsis-induced lung inflammation in vivo [17, 18].
The above study pointed out that TWIK2 channel could be a potential drug target for NLRP3 related inflammatory diseases. However, TWIK2 channel has long been considered to mainly locate on lysosome or endosome with little or no plasma membrane expression, and there is a lack of pharmacological study regarding TWIK2 channel till now [19, 20]. Thus, it is still puzzling whether it is feasible to target TWIK2 channel to identify channel inhibitors to further modulate NLRP3 inflammasome.
In the present study, we examined known K2P channels modulators for their effects on TWIK2 using a mutant channel with three mutations (TWIK2I289A/L290A/Y308A) in the C-terminus to increase plasma membrane expression and thus currents in heterologous system [21]. Among K2P modulators tested, including quinine, fluoxetine, DCPIB, ML365, ML335, and TKDC, only ML365 showed potent inhibitory effect on TWIK2 with an IC50 of 4.07 μM. The data showed that ML365 specifically inhibited ATP-induced NLRP3 inflammasome in macrophages, while silencing TWIK2 channel dramatically impaired the inhibition of ML365 on NLRP3 inflammasome. Furthermore, pretreatment with ML365 significantly reduced IL-1β level in the serum and inflammatory lung injury induced by LPS endotoxic shock in vivo. Collectively, our data demonstrate that ML365 provides a potential lead compound for future TWIK2 channel inhibitors design, which can be an important drug target for future ATP/NLRP3 inflammasome-related inflammatory diseases.
Materials and methods
Animals
C57BL/6 mice (7–8 weeks old weighing 20 ± 2 g) and SD rats (200 ± 20 g) were purchased from the Laboratory Animal Centre of Southern Medical University (Guangzhou, China). Animals were housed in a standard feeding environment (50%–70% humidity, 23–25 °C, 12–12 h light/dark cycle) and had free access to water and standard chow. All the procedures conducted on animals were complied with the ARRIVE guidelines and approved by the Ethics Committee of Southern Medical University.
Reagents and antibodies
LPS (Cat# L2630), ATP (Cat# A2383), and PMA (Cat# P8139) were purchased from Sigma (Burlington, MI, USA). Nigericin (Cat# tlrl-nig), poly(dA:dT) (Cat# tlrl-patn), LPS-EB Ultrapure (Cat# tlrl-3pelps), Pam3CSK4(Cat# tlrl-pms) and flagellin (Cat# tlrl-pafla) were purchased from Invivogen (Vista Sorrento Pkwy San Diego, CA, USA). ML365 (Cat#HY-12345), MCC950 (Cat# HY-12815), and M-CSF (Cat# HY-P7085) were purchased from MCE (Monmouth Junction, NJ, USA). Quinine (Cat# Q105031) was purchased from Aladdin (Shanghai, China). Fluoxetine (Cat# T0450) was purchased from Targetmol (Shanghai, China). DCPIB (Cat# B6780) was purchased from Apexbio (Houston, TX, USA) and TKDC was kindly provided by Prof Huai-yu Yang (East China Normal University, Shanghai, China). The primary antibodies used in this study were as followed: anti-IL-1β (Cat# AF401-NA; R&D System, Minneapolis, MN, USA; 1:2500), anti-CASP1 (Cat# A28881708; Adipogen, San Diego, CA, USA; 1:1000) and anti-β-actin (Cat# abs830031; Absin, Shanghai, China; 1:2500). The secondary antibodies were as followed: goat anti-mouse-HRP (Cat# FDM007; Fudebio-tech, Hangzhou, China; 1:2000) and rabbit anti-goat-HRP (Cat# KGAA38; Keygen, Nanjing, China; 1:2000).
Cell culture
COS-7 cells (ATCC: CRL-1651), RAW264.7 cells (ATCC: SC-6003), and BV-2 cells (ATCC: CRL-2468) were cultured in DMEM (Gibco, Grand Island, NY, USA) with 10% FBS (Gibco) and 1% penicillin and streptomycin. THP-1 cells (ATCC: TIB-202) were cultured in RPMI-1640 medium (Gibco) with 10% FBS and 1% penicillin and streptomycin. Primary murine BMDMs were isolated from bone marrow of 7–8 weeks old C57BL/6 mice and differentiated in DMEM with 10% FBS and recombinant M-CSF (20 ng/mL) and used between 7 and 10 days after isolation. All cells were cultured at 37 °C and 5% CO2.
Plasmids and transfection
Plasmids expressing wild type and mutant mouse TWIK1 (NM_008430.2) and TWIK2 (NM_001033525.3) channels in pIRES-EGFP vector were kindly provided from Prof. Florian Lesage (Institute of Molecular and Cellular Pharmacology, Paris, France). Plasmids expressing mouse THIK1 (NM_001164426.1) were subcloned into pcDNA3.1 vector and further confirmed by sequencing the whole open reading frame.
To record currents of TWIK1 and TWIK2 channels, COS-7 cells were plated in 12-well plates. At 60%–70% confluence, 1500 ng plasmids expressing TWIK1 or TWIK2 were transfected into COS-7 cells using 1.8 μL Lipofectamine 3000 (Cat# L3000015; Invitrogen) according to the manufacturer’s instructions. For THIK1 channel, plasmids expressing EGFP as a marker were cotransfected with cDNA expressing THIK1 into the COS-7 cells with a ratio of 1:10 using 1.8 μL Lipofectamine 3000. Then, 24 h after transfection, the cells were split by trypsin-EDTA and replated onto coverslips coated with 0.1 mg/mL poly-L-lysine (Sigma) for subsequent electrophysiology experiments.
Electrophysiological recording
Currents of TWIK or THIK1 channels were recorded in the whole-cell configuration as described earlier [22]. The extracellular solution was comprised of 145 mM NaCl, 2.5 mM KCl, 1 mM CaCl2, 3 mM MgCl2, and 10 mM HEPES (pH adjusted to 7.4 with NaOH). The intracellular solution was comprised of 140 mM KCl, 1 mM MgCl2, 10 mM HEPES, and 5 mM EGTA (pH adjusted to 7.4 with KOH). All electrophysiological experiments were performed at room temperature (25 ± 2 °C) using a MultiClamp 700B patch-clamp amplifier/Digidata 1550B digitizer and the pClamp 10 software (Molecular Devices, Sunnyvale, CA, USA). All data were digitized at 20 kHz and filtered at 2 kHz. Series resistance was compensated to 60%–80%.
Inflammasomes activation
Inflammasome activation and analysis were performed as described earlier [23]. For NLRP3 inflammasome activation, 5 × 105 BMDM cells were plated in six-well plates and the medium was changed to pre-warmed opti-MEM, then the cells were primed with LPS (1 μg/mL) overnight. Next morning, ML365 was added into the culture for another 2 h followed by stimulating with ATP (5 mM) or nigericin (10 μM) for 1 h. Cell culture supernatants were further analyzed by ELISA. Cell extracts and precipitated supernatants were analyzed by immunoblotting.
For other inflammasomes activation, 2.5 × 105 BMDMs were plated in 12-well plates and the medium was changed to opti-MEM. BMDMs were primed with LPS (1 μg/mL) for 12 h and the cells were pretreated with ML365 for 2 h. Then BMDMs were transfected with poly(dA:dT) (2.5 μg) using 2.5 μL Lipofectamine 3000 for 4 h to activate AIM2 inflammasome. To activate non-canonical NLRP3 inflammasome and NLRC4 inflammasome, the cells were primed with Pam3CSK4 (2 μg/mL) for 6 h. Subsequently, BMDMs were pretreated with ML365 for 2 h and then stimulated for 16 h with ultrapure LPS (2.5 μg/mL) with 7.5 μL DOTPA (Cat# 38254100; Roche, Basel, Switzerland). For bacterial protein flagellin transfection, ultrapure flagellin from P. aeruginosa (1 μg/mL) was mixed with 40 μL DOTAP for each reaction. The reaction mixture was incubated for 45–60 min then added to BMDMs for 16 h.
To differentiate THP-1 cells into macrophages, 5 × 105 THP-1 cells were cultured with PMA (100 ng/mL) for 12 h in six-well plates, then cells were washed in PBS and cultured for 36 h in RPMI-1640 medium. After that, LPS (1 μg/mL) was added into the culture for another 12 h, and then, the cells were pretreated with ML365 for 2 h and then stimulated with ATP (5 mM) for 6 h or nigericin (10 μM) for 1 h to activate NLRP3 inflammasome.
Immunoblotting
All immunoblotting procedures and analysis were complied with APS guidelines [24]. Cell lysates were homogenized in ice-cold lysis RIPA buffer (Cat# KGP702; KeyGen, Nanjing, China) containing 0.1% protease inhibitors (Cat# KGP602; KeyGen) and 1% phosphatase inhibitors (Cat# KGP602; KeyGen) and centrifuged at 15,000 × g for 15 min at 4 °C. Protein concentration was quantified using a BCA protein assay kit (Cat# 23227; Thermo Fisher, Waltham, MA, USA). Equal amounts of protein were separated by electrophoresis in SDS-PAGEs and then transferred onto PVDF membranes (Cat# 3010040001; Roche). The membranes were blocked with 5% non-fat milk (w/v) in 0.05% Tris-buffered saline with Tween-20 for 1 h at room temperature and then incubated with respective primary antibodies overnight at 4 °C. Afterwards, the membranes were incubated with respective secondary antibodies for 1 h at room temperature. Then, the membranes were visualized by ECL Luminous solution (Cat# G3308; Gbcbio, Guangzhou, China).
The supernatant proteins were extracted as described earlier [25]. After inflammasomes activation, supernatants were collected into 1.5 mL tubes, centrifuged for 5 min at maximum speed, and then transferred into fresh 1.5 mL tubes. Subsequently, chloroform and methanol were added into the tube with a ratio of 3:10:10. After being vortexed, the mixture was spun down in a microcentrifuge at maximum speed for 10 min at room temperature. Then, 800 μL of methanol was added and spun again after discarding the aqueous phase (at the top). The supernatants were removed and the pellets were dried for 10 min at 37 °C. After that, 25–50 μL of SDS sample buffer was added, mixed, and incubated for 5 min at 95 °C for immunoblotting.
Quantitative real-time PCR
Total RNA was extracted using TRIZOL (TaKaRa, Kyoto, Japan). RNA concentrations were measured using NanoDrop 2000c (Thermo Fisher) and normalized among the experimental sample set. cDNA was reverse transcribed using the PrimeScriptTM RT reagent Kit with gDNA Eraser (TaKaRa) according to the manufacturer’s instructions. Quantitative PCR was performed with SYBR® Green PCR Master Mix Kit (Thermo Fisher) on the LightCycler 480 System (Roche). The primers used in quantitative PCR were listed in Table 1. The levels of target genes were normalized to the levels of β-actin mRNA and relative expression was determined according to the ΔΔCt method [26].
Small-interfering RNA transfection
BMDM cells were plated in six-well plates (2.5 × 105 cells per well) and then transfected with 50 nM siRNA using 7.5 μL Lipofectamine RNAiMAX (Thermo Fisher) for 48 h according to the manufacturer’s instructions. The siRNA sequence for mouse TWIK2 (5’-TCACAGAACTCATCTTGCT-3’) and scrambled siRNA were chemically synthesized by RiboBio Co. (Guangzhou, China).
LPS-induced endotoxic shock
C57BL/6 mice (aged 8–12 weeks) were injected intraperitoneally (i.p.) with ML365 (1, 10, 25 mg/kg) or MCC950 (50 mg/kg) at 1 h prior to stimulation with 10 mg/kg LPS (Escherichia coli O111:B4; Sigma). After 7 h, mice were sacrificed and serum levels of IL-1β and TNF-α were measured by ELISA. Lung tissues were harvested for H&E staining.
Pharmacokinetic study of ML365
SD rats were dosed i.v. (7 mg/kg) or p.o. (17.5 mg/kg) and blood samples were taken via obital vein at the following time points post dosing: 0.03 (i.v. only), 0.083, 0.25, 0.5, 0.75 (only p.o.), 1, 1.5 (only p.o.), 2, 4, 6, 8, 12, 24 and 36 h. Plasma was separated from whole blood by centrifugation and stored at −20 °C until analysis. Compound concentrations were determined using an Agilent 1260 + 6460 LC/MS/MS system. The Cmax, Tmax, t1/2, and AUC were evaluated using WinNonlin 4.0.
ELISA analysis
The amounts of IL-1β and TNF-α in cell culture supernatants and serum were measured using ELISA kits (Cat# 88-7013-88 for mouse IL-1β and Cat# 88-7324-88 for mouse TNF-α; Invitrogen) according to the manufacturer’s instructions.
Data analysis and statistics
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology [27]. Electrophysiological data were processed using Clampfit 10.6 (Molecular Devices, USA). Dose-response curves were fitted to the Hill equation, f = 1/[1 + (C/IC50)n], where f = I/Icontrol, IC50 corresponds to drug concentration producing half of the maximum response, [C] corresponds to the drug concentration, and n is the Hill coefficient. Each experiment in this study was at least performed five times. Statistical analyses were performed by a t-test using GraphPad Prism 6 and data were reported as the means ± SEM (standard error of the mean). A P value < 0.05 was considered as statistically significant.
Results
Identification of ML365 as a potent TWIK2 channel inhibitor
Wild type TWIK2 channel has been reported to produce no or little current in heterologous systems [21, 28]. A mutant TWIK2 (muTWIK2) with three mutations introduced in the C-terminus (I289A/L290A/Y308A) has been reported to increase the plasma membrane expression and thus produce detectable currents in transfected cells [21]. Similar to earlier studies, introducing the three mutations into wtTWIK2 led to a ~14 fold current increase (24.3 ± 0.74 nA for muTWIK2 vs. 1.72 ± 0.36 nA for wtTWIK2) in transfected COS-7 cells (Supplementary Figure S1), which makes further TWIK2 modulators screening feasible.
We then explored several known K2P modulators (chemical structures in Fig. 1a) for their effects on muTWIK2 channel. Among all compounds tested, only ML365 showed potent inhibition on muTWIK2, with an I/I0 of 0.35 ± 0.02 with 10 μM (Fig. 1b-d). To further examine the electrophysiological characteristics of ML365 inhibition on muTWIK2, we tested the effects of ML365 on TWIK2 currents under different voltages. TWIK2 channel was slightly inactivated at positive potentials and the inhibitory effects of ML365 on TWIK2 had no voltage dependence, namely, ML365 inhibited TWIK2 channel to similar extent at different voltages (Fig. 2a, b). Furthermore, the concentration-response curve showed that the IC50 of ML365 on muTWIK2 was 4.07 ± 1.5 μM (Fig. 2c).

a Chemical structures of six K2P channel modulators used in this study. b Example traces of muTWIK2 currents in the absence or presence of 10 μM ML365. To record K2P currents, a “step-ramp” voltage protocol was used as described earlier. For the “step” component, cells were hyperpolarized from a holding potential of −60 mV to −80 mV for 100 ms and then stepped to −40 mV for 500 ms. For the “ramp”, cells were then stepped to −100 mV for 100 ms, followed by a 500 ms voltage ramp to +20 mV, and then stepped back to −80 mV for 100 ms prior to hold at −60 mV. The protocol was applied every 15 s until reaching stable modulation by ML365. Left: representative time course of currents with the application of ML365 as indicated. Currents were measured at −40 mV in the step component. I/I0 means currents in the step component before and after the application of ML365. Right: exemplar current traces in the absence (black) or presence (red) of ML365, respectively. c Summary of effects of K2P modulators on muTWIK2 channel. The concentrations for each compound were as followed: DCPIB:10 μM; Quinine: 100 μM; Fluoxetine: 10 μM; ML365: 10 μM; ML335: 10 μM; TKDC: 10 μM. The concentrations were chosen depending on their reported IC50/EC50 on related subtypes of K2P channels. The number of cells recorded for each experiment is indicated in the parentheses at the top of the graphs. Data are represented as mean ± SEM.

a Representative current traces of muTWIK2 in the absence or presence of 10 μM ML365. b Summary of the inhibitory effect of ML365 (10 μM) on muTWIK2 at voltages ranging from −60 to +80 mV. c Dose–response curve of the inhibition of muTWIK2 by ML365.
ML365 selectively inhibits TWIK2, but not TWIK1 or THIK1 channels
TWIK2 belongs to TWIK subfamily of K2P channels, with two members, TWIK1 and TWIK2 [29]. We next examined the effect of these compounds on TWIK1 channel. Similar to TWIK2, wild-type TWIK1 channel gave no current in transfected cells. Similarly, introducing a mutant channel (muTWIK1) with three mutations (I293A/I294A/K274E) significantly increased TWIK1 currents (20.4 ± 1.06 nA for muTWIK1 vs. 6.4 ± 1.02 nA for wtTWIK1, respectively) (Supplementary Fig. S2). ML365 (10 μM) showed no inhibitory effect on muTWIK1 channel (Fig. 3), even at higher dosage (20 μM) (Supplementary Fig. S3a, c). Besides, quinine and fluoxetine demonstrated modest inhibition on muTWIK1 (Fig. 3).

a Example traces of muTWIK1 in the absence or presence of 10 μM ML365. Left: Representative time course of currents with the application of ML365 as indicated. Currents were measured at −40 mV in the step component. Right: exemplar current traces in the absence (black) or presence (red) of ML365, respectively. The protocol to record muTWIK1 was similar to that described in Fig. 1 with slight change shown at the right bottom. b Summary of the modulatory effect of tested K2P modulators on muTWIK1 channel. The concentrations applied for each compound were the same as in Fig. 1.
Recently, another member of K2P channels, THIK1, has been reported to play an essential role in microglia (the macrophage resides in the central nervous system) [30, 31]. THIK1 is responsible for the maintenance of the microglial resting membrane potential and thus is essential for microglial ramification, surveillance, and IL-1β release. Thus, we examined the expression level of THIK (THIK1 and THIK2) and TWIK (TWIK1 and TWIK2) subfamilies channels by quantitative real-time PCR in different types of macrophages, including mouse BMDMs, RAW264.7, and microglia BV-2 cell lines. The results showed mRNA levels of TWIK2 and THIK1 were both significant high in all three macrophages tested, while little or no mRNA level was examined for TWIK1 or THIK2 (Fig. 4a). Therefore, we tested K2P modulators for their effects on THIK1 channel. As shown in Fig. 4, ML365, DCPIB, fluoxetine, ML335 and TKDC had no effect on THIK1, while quinine demonstrated a modest inhibitory effect. Besides, high concentration of ML365 (20 μM) also showed no obvious modulation on THIK1 (Supplementary Fig. S4). Collectively, the above results showed that quinine and fluoxetine were non-selective and weak blockers for TWIK1, TWIK2 and THIK1 channels among all K2P modulators examined. Most interestingly, the data demonstrated that ML365 was a potent inhibitor of TWIK2 channel with no obvious modulation on TWIK1 or THIK1 channel.

a RT-qPCR analysis of TWIK and THIK subfamilies in BMDMs, RAW264.7 and BV-2 cells. b Example traces of THIK1 in the absence or presence of 10 μM ML365. Left: Representative time course of currents with the application of ML365 as indicated. Currents were measured at −40 mV in the step component. Right: exemplar current traces in the absence (black) or presence (red) of ML365, respectively. The protocol to record THIK1 was inserted at the top of the panel with slight modification from that described in Fig. 1. c Summary of the modulatory effect of tested K2P modulators on muTWIK1 channel. The concentrations applied for each compound were the same as in Fig. 1.
ML365 suppresses ATP-induced NLRP3 activation
Given the critical role of TWIK2 channel in NLRP3 inflammation activation [17], we next examined the effect of ML365 on NLRP3 inflammasome. As shown in Fig. 5a–c, ML365 concentration-dependently inhibited ATP-induced NLRP3 inflammasome in BMDMs, demonstrated by reduced mature IL-1β and the generation of the p20 subunit of active caspase-1 level in the cell supernatants by Western blotting (Fig. 5a) and reduced IL-1β release examined using ELISA (Fig. 5b). In the same supernatants, the level of TNF-α, an inflammasome unrelated cytokine, was preserved with ML365 (Fig. 5c). Furthermore, ML365 had no effect on nigericin-induced NLRP3 inflammasome activation (Fig. 5d–f). The distinct effects of ML365 on ATP or nigericin-induced NLRP3 inflammasome were also confirmed in THP-1 differentiated macrophages (Supplementary Fig. S4).

Western blots of cell lysates or supernatants from BMDMs primed with LPS (1 μg/mL) for 12 h and subsequently challenged with 5 mM ATP (a) or 10 μM nigericin (d) for 1 h with the application of different concentrations of ML365 as indicated. b, c, e, f Production of IL-1β and TNF-α in the supernatants from BMDMs stimulated with ATP or nigericin in combination with ML365 measured using ELISA. g Production of IL-1β from BMDMs stimulated with Pam3CSK4 and ultrapure LPS with the application of ML365 (5 μM) as indicated. h Production of IL-1β from BMDMs stimulated with LPS and transfected poly(dA:dT) in the absence or presence of ML365 (5 μM). i Production of IL-1β from BMDMs primed with Pam3CSK4 and transfected ultrapure flagellin and in the absence or presence of with ML365 (5 μM). All results are representative of five independent experiments. Data are demonstrated as mean ± SEM. *P < 0.05 and ***P < 0.001 vs. control group (LPS + ATP).
We next investigated the effects of ML365 on other inflammasomes such as AIM2, NLRC4 or noncanonical inflammasomes. Given that NLRP3 inflammasome can also be activated by LPS through caspase-11 activation independently of TLR4 [32, 33], we evaluated the effect of ML365 on non-canonical inflammasome. The data showed that ML365 did not inhibit IL-1β release followed by non-canonical inflammasome activation (Fig. 5g). Besides, BMDM cells were transfected with poly(dA:dT) to activate the AIM2 inflammasome or flagellin to activate NLRC4 inflammasome. ML365 did not inhibit either AIM2 or NLRC4 inflammasome-induced IL-1β production (Fig. 5h, i). These results suggested that ML365 specifically impaired ATP-induced NLRP3 inflammasome, which was consistent with the critical role of TWIK2 channel for NLRP3 inflammasome.
TWIK2 deficiency impairs the inhibition of ATP-induced NLRP3 inflammasome by ML365
To examine whether the impairment of ML365 on NLRP3 inflammasome targeting TWIK2 channel, we knocked down TWIK2 channel using small interfering RNAs (siRNA) (Fig. 6a). Silencing TWIK2 significantly reduced IL-1β production in response to ATP-activated BMDMs (Fig. 6b, c). Furthermore, we found that the inhibition by ML365 on ATP-induced NLRP3 inflammasome was significantly weakened by TWIK2 knockdown (Fig. 6b, c). This result further strengthened that ML365 blocked ATP-induced NLRP3 inflammasome by targeting TWIK2 channel.

a RT-PCR demonstrated the expression was significantly decreased by TWIK2 siRNA. b Western blots of cell lysates or supernatants from BMDMs transfected with TWIK2 siRNA or scrambled siRNA further activated by ATP in combination with ML365 (5 μM) as indicated. c ELISA analysis of the production of IL-1β in the supernatants from BMDMs stimulated with ATP in combination of ML365 with transfected TWIK2 siRNA or scrambled siRNA. All results are representative of five independent experiments. Data are demonstrated as mean ± SEM. ***P < 0.001 vs. control group (LPS + ATP).
ML365 ameliorates LPS-induced endotoxic shock in vivo
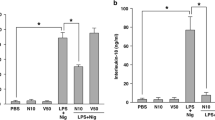
Dysregulation of NLRP3 inflammasome and overload of IL-1β have been shown to play critical roles in the pathogenesis of endotoxic shock [34]. To assess the role of ML365 in vivo, we investigated a LPS-induced endotoxic shock model in mice with ML365 and the reported NLRP3 inhibitor, MCC950 [35] (Fig. 7a). After 7 h injection of LPS, serum concentrations of IL-1β and TNF-α increased dramatically compared to the control group (Fig. 7b, c). Pretreatment with ML365 reduced serum concentrations of IL-1β but did not considerably decrease the amount of TNF-α (Fig. 7b, c). We also observed a marked decrease in endotoxemic inflammatory lung injury in ML365 pretreated mice as evaluated by the histopathological examination of lungs (Fig. 7d). Taken together, our finding indicated the therapeutic potential of ML365 in septic shock and other NLRP3 inflammasome-driven diseases.

a Schematic of LPS-induced endotoxic shock model. Serum levels of IL-1β (b) and TNF-α (c) from C57BL/6 mice pretreated with ML365 or MCC950 or vehicle control post i.p. LPS injection. d Histological sections of lungs were stained with hematoxylin and eosin (H&E) 7 h after LPS injection to assess immune cell infiltration. The representative H&E images are shown as followed: PBS; LPS; LPS/ML365 (25 mg/kg) and LPS/MCC950 (50 mg/kg). Scale bars: 10 μm. Data are demonstrated as mean ± SEM. n = 6, *P < 0.05 and **P < 0.01 vs. LPS group.
In vivo pharmacokinetics of ML365
To assess the therapeutic potential of ML365, we examined the pharmacokinetic profile of this compound in vivo. A single-dose pharmacokinetic profile of ML365 in SD rats was determined via intravenous (7 mg/kg) and oral (17.5 mg/kg) administration of ML365. The area under the curve was 1618.95 ng· h ·mL−1, with a Cmax of 2005.90 ng/mL and a half-life of 13.58 h. In addition, ML365 showed fair absolute oral bioavailability in rat with F values of 22.49% (Table 2). These data suggested that ML365 constitutes a reasonable starting point for further drug development to treat NLRP3-related diseases.
Discussion
Potassium channels have been extensively proven to be attracting drug targets for the treatment of cardiovascular and neurological diseases [36,37,38]. Emerging evidence has demonstrated that intact potassium transport is required for both innate and adaptive immunity [17, 31, 39, 40]. Two-pore domain potassium (K2P) channels comprise a major and structurally distinct subset of the mammalian K+ channel superfamily, including 15 K2P subtypes that form six subfamilies (TWIK, TASK, TRESK, TALK, THIK, and TRESK) [18, 41, 42]. They are regulated by a wide variety of physical, chemical, and biological stimuli and implicated in multiple physiological processes [43]. In recent years, critical roles of TWIK and THIK subfamily channels in innate immunity have been gradually revealed [17, 31]. TWIK2 channel was demonstrated to work together with P2X7 receptor to mediate potassium efflux, which is essential for ATP-induced NLRP3 inflammasome and the depletion of TWIK2 could prevent sepsis-induced lung inflammation [17]. However, there is no pharmacological study regarding TWIK channel given that this channel has long been considered to locate on lysosome or endosome with low or no plasma membrane expression [21, 44]. Thus, it is important to identify TWIK2 channel modulators and further examine their modulation on NLRP3 inflammasome.
We screened six known K2P channels modulators, including quinine (a non-selective K2P inhibitor), fluoxetine (an inhibitor of TREK1 channel), DCPIB (a non-selective K2P channels modulator), ML365 (an inhibitor of TASK1 and TASK3), ML335 (an activator of TREK1 and TREK2) and TKDC (an inhibitor of TREK1 and TREK2), for their modulation on TWIK2 channel [45,46,47,48,49]. Our study shows that ML365 was a potent TWIK2 channel inhibitor with an IC50 of 4.07 μM. Besides, ML365 had no effect on another TWIK subfamily member TWIK1 or immune-related channel THIK1. It would be valuable to study the molecular mechanism of ML365 inhibition on TWIK2 but not TWIK1/THIK1 in the future.
It has been well accepted that potassium efflux is both necessary and sufficient for NLRP3 inflammasome activation in most cases [12, 50,51,52]. AIM2 and NLRC4 inflammasomes are not affected by high concentrations of extracellular K+, indicating the specific role of potassium in modulating NLRP3 inflammasome [12, 52]. Structurally diverse DAMPs/PAMPs employ distinct mechanisms to cause potassium efflux to activate NLRP3 inflammasome. Firstly, existing research mainly focuses on the molecular mechanism of potassium efflux during ATP-induced NLRP3 inflammasome activation. P2X7 receptor and TWIK2 channel have been reported to co-operate in the process above [17]. Secondly, toxins such as nigericin directly promote potassium efflux by forming pores on the plasma membrane. Besides, the NLRP3 inflammasome can also be activated followed by the noncanonical inflammasome, which also depends on potassium efflux [53, 54]. GSDMD and pannexin-1 have been proposed to mediate potassium efflux in the above process to activate NLRP3 inflammasome [6, 55]. In our study, we found ML365 concentration-dependently impaired ATP-induced NLRP3 inflammasome, while the compound had no effect on nigericin-induced NLRP3 inflammasome, non-canonical, AIM2 or NLRC4 inflammasome activation. ML365 has been previously reported to block TASK1 and TASK3 channels, however, no or little TASK1 and TASK3 expression was observed in macrophage or microglia by RT-PCR or RNA sequencing [17, 56], which indicates the inhibition of NLRP3 inflammasome is not related to their inhibitory effects on TASK1 or TASK3. Furthermore, silencing TWIK2 channel dramatically impaired the inhibition by ML365 on ATP-induced NLRP3 inflammasome. Collectively, the results suggested ML365 specifically blocked ATP-induced NLRP3 inflammasome by inhibiting TWIK2 channel.
In conclusion, we firstly found a potent TWIK2 channel inhibitor—ML365. Further experiments showed that ML365 could specifically inhibit ATP-induced NLRP3 inflammation in a concentration-dependent manner. Lastly, our study demonstrated that ML365 ameliorated LPS-induced endotoxic shock in vivo. Therefore, ML365 not only can be a very powerful tool for further exploration of the physiological and pathological functions of TWIK2 channel but also provides a structural reference for future design of selective inhibitors of TWIK2 channel, which could be beneficial for the treatment of sepsis-induced lung inflammation and other NLRP3- related inflammatory diseases.
References
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2018;19:610–21.
de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol. 2014;6:a016287.
Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–89.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8.
Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med. 2018;10:eaah4066.
Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10:128.
Xu C, Lu Z, Luo Y, Liu Y, Cao Z, Shen S, et al. Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat Commun. 2018;9:4092.
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez GK. + efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–53.
Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–9.
Xu Z, Chen ZM, Wu X, Zhang L, Cao Y, Zhou P. Distinct molecular mechanisms underlying potassium efflux for NLRP3 inflammasome activation. Front Immunol. 2020;11:609441.
Pelegrin P, Surprenant A. The P2X(7) receptor-pannexin connection to dye uptake and IL-1beta release. Purinergic Signal. 2009;5:129–37.
Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. 2017;47:15–31.
Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, et al. The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity. 2018;49:56–65.e54.
Enyedi P, Czirják G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90:559–605.
Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SA. Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell. 2005;121:37–47.
Feliciangeli S, Tardy MP, Sandoz G, Chatelain FC, Warth R, Barhanin J, et al. Potassium channel silencing by constitutive endocytosis and intracellular sequestration. J Biol Chem. 2010;285:4798–805.
Bobak N, Feliciangeli S, Chen CC, Ben Soussia I, Bittner S, Pagnotta S, et al. Recombinant tandem of pore-domains in a weakly inward rectifying K+ channel 2 (TWIK2) forms active lysosomal channels. Sci Rep. 2017;7:649.
Lv J, Liang Y, Zhang S, Lan Q, Xu Z, Wu X, et al. DCPIB, an inhibitor of volume-regulated anion channels, distinctly modulates K2P channels. ACS Chem Neurosci. 2019;10:2786–93.
Gross O. Measuring the inflammasome. Methods Mol Biol. 2012;844:199–222.
Tie L, Xiao H, Wu DL, Yang Y, Wang P. A brief guide to good practices in pharmacological experiments: Western blotting. Acta Pharmacol Sin. 2021;42:1015–7.
Schneider KS, Thomas CJ, Groß O. Inflammasome activation and inhibition in primary murine bone marrow-derived cells, and assays for IL-1α, IL-1β, and caspase-1. Methods Mol Biol. 2013;1040:117–35.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8.
Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA, et al. Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol. 2018;175:987–93.
Patel AJ, Maingret F, Magnone V, Fosset M, Lazdunski M, Honoré E. TWIK-2, an inactivating 2P domain K+ channel. J Biol Chem. 2000;275:28722–30.
Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Ren Physiol. 2000;279:F793–801.
Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18:225–42.
Madry C, Kyrargyri V, Arancibia-Cárcamo IL, Jolivet R, Kohsaka S, Bryan RM, et al. Microglial ramification, surveillance, and interleukin-1β release are regulated by the two-pore domain K+ channel THIK-1. Neuron. 2018;97:299–312.e296.
Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA, Cytoplasmic LPS. activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–3.
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–9.
Danielski LG, Giustina AD, Bonfante S, Barichello T, Petronilho F. The NLRP3 inflammasome and its role in Sepsis development. Inflammation. 2020;43:24–31.
Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55.
Cao Y, Chen S, Liang Y, Wu T, Pang J, Liu S, et al. Inhibition of hyperpolarization-activated cyclic nucleotide-gated channels by β-blocker carvedilol. Br J Pharmacol. 2018;175:3963–75.
Hu HN, Zhou PZ, Chen F, Li M, Nan FJ, Gao ZB. Discovery of a retigabine derivative that inhibits KCNQ2 potassium channels. Acta Pharmacol Sin. 2013;34:1359–66.
Zhou PZ, Babcock J, Liu LQ, Li M, Gao ZB. Activation of human ether-a-go-go related gene (hERG) potassium channels by small molecules. Acta Pharmacol Sin. 2011;32:781–8.
Mackenzie AB, Chirakkal H, North RA. Kv1.3 potassium channels in human alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2003;285:L862–868.
Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol. 2015;33:291–353.
Feliciangeli S, Chatelain FC, Bichet D, Lesage F. The family of K2P channels: salient structural and functional properties. J Physiol. 2015;593:2587–603.
Loucif AJC, Saintot PP, Liu J, Antonio BM, Zellmer SG, Yoger K, et al. GI-530159, a novel, selective, mechanosensitive two-pore-domain potassium (K2P) channel opener, reduces rat dorsal root ganglion neuron excitability. Br J Pharmacol. 2018;175:2272–83.
Liao P, Qiu Y, Mo Y, Fu J, Song Z, Huang L, et al. Selective activation of TWIK-related acid-sensitive K+ 3 subunit-containing channels is analgesic in rodent models. Sci Transl Med. 2019;11:eaaw8434.
Nematian-Ardestani E, Abd-Wahab F, Chatelain FC, Sun H, Schewe M, Baukrowitz T, et al. Selectivity filter instability dominates the low intrinsic activity of the TWIK-1 K2P K+ channel. J Biol Chem. 2020;295:610–8.
Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, et al. TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J. 1996;15:1004–11.
Kennard LE, Chumbley JR, Ranatunga KM, Armstrong SJ, Veale EL, Mathie A. Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br J Pharmacol. 2005;144:821–9.
Flaherty DP, Simpson DS, Miller M, Maki BE, Zou B, Shi J, et al. Potent and selective inhibitors of the TASK-1 potassium channel through chemical optimization of a bis-amide scaffold. Bioorg Med Chem Lett. 2014;24:3968–73.
Luo Q, Chen L, Cheng X, Ma Y, Li X, Zhang B, et al. An allosteric ligand-binding site in the extracellular cap of K2P channels. Nat Commun. 2017;8:378.
Lolicato M, Arrigoni C, Mori T, Sekioka Y, Bryant C, Clark KA, et al. K2P2.1 (TREK-1)-activator complexes reveal a cryptic selectivity filter binding site. Nature. 2017;547:364–8.
Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J. 1995;14:1607–14.
Hafner-Bratkovič I, Pelegrín P. Ion homeostasis and ion channels in NLRP3 inflammasome activation and regulation. Curr Opin Immunol. 2018;52:8–17.
Gong T, Yang Y, Jin T, Jiang W, Zhou R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol. 2018;39:393–406.
Rivers-Auty J, Brough D. Potassium efflux fires the canon: potassium efflux as a common trigger for canonical and noncanonical NLRP3 pathways. Eur J Immunol. 2015;45:2758–61.
Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20:143–57.
Chen KW, Demarco B, Broz P. Pannexin-1 promotes NLRP3 activation during apoptosis but is dispensable for canonical or noncanonical inflammasome activation. Eur J Immunol. 2020;50:170–7.
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–47.
Acknowledgements
We would like to thank the following foundations: the National Natural Science Foundation of China (81973333), Young Scholars of Pearl River in Guangdong Province, and Natural Science Foundation of Guangdong Province (2019A1515010964; 2114050001716) to PZZ.
Author information
Authors and Affiliations
Contributions
PZZ, SWL, and KKLY designed the research; XYW, JYL, SQZ, XY, ZWX, and YXZ performed the experiments; JXP, KKLY, and BXZ provided reagent; XYW analyzed the data; XYW, SWL, and PZZ wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Wu, Xy., Lv, Jy., Zhang, Sq. et al. ML365 inhibits TWIK2 channel to block ATP-induced NLRP3 inflammasome. Acta Pharmacol Sin 43, 992–1000 (2022). https://doi.org/10.1038/s41401-021-00739-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00739-9
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
ML335 inhibits TWIK2 channel-mediated potassium efflux and attenuates mitochondrial damage in MSU crystal-induced inflammation
Journal of Translational Medicine (2024)
-
The role of inflammasomes in human diseases and their potential as therapeutic targets
Signal Transduction and Targeted Therapy (2024)
-
Pharmacological inhibition of Kir4.1 evokes rapid-onset antidepressant responses
Nature Chemical Biology (2024)
-
Extracellular histones promote TWIK2-dependent potassium efflux and associated NLRP3 activation in alveolar macrophages during sepsis-induced lung injury
Inflammation Research (2024)