
Abstract
Intracerebral hemorrhage (ICH) is a devastating disease, in which neuroinflammation substantially contributes to brain injury. Uncoupling protein 2 (UCP2) is a member of the mitochondrial anion carrier family, which uncouples oxidative phosphorylation from ATP synthesis by facilitating proton leak across the mitochondrial inner membrane. UCP2 has been reported to modulate inflammation. In this study we investigated whether and how UCP2 modulated neuroinflammation through microglia/macrophages following ICH in vitro and in vivo. We used an in vitro neuroinflammation model in murine BV2 microglia to mimic microglial activation following ICH. ICH in vivo model was established in mice through collagenase infusion into the left striatum. ICH mice were treated with anetholetrithione (ADT, 50 mg· kg−1 ·d−1, ip) or the classical protonophoric uncoupler FCCP (injected into hemorrhagic striatum). We showed that the expression and mitochondrial location of microglial UCP2 were not changed in both in vitro and in vivo ICH models. Knockdown of UCP2 exacerbated neuroinflammation in BV2 microglia and mouse ICH models, suggesting that endogenous UCP2 inhibited neuroinflammation and therefore played a protective role following ICH. ADT enhanced mitochondrial ROS production thus inducing mitochondrial uncoupling and activating UCP2 in microglia. ADT robustly suppressed neuroinflammation, attenuated brain edema and improved neurological deficits following ICH, and these effects were countered by striatal knockdown of UCP2. ADT enhanced AMP-activated protein kinase (AMPK) activation in the hemorrhagic brain, which was abrogated by striatal knockdown of UCP2. Moreover, striatal knockdown of AMPK abolished the suppression of neuroinflammation by ADT following ICH. On the other hand, FCCP-induced mitochondrial uncoupling was independent of UCP2 in microglia; and striatal knockdown of UCP2 did not abrogate the suppression of neuroinflammation by FCCP in ICH mice. In conclusion, the uncoupling activity is essential for suppression of neuroinflammation by UCP2. We prove for the first time the concept that activators of endogenous UCP2 such as anetholetrithione are a new class of uncouplers with translational significance.
Similar content being viewed by others


Introduction
Uncoupling protein 2 (UCP2), a member of the mitochondrial anion carrier family, uncouples oxidative phosphorylation from ATP synthesis by facilitating proton leak across the mitochondrial inner membrane [1, 2]. UCP2 is abundantly expressed in innate immune cells, including macrophages and microglia [3,4,5]. Unlike canonical UCP1, UCP2 is not involved in adaptive thermogenesis and its roles in pathophysiological settings remain enigmatic [6]. Indeed, although considered as an uncoupling protein, UCP2 has been shown to be an important metabolic switch and regulator of lipid synthesis. Emerging evidence suggests that UCP2 plays an important but paradoxical role in regulating inflammation [7, 8]. For instance, UCP2 exacerbates LPS-mediated inflammation by inducing fatty acid synthesis during sepsis [5]. Moreover, UCP2 is responsible for microglial neuroinflammation in mice fed with high-fat diet, and the effect is independent of the uncoupling activity of UCP2 [4]. On the other hand, UCP2 is also reported to inhibit inflammation. For instance, UCP2 promotes anti-inflammatory polarization of microglia, while down-regulation of UCP2 is required for inflammation induced by LPS [9].
UCP2 is an uncoupling protein. Surprisingly, no direct evidence currently exists to suggest how the uncoupling activity of UCP2 modulates inflammation. Notably, UCP2 is a weak mitochondrial uncoupling protein [10]. The uncoupling activity of UCP2 is remarkably elevated upon activation by endogenous factors, such as endogenously produced radical oxygen species (ROS) from mitochondria [11, 12]. No activators of UCP2 have been identified yet. This represents a major obstacle to investigate the role of the uncoupling activity of UCP2 in inflammation. In addition, mitochondrial uncoupling is considered a promising therapy for treating various diseases, including obesity, diabetes, stroke, and inflammatory diseases [13, 14]. However, the clinical application is prevented by the currently identified chemical uncouplers, which exclusively act as protonophores to induce uncontrolled uncoupling and consequently display intolerable toxicity [14, 15]. To explore whether UCP2 modulated neuroinflammation via its uncoupling activity, we aimed to identify activators of endogenous UCP2. Moreover, activators of endogenous UCP2 hold the potential to treat various diseases.
Anethole trithione [5-(4-methoxyphenyl) dithiole-3-thione, ADT] is a clinical choleretic and hepatoprotective drug with a high safety profile [16]. The precise mechanisms underlying the therapeutic action of ADT are unclear, although a recent publication suggested that ADT prevented mitochondrial complex I from generating ROS [16]. Notably, ADT is a derivative of the widely used slow-releasing hydrogen sulfide (H2S) donor, 5-(4-hydroxyphenyl) dithiole-3-thione [17]. We recently reported that sulfide-quinone oxidoreductase (SQR)-mediated oxidation of H2S released from some H2S donors induced mitochondrial production of ROS via reverse electron transfer at mitochondrial complex I (Complex I RET), and mitochondrial ROS, in turn, activated UCP2 to induce mitochondrial uncoupling [18]. In the study, we tested the hypothesis that ADT conferred therapeutic effects by activating UCP2 to modulate neuroinflammation.
Intracerebral hemorrhage (ICH) is initiated by the rupture of cerebral vessels and is characterized by the entry of blood components into the cerebral parenchyma to elicit multiple pathogenic cascades, including microglia/macrophage-mediated neuroinflammation [19, 20]. Currently, drug therapies are not available for ICH [21]. In the study, we investigated whether UCP2 acted through microglia/macrophages to modulate neuroinflammation following ICH. To further explore whether UCP2 modulated neuroinflammation via its uncoupling activity, we characterized ADT as a clinically used activator of UCP2. By using ADT, we showed that activating the uncoupling activity of UCP2 robustly inhibited neuroinflammation following ICH. In addition, for the first time we proved the concept that activators of endogenous UCP2 are a new class of uncouplers with translational significance.
Materials and methods
Establishment of the in vitro neuroinflammation model to mimic microglial activation following ICH
Murine BV2 microglia were cultured in Dulbecco’s modified Eagle’s medium (DMEM, HyClone, Logan City, UT, USA, SH30243.01) supplemented with fetal bovine serum (LONSERA, Shanghai, China, S711-001), penicillin and streptomycin. BV2 microglia were used at 2–3 passages. To mimic ICH-induced microglial activation in vitro, BV2 microglia were treated with the lysate of red blood cell (RBC) as reported previously [20, 22]. In brief, blood was drawn from the mouse heart and centrifuged at 300 × g for 5 min. The plasma and buffy coat were discarded. To lyse packed RBCs, cells were frozen in liquid nitrogen for 10 min and then allowed to thaw at 37 °C for 5 min. The freeze-thaw cycle was repeated three times [22]. BV2 microglia seeded on 24-well plates were treated with RBC lysate (10 μL/well). Control cells were treated with an equal volume of saline.
Western blot
Tissue or cell samples were lysed in RIPA lysis buffer (NCM Biotech, Soochow, China, WB3100), which was supplemented with a cocktail of phosphatase inhibitors and protease inhibitors. Lysates were centrifuged and the supernatants were collected. Mitochondrial and cytoplasmic proteins were isolated from BV2 microglia using a mitochondrial isolation kit according to manufacturer’s manual (Boster, Wuhan, China, AR0156). A commercial kit (Beyotime, Shanghai, China, P0011) was used to determine the protein concentrations of lysates. Lysates containing 40 μg protein/sample were mixed with loading buffer and boiled for 10 min. Proteins were subjected to SDS-PAGE and then transferred to polyvinylidene difluoride (PVDF) membranes. After incubation in phosphate buffered saline (PBS) containing 5% non-fat milk and 0.1% (v/v) Tween-20, PVDF membranes were incubated with primary antibodies against UCP2 (1:1000, Santa Cruz Biotechnology, Dallas, TX, USA, sc-390189), phosphorylated AMPK (1:1000, Cell Signaling, Danvers, MA, USA, 2535), total AMPK (1:1000, Cell Signaling, Danvers, MA, USA, 2532), TOMM20 (1:10000, Proteintech, Wuhan, China, 11802-1-AP), β-tubulin (1:2000, Abmart, Shanghai, China, M30109s) or β-actin (1:10000, Abmart, Shanghai, China, M20011XS), followed by incubation with HRP-conjugated goat anti-rabbit IgG (1:5000, ABclonal, Wuhan, China, AS014) or HRP-conjugated goat anti-mouse IgG (1:5000, ABclonal, Wuhan, China, AS003). Enhanced chemiluminescent reagent (SuperSignal West Pico, Pierce, Rockford, IL, USA) was used to visualize protein bands, and images were captured with Chemiluminescence Imaging System (Bio-Rad, Hercules, CA, USA). The optical densities of protein bands were semi-quantified with ImageJ software, and expressed as the ratio of target proteins to an internal loading control or total proteins.
Analysis of the subcellular location of UCP2 in microglia
BV2 microglia were seeded on 24-well plates (105 cells/well) and cultured overnight. Then, cells were treated with RBC lysate or vehicle (saline) for 6 h. After washing with PBS, cells were fixed with paraformaldehyde for 20 min. Cells were incubated in the buffer containing the antibodies against UCP2 (1:400, Santa Cruz Biotechnology, sc-390189) and the antibodies against the mitochondrial marker TOMM20 (1:500, Abcam, ab186735), followed by the incubation in the buffer containing the appropriate secondary antibodies conjugated with Alexa Flour 488 (1:500, Invitrogen, Carlsbad, CA, USA, A32731) or Alexa Flour 555 (1:200, Invitrogen, Carlsbad, CA, USA, A21432). After washing, cells were further stained with 10 μM Hoechst 33342 (Sigma-Aldrich, St. Louis, MO, USA) for 10 min to label nuclei. Images of fluorescence were captured under confocal microscopy (Zeiss LSM700, Germany).
RNA interference
Small interference RNAs (siRNAs) targeting murine AMPK α1/α2 (5′-GAGAAGCAGAAGCACGACGTT-3′), murine UCP2 (5′-CGUAGUGAUGUUUGUCACCTT-3′) and murine SQR (5′-GACGAGAACUGUAUCCGCATT-3′) have been validated [23] and used in our published studies [18, 24,25,26]. A nonsense siRNA (Ctrl-siRNA), which does not target any mammalian DNA sequence, served as the negative control. All siRNAs and Ctrl-siRNA were synthesized by Genepharma (Shanghai, China). BV2 microglia were seeded on 24-well plates or 96-well plates. siRNA (0.5 μg/mL) or Ctrl-siRNA was transfected into BV2 microglia using Lipofectamine RNAiMAX per manufacturer’s instruction (Invitrogen, Carlsbad, CA, USA, 13778-150). Cells were harvested for the assessment of the knockdown efficiency with Western blot or used for further experiments at 48 h after transfection.
Measurement of ATP levels and ADP/ATP ratios
The commercial kit from Sigma-Aldrich (St. Louis, MO, USA, MAK135) was used to assess cellular ATP and ADP levels. BV2 microglia were seeded on 96-well plates (104 cells/well). To investigate whether ADT induced uncoupling required endogenous UCP2 and SQR, BV2 microglia were transfected with siRNA against UCP2 or SQR for 48 h. Then, cells were treated with ADT (50 μM), FCCP (0.5 μM), or vehicle for 30 min. Finally, the ADP/ATP ratios were measured according to the manufacturer’s manual. For some experiments, BV2 microglia seeded on 96-well plates were co-treated with ADT (50 μM) and ZnCl2 (800 μM) or rotenone (Rot) (10 nM) for 30 min. Then, ADP/ATP ratios were measured. Cellular ATP levels were expressed as the percentages of ATP levels to that of control cells treated with vehicle.
Measurement of the mitochondrial membrane potential (ΔΨm)
The mitochondrial membrane potential (ΔΨm) was assessed with the fluorescent probe tetramethylrhodamine ethyl ester (TMRM, Sigma-Aldrich, St. Louis, MO, USA, T5428). BV2 microglia were transfected with siRNAs against UCP2 or SQR for 48 h. Then, cells were treated with ADT (50 μM), FCCP (0.5 μM), or vehicle for 30 min. For some experiments, BV2 microglia were co-treated with ADT (50 μM) and ZnCl2 (800 μM) or Rot (10 nM) for 30 min. Then, BV2 cells were washed with PBS and stained with 100 nM TMRM for 10 min. Images of fluorescence were captured under confocal microscopy (Zeiss LSM700, Germany). All images were acquired with constant parameters.
Cellular oxygen consumption rate (OCR) assay
OCR was assayed as we described in detail in the previous publication [18]. Briefly, BV2 microglia were seeded on 24-well plates (1 × 105 cells/well). BV2 microglia were washed and incubated with XF assay buffer before the assay. Cells was then kept in a CO2-free incubator for 60 min. ADT, FCCP (0.5 μM), MitoTEMPO (a mitochondria-targeted ROS scavenger, 1.5 mM), genipin (Gen, 20 nM), ZnCl2 (800 μM), oligomycin (Oligo), Rot, and antimycin A (AA) were added to the cells as indicated. One well per row containing only medium served as the background control. OCR (pmol O2/min) was measured in real time with Seahorse XF-24. OCR in the presence of Rot and AA represents the non-mitochondrial OCR, which was subtracted from the raw values of basal OCR or OCR in the presence of Oligo and/or ADT or FCCP. To minimize the confounding effect of the variation in basal OCR, OCR values in the presence of Oligo and/or ADT (FCCP) were normalized with the basal OCR values, and expressed as % of basal OCR for each well of cells for statistical analysis. To explore whether ADT-induced uncoupling required endogenous UCP2 and SQR, BV2 microglia were transfected with siRNA against UCP2 or SQR 2 days before the assay of OCR.
Assessment of mitochondrial ROS generation in cultured cells
BV2 microglia plated on 24-well plates were treated with ADT (50 μM), Rot (10 nM), MitoTEMPO (1.5 mM) and/or ZnCl2 (800 μM). Then, cells were incubated with MitoSOX (5 μM, Thermo Fisher scientific, Carlsbad, CA, USA, M36008) and MitoTracker Green FM (50 nM, Thermo Fisher scientific, Carlsbad, CA, USA, M7514). After cells were washed with PBS, they were incubated with Hoechst 33342 (10 μM, Sigma-Aldrich). Fluorescence was captured under confocal microscopy (Zeiss LSM700, Germany). All images were acquired with the constant parameters.
Mitochondrial swelling assay
Mitochondria isolated from the liver of adult C57BL/6J mice were used for the assay of mitochondrial swelling induced by protonophoric uncouplers, as we described in detail in the previous publication [18]. Liver tissue (~2000 mg) was minced on ice and collected with ice-cold isolation buffer (250 mM sucrose, 0.5 mM EGTA, 10 mM HEPES, 1 mg/mL bovine serum albumin, pH 7.4). Following procedures were performed at 4 °C. Liver pieces were centrifuged at 300 × g for 1 min. The pellet was collected, re-suspended in isolation buffer, and homogenized with a Downs homogenizer. The homogenate was centrifuged at 2000 × g for 10 min. The supernatant was collected and centrifuged at 8000 × g for 10 min. The pellet was resuspended in the buffer containing 250 mM sucrose and 10 mM HEPES (pH 7.4). The procedure was repeated and the pellet was finally suspended with 30–50 μL buffer containing 250 mM sucrose and 10 mM HEPES (pH 7.4). Protein concentrations of the mitochondrial suspension were determined using a BCA Protein Assay Kit (Beyotime, Shanghai, China, P0011). To start mitochondrial swelling assay, isolated mitochondria were added into the isotonic acetate buffer, which contained 5 mM Tris-HCl, 145 mM potassium acetate, 0.5 mM EDTA, 3 μM valinomycin, and 1 μM Rot (pH 7.4). Then, ADT (50 μM), FCCP (0.5 μM) or vehicle was added to the mitochondrion solution. The absorbance at 600 nm was continually measured with a microplate reader (TECAN, Grodig, Austria). The swelling of mitochondria was indicated by the decrease in the absorbance at 600 nm.
Primary microglia/astrocyte culture and lentiviral infection
To culture primary microglia and astrocytes, mouse cortices were harvested from 1-day-old newborn mice (C57BL/6J) and kept in icy DMEM/F12 medium. The cortices were digested with EDTA-trypsin for 10 min and digestion was terminated with DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS). To remove tissue debris, cell suspension was filtered through a 50 µm cell strainer. After centrifugation, cells were collected and plated in flasks pre-coated with poly-D-lysine. The mixed glia were cultured at 37 °C in 5% CO2 and 95% oxygen. Medium was replaced every 3 days. After 10–12 days, flasks were shaken at 180 rpm for 30 min. Floating microglia were collected and cultured in DMEM supplemented with 10% FBS and penicillin and streptomycin. To obtain primary astrocytes, flasks were shaken for 6 h at 240 rpm followed by beating flasks with hand for 1 min. Adherent astrocytes were digested with EDTA-trypsin and cultured in DMEM medium supplemented with 10% FBS. After primary microglia and astrocytes seeded on 24-well plates reached 90% convergence, they were infected with lentivirus co-expressing green fluorescence protein (GFP) and UCP2-shRNA or no-sense shRNA at the multiplicity of infection of 4. The UCP2-shRNA sequence was designed based on the sequence of UCP2 siRNA that has been validated in our previous publication [18]. Lentiviruses were generated by GeneChem (Shanghai, China). After 12 h after lentiviral infection, medium was replaced. The expression of GFP in primary microglia or astrocytes was observed under fluorescence microscopy or the knockdown efficiency was analyzed with Western blot at 4 days after infection. Infected cells were used for further experiments at 4 days after infection.
Enzyme-linked immunosorbent assay (ELISA)
Commercial ELISA kits (Invitrogen, San Diego, California, USA, 88-7013-88) were used to measure concentrations of IL-1β, TNF-α and IL-6 in the culture medium or mouse striatal tissue. The culture medium was collected from the BV2 microglia for ELISA assay, and the cells were harvested for the measurement of protein concentrations. Striatal tissues were harvested from ipsilateral, hemorrhagic striatum as well as the contralateral striatum at 3 days after mice were subjected to ICH. Protein concentrations were measured using the BCA kit. IL-1β, IL-6 and TNF-α protein levels were assayed per the manufacturer’s instructions.
Induction of ICH in the left striatum in mice and drug administration
Experimental animal procedures were performed according to the protocols approved by the Animal Care and Use Committee of Soochow University. Male adult (2-month-old) ICR mice were used in the study. They had free access to food and water, and were housed on a 12 h circadian cycle with lights on at 7:00 a.m. ICH was induced by injecting collagenase into the left striatum as we described in previous publications [18, 25]. In brief, mice were anesthetized and placed onto a stereotaxic frame (Model 500, Kopf Instruments, Tujunga, CA, USA). After a cranial burr hole was drilled, a 26 s-gauge needle was inserted into the left striatum. The coordinates were: 0.5 mm anterior to the bregma, 2 mm lateral to the midline and 3.5 mm below the skull. Infusion of collagenase VII-S (0.03 Units; Sigma-Aldrich, St. Louis, MO, USA) was performed at 0.1 μL/min using a microinfusion pump (Harvard Apparatus Inc., South Natick, MA, USA). For sham-operation, 1 μL of saline was infused into the left striatum. To prevent leak of the solution, the needle was kept in place for 10 min after infusion. The skin incision was closed with a suture after the removal of the needle. Mice were randomized to receive a daily intraperitoneal injection of ADT (50 mg· kg−1 ·d−1) or vehicle (10% dimethyl sulfoxide in corn oil) for 3 days, starting at 3 h after collagenase infusion. In some experiments, mice were randomized to receive intrastriatal infusion of (FCCP, 50 μM, 2 μL) or vehicle (DMSO, 2 μL). No animal was excluded from the analysis. We have reported that ADT at 50 mg· kg−1 ·d−1 significantly reduced acute infarct damage [27] and that intrastriatal infusion of FCCP (50 μM, 2 μL) conferred therapeutic effects in the mouse ICH model [25]. The doses of ADT and FCCP were chosen based on the publications.
Lentiviral knockdown of target genes in the striatum
Lentiviruses were injected into the left striatum as we reported previously [18, 25]. Briefly, mice were anesthetized and placed onto a stereotaxic apparatus. After a small hole was drilled through the skull, lentiviruses expressing UCP2-shRNA, AMPK-shRNA, SQR-shRNA or non-targeted control short hairpin RNA (Ctrl-shRNA) were injected into the two sites of the left striatum at a rate of 0.5 μL/min with a 30-gauge needle (1.5 μL of 1 × 108 TU/mL lentivirus per injection site). The UCP2-shRNA sequence was designed based on the sequence of UCP2 siRNA that has been validated in our previous publication, and SQR-shRNA and AMPK-shRNA have also been validated and successfully used to knockdown the target genes in the striatum by us [18, 25]. Lentiviruses were generated by GeneChem (Shanghai, China). The coordinates of the two injection sites were: 0.5 mm anterior to the bregma, 2.0 mm lateral to the midline and 3.5 mm below the skull; 1 mm anterior to the bregma, 1.5 mm lateral to the midline and 3.2 mm below the skull. In vivo knockdown efficiency was analyzed on striatal tissue isolated from mice at 14 days after lentiviral injection. Mice were subjected to ICH at 14 days after lentiviral injection.
Histochemistry
At 3 days following ICH, mice were perfused transcardially with 0.1 mol/L PBS, followed by perfusion with 4% PFA. Brains were harvested and cut into coronal sections (25 μm) on a cryostat. From the onset of hemorrhage, one of every 11 sections was collected, and a total of 4 sections were harvested from the entire striatum. Sections were rinsed with PBS supplemented with triton X-100 (PBST), followed by incubation with the blocking buffer containing 3% normal goat serum and 0.2% Triton X-100 at room temperature for 30 min. After washing with PBST, sections were incubated with primary antibodies against Iba1 (1:500, Wako, Chuo-Ko, Osaka, Japan, 019-19741) and CD86 (1:100, BD, Franklin Lake, NJ, USA, 550542), or incubated with primary antibodies against GFAP (1:200, Santa Cruz Biotechnology, Dallas, TX, USA, sc-33673) and S100 (1:300, Proteintech, Wuhan, China, 15146-AP) for 12 h. Then, sections were incubated in the buffer containing the appropriate secondary antibodies conjugated with Alexa Flour 488 (1:500, Invitrogen, CA, USA, A32731) or Alexa Flour 555 (1:200, Invitrogen, CA, USA, A21432) for 12 h at 4 °C. To assess brain injury histologically, the sections collected from ICH mice were incubated with PBST at 37 °C for 1 h, followed by incubation with the solution containing FluoroMyelin (1:300, Invitrogen, CA, USA, F34652) to label myelin. Hemorrhagic lesions, as indicated by the destruction of myelin architecture, were assessed under fluorescence microscopy.
Assessment of brain edema following ICH
Brains were harvested, and the left and right striatum were dissected and collected at 3 days after ICH. Wet weight of the striatal tissue was measured. The dry weight was measured after the tissues were kept in an oven at 95 °C for 48 h. The edema was calculated according to the formula: [(wet weight − dry weight)/wet weight] × 100% [28, 29].
Assessment of neurological deficits following ICH
Neurological deficits were blindly assessed with the neurological score test, the corner turn test and the vibrissa-elicited forelimb placement test [30, 31], as we described in detail before [25]. For the assessment of neuroscores, each mouse was given points based on the summation of the following tests: gait, body symmetry, climbing, circling behavior, symmetry in front limbs, compulsory circling, and whisker response. The maximum score indicated the most severe deficit. For the forelimb placement test, mouse vibrissae were gently stimulated by brushing them on the corner edge of a countertop. Healthy mice quickly placed the forelimb ipsilateral to vibrissae stimulation on the countertop. Placement of the forelimb contralateral to the hemorrhagic brain was impaired in response to vibrissae stimulation, depending on the injury severity. Each mouse was tested for ten times. Results were presented as the percentages of the number of trials that mice appropriately placed their forelimb compared to total trials. For the corner turn test, mice were forced into a corner with a 30° angle. Normal mice turned either left or right without preference, whereas the ICH mice preferentially turned toward the non-impaired (left) side [23]. Each mouse was tested for ten times. Results were presented as the percentages of the number of left turns compared to total trials.
Statistical analysis
Statistical differences were analyzed using SPSS Statistics 17.0. One-way ANOVA was applied for multiple comparisons, while two-tailed Student’s t tests were used for pairwise comparisons. Data from behavioral tests were analyzed by two-way ANOVA. P < 0.05 was considered statistically significant.
Results
Endogenous UCP2 inhibits microglia-mediated inflammation following ICH
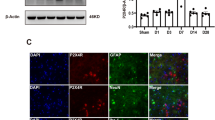
We first investigated whether endogenous UCP2 modulated microglia-mediated neuroinflammation following ICH. To mimic microglial activation following ICH in vitro, cultured BV2 microglia were treated with RBC lysate as previously reported [18, 20]. The total protein levels of UCP2 was not altered in microglia treated with RBC lysate vs. control microglia treated with vehicle (Fig. 1a, b). At 3 days following ICH, the expression of UCP2 also remained unchanged in the hemorrhagic striatum compared to that in the contralateral striatum, or that in the striatum harvested from sham-operated mice (Fig. 1c, d). Moreover, immunohistochemistry results showed that UCP2 was located in the mitochondria of BV2 microglia treated with RBC lysate or vehicle (Fig. 1e). We further extracted mitochondrial proteins from BV2 microglia and assessed the mitochondrial levels of UCP2. There was no change in mitochondrial levels of UCP2 in BV2 microglia treated with RBC lysate vs. control microglia treated with vehicle (Fig. 1f, g). Collectively, these results implicated that the expression and mitochondrial location of UCP2 in microglia were not altered following ICH.

a, b Immunoblot analysis and semi-quantification of UCP2 expression in BV2 microglia treated with RBC lysate for 2–6 h, n = 3. c, d Immunoblot analysis and semi-quantification of UCP2 expression in the striatal tissues collected from mice at 24 h after ICH or sham-operation (Sham). Ipsil: the ipsilateral (hemorrhagic) sriatum; Contra: the contralateral striatum; n = 3. e Representative images of the subcellular location of UCP2 in BV2 microglia following the treatment with RBC lysate for 6 h (three independent replicates). Ctrl: cells treated with vehicle. Lysate: cells treated with RBC lysate. Mitochondrial marker: TOMM20. f, g Western blot analysis and semi-quantification data of UCP2 in mitochondrial fractions extracted from BV2 cells with/without RBC lysate treatment, n = 3. h–j Protein levels of pro-inflammatory mediators IL-6, IL-1β, and TNF-α in the culture medium collected from BV2 microglia (n = 3). BV2 microglia were transfected with nonsense siRNA (Ctrl-siRNA) or UCP2-siRNA for 2 days. Then, cells were treated with RBC lysate for 24 h. Ctrl: control cells without siRNA transfection and RBC lysate treatment. k, l Immunoblot analysis and semi-quantification of the efficiency of knockdown of UCP2 in BV2 microglia transfected with UCP2-siRNA for 2 days (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001. NS: not significant.
To investigate the role of endogenous UCP2 in microglia-mediated neuroinflammation, we examined the effects of UCP2 knockdown on neuroinflammation in the in vitro and in vivo models of ICH. RBC lysate-induced neuroinflammation in BV2 microglia, as evidenced by the elevated expression of pro-inflammatory mediators IL-1β, IL-6 and TNF-α (Fig. 1h, l). RBC lysate induced neuroinflammation was further exacerbated in BV2 microglia transfected with UCP2 siRNA vs. BV2 microglia transfected with control siRNA (Fig. 1h, l). Given ICH was induced in the left striatum in mice, we injected lentiviruses expressing shRNA into the left striatum to knockdown striatal UCP2 in vivo. Compared to a control lentivirus (Ctrl-shRNA), the lentivirus expressing UCP2-shRNA significantly decreased endogenous UCP2 expression in the striatum ipsilateral to lentiviral injection at 14 days after injection (Fig. 2a). According to the results, mice were subjected to collagenase-induced ICH at 14 days after lentiviral injection. Consistent with the in vitro results, striatal knockdown of UCP2 exacerbated neuroinflammation in the hemorrhagic striatum at 3 days following ICH, as indicated by the elevated expression of pro-inflammatory mediators IL-1β, IL-6 and TNF-α (Fig. 2b–d). The results implicated that endogenous UCP2 attenuated neuroinflammation following ICH. Consequently, striatal knockdown of UCP2 exacerbated brain edema and neurological deficits at 3 days following ICH in mice (Fig. 2e–h). Taken together, both the in vitro and in vivo results suggested that endogenous UCP2 attenuated neuroinflammation and therefore played a protective role following ICH.

a Immunoblot analysis of UCP2 expression in the striatum transfected with a lentivirus expressing UCP2-shRNA or Ctrl-shRNA for 14 days. b–d Protein levels of pro-inflammatory mediators IL-1β, IL-6 and TNF-α in the ipsilateral (Ipsil, hemorrhagic) and the contralateral (Contra) striatum assessed with ELISA at 3 days following ICH (n = 4). Lentivirus expressing Ctrl-shRNA or UCP2-shRNA was injected into the left striatum. At 14 days after lentiviral injection, ICH was induced by injecting collagenase into the left striatum. Sham: Sham-operated mice without lentiviral injection and collagenase injection. e Brain edema at 3 days following ICH was indicated by the water content in the ipsilateral (Ipsil) and contralateral (Contra) striatum (n = 6). f–h Neurological deficits of the mice treated with Ctrl-shRNA or UCP2-shRNA. Neurological deficits were assessed with neuroscores (f, n = 6), the corner test (g, n = 6), and the forelimb placement test (h, n = 6) before ICH (Baseline), immediately before the injection of ADT (3 h after ICH, 0 day), 1 day, and 3 days after ICH. *P < 0.05, **P < 0.01, NS: not significant.
ADT induces mitochondrial uncoupling by activating UCP2 in microglia
Next, we aimed to identify activators of UCP2 with translational significance. Based on our previous finding [18], we hypothesized that the clinical drug ADT might function as a mitochondrial uncoupler by activating UCP2. The hallmarks for mitochondrial uncoupling are enhanced oxygen consumption rate (OCR) in the presence of ATP synthase inhibitor Oligo, reduced cellular ATP levels and mitochondrial membrane potential (ΔΨm). By assessing these properties, we investigated whether ADT induced mitochondrial uncoupling in a UCP2-dependent manner. ADT enhanced OCR in the presence Oligo, reduced mitochondrial membrane potential as indicated by TMRM red fluorescence, decreased cellular ATP levels and increased ADP/ATP ratio in BV2 microglia (Fig. 3a–f). Importantly, siRNA knockdown of UCP2 in BV2 microglia abolished all these effects conferred by ADT. In contrast, as indicated by OCR in the presence of Oligo, mitochondrial membrane potential and cellular ATP levels, the uncoupling induced by the classic protonophoric uncoupler FCCP was not abrogated by siRNA knockdown of UCP2 in microglia (Fig. 3g–l). These results showed that, unlike protonophoric uncouplers, ADT acted through endogenous UCP2 to induce mitochondrial uncoupling. Genipin specifically blocks mitochondrial ROS-mediated activation of UCP2 [32]. Genipin also blocked the uncoupling induced by ADT, as evidenced by OCR in the presence of Oligo, mitochondrial membrane potential and cellular ATP levels (Fig. 4a–f). Collectively, by using both pharmacological and gene-manipulation approaches, we found that ADT-induced uncoupling in microglia was dependent on the activation of UCP2.

a–f Knockdown of endogenous UCP2 in BV2 microglia abolished ADT-induced mitochondrial uncoupling, as indicated by real-time change and statistical analysis of OCR (a, b, n = 9), cellular ATP levels and ADP/ATP ratios (c, d, n = 3) and ΔΨm (e: representative images of TMRM staining; f: quantification data, n = 3). Ctrl: control cells without siRNA transfection. Veh: vehicle. BV2 microglia were transfected with UCP2-siRNA or control siRNA (Ctrl-siRNA) for 2 days and then used for the assays of OCR, cellular ATP levels and ΔΨm. For OCR assay, the reagents were added at the indicated timepoints. Oligo: oligomycin, Rot: rotenone, AA: antimycin A. g–l Knockdown of endogenous UCP2 failed to abrogate mitochondrial uncoupling induced by the protonophoric uncoupler FCCP in BV2 microglia, as indicated by real-time change and statistical analysis of OCR (g, h, n = 9), cellular ATP levels and ADP/ATP ratios (i, j, n = 3), ΔΨm (k: representative images of TMRM staining; three independent replicates, l: quantification data, n = 3). **P < 0.05, ***P < 0.01.

a–f The UCP2 inhibitor genipin (Gen) abolished ADT-induced mitochondrial uncoupling in BV2 microglia, as indicated by real-time change and statistical analysis of OCR (a, b, n = 9), cellular ATP levels and ADP/ATP ratios (c, d, n = 3), ΔΨm (e: representative images of TMRM staining; f: quantification data, n = 3). g The protonophoric action was assessed by assaying mitochondrial swelling in the presence of rotenone. Mitochondrial swelling was indicated by a decrease in the absorbance at 600 nm (Representative results from four independent replicates). Mitochondria and reagents were added to the assay buffer at the indicated timepoints. **P < 0.01, ***P < 0.001. NS: not significant.
The protonophoric activity of classical uncouplers can be directly tested by the induction of swelling of non-respiring liver mitochondria treated with Rot [33]. Rotenone (Rot) is the inhibitor of mitochondrial complex I. Since we hypothesized that the function of complex I was required for ADT-induced uncoupling, we expected that ADT would not induce swelling of mitochondria in the presence Rot. As expected, the well-established protonophore uncoupler FCCP, but not ADT, induced swelling of Rot-treated mitochondria (Fig. 4g). The results excluded the possibility that ADT directly functioned as a protonophore to induce mitochondrial uncoupling. For the first time, we showed that the clinical drug ADT induced mitochondrial uncoupling by activating UCP2.
ADT activates UCP2 to induce uncoupling in a mitochondrial ROS-dependent manner
UCP2 is a weak uncoupling protein. Its uncoupling activity is remarkably elevated upon activation by endogenously produced mitochondrial ROS [11, 12]. Thus, ADT might induce uncoupling via ROS-mediated activation of UCP2. In contrast, a recent publication reported that ADT prevented mitochondrial complex I from producing ROS [16]. To explore the mechanism underlying ADT-induced uncoupling, we investigated whether ADT enhanced mitochondrial ROS generation and whether mitochondrial ROS was required for the uncoupling induced by ADT. BV2 microglia were stained with MitoSOX to specifically label mitochondrial ROS. Furthermore, Mitotracker Green and Hoechst were used to visualize mitochondria and nuclei respectively. As indicated by the co-localization of MitoSOX red fluorescence and MitoTracker green fluorescence, mitochondrial ROS levels were remarkably enhanced in ADT-treated BV2 microglia and the enhancement was abolished by the mitochondrial ROS scavenger MitoTempo (Fig. 5a). Moreover, MitoTempo abrogated the uncoupling effects conferred by ADT, as evidenced by mitochondrial membrane potential, OCR in the presence of oligomycin and cellular ATP levels (Fig. 5b–g). The results suggested that ADT enhanced mitochondrial ROS and that mitochondrial ROS was required for ADT-induced mitochondrial uncoupling in microglia.

a Mitochondrial ROS levels in BV2 microglia treated with ADT and/or the mitochondrial ROS scavenger MitoTempo (MitoT), representative images from three independent replicates. b–g MitoT abolished ADT-induced mitochondrial uncoupling in BV2 microglia, as indicated by ΔΨm (b: representative images of TMRM staining; c: quantification data, n = 3), real-time change and statistical analysis of OCR (d, e, n = 9), cellular ATP levels and ADP/ATP ratios (f, g, n = 3). **P < 0.01, ***P < 0.001.
ADT structurally resembles to a widely used organic H2S donor [16]. We report that SQR-mediated oxidation of H2S drives RET at complex I, which enhances production of mitochondrial ROS [18]. To investigate whether ADT enhanced production of mitochondrial ROS by acting as a H2S donor to drive complex I RET, ZnCl2 was used to block the effects of free H2S and Rot was used to inhibit complex I RET [18, 34,35,36]. As expected, ADT-enhanced production of mitochondrial ROS was inhibited by ZnCl2 or Rot (Fig. 6a, b). This suggested that ADT functioned as a H2S donor to drive complex I RET and consequently enhanced production of mitochondrial ROS. Moreover, ZnCl2 and Rot abolished uncoupling effects conferred by ADT in BV2 microglia, as evidenced by mitochondrial membrane potential, OCR in the presence of oligomycin, and cellular ATP levels (Fig. 6c–i). Finally, we showed that siRNA knockdown of SQR in BV2 microglia blocked ADT-induced uncoupling effects (Supplementary Fig. S1a–e). Collectively, these results suggested that oxidation of ADT-released H2S by SQR drove complex I RET to enhance mitochondrial ROS production, and mitochondrial ROS in turn activated UCP2 to induce uncoupling.

a Mitochondrial ROS levels in BV2 microglia treated with ADT and/or ZnCl2 (800 μM), representative MitoSOX images from three independent replicates. b Mitochondrial ROS levels in BV2 microglia treated with ADT and/or rotenone (Rot, 10 nM), representative MitoSOX images from three independent replicates. c, d ZnCl2 and rotenone abolished ADT-induced reduction in ΔΨm in BV2 microglia (c: representative images of TMRM staining; d: quantification data, n = 3). e, f Real-time change and statistical analysis of OCR (n = 9) indicated that ZnCl2 blocked ADT-induced increase in OCR in the presence of Oligo in BV2 microglia. g, h Real-time change and statistical analysis of OCR (n = 9) indicated that rotenone blocked ADT-induced increase in OCR in the presence of Oligo in BV2 microglia. i ZnCl2 and rotenone abolished ADT-induced reduction in cellular ATP levels in BV2 microglia. **P < 0.01, ***P < 0.001. NS: not significant.
ADT acts through UCP2 to inhibit neuroinflammation and to confer therapeutic effects following ICH
Next, we investigated whether ADT acted through UCP2 to suppress neuroinflammation and to confer therapeutic effects following ICH. In vitro results showed that ADT robustly inhibited RBC lysate-induced neuroinflammation in BV2 microglia, and siRNA knockdown of UCP2 abrogated the suppressive effects of ADT on neuroinflammation in BV2 microglia treated with RBC lysate, as evidenced by the protein levels of pro-inflammatory cytokines (IL-1β, TNF-α and IL-6) secreted from microglia (Fig. 7a–c). Consistently, ADT also inhibited neuroinflammation in mouse primary microglia treated with RBC lysate (Fig. 7d–i). Moreover, lentivirus-mediated knockdown of UCP2 in primary microglia not only exacerbated neuroinflammation induced by RBC lysate, but also abolished the inhibition of neuroinflammation by ADT in primary microglia treated with RBC lysate (Fig. 7d–i).

a–c Knockdown of UCP2 abolished the inhibition of neuroinflammation by ADT in BV2 microglia treated with RBC lysate (lysate). BV2 microglia were transfected with Ctrl-siRNA or UCP2-siRNA for 2 days. Cells were then treated with RBC lysate and/or vehicle or ADT for 24 h. The protein levels of pro-inflammatory mediators IL-1β, IL-6 and TNF-α in the culture medium were measured with ELISA, n = 3. d–f Knockdown of UCP2 abolished the inhibition of neuroinflammation by ADT in mouse primary microglia treated with RBC lysate. Primary microglia were infected with lentivirus expressing Ctrl-shRNA or UCP2-shRNA for 4 days, followed by the treatment with RBC lysate and/ADT or vehicle for 24 h. The protein levels of pro-inflammatory mediators IL-1β, IL-6 and TNF-α in the culture medium were measured with ELISA, n = 3. g–i The knockdown efficiency of UCP2 in microglia at 4 days after infection with lentivirus co-expressing UCP2-shRNA and GFP (g: representative images of Western blot analysis of UCP2. h: lentiviral infection as indicated by GFP expression in microglia; left panel: the representative image of GFP expression under fluorescence microscopy; middle panel: the representative image of microglia under light microscopy; right panel: the merge image. i: semi-quantification data of Western blot analysis of UCP2, n = 3). j, k Semi-quantification data and representative images of double-staining immunofluorescence of the inflammatory marker CD86 and microglia/macrophage marker Iba1 on brain sections obtained from mice at 3 days after ICH or from sham-operated animals (3 mice for each group and 4 slides for each animal). Lentiviruses expressing Ctrl-shRNA or UCP2-shRNA were injected into the left striatum. At 14 days after injection, ICH was induced in the left striatum. Ipsil: ipsilateral, Contra: contralateral, Veh: vehicle. l–n The inhibition of ICH-induced neuroinflammation by ADT in mice was abolished by striatal knockdown of UCP2. Protein levels of IL-1β, IL-6 and TNF-α in the ipsilateral and contralateral striatum were assessed with ELISA at 3 days following ICH, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001.
We further investigated whether cerebral knockdown of UCP2 abolished the suppression of neuroinflammation by ADT in the mouse ICH model. We injected lentivirus expressing UCP2-shRNA or control-shRNA into the left striatum, and ICH was induced in the left striatum at 14 days after lentiviral injection. Inflammatory microglia/macrophages are commonly characterized by the expression of signature genes for surface markers. To examine the effects of ADT on neuroinflammation, we used CD86 as the marker for inflammatory microglia/macrophages following brain injury, as reported previously [37]. A significant increase of CD86 expression in microglia/macrophages (CD86+/Iba1+ cells) was observed in the hemorrhagic lesions at 3 days following ICH, which was markedly inhibited by ADT (Fig. 7j, k). The inhibition of inflammatory activation of microglia/macrophages by ADT was attenuated by striatal knockdown of UCP2 (Fig. 7j, k). The results suggested that ADT inhibited microglia/macrophage-mediated neuroinflammation via a UCP2-dependent mechanism following ICH. We further assessed ICH-induced neuroinflammation by measuring the levels of the pro-inflammatory mediators IL-1β, TNF-α, and IL-6 in the hamorrhagic striatum at 3 days post-ICH (Fig. 7l–n). ICH-enhanced expression of IL-1β, TNF-α, and IL-6 was robustly inhibited by ADT, which was countered by striatal knockdown of UCP2 (Fig. 7l–n). The results supported that ADT acted through UCP2 to inhibit post-ICH neuroinflammation.
To further show that ADT acted through UCP2 to confer therapeutic effects in mice following ICH, We investigated whether ADT reduced hemorrhagic lesions histologically. Brain sections were stained with FluoroMyelin to label myelin in the striatum, and lesions were identified by the destruction of the myelin architecture as previously reported [38]. The lesions were smaller in mice receiving ADT (50 mg· kg−1 ·d−1) than in those receiving vehicle (Fig. 8a, b). However, the reduction in lesion sizes induced by ADT was reversed by striatal knockdown of UCP2 (Fig. 8a, b). We further assessed ICH-induced brain edema by measuring the water content of the striatum, and assessed neurological deficits with the neuroscores, the vibrissae-elicited forelimb placement test and the corner test. ADT (50 mg· kg−1 ·d−1) suppressed ICH-induced brain edema (Fig. 8c) and improved neurological deficits in the behavioral tests at 3 days following ICH (Fig. 8d–f). All the therapeutic effects conferred by ADT were abrogated by UCP2 knockdown in the hemorrhagic striatum (Fig. 8c–f). The results suggested that ADT acted through UCP2 to confer therapeutic effects. Finally, we examined the dose-dependent effects of ADT by assessing the inhibition of brain edema by ADT following ICH. ADT at the dose of 20 mg· kg−1 ·d−1, but not at the dose of 5 mg· kg−1 ·d−1, reduced brain edema at 3 days following ICH (Fig. 8g).

a, b Representative images and quantification data of FluoroMyelin-stained brain sections at 3 days post-ICH. Hemorrhagic areas lacked red fluorescence-labelled myelin architecture and were circled in white. c–f Striatal knockdown of UCP2 abolished the therapeutic effects of ADT in mice subjected to ICH. Brain edema was indicated by the water content in the hemorrhagic, ipsilateral (Ipsil) and contralateral (Contra) striatum at 3 days following ICH (c, n = 6). Neurological deficits were assessed with neuroscores (d, n = 6), the forelimb placement test (e, n = 6), and the corner test (f, n = 6) before ICH (Baseline), immediately before the injection of ADT (3 h after ICH, 0 day), 1 day, 2 and 3 days after ICH. g Dose-dependent effects of ADT on brain edema following ICH. *P < 0.05, **P < 0.01, ***P < 0.001. NS: not significant.
UCP2 may also play roles in astrocyte-mediated neuroinflammation after brain injury. We examined the effects of UCP2 and ADT on astrocyte-mediated neuroinflammation following ICH. Unlike what we observed in microglia, ADT did not inhibit inflammation in primary astrocytes treated with RBC lysate, and lentivirus-mediated knockdown of UCP2 had no effect on neuroinflammtion in astrocytes treated with RBC lysate (Fig. 9a–f). S100 is the marker of activated astrocytes. Consistent with the in vitro results, ADT did not inhibit activation of astrocytes (GFAP+S100+ cells) in the hemorrhagic lesions at 3 days following ICH (Fig. 9g, h). Moreover, striatal knockdown of UCP2 had no effect on astrocyte activation following ICH in the mice with/without ADT treatment (Fig. 9g, h). Collectively, the results suggested that UCP2 and ADT unlikely acted through astrocytes to modulate neuroinflammation following ICH.

a–c Knockdown of UCP2 did not exacerbate neuroinflammation in primary astrocytes treated with RBC lysate, and ADT had no effect on astrocyte-mediated neuroinflammation induced by RBC lysate. Primary astrocytes were infected with lentivirus expressing Ctrl-shRNA or UCP2-shRNA for 4 days, followed by the treatment with RBC lysate and/ADT or vehicle for 24 h. Astrocyte-mediated neuroinflammation was indicated by the protein levels of pro-inflammatory mediators IL-1β, IL-6 and TNF-α in culture medium, which were measured with ELISA at 24 h after the treatment with RBC lysate, n = 3. d–f The knockdown efficiency of UCP2 in primary astrocyte at 4 days after infection with lentivirus co-expressing UCP2-shRNA and GFP (d: representative images of Western blot analysis of UCP2. e: lentiviral infection as indicated by GFP expression in astrocytes; left panel: the representative image of GFP expression under fluorescence microscopy; middle panel: the representative image of microglia under light microscopy; right panel: the merge image. f: semi-quantification data of Western blot analysis of UCP2, n = 3). g, h Semi-quantification data and representative images of double-staining immunofluorescence of the activation marker S100 and the astrocytic marker GFAP on brain sections collected from mice at 3 days after ICH or from sham-operated animals (3 mice for each group and 4 slides for each animal). *P < 0.05, **P < 0.01.
If ADT inhibited neuroinflammation and conferred therapeutic effects by activating the uncoupling activity of UCP2 following ICH, we expected that protonophoric uncouplers would also inhibit neuroinflammation and confer therapeutic effects following ICH. However, since the uncoupling activity of protonophores is independent of the uncoupling activity of endogenous UCP2, the suppression of neuroinflammation by protonophoric uncouplers would not be blocked by the knockdown of UCP2. We tested the hypothesis. To avoid the toxicity that may result from the systemic administration of protonopheric uncouplers, FCCP was directly injected into the hemorrhagic (left) striatum following ICH. Consistent with the expectation, FCCP inhibited neuroinflammation, reduced brain edema and improved neurological deficits at 3 days after ICH (Fig. 10a–g). Importantly, striatal knockdown of UCP2 with lentiviral shRNA did not abolish the suppression of neuroinflammation and therapeutic effects conferred by FCCP (Fig. 10a–g). Collectively, these results suggested that ADT inhibited neuroinflammation by activating the uncoupling activity of UCP2 following ICH, and supported that uncoupling activity was essential for the suppression of neuroinflammation by UCP2.

a–c The inhibition of ICH-induced neuroinflammation by FCCP was not abolished by striatal knockdown of UCP2 in mice. Lentiviruses expressing Ctrl-shRNA or UCP2-shRNA were injected into the left striatum of mice. At 14 days after injection, ICH was induced in the left striatum. Vehicle (Veh) or FCCP was administered directly into the striatum daily for 3 days, starting at 3 h after ICH. Protein levels of IL-1β, IL-6 and TNF-α in the ipsilateral (Ipsil) and contralateral (Contra) striatum were assessed with ELISA at 3 days following ICH, n = 3. d–g Striatal knockdown of UCP2 did not abolish the therapeutic effects of FCCP following ICH in mice. Brain edema was indicated by the water content in the striatum at 3 days following ICH (d, n = 6). Neurological deficits were assessed with neuroscores (e, n = 6), the forelimb placement test (f, n = 6), and the corner test (g, n = 6) before ICH (Baseline), immediately before the injection of ADT (3 h after ICH, 0 day), 1 day, 2 and 3 days after ICH. **P < 0.01, ***P < 0.01. NS: not significant.
Since SQR was required for ADT-induced mitochondrial uncoupling in microglia (Supplementary Fig. S1), we asked whether SQR was also indispensable for the suppression of neuroinflammation by ADT following ICH. As expected, the suppressive effects of ADT on neuroinflammation in the ipsilateral, hemorrhagic striatum was countered by striatal knockdown of SQR, as evidenced by the expression levels of IL-1β and TNF-α in the hemorrhagic striatum at 3 days post-ICH (Supplementary Fig. S2a, b). Moreover, striatal knockdown of SQR also abolished the therapeutic effects of ADT on brain edema and neurologic deficits at 3 days following ICH (Supplementary Fig. S2c–f).
UCP2 mediates the suppressive effects of ADT on neuroinflammation via AMPK activation
AMPK activation, a well-established signaling pathway downstream of mitochondrial uncoupling, has been shown to mediate the effects of mitochondrial uncouplers [14]. Moreover, we have shown that the suppressive effect of the protonophore FCCP on neuroinflammation was mediated by AMPK activation [25]. Since ADT induced mitochondrial uncoupling by activating UCP2, we hypothesized that activation of UCP2 mediated the suppression effects of ADT on neuroinflammation by activating AMPK following ICH. In the striatum transfected with a lentivirus expressing control shRNA, ADT enhanced AMPK activation compared to vehicle following ICH (Fig. 11a). Compared to control shRNA, UCP2 shRNA abolished ADT-induced AMPK activation in the ipsilateral (hemorrhagic) striatum (Fig. 11a). These results suggested that ADT enhanced AMPK activation via UCP2 following ICH. To investigate whether AMPK activation was required for suppressive effects of ADT on post-ICH neuroinflammation, we injected a lentivirus expressing AMPK-shRNA into the left striatum. Compared to the control lentivirus, the lentivirus expressing AMPK-shRNA decreased endogenous expression of AMPK in the striatum ipsilateral to lentiviral injection at 14 days after injection (Fig. 11b). Striatal knockdown of AMPK abolished the suppressive effects of ADT on neuroinflammation in the hemorrhagic brain at 3 days following ICH (Fig. 11c–e). Moreover, the therapeutic effects of ADT were also abolished by cerebral knockdown of AMPK following ICH, as evidenced by neurological deficits and brain edema (Fig. 11f–i). The results suggested that UCP2 mediated the suppressive effects of ADT on neuroinflammation via AMPK activation. In conclusion, oxidation of H2S released from ADT by SQR drove reverse electron transfer at mitochondrial complex I and consequently enhanced production of mitochondrial ROS. Mitochondrial ROS, in turn, resulted in the activation of UCP2 and subsequent activation of AMPK, which was a key mechanism underlying the inhibition of microglia-mediated neuroinflammation by ADT. In contrast, the classic protonophoric uncoupler FCCP inhibited neuroinflammation independent of UCP2 (Fig. 11j).

a Cerebral knockdown of UCP2 abolished AMPK activation enhanced by ADT in the hemorrhagic striatum. Lentiviruses expressing Ctrl-shRNA or UCP2-shRNA were injected into the left striatum of mice. AMPK activation was assessed with immunoblot analysis of AMPK phosphorylation (p-AMPK) in the striatum at 3 days after ICH (a: representative Immunoblot images from three independent replicates). Ipsil: the ipsilateral (hemorrhagic) striatum, Contra: the contralateral striatum, Veh: vehicle. b Immunoblot analysis of the efficiency of AMPK knockdown in the striatum. Lentiviruses expressing Ctrl-shRNA or AMPK-shRNA were injected into the left striatum of mice for 14 days. Ipsil: the striatum ipsilateral to lentiviral injection, Contra: the striatum contralateral to injection. c–e The inhibition of ICH-induced neuroinflammation by ADT in mice was abolished by striatal knockdown of AMPK, as indicated by the protein levels of IL-1β, IL-6 and TNF-α in the ipsilateral (Ipsil) and contralateral (Contra) striatum assessed with ELISA at 3 days following ICH, n = 3. f–i Striatal knockdown of AMPK abolished therapeutic effects of ADT in the mouse ICH model, as evidenced by neurological deficits assessed with neuroscores (f, n = 6), the corner test (g, n = 6), and the forelimb placement test (h, n = 6) before ICH (Baseline), immediately before the injection of ADT (3 h after ICH, 0 day), 1 day, and 3 days after ICH. Brain edema was indicated by the water content in the striatum at 3 days following ICH (i, n = 6). j Proposed mechanisms underlying the activation of UCP2 by ADT and therapeutic actions of ADT. **P < 0.01, ***P < 0.01. NS: not significant.
Discussion
We reported three important findings in the study. First, we showed that endogenous UCP2 inhibited microglia-mediated neuroinflammation following ICH. Second, we found that the clinical drug ADT induced mitochondrial uncoupling by enhancing mitochondrial ROS production to activate UCP2. Third, by using ADT, we showed that activating the uncoupling activity of UCP2 inhibited neuroinflammation following ICH. Moreover, UCP2 activation mediated the suppressive effects of ADT on neuroinflammation via uncoupling-enhanced activation of AMPK. The study provided first line of evidence that the uncoupling activity of UCP2 play an inhibitory role in modulating neuroinflammation.
Among five UCPs that have been identified in mammals, UCP1, UCP2, and UCP3 are evolutionarily related, while UCP4 and UCP5 are more divergent [1, 39]. The canonical UCP1 is expressed only in the brown adipose tissue, and serves as the mediator of adaptive thermogenesis in mammals [1]. UCP3 expression is mainly restricted in the skeleton muscle. UCP2 is predominantly expressed in innate immune cells, including macrophages and microglia [3, 4, 9]. In addition to functioning as an uncoupling protein, UCP2 is an important metabolic switch and a regulator of lipid synthesis [40,41,42,43,44,45]. Indeed, UCP2 has been reported to promote inflammation by inducing fatty acid synthesis [4, 5]. On the contrary, UCP2 has also been shown to suppress innate inflammation. For instance, UCP2 promotes anti-inflammatory polarization of microglia, and down-regulation of UCP2 is required for macrophage-mediated inflammation induced by LPS [9]. Moreover, overexpressing UCP2 inhibits the production of inflammatory cytokines following cerebral ischemia [46], and UCP2 plays a protective role in the inflammatory brain following cerebral ischemia [47]. To conclude, the roles of UCP2 in modulating inflammation are controversial.
ICH is characterized by the entry of blood components into the cerebral parenchyma to elicit multiple pathogenic cascades, including microglia/macrophage-mediated neuroinflammation [19, 20, 48]. UCP2 is abundantly expressed in microglia [4, 9]. The role of UCP2 in post-ICH neuroinflammation has not been investigated. We found that the expression levels of UCP2 were not altered in the hemorrhagic brain tissue following ICH. Moreover, by using the cellular ICH model, we further showed that mitochondrial levels of UCP2 were not altered in microglia following ICH. However, knockdown of UCP2 exacerbated neuroinflammation both in the mouse ICH model and the microglia ICH model, suggesting that endogenous UCP2 inhibited neuroinflammation following ICH. Collectively, these results suggested that modulation of post-ICH neuroinflammation by UCP2 was not dependent on the changes in the expression or subcellular location of UCP2. Given that UCP2 exacerbates microglia/macrophage inflammation induced by sepsis and high-fat diet [4, 5], UCP2 possibly displays differential effects on inflammation in different pathophysiological settings. Besides different stimulus, different approaches were used. Our study used the technique of lentiviral shRNA to transiently knockdown UCP2 in the adult brain, while germline deletion of UCP2 in microglia or macrophages was shown to exacerbate inflammation in adult mice [4, 5].
UCP2 is considered an uncoupling protein based on its homology to UCP1 [1, 6]. However, no direct evidence currently exists to suggest how the uncoupling activity of UCP2 modulates inflammation. It is widely accepted that mitochondrial uncoupling impacts microglia/macrophage inflammation [6]. However, UCP2 is a weak uncoupling protein [6] and no activators of the uncoupling activity of UCP2 have been identified. To investigate the involvement of the uncoupling activity of UCP2 in inflammation, we identified a clinically used UCP2 activator. We provided the following lines of evidence suggesting that the clinic drug ADT functioned as a UCP2 activator rather than a protonophoric uncoupler to induce mitochondrial uncoupling. First, in contrast to the uncoupling induced by the classic protonophoric uncoupler FCCP, ADT-induced mitochondrial uncoupling was critically dependent on UCP2. Second, unlike FCCP, ADT did not induce the swelling of the mitochondria treated with Rot, which is characteristic of classic protonophoric uncouplers. Third, ADT-induced mitochondrial uncoupling required mitochondrial ROS, which was likely generated via SQR-mediated oxidation of H2S released from ADT. To the best of our knowledge, for the first time we identified a clinical drug that activated UCP2.
ADT led to mitochondrial uncoupling via ROS-mediated activation of UCP2. This represented a possible mechanism underlying the therapeutic action of ADT. In contrast, a recent publication showed that ADT likely conferred therapeutic effects by inhibiting ROS generated at mitochondrial complex I [16]. However, our results were consistent with the long-held view that the uncoupling activity of UCP2 is remarkably elevated upon activation by ROS [11, 12]. Genipin specifically suppresses activation of UCP2 by ROS [32]. Relevantly, genipin also abolished ADT-induced uncoupling. Collectively, we showed that induction of mitochondrial ROS was a practical approach to activate the uncoupling activity of UCP2. Nevertheless, the mechanisms through which mitochondrial ROS activates UCP2 need further investigation. We did not address the issue, which was a limitation of the study.
By using ADT, we further showed that uncoupling activity was essential to suppression of inflammation by UCP2 following ICH. We observed that ADT suppressed neuroinflammation and conferred therapeutic effects in a UCP2-depedent manner following ICH both in vivo and in vitro. To confirm that ADT conferred therapeutic effects following ICH in mice, we assessed brain edema and examined hemorrhagic injury histologically. Collagenase-induced hemorrhagic brain injury reaches a maximum at 3 days after ICH [38]. Moreover, we have reported that protonophoric uncouplers inhibited brain edema and neuroinflammation at 3 days following ICH [25]. Therefore, we assessed the effects of ADT on brain edema, hemorrhagic injury and neuroinflammation at 3 days following ICH. To do this, ADT was administered daily, starting at 3 h following ICH. Notably, in contrast to the inhibition of neuroinflammation and brain edema by the classic protonophore FCCP, the inhibition by ADT was critically dependent on UCP2. These results suggested that mitochondrial uncoupling induced either by UCP2 activation or protonophoric uncouplers suppressed neuroinflammation following ICH. Moreover, the results further supported that UCP2 inhibited neuroinflammation via its uncoupling activity.
Microglia are the primary type of cells that are responsible for neuroinflammation following brain injury [49,50,51,52,53]. The following lines of evidence suggested that UCP2 acted through microglia/macrophages to modulate neuroinflammation following ICH. In the in vitro models, we treated microglia and astrocytes with RBC lysate to mimic ICH-induced activation of microglia or astrocytes. We observed that knockdown of UCP2 exacerbated neuroinflammation in primary microglia or BV2 microglia treated with RBC lysate, but not in primary astrocytes treated with RBC lysate. By using the cellular models, we further showed that ADT inhibited neuroinflammation in primary microglia or BV2 microglia that were treated with RBC lysate, and the effects were abrogated by microglial knockdown of UCP2. However, ADT did not inhibit neuroinflammation in primary astrocytes. Consistently, in vivo results also showed that the number of inflammatory microglia/macrophages was enhanced flowing ICH, which was markedly attenuated by ADT. Moreover, cerebral knockdown of UCP2 abolished the inhibitory effects of ADT on inflammatory activation of microglia/macrophages. ICH also enhanced inflammatory activation of astrocytes in the mouse ICH model. However, ADT had no effects on inflammatory activation of astrocytes following ICH in vivo. Collectively, the results suggested that UCP2 mainly acted through microglia/macrophages to modulate neuroinflammation following ICH.
The downstream mechanisms through which UCP2 suppressed neuroinflammation via the uncoupling action might be complex. For instance, inhibition of nuclear factor-kappa B pathway is a well-established signaling cascade that inhibits neuroinflammation [54]. Reduction in cellular ATP levels, which can be induced by mitochondrial uncoupling, is a major mechanism underlying activation of AMPK [14]. AMPK activation protects neurons [55]. Moreover, we and others find that AMPK activation is an important mechanism underlying the inhibition of macrophage/microglia-mediated inflammation [24, 56]. Consistent with these published results, ADT enhanced AMPK activation in the hemorrhagic brain of the mice subjected to ICH, and the enhancement was abrogated by cerebral knockdown of UCP2. More relevantly, cerebral knockdown of AMPK abolished the suppressive effects of ADT on neuroinflammation in mice following ICH, suggesting that uncoupling-induced activation of AMPK was required for the inhibition of neuroinflammation by UCP2 following ICH. In support of this, we recently reported that the protonophoric uncoupler FCCP also inhibit neuroinflammation by enhancing AMPK activation following ICH [25].
This study has important translational significance. Mitochondrial uncoupling is considered a promising therapy for treating various diseases, including obesity, diabetes, stroke and neuroinflammatory diseases [13, 14]. However, no mitochondrial uncouplers have been approved for clinical use. Historically, 2,4-dinitrophenol (DNP), a well-known protonophoric uncouplers, was approved by US Food and Drug Administration to treat obesity, but was quickly withdrawn from the market due to intolerable toxicity [14]. Notably, like DNP, currently identified chemical uncouplers exclusively act as protonophores to induce mitochondrial uncoupling and display high toxicity. To reduce toxicity, current research mainly focuses on chemical modification or design of controlled release formulations of protonophoric uncouplers [14, 15]. The uncoupling activity of endogenous UCP2 was remarkably enhanced upon activation [11, 12], and activators of endogenous UCP2 may represent a new class of uncouplers with high safety due to the limited uncoupling capacity of endogenous UCP2. We proved the concept in this study.
Currently, no clinical drug has been characterized as the activators of UCPs. ADT is a clinical choleretic and hepatoprotective drug with a high safety profile (oral lethal dose: 3500 mg/kg). It is also indicated as an adjunctive therapy for cholecystitis, indigestion, and acute/chronic hepatitis. ADT has been approved in human therapy in many countries, including China, France, and Germany [16]. Until now, no precise mechanism of action has been characterized for ADT [16]. Here, for the first time, we identified ADT as the clinically used activator of UCP2, which induced mitochondrial uncoupling via ROS-dependent activation of UCP2. Moreover, ADT conferred therapeutic effects in a UCP2-dependent manner. Since mitochondrial uncoupling is a promising therapy for treating various diseases, ADT, as a novel activator of UCP2 with a profile of high safety, has the potential to treat various diseases, including ICH.
To conclude, endogenous UCP2 of microglia attenuated neuroinflammation following ICH. By using ADT, the activator of uncoupling activity of UCP2, we further showed that uncoupling activity was essential for the suppression of neuroinflammation by UCP2. In addition, we provided a potentially new and practical pharmacological approach to treat diseases by activating UCP2 with a clinical drug.
References
Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-Doulcier AM, Bouillaud F, et al. The biology of mitochondrial uncoupling proteins. Diabetes. 2004;53:S130–5.
Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26:435–9.
Arsenijevic D, Clavel S, Sanchis D, Plamondon J, Huang Q, Ricquier D, et al. Induction of Ucp2 expression in brain phagocytes and neurons following murine toxoplasmosis: an essential role of IFN-gamma and an association with negative energy balance. J Neuroimmunol. 2007;186:121–32.
Kim JD, Yoon NA, Jin S, Diano S. Microglial UCP2 mediates inflammation and obesity induced by high-fat feeding. Cell Metab. 2019;30:952–62.e5
Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2015;125:665–80.
Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiol (Bethesda). 2011;26:192–205.
Emre Y, Nubel T. Uncoupling protein UCP2: when mitochondrial activity meets immunity. FEBS Lett. 2010;584:1437–42.
Rousset S, Emre Y, Join-Lambert O, Hurtaud C, Ricquier D, Cassard-Doulcier AM. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine. 2006;35:135–42.
De Simone R, Ajmone-Cat MA, Pandolfi M, Bernardo A, De Nuccio C, Minghetti L, et al. The mitochondrial uncoupling protein-2 is a master regulator of both M1 and M2 microglial responses. J Neurochem. 2015;135:147–56.
Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, et al. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat Genet. 1997;15:269–72.
Echtay KS, Murphy MP, Smith RA, Talbot DA, Brand MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem. 2002;277:47129–35.
Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, et al. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–9.
Khan RS, Dine K, Geisler JG, Shindler KS. Mitochondrial uncoupler prodrug of 2,4-dinitrophenol, MP201, prevents neuronal damage and preserves vision in experimental optic neuritis. Oxid Med Cell Longev. 2017;2017:7180632.
Tao H, Zhang Y, Zeng X, Shulman GI, Jin S. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat Med. 2014;20:1263–9.
Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science. 2015;347:1253–6.
Detaille D, Pasdois P, Semont A, Dos Santos P, Diolez P. An old medicine as a new drug to prevent mitochondrial complex I from producing oxygen radicals. PLoS One. 2019;14:e0216385.
Lee M, Tazzari V, Giustarini D, Rossi R, Sparatore A, Del Soldato P, et al. Effects of hydrogen sulfide-releasing L-DOPA derivatives on glial activation: potential for treating Parkinson disease. J Biol Chem. 2010;285:17318–28.
Jia J, Wang Z, Zhang M, Huang C, Song Y, Xu F, et al. SQR mediates therapeutic effects of H2S by targeting mitochondrial electron transport to induce mitochondrial uncoupling. Sci Adv. 2020;6:eaaz5752.
Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. 2014;115:25–44.
Wang YC, Zhou Y, Fang H, Lin S, Wang PF, Xiong RP, et al. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann Neurol. 2014;75:876–89.
Tian X, Liu C, Shu Z, Chen G. Review: therapeutic targeting of HMGB1 in Stroke. Curr Drug Deliv. 2017;14:785–90.
Bhasin RR, Xi G, Hua Y, Keep RF, Hoff JT. Experimental intracerebral hemorrhage: effect of lysed erythrocytes on brain edema and blood-brain barrier permeability. Acta Neurochir Suppl. 2002;81:249–51.
Modis K, Coletta C, Erdelyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27:601–11.
Zhou X, Cao Y, Ao G, Hu L, Liu H, Wu J, et al. CaMKKbeta-dependent activation of AMP-activated protein kinase is critical to suppressive effects of hydrogen sulfide on neuroinflammation. Antioxid Redox Signal. 2014;21:1741–58.
Pan X, Song Y, He M, Yan X, Huang C, Li J, et al. Mitochondrial uncouplers confer protection by activating AMP-activated protein kinase to inhibit neuroinflammation following intracerebral hemorrhage. Biol Pharm Bull. 2020;43:1210–9.
Zhang M, Wu X, Xu Y, He M, Yang J, Li J, et al. The cystathionine beta-synthase/hydrogen sulfide pathway contributes to microglia-mediated neuroinflammation following cerebral ischemia. Brain Behav Immun. 2017;66:332-46.
Wang Y, Jia J, Ao G, Hu L, Liu H, Xiao Y, et al. Hydrogen sulfide protects blood-brain barrier integrity following cerebral ischemia. J Neurochem. 2014;129:827–38.
Hu HM, Li B, Wang XD, Guo YS, Hui H, Zhang HP, et al. Fluoxetine is neuroprotective in early brain injury via its anti-inflammatory and anti-apoptotic effects in a rat experimental subarachnoid hemorrhage model. Neurosci Bull. 2018;34:951–62.
Liu H, Wang Y, Xiao Y, Hua Z, Cheng J, Jia J. Hydrogen sulfide attenuates tissue plasminogen activator-induced cerebral hemorrhage following experimental stroke. Transl Stroke Res. 2016;7:209–19.
Liu RY, Wang JJ, Qiu X, Wu JM. Acute hyperglycemia together with hematoma of high-glucose blood exacerbates neurological injury in a rat model of intracerebral hemorrhage. Neurosci Bull. 2014;30:90–8.
Ni W, Mao S, Xi G, Keep RF, Hua Y. Role of erythrocyte CD47 in intracerebral hematoma clearance. Stroke. 2016;47:505–11.
Zhang CY, Parton LE, Ye CP, Krauss S, Shen R, Lin CT, et al. Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced beta cell dysfunction in isolated pancreatic islets. Cell Metab. 2006;3:417–27.
Kanemoto N, Okamoto T, Tanabe K, Shimada T, Minoshima H, Hidoh Y, et al. Antidiabetic and cardiovascular beneficial effects of a liver-localized mitochondrial uncoupler. Nat Commun. 2019;10:2172.
Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–5.
Scialo F, Sriram A, Fernandez-Ayala D, Gubina N, Lohmus M, Nelson G, et al. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 2016;23:725–34.
Mimoun S, Andriamihaja M, Chaumontet C, Atanasiu C, Benamouzig R, Blouin JM, et al. Detoxification of H2S by differentiated colonic epithelial cells: implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid Redox Signal. 2012;17:1–10.
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–70.
Chang CF, Cho S, Wang J. (-)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways. Ann Clin Transl Neurol. 2014;1:258–71.
Yoshitomi H, Yamazaki K, Tanaka I. Cloning of mouse uncoupling protein 3 cDNA and 5’-flanking region, and its genetic map. Gene. 1998;215:77–84.
Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–51.
Horvath TL, Andrews ZB, Diano S. Fuel utilization by hypothalamic neurons: roles for ROS. Trends Endocrinol Metab. 2009;20:78–87.
Sheets AR, Fulop P, Derdak Z, Kassai A, Sabo E, Mark NM, et al. Uncoupling protein-2 modulates the lipid metabolic response to fasting in mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1017–24.
Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D, et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci USA. 2014;111:960–5.
Samudio I, Fiegl M, McQueen T, Clise-Dwyer K, Andreeff M. The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. 2008;68:5198–205.
Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30:4860–73.
Haines B, Li PA. Overexpression of mitochondrial uncoupling protein 2 inhibits inflammatory cytokines and activates cell survival factors after cerebral ischemia. PLoS One. 2012;7:e31739.
Mehta SL, Li PA. Neuroprotective role of mitochondrial uncoupling protein 2 in cerebral stroke. J Cereb Blood Flow Metab. 2009;29:1069–78.
Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908.
Zhao Q, Wang Q, Wang J, Tang M, Huang S, Peng K, et al. Maternal immune activation-induced PPARgamma-dependent dysfunction of microglia associated with neurogenic impairment and aberrant postnatal behaviors in offspring. Neurobiol Dis. 2019;125:1–13.
Han Y, Wang J, Zhao Q, Xie X, Song R, Xiao Y, et al. Pioglitazone alleviates maternal sleep deprivation-induced cognitive deficits in male rat offspring by enhancing microglia-mediated neurogenesis. Brain Behav Immun. 2020;87:568–78.
Gan P, Zhang L, Chen Y, Zhang Y, Zhang F, Zhou X, et al. Anti-inflammatory effects of glaucocalyxin B in microglia cells. J Pharmacol Sci. 2015;128:35–46.
Wu J, Du J, Gu R, Zhang L, Zhen X, Li Y, et al. Inhibition of neuroinflammation by synthetic androstene derivatives incorporating amino acid methyl esters on activated BV-2 microglia. ChemMedChem. 2015;10:610–6.
Zhang Y, Gu R, Jia J, Hou T, Zheng LT, Zhen X. Inhibition of macrophage migration inhibitory factor (MIF) tautomerase activity suppresses microglia-mediated inflammatory responses. Clin Exp Pharmacol Physiol. 2016;43:1134–44.
Tao L, Zhang F, Hao L, Wu J, Jia J, Liu JY, et al. 1-O-tigloyl-1-O-deacetyl-nimbolinin B inhibits LPS-stimulated inflammatory responses by suppressing NF-kappaB and JNK activation in microglia cells. J Pharmacol Sci. 2014;125:364–74.
Yan Q, Han C, Wang G, Waddington JL, Zheng L, Zhen X. Activation of AMPK/mTORC1-mediated autophagy by metformin reverses Clk1 deficiency-sensitized dopaminergic neuronal death. Mol Pharmacol. 2017;92:640–52.
Zhu YP, Brown JR, Sag D, Zhang L, Suttles J. Adenosine 5′-monophosphate-activated protein kinase regulates IL-10-mediated anti-inflammatory signaling pathways in macrophages. J Immunol. 2015;194:584–94.
Acknowledgements
This work was supported by following grants: National Natural Science Foundation of China (81971119, 82071469, 81571124, 81671310), Priority Academic Program Development of the Jiangsu Higher Education Institutions (PAPD), Suzhou Clinical Research Center of Neurological Disease (Szzx201503) and the Jiangsu key laboratory grant (BM2013003).
Author information
Authors and Affiliations
Contributions
JJ, JC, and ZCH conceived the concept, designed the research, interpreted data, and wrote the paper. XLY, FYX and JJJ carried out the cell culture, Western blotting, Histochemistry, animal model and behavior tests. PS, YQP, MJH, ZCW and SJY participated in the above work. XLY, FYX and JJJ acquired and analyzed data. FYX drafted all figures. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary Infromation
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Yan, Xl., Xu, Fy., Ji, Jj. et al. Activation of UCP2 by anethole trithione suppresses neuroinflammation after intracerebral hemorrhage. Acta Pharmacol Sin 43, 811–828 (2022). https://doi.org/10.1038/s41401-021-00698-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00698-1
- Springer Nature Singapore Pte Ltd.