
Abstract
Ischemic stroke is a devastating disease in which mitochondrial damage or dysfunction substantially contributes to brain injury. Mitochondrial uncoupling protein-2 (UCP2) is a member of the UCP family, which regulates production of mitochondrial superoxide anion. UCP2 is reported to be neuroprotective for ischemic stroke–induced brain injury. However, the molecular mechanisms of UCP2 in ischemic stroke remain incompletely understood. In this study, we investigated whether and how UCP2 modulates neuroinflammation and regulates neuronal ferroptosis following ischemic stroke in vitro and in vivo. Wild-type (WT) and UCP2 knockout (Ucp2−/−) mice were subjected to middle cerebral artery occlusion (MCAO). BV2 cells (mouse microglial cell line) and HT-22 cells (mouse hippocampal neuronal cell line) were transfected with small interfering (si)-RNA or overexpression plasmids to knockdown or overexpress UCP2 levels. Cells were then exposed to oxygen–glucose deprivation and reoxygenation (OGD/RX) to simulate hypoxic injury in vitro. We found that UCP2 expression was markedly reduced in a time-dependent manner in both in vitro and in vivo ischemic stroke models. In addition, UCP2 was mainly expressed in neurons. UCP2 deficiency significantly enlarged infarct volumes, aggravated neurological deficit scores, and exacerbated cerebral edema in mice after MCAO. In vitro knockdown of Ucp2 and in vivo genetic depletion of Ucp2 (Ucp2−/− mice) increased neuronal ferroptosis-related indicators, including Fe2+, malondialdehyde, glutathione, and lipid peroxidation. Overexpression of UCP2 in neuronal cells resulted in reduced ferroptosis. Moreover, knockdown of UCP2 exacerbated neuroinflammation in BV2 microglia and mouse ischemic stroke models, suggesting that endogenous UCP2 inhibits neuroinflammation following ischemic stroke. Upregulation of UCP2 expression in microglia appeared to decrease the release of pro-inflammatory factors and increase the levels of anti-inflammatory factors. Further investigation showed that UCP2 deletion inhibited expression of AMPKα/NRF1 pathway-related proteins, including p-AMPKα, t-AMPKα, NRF1, and TFAM. Thus, UCP2 protects the brain from ischemia-induced ferroptosis by activating AMPKα/NRF1 signaling. Activation of UCP2 represents an attractive strategy for the prevention and treatment of ischemic stroke.
Graphical Abstract
Ischemic stroke suppresses UCP2 induction of lipid peroxidation, leading to neuronal ferroptosis via direct interaction with GPX4. In contrast, low levels of UCP2 activate the AMPKα/NRF1 signaling axis and lead to microglia release of IL-1β, TNF-α, and IL-6. UCP2, mitochondrial uncoupling protein 2; GPX4, glutathione peroxidase 4; MDA, malondialdehyde; GSH, glutathione; LIPID ROS, lipid peroxide reactive oxygen species; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; IL-4, interleukin-4; IL-10, interleukin-10; IL-13, interleukin-13; AMPK, adenosine 5′-monophosphate (AMP)-activated protein kinase; NRF1, nuclear respiratory factor 1.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke is a health issue that has high mortality and disability rates worldwide and leads to irreversible neurological impairment [1]. Current therapies for ischemic stroke are still limited due to complexity of the underlying mechanisms and the narrow treatment window [2]. It is increasingly understood that oxygen consumption leads to mitochondrial dysfunction or damage, increased reactive oxygen species (ROS) production, and neuronal death. Indeed, neuronal death immediately after ischemic stroke and the secondary inflammatory cascade induced by ischemic stroke are two crucial features currently being intensively investigated [3]. It remains unclear how these two events are coordinated and regulated after ischemia.
The inflammatory response and oxidative stress play integral roles in ischemic stroke [4]. The rapid immunological response following stroke causes immediate brain injury, while early activation of microglia leads to increased blood–brain barrier permeability and infiltration of circulating neutrophils and lymphocytes [5]. Immune cells release cytokines and chemokines (e.g., tumor necrosis factor [TNF]-α, interleukin [IL]-1β, IL-17, and IL-23) that aggravate inflammation following ischemic stroke. Recanalization with delayed recombinant tissue-type plasminogen activator treatment produces large amounts of ROS, forms a stressful microenvironment, activates adhesion molecules, and further aggravates the inflammatory response and immunocyte infiltration [6,7,8]. Mitochondria are the main source of intracellular ROS. Mitochondrial uncoupling proteins (UCPs) are metabolite transporters involved in the maintenance of intracellular ROS homeostasis. They are mainly expressed in the inner membrane of mitochondria and protect cells against oxidative stress injury [9]. Uncoupling protein-2 (UCP2) is a UCP family member that regulates the production of mitochondrial superoxide anion. Activation of UCP2 can prevent neurons from ischemia/reperfusion (I/R)-induced oxidative stress and preserves mitochondrial function [10]. Lu et al. [11] found that deletion of UCP2 aggravates neuroinflammation by activating nod-like receptor protein 3 in astrocytes following Parkinson’s disease. However, it is not clear whether UCP2 also regulates neuroinflammation in the setting of ischemic stroke.
Ferroptosis is an iron-dependent form of non-apoptotic cell death that was first discovered in 2012 by Brent R. Stockwell. Distinguished from other forms of programmed cell death, such as apoptosis and necrosis, ferroptosis has its own unique morphological, genetic, immunological, and biochemical features [12]. Ferroptosis is caused by iron-dependent phospholipid peroxidation, and is regulated by multiple cellular events, including iron handling, redox homeostasis, and mitochondrial balance [13,14,15]. In the past decade, knowledge of the physiological roles of ferroptosis have exponentially increased. Recent studies have shown that ferroptosis plays a role in several neurological diseases, including haemorrhagic stroke, neurodegenerative diseases, and ischemic stroke [16,17,18]. Mitochondrial dysfunction is implicated as a critical step towards ferroptosis. However, the exact mechanism of ferroptosis and mitochondrial dysfunction in ischemic stroke is poorly understood. Previous studies have suggested that UCP2 is a key regulator of mitochondrial redox status and lipid signaling by decreasing superoxide production and increasing hydrogen peroxide generation [19]. It is known that hydrogen peroxide is responsible for the induction of lipid peroxidation [20]. However, the role of UCP2 in ferroptosis has not been studied. The objective of this study was to investigate the effect of UCP2 deletion on ferroptosis and neuroinflammation following cerebral I/R injury and explore the underlying mechanisms.
Materials and Methods
Animals
Male C57BL/6 J wild-type mice (WT) (8 weeks old, n = 190, 25–30 g) were purchased from the Hubei Experimental Animal Research Center (Hubei, China; Nos. 42000600035350, 42000600032450). Ucp2 knockout (Ucp2−/−) mice (n = 52) were obtained from the Model Animal Research Center of Nanjing University. All Ucp2−/− male mice were raised to 6–8 weeks of age at the People’s Medical Animal Experimental Center of Wuhan University. All animal experimentation protocols were approved by the Animal Experimentation Ethics Committee of Wuhan University and were performed according to the guidelines of the Animal Welfare and Use Committee of Renmin Hospital of Wuhan University. All mice were reared in a room with controlled humidity (65 ± 5%) and temperature (25 ± 1 °C) on a 12/12-h light/dark cycle for 1 week before the experiment.
Drug Administration and Experimental Groups
Ferrostatin-1 (Fer-1) (5 mg/kg; SML0583-25MG, Sigma–Aldrich, St. Louis, MO, USA) or vehicle (2% dimethylsulfoxide + 2% Tween 80 + ddH2O) was delivered intraperitoneally (i.p.) to mice immediately after middle cerebral artery occlusion (MCAO) and 6 h post-reperfusion. The optimal drug concentration was selected in accordance with previous studies [21].
Middle Cerebral Artery Occlusion Model
Mice were anaesthetized with 5% isoflurane in O2 via a face mask. Intraoperative oxygen flow was maintained at 4–6 L/min with 1–1.5% isoflurane. Transient MCAO was performed. The MCAO model was slightly adjusted in accordance with a previously described method [22]. Briefly, the left internal carotid artery was exposed, and a 6–0 monofilament (Doccol Corp., Redlands, CA, USA) was inserted to ligate the left common carotid artery and external carotid artery, thereby opening the cephalic artery. The suture was inserted along the internal carotid artery and then removed after 1 h of occlusion to allow reperfusion. A thermostatic heating pad was used to monitor and stabilize the body temperature of the mice at 37 ± 0.5 °C. The same procedure was performed on sham mice but without monofilament ligation.
2,3,5-Triphenyltetrazolium Chloride Staining and Infarct Volume Measurement
Mice were heavily sedated before being administered an overdose of isoflurane and then decapitated. Brain tissue was cut into 2-mm coronal pieces and stained with 2,3,5-triphenyltetrazolium chloride (TTC) (17,779, Sigma–Aldrich). The collected brain slices were incubated in 4% paraformaldehyde (G1101-500ML, Servicebio, Wuhan, China) for 48 h. A blinded observer used ImageJ v1.37 (NIH, Bethesda, MA, USA) to quantify and assess the infarct volume [23], which was subsequently normalized and expressed as a percentage of the nonischemic hemisphere to account for oedema.
Neurological Function Assessment
Before evaluation of neurological function, any dead animals or unsuccessful models (including those without infarction or infarction with hemorrhage) were excluded. Neurological assessment was scored by the same examiner as a single-blind observer. Mice in the MCAO group at 0 h, 12 h, and 72 h after reperfusion were examined. The Longa scoring system [24] was used for evaluation: 0, no symptoms of nerve injury; 1, the right forepaw could only be slightly straightened when the mouse tail was lifted; 2, turned right when walking (moderate); 3, barely walking, right paralysis (severe); and 4, unable to walk spontaneously.
Brain Water Content
After 3 days of MCAO, the mice were deeply anaesthetized and euthanized with an overdose of isoflurane. Brain tissue was then removed and coronal brain sections (approximately 3-mm thick) were cut 4 mm from the frontal pole. Sections were divided into ipsilateral and contralateral hemispheres. Brain samples were immediately weighed on an electronic balance to determine the wet weight, and then dried in an oven at 120 °C for 8 h to determine the dry weight. The water content of the brain was calculated as (wet weight—dry weight)/wet weight × 100%.
Cell Culture, Oxygen–Glucose Deprivation/Reperfusion, and Transfection
HT-22 and BV2 cells were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China) and grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (#13,011–8611, Zhejiang Tianxing Biotechnology Co., Ltd., Zhejiang, China). Prior to induction of oxygen–glucose deprivation/reperfusion (OGD/RX) injury, cultured cells were in the logarithmic growth phase. They were washed twice with phosphate-buffered saline (PBS) and maintained in glucose-free DMEM. Cells were then placed in a hypoxic incubator (CB-210 Hypoxia Workstation, Binder, Tuttlingen, Germany) containing 1% O2, 5% CO2, and 94% N2 at 37 °C for the appropriate time to replicate OGD damage [6]. After that, cultures were recovered with glucose in DMEM and placed under normoxia (37 °C, 5% CO2) for 12 h (reoxygenation). To induce UCP2 overexpression and knockdown in vitro, cells were transfected with plasmid (10 nmol/l) or si-Ucp2 (50 nmol/L) for 24 h using Lipo6000™ transfection reagent (C0526, Beyotime, Shanghai, China) according to the manufacturer’s protocol. At 12 h post-transfection, the medium was changed and cells were incubated for a further 2 days. After confirmation of successful UCP2 overexpression and knockdown, cells were harvested for the subsequent experiments. Cell Counting Kit – 8 (C0039, Beyotime) was used to assess cell viability.
Measurement of Fe2+, Malondialdehyde, and Glutathione Levels
After 3 days of MCAO, the mice were deeply anaesthetized and euthanized with an overdose of isoflurane. Fresh brain tissue was obtained and kept on ice. Then, Fe2+, glutathione (GSH), and malondialdehyde (MDA) levels were determined using relevant kits (A006-2–1 for GSH, A003-1–2 for MDA, and A039-2–1 for Fe2+, Nanjing Jiancheng, Nanjing, China).
TdT-Mediated dUTP Nick End-Labeling Staining
A commercially available TdT-mediated dUTP nick end-labeling (TUNEL) staining kit (C1088, Beyotime) was used to detect apoptosis, as described previously [25]. Images were acquired using an automated fluorescence microscope (BX63, Olympus, Tokyo, Japan).
Enzyme-Linked Immunosorbent Assay
The infarct area of brain tissue homogenates containing IL-6, IL-1β, TNF-α, IL-10, IL-13, and IL-4 was centrifuged at 1200 × g, 10 min, 4 °C. In addition, concentration of 12(S)-hydroxyeicosatetraenoic acid (HETE) in supernatants was examined. All substances were measured with enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions (MLB00C for IL-1β, MTA00B for TNF-α, M1000B for IL-10, and M4000B for IL-4, R&D Systems, Minneapolis, MN, USA; abs520012 for IL-13, ab132023 for IL-6, and ab133034 for 12[S]-HETE, Absin Bioscience, Shanghai, China).
Lipid Peroxidation Assessment with BODIPY
First, cells in the logarithmic growth phase were seeded into 24-well plates and incubated with BODIPY (1 μM, D3861, Thermo Fisher Scientific) for 1 h at 37 °C. After incubation, cells were collected, washed with PBS, and then resuspended in 500 μl fresh PBS. Cell fluorescence was detected using a BD Accuri™ C6 flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed using FlowJo software (BD Biosciences).
Immunofluorescence Staining
First, mice were deeply anesthetized with sodium pentobarbital (20 mg/kg, i.p.), then infused with PBS (0.01 M, pH 7.4, 4 °C), followed by 4% paraformaldehyde solution (pH 7.4, 4 °C). The brain was fixed by immersion in the same paraformaldehyde solution for 48 h at 4 °C. Next, the brain was cut into 4-µm coronal sections using a freezing microtome (Cryotome FSE, Thermo Fisher Scientific). After washing with fresh PBS, the sections were blocked with 0.1 M PBS containing 5% fetal bovine serum and 0.05% Triton X-100 for 1 h at room temperature. After washing five times with PBS, primary antibody was added and incubated overnight for 12 h at 4 °C. The next day, the corresponding secondary antibody was added and incubated for 2 h at 25 °C. Sections were cleaned and sealed with 4′,6-diamidino-2-phenylindole (DAPI) (GDP1024, G1101-500ML, Servicebio) before examination under an automatic fluorescence microscope (BX63, Olympus). ImageJ software was used to count the number of immunoreactive cells (Media Cybernetics Inc., Rockville, MD, USA). Each mouse was counted in four different fields, with three mice in each group. Single-blind observers counted the data. Anti-UCP2 (1:200; ab247184, Abcam, Cambridge, UK), anti-NeuN (1:200; ab104224, Abcam), anti-myeloperoxidase (MPO) (1:100, GB11224, Servicebio), anti-CD68 (1:200; MCA1957, AbD Serotec, Oxford, UK), anti-glial fibrillary acidic protein (GFAP) (1:50; ab7260, Abcam), and anti-glutathione peroxidase 4 (GPX4) (1:200; ab125066, Abcam). The secondary antibodies used were Alexa 594-conjugated antibody (1:200 for NeuN, CD68, GFAP, and MPO, ANT030, Millipore, Billerica, MA, USA) or Alexa 488-conjugated antibody (1:200 for UCP2, ANT024, Millipore). Images were captured by a fluorescence microscope (DX51, Olympus).
Coimmunoprecipitation
Coimmunoprecipitation was performed using a commercial kit (abs955, Absin). First, HT-22 cells were transfected to overexpress Flag-tagged GPX4 plasmid or HA-tagged UCP2 plasmid. After removing the medium, cells were harvested and washed three times with ice-cold fresh 1 × PBS. Then, PBS was removed and 0.5 ml of ice-cold lysis buffer (1% NP40 lysis buffer with protease-inhibitor cocktail) was added to each cell plate (10 cm) and incubated for at least 5 min on ice. Cell lysates were collected in microcentrifuge tubes and centrifuged at 14,000 × g for 10 min at 4 °C. The supernatant was retained. Cell lysates were incubated with protein A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4 °C for 30–60 min with rotation. After centrifugation at 12,000 × g, for 1 min at 4 °C, the supernatants were collected into a new microcentrifuge tube. Cell lysates were incubated with anti-Flag or anti-HA antibodies overnight at 4 °C with rotation. The next day, 5 μl protein A/G agarose beads were added and mixed gently for 1–3 h. Immunocomplexes were then centrifuged at 12,000 × g for 1 min at 4 °C and washed three times with 200 µl PBS. Immunocomplexes were subjected to immunoblotting using the indicated primary and corresponding secondary antibodies.
Real-Time RT-PCR
PrimerBank 1 (https://pga.mgh.harvard.edu/primerbank/) was used to obtain primer sequences, which are listed in Table 1. On the third day post-MCAO or -OGD/RX, the penumbral region of the ischemic hemisphere and treated cells were extracted using RNAisoPlus (#9109, Takara, Shiga, Japan) and chloroform. The concentration and purity of total RNA were determined, and then 1 g of total RNA was reverse-transcribed using the PrimerScript RT reagent kit containing gDNA Eraser (RR820a, Takara) to remove genomic DNA. For quantitative PCR (qPCR), cDNA was mixed with synthetic primers from the Beijing Genomics Institute and SYBR Premix Ex Taq2 (RR047a, Takara). The following thermocycling parameters were used: 50 °C for 2 min, 95 °C for 10 min, followed by 45 cycles of 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 15 s. Finally, t values were normalized to the internal reference glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Western Blot Analysis
Western blotting was performed as previously described [26]. Ipsilateral brain and cell lysates were used to extract total protein. A bicinchoninic acid kit (G2026, Servicebio) was used to determine protein content. Protein samples (20 µl/well) were separated by electrophoresis on 4–15% sodium dodecyl sulfate–polyacrylamide gels and then transferred to polyvinylidene fluoride membranes (SF1J090I08, Millipore). Membranes were blocked for 1 h in 5% skim milk containing PBS/0.1% Tween, followed by overnight incubation with mouse anti-GPX4 (1:200; ab125066, Abcam), anti-Ig (1:1000, Proteintech, Wuhan, China), anti-UCP2 (1:1,000; ab97931, Abcam), anti-phosphorylated (p)-AMPK (1:1.000; ab133448, Abcam), and anti-total (t)-AMPK. After washing with PBS/0.1% Tween, membranes were incubated at room temperature for 1–2 h with IRDye-labeled secondary antibody (1:10,000; c60405-05, Li-Cor Bioscience, Lincoln, NE, USA). Images were captured using the Odyssey Western blot analysis system (LI-COR). Quantity One v4.6.2 software (Bio-Rad Laboratories, Hercules, CA, USA) was used to calculate relative band intensities, which were then normalized to the tubulin control. All these experiments were performed three times.
Statistical Analysis
GraphPad Prism 8 (GraphPad Software, Boston, MA, USA, www.graphpad.com) and SPSS 26.0 statistical software (IBM Corp., Armonk, NY, USA) were used for analyses. All experimental data were analyzed using mean ± SD. A completely randomized one-way analysis of variance (ANOVA) design was used to compare groups. Homogeneity of variance was tested using the least significant difference (LSD) t-test. p < 0.05 indicated a significant difference.
Results
Time-Course Expression and Cellular Localization of UCP2 After Ischemic Stroke
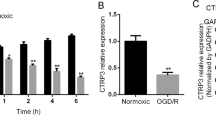
We first investigated expression of UCP2 levels and its cellular location following ischemic stroke in mice. Protein and gene expression levels of UCP2 were measured by Western blotting and RT-PCR in ischemic cortical tissue from groups at different time points (6 h, 12 h, 24 h, 48 h, and 72 h) after MCAO and the Sham group. The results showed that mRNA and protein expression of UCP2 were markedly reduced in a time-dependent manner after MCAO (p < 0.05 compared with the Sham group; Fig. 1a–c). Next, we detected protein and gene expression of UCP2 in a mouse hippocampal cell line (HT-22) following OGD/RX. Again, Western blotting and RT-PCR were used to examine protein and gene expression of UCP2 in HT-22 cells at different time points (0 h, 4 h, 8 h, and 10 h) after OGD/RX; the results showed that mRNA and protein expression of UCP2 were markedly reduced in a time-dependent manner after OGD/RX (p < 0.05 compared with 0 h group; Fig. 1d–f). To confirm which brain cell types showed high expression of UCP2, double immunofluorescence staining was performed. Our results showed that UCP2-positive cells mainly co-localized with NeuN-positive cells (a neuronal marker) in both the Sham and MCAO groups (Fig. 2a). Moreover, immunofluorescence staining revealed the significantly reduced proportion of UCP2+ and NeuN+ neuronal cells after ischemia (p < 0.05 compared with Sham group; Fig. 2b). These results indicate that UCP2 expression is suppressed in ischemic stroke and that UCP2 is highly expressed in neuronal cells.

Reduction in UCP2 coincides with cerebral I/R injury. a Representative image of UCP2 in tissue extracts of mouse brain after 60 min of middle cerebral artery occlusion (MCAO) followed by reperfusion for various durations. Tubulin was used as a loading control. b Quantitative analysis of UCP2 protein expression levels. c mRNA levels of UCP2 measured by qPCR. RNA was extracted from mouse brains subjected to 60 min of MCAO followed by reperfusion for various durations. d Representative image of UCP2 in HT-22 cells after 0 h, 4 h, 8 h, and 10 h of oxygen–glucose deprivation and then 12 h of reoxygenation (OGD/RX). e Quantitative analysis of UCP2 protein expression levels in HT-22 cells. f mRNA levels of UCP2 were determined by qPCR of RNA extracted from OGD/RX-treated HT-22 cells. Data are mean ± SD for all panels. *p < 0.05, **p < 0.01 by one-way ANOVA. All data are representative of five independent experiments unless otherwise indicated

Cellular location of UCP2 in brain cell types. a Representative imaging of double immunofluorescent labeling for Iba-1 with UCP2, NeuN with UCP2, and GFAP with UCP2 in the cerebral cortex of the Sham group and MCAO groups. Sections were counterstained with DAPI. b Quantitative analysis of UCP2-positive and NeuN-positive cells in the Sham and MCAO groups. Scale bar = 25 µm. Yellow arrows to indicate co-labeled cells. Data are mean ± SD for all panels: *p < 0.05, **p < 0.01 by one-way ANOVA. All data are representative of five independent experiments unless otherwise indicated
UCP2 Deficiency Aggravates Ischemic Brain Injury
Next, we explored the effects of UCP2 deficiency on ischemic stroke. Infarct volume and brain water content were measured at 3 days after MCAO. Neurological deficits were measured at 0 h, 12 h, and 72 h after MCAO. Our results showed that compared with WT mice, Ucp2−/− mice had increased infarct volumes and aggravated brain oedema at 3 days after MCAO (p < 0.05; Fig. 3a, b, and d). Moreover, compared with WT mice, UCP2 deficiency also increased neurological deficit scores after MCAO (p < 0.05; Fig. 3c). Ferrostatin-1, a highly potent and selective small molecular inhibitor of ferroptosis, was used to examine the neuroprotective effect of ferroptosis inhibition. We also verified the neuroprotective effect of Ferrostatin-1 after MCAO, compared with Sham + Fer group and MCAO + WT group, respectively (p < 0.05; Fig. 3). Additionally, by treating Ucp2−/− mice with Fer-1, we found that infarct volume, brain oedema, and neurological deficit scores could be partially reversed in treated Ucp2−/− mice compared with untreated Ucp2−/− mice (p < 0.05; Fig. 3a–d). TUNEL staining labels DNA breakpoints with 3′-OH terminals and is well known to detect DNA damage and cell apoptosis. The levels of DNA fragmentation and cell apoptosis were increased in MCAO mice and significantly increased by UCP2 deficiency (Fig. 3e, f). Fer-1 treatment partially reversed this increase.

UCP2 deficiency aggravates ischemic brain damage. a 2,3,5-Triphenyltetrazolium chloride staining of brain tissue from mice at 3 days after MCAO. Quantification of infarct sizes are shown (b). c Neurological deficit scores in mice at 0 h, 24 h, and 72 h after MCAO. d Brain water content in mice at 3 days after MCAO. e, f Representative image and quantification of TUNEL-positive cells in ischemic cortical tissue from mice at 3 days after MCAO. g, h Representative image and quantification of CD68-positive cells in ischemic cortical tissue from mice at 3 days after MCAO. Scale bar = 50 µm. Mean ± SD. *p < 0.05, **p < 0.01 by one-way ANOVA. n = 5/group. WT, wild-type; UCP2−/−, Ucp2.−/− mice; Fer, ferrostatin-1
UCP2 Deficiency Accelerates Neuroinflammation After MCAO
Given that neuroinflammation plays an important role in ischemic stroke, we investigated the neuroinflammatory effect of UCP2 deficiency on ischemic stroke. We detected activation of local resident cells (microglia) and recruitment of peripheral immune cells (neutrophils) by immunofluorescence staining. The results showed that compared with WT mice, UCP2 deficiency significantly increased microglia (CD68-positive cells) activation (Fig. 3g, h) and neutrophil (MPO-positive cells) infiltration (Fig. 4a, b) in the brain on day 3 after MCAO. In addition, we also found that mRNA expression of proinflammatory cytokines (TNF-α, IL-1β, and IL-6) was increased in Ucp2−/− mice compared with WT mice after MCAO at 3 days (Fig. 4c–e). In contrast, mRNA levels of anti-inflammatory cytokines (IL-10, IL-13, and IL-4) were reduced (Fig. 4f–h). Quality of the RNA is shown in Supplemental Fig. 1a. Taken together, these results suggest that UCP2 silencing enhanced the inflammatory response and aggravated brain injury following ischemic stroke in mice.

UCP2 inhibits post-ischemic neuroinflammation. a, b Representative image and quantification of myeloperoxidase (MPO)-positive cells in cerebral tissue from mice at 3 days after MCAO. c–e mRNA levels of proinflammatory cytokines (TNF-α, IL-6, and IL-1β) in ischemic cortical tissue from mice at 3 days after MCAO. f–h mRNA levels of proinflammatory cytokines (IL-13, IL-10, and IL-4) in ischemic cortical tissue from mice at 3 days after MCAO. Scale bar = 50 µm. Mean ± SD. *p < 0.05, **p < 0.01 by one-way ANOVA. n = 3/group. WT, wild-type; UCP2−/−, Ucp2.−/− mice; Fer, ferrostatin-1
UCP2 Inhibits Neuronal Ferroptosis Post-Ischemia
UCP2 is reported to increase the rate of calcium uptake, reduce ROS production, and suppress lipid peroxidation. Lipid peroxidation, degradation of GSH, and accumulation of redox-active iron are three key events in ferroptosis. Thus, we further explored the effects of UCP2 on neuronal ferroptosis. We measured the expression of ferroptosis-related proteins, including GPX4, ACSL4, and SLC7A11. Western blotting showed increased expression of ACSL4 and decreased expression of GPX4 and SLC7A11 in Ucp2−/− mice compared with WT mice after MCAO (Fig. 5a–d). Fer-1 co-treatment partially reversed these changes (Fig. 5a–d). We next measured MDA and GSH content in fresh brain tissue from mice in each group. Compared with WT mice, UCP2 deficiency increased MDA levels and reduced GSH content in MCAO mice (p < 0.05; Fig. 5e, f). These expression levels were partially reversed in Ucp2−/− mice by co-treating with Fer-1, compared with Fer-1 alone. As there is no specific marker to label ferroptosis, we also measured other ferroptosis-related indicators, specifically, 12(S)-HETE by ELISA to detect downstream metabolites of ferroptosis in MCAO mice. Our results showed that 12(S)-HETE levels were increased after MCAO and further increased in Ucp2−/− mice compared with WT mice (p < 0.05; Fig. 5g). Moreover, 12(S)-HETE levels increased in Ucp2−/− mice with Fer-1 co-treatment compared with Fer-1 only. GPX4 expression was also detected by immunofluorescence staining. Consistent with our Western blot results, UCP2 deficiency decreased the number of GPX4-positive cells in ischemic cortical tissue after MCAO.

UCP2 inhibits post-ischemic neuronal ferroptosis. a Representative image and quantification of glutathione peroxidase 4 (GPX4) expression in mouse brain after 3 days of MCAO. Tubulin was used as a loading control. Ferroptosis-related indicators, including malondialdehyde (MDA) (b), glutathione (GSH) (c), and 12(S)-hydroxyeicosatetraenoic acid (12[S]-HETE) (d) were measured using commercial kits. e, f Representative image and quantification of GPX4-positive cells in cerebral tissue from mice at 3 days after MCAO. Scale bar = 50 µm. Mean ± SD. **p < 0.01, ***p < 0.001 by one-way ANOVA. n = 5/group. WT, wild-type; UCP2−/−, Ucp2.−/− mice; Fer, ferrostatin-1
UCP2 Attenuates Ferroptosis in HT-22 Cells Following OGD/RX
To determine whether UCP2 also has a neuroprotective effect via inhibition of neuronal ferroptosis in in vitro models of ischemic stroke, we first used siRNA and vehicle vector to downregulate HT-22 cell expression of UCP2. OGD/RX was used to simulate I/R injury in vitro. Western blotting confirmed that si-Upc2 significantly decreased levels of UCP2 protein (Supplemental Fig. 1b, c). OGD/RX treatment significantly suppressed HT-22 cell viability and increased lactate dehydrogenase (LDH) release compared with controls, which was further reduced by Ucp2 silencing compared with the vector control group (Fig. 6a, b). Subsequently, Fe2+, MDA, and GSH concentrations were measured in each group. Consistent with our in vivo results, treatment of HT-22 cells by OGD/RX also led to a visible decrease in GSH concentration and increase in Fe2+ and MDA levels in vitro. Furthermore, Ucp2 silencing in HT-22 cells exacerbated OGD/RX-induced ferroptosis by further reducing GSH concentration and increasing Fe2+ and MDA levels (Fig. 6c–e). Consequently, we detected expression of ferroptosis-related proteins, including GPX4, ACSL4, and SLC7A11, by Western blot analysis in HT-22 cells following OGD/RX. Consistent with our in vivo results, the treatment of HT-22 cells by OGD/RX reduced GPX4 and SLC7A11 expression levels and increased ACSL4 levels. Ucp2 silencing further decreased GPX4 and SLC7A11 levels and increased ACSL4 levels (Fig. 6f–i). Lipid peroxidation measured using the C11-BODIPY 581/591 probe is the gold standard for detection of ferroptosis [27], but is not suitable for in vivo experiments. Thus, we used the lipid peroxidation-sensitive dye, BODIPY 581/591 C1, for in vitro HT-22 cells. We found that OGD/RX increased lipid peroxidation levels in HT-22 cells, and that UCP2 downregulation aggravated OGD/RX-induced lipid peroxidation (Fig. 6j, k). To further determine the critical role of UCP2 in lipid peroxidation following ischemic stroke in vitro, we used Fer-1 to rescue neurons exposed to OGD/RX. Similar to our in vivo results, Fer-1 treatment in Ucp2-silenced HT-22 cells led to decreased lipid peroxidation levels compared with the untreated Ucp2-silenced group (Fig. 6i, m).

UCP2 knockdown aggravates neuronal ferroptosis after OGD/RX. a Cell viability and b lactate dehydrogenase (LDH) release were measured and quantified in transfected HT-22 cells subjected to oxygen–glucose deprivation and reoxygenation (OGD/RX) or untreated (Ctrl). Levels of Fe2+ (c), glutathione (GSH) (d), and malondialdehyde (MDA) (e) were measured in transfected HT-22 cells subjected to OGD/RX. f, i Representative and quantitative images of GPX4, ACSL4, and SLC7A11 in HT-22 cells transfected with NC or si-Ucp2 and subjected to OGD/RX or normal conditions. j, k Representative histograms and mean fluorescence intensity (MFI) of BODIPY oxidation in HT-22 cells transfected with NC or si-Ucp2 and subjected to OGD/RX or normal conditions. l, m Representative histograms and MFI of BODIPY oxidation in HT-22 cells transfected with NC or si-Ucp2 and subjected to OGD/RX with or without Fer-1. Mean ± SD. *p < 0.05, **p < 0.01 by two-way ANOVA. All data are representative of three independent experiments unless otherwise indicated. Ctrl, control group without Ucp2 knockdown; NC, negative control group without OGD/RX treatment
Next, to further confirm the neuroprotective effect of UCP2 in ischemic stroke, we transfected HT-22 cells with an overexpression plasmid for UCP2. Western blotting was used to determine the transfection efficiency (Supplemental Fig. 1b, c). Expectedly, UCP2 overexpression prominently enhanced HT-22 cell viability and decreased LDH release under OGD/RX conditions (Fig. 7a, b). Next, we determined the concentrations of Fe2+, MDA, and GSH in each group. The levels of Fe2+ and MDA in the OGD/RX group were significantly higher compared with the control group, while UCP2 overexpression decreased Fe2+ and MDA concentrations in HT-22 cells (Fig. 7c, d). The levels of GSH were considerably reduced in the OGD/RX group, and significantly reversed by UCP2 overexpression. Moreover, we also investigated the effects of UCP2 overexpression on ferroptosis-related proteins, including GPX4, ACSL4, and SLC7A11. Western blot results showed reduced expression of GPX4 and SLC7A11 levels, and increased ACSL4 expression in HT-22 cells following OGD/RX. UCP2 overexpression reversed the levels of GPX4 and SLC7A11 and reduced ACSL4 expression. Taken together, these results suggest that endogenous UCP2 attenuates ferroptosis and therefore plays a protective role following ischemic stroke.

UCP2 overexpression inhibits neuronal ferroptosis after OGD/RX. a Cell viability and b lactate dehydrogenase (LDH) release were measured and quantified in transfected HT-22 cells subjected to oxygen–glucose deprivation and reoxygenation (OGD/RX) or untreated (Ctrl). Levels of Fe2+ (c), glutathione (GSH) (d), and malondialdehyde (MDA) (e) were measured in transfected HT-22 cells subjected to OGD/RX. f, i Representative and quantitative images of GPX4, ACSL4, and SLC7A11 in HT-22 cells transfected with NC or UCP2-OE and subjected to OGD/RX or normal conditions. Mean ± SD. *p < 0.05, **p < 0.01 by one-way ANOVA. All data are representative of three independent experiments unless otherwise indicated. Ctrl, control group without UCP2 overexpression; NC, negative control group without OGD/RX treatment
UCP2 Reduces Microglia-Mediated Inflammation
Microglia are major contributors to the neuroinflammation of ischemic stroke. To simulate I/R injury, we performed OGD/RX in BV2 cells and then knocked-down Ucp2 with siRNA and vehicle vectors individually transfected into BV2 cells. We measured the levels of proinflammatory cytokines (including TNFα, IL-6, and IL-1β), and anti-inflammatory cytokines (including IL-13, IL-10, and IL-4) by ELISA analysis. Our results show that Ucp2 silencing increased TNFα, IL-6, and IL-1β levels, and reduced IL-13, IL-10, and IL-4 levels in BV2 cells after OGD/RX (Fig. 8a–i). We also detected lipid peroxidation levels in BV2 cells using BODIPY 581/591 C1. We found no significant difference in lipid peroxidation between control groups and Ucp2 knockdown groups (Fig. 8h). To further confirm that UCP2 attenuates neuroinflammation following ischemic stroke, we transfected BV2 cells with an overexpression plasmid for UCP2. We again detected the levels of proinflammatory cytokines and anti-inflammatory cytokines by ELISA analysis after OGD/RX. Our results showed that UCP2 overexpression reduced the levels of TNFα, IL-6, and IL-1β, but increased the levels of IL-13, IL-10, and IL-4 in BV2 cells after OGD/RX (Fig. 9a–i).

Silencing of UCP2 enhances microglia-mediated inflammation. a–c The levels of anti-inflammatory cytokines (IL-13, IL-10, and IL-4) in microglial BV2 cells transfected with NC or si-Ucp2 and subjected to OGD/RX or normal conditions; determined by ELISA. d–f The levels of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) in microglial BV2 cells transfected with NC or si-Ucp2 and subjected to OGD/RX or normal conditions; determined by ELISA. g, h Flow cytometry analysis of representative histograms and mean fluorescence intensity (MFI) of BODIPY oxidation in microglial BV2 cells transfected with NC or si-Ucp2 and subjected to OGD/RX. Mean ± SD. *p < 0.05, **p < 0.01 by one-way ANOVA. n = 5/group. Ctrl, control group without Ucp2 knockdown; NC, negative control group without OGD/RX treatment

Overexpression of UCP2 reduces microglia-mediated inflammation. a–c The levels of anti-inflammatory cytokines (IL-13, IL-10, and IL-4) in microglial BV2 cells transfected with NC or UCP2-OE and subjected to OGD/RX or normal conditions; determined by ELISA. d–f The levels of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) in microglial BV2 cells transfected with NC or UCP2-OE and subjected to OGD/RX or normal conditions; determined by ELISA. Mean ± SD. *p < 0.05, **p < 0.01 by one-way ANOVA. n = 5/group. Ctrl, control group without UCP2 overexpression; NC, negative control group without OGD/RX treatment
Ucp2 Knockdown Suppresses the AMPKα/NRF1 Pathway After I/R Injury
5′ AMP-activated protein kinase (AMPK)-α activation is responsible for maintaining mitochondrial homeostasis under various stress perturbations. We used Western blot analysis to measure the expression of AMPKα/nuclear respiratory factor 1 (NRF1) pathway-related proteins, including p-AMPKα, t-AMPKα, NRF1, and mitochondrial transcription factor A (TFAM). Our results showed reduced expression of UCP2, p-AMPKα, NRF1, and TFAM in Ucp2−/− mice at 3 days after MCAO. Genetic depletion of Ucp2 reduced the expression of p-AMPKα, NRF1, and TFAM (Fig. 10d–h). These results indicate that UCP2 may exert its physiological effect by regulating the AMPK/NFR1 signaling pathway. Moreover, GPX4 is a unique lipid hydroperoxidase enzyme that prevents ferroptosis by disrupting the lipid peroxidation chain reaction. Interestingly, in our study, we found that UCP2 suppressed the process of neuronal ferroptosis by directly interacting with GPX4. First, we detected UCP2 and GPX4 expression in the cytoplasm of HT-22 cells by immunofluorescence analysis after OGD/RX (Supplemental Fig. 2a). Then, HA-tagged UCP2 or Flag-tagged GPX4 were overexpressed in HT-22 cells after OGD/RX. Immunoprecipitation results showed that UCP2 co-immunoprecipitated GPX4 and vice versa (Supplemental Fig. 2b, c).

UCP2 knockdown suppresses the AMPKα/NRF1 pathway after I/R injury. a Western blotting to measure protein expression levels of UCP2, p-AMPK, t-AMPK, NRF1, and TFAM in the ischemic cortical penumbra region at 3 days after cerebral ischemia. b–e Quantitative analysis of the protein levels ofof UCP2, p-AMPK, t-AMPK, NRF1, and TFAM. Mean ± SD. *p < 0.05, **p < 0.01 by one-way ANOVA. n = 3/group. WT, wild-type; UCP2−/−, Ucp2−/− mice
Discussion
Based upon the results of our present study, we hypothesize a dual neuroprotective role for UCP2 in ischemic stroke, including the alleviation of ferroptosis and inhibition of the neuroinflammatory response. We first found that UCP2 deficiency aggravated I/R injury by promoting lipid peroxidation, accelerating neuronal ferroptosis, and stimulating microglial activation and neutrophil infiltration after ischemic stroke in vivo and in vitro. Importantly, we also intimate that UCP2 may exert its physiological effect by regulating the AMPK/NFR1 signaling pathway following ischemic stroke. Additionally, we found a protective role of UCP2 in the progression of lipid peroxidation and ferroptosis may by direct interaction with GPX4.
Previous research has suggested that UCP2 regulates the cellular redox state by promoting mitochondrial hydrogen peroxide release and transporting intramitochondrial protons to an extramitochondrial site [28, 29]. In the central nervous system, UCP2 is widely expressed in neurons, residing and invading microglia, neutrophils, and endothelial cells [30]. Recent investigations have implicated UCP2 in diverse diseases, including obesity and diabetes [31, 32], atherosclerosis [33], neurodegenerative diseases [34,35,36], and cancer [37]. Our findings show that UCP2 appears to protect the brain against cerebral I/R injury. After MCAO in mice and OGD/RX in neuronal cell lines, expression of UCP2 was decreased with increasing reperfusion time. Immunofluorescence results showed that UCP2 labeling co-localized predominantly with NeuN (a neuronal marker), compared with CD68 (a microglia/macrophage marker) and GFAP (an astrocyte marker), suggesting that UCP2 is mainly expressed in neurons. As ischemic stroke induced a reduction in UCP2, we used Ucp2−/− mice to test its effects on brain ischemia. We found that Ucp2−/− mice had increased infarct volumes, neurological deficit scores, and cerebral oedema compared with WT mice. Consistent with these in vivo results, Ucp2 knockdown also reduced the viability of HT-22 cells following OGD/RX.
Ferroptosis was first identified by Stockwell et al., in 2012 [12] as a new form of regulated cell death, and has since been closely correlated with many diseases [38, 39]. There are four key factors in the process of ferroptosis induction: iron, polyunsaturated fatty acids, oxygen, and a reduction in antioxidants [40]. Owing to its high lipid abundance and possibly fewer antioxidant properties, the brain is extremely susceptible to lipid peroxidation-induced injury during cerebral ischemia and reperfusion. Moreover, previous studies have shown that ferroptosis is implicated in ischemic stroke, while ferroptosis inhibitors (such as liproxstatin-1 and Fer-1) prevent brain injury in MCAO mice MCAM [21]. Recently, as a crucial regulator of mitochondrial ROS, UCP2 was shown to play a critical role in the antioxidant defense mechanism. However, whether UCP2 protects from I/R injury by inhibiting ferroptosis is unclear. In this study, we found that Ucp2 knockdown aggravated levels of lipid peroxidation, increased accumulation of Fe2+ and MDA, and reduced GPX4 expression in MCAO and OGD/RX ischemia stroke models. Moreover, genetic depletion of Ucp2 worsened ferroptosis in vivo and in vitro, which was efficiently rescued by the ferroptosis inhibitor, Fer-1.
Ferroptosis is influenced by a variety of critical factors, including SLC7A11, a vital cysteine transporter that transports extracellular cystine into cells. Cysteine is converted into GSH, which can be used by GPX4 to reduce lipid hydroperoxides for suppressing ferroptosis. Once system xc− is inhibited, cell ferroptosis is induced. SLC7A11 has been shown to modulate ferroptosis in multiple studies. Liu et al. found that pumilio RNA binding family member 2 suppressed SLC7A11 by inhibiting sirtuin 1 to aggravate brain injury in MCAO mice [41]. Moreover, Zhu et al. also reported that miR-27a upregulated ferroptosis via SLC7A11, which aggravated cerebral I/R injury [42]. Consistent with previous studies, we also found that SLC7A11 expression was downregulated in our ischemic stroke model. UCP2 deficiency further reduced SLC7A11 expression and aggravated ferroptosis. Importantly, we found that UCP2 suppressed the process of neuronal ferroptosis by directly interacting with GPX4. It is well known that UCP2 is mainly expressed in mitochondria, while GPX4 is mostly expressed in the cytoplasm and to a lesser extent in mitochondria. We hypothesized that following ischemic stroke, ischemia and hypoxia lead to mitochondrial dysfunction or even rupture, which releases UCP2 into the cytoplasm where it binds to GPX4. Recent research has found that FUN14 domain containing 1, a mitophagy receptor located in mitochondria, directly binds to GPX4 to govern hepatic ferroptosis [43]. However, due to experimental conditions, we did not explore whether UCP2 can modify GPX4, how UCP2 is released into the cytoplasm, and whether UCP2 can bind to GPX4 in mitochondria in our present study.
The over-activated inflammatory response in the brain after ischemic stroke is considered a radical factor in brain injury, and blamed for the poor outcomes. Anti-inflammatory medications or nervous system treatments can be effective in alleviating brain injury and improving neurological outcomes [44]. After ischemia, primary immune cells, such as microglia and astrocytes, become dramatically activated, resulting in the release of inflammatory mediators and the invasion of peripheral inflammatory cells [45, 46]. These cells, in turn, may further aggravate neuronal damage in the cerebral ischemic penumbra and exacerbate damage in surrounding regions [46]. Several studies have suggested an important role of UCP2 in inflammatory responses [47]. UCP2 deletion in microglia can prevent changes in mitochondrial dynamics and function, microglial activation, and hypothalamic inflammation [48]. Thus, we assumed that Ucp2 silencing would enhance neuroinflammation during ischemic stroke. Our immunofluorescence and RT-PCR results confirmed this hypothesis. The levels of microglial activation and neutrophil infiltration were increased in Ucp2−/− mice. Moreover, Ucp2 deletion enhanced the levels of proinflammatory factors, including TNFα, IL-6, and IL-1β, and reduced the levels of anti-inflammatory cytokines, including IL-13, IL-10, and IL-4 in our in vitro and in vivo I/R models.
In conclusion, we demonstrate the importance of UCP2-regulated ferroptosis and neuroinflammation in ischemic stroke. Given the importance of ferroptosis and inflammatory responses in ischemia, UCP2 may be a new target for treating brain damage caused by ischemia stroke.
Our research has limitations. First, as previously stated, cerebral I/R injury encompasses multiple pathological processes; we only studied the relationship between mitochondrial dysfunction (using a related protein, UCP2), inflammation, and ferroptosis after ischemic stroke. Thus, we have not determined whether UCP2 can alleviate cerebral I/R injury via other pathways. Second, according to cerebral I/R injury, several signaling pathways are involved in mitochondrial dysfunction. We acknowledge the importance of the AMPK/NFR1 pathway but cannot exclude other possibilities. Finally, although we demonstrate that UCP2 is interacts directly with GPX4, the details of how UCP2 and GPX4 are altered remain unknown.
Data Availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request in compliance with ethical standards.
References
Campbell BCV, De Silva DA, Macleod MR, Coutts SB, Schwamm LH, Davis SM et al (2019) Ischaemic stroke. Nat Rev Dis Primers 5:70
Shi K, Tian DC, Li ZG, Ducruet AF, Lawton MT, Shi FD (2019) Global brain inflammation in stroke. Lancet Neurol 18:1058–1066
Han B, Jiang W, Liu H, Wang J, Zheng K, Cui P et al (2020) Upregulation of neuronal pgc-1alpha ameliorates cognitive impairment induced by chronic cerebral hypoperfusion. Theranostics 10:2832–2848
Dabrowska S, Andrzejewska A, Lukomska B, Janowski M (2019) Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J Neuroinflammation 16:178
Tao T, Liu M, Chen M, Luo Y, Wang C, Xu T et al (2020) Natural medicine in neuroprotection for ischemic stroke: challenges and prospective. Pharmacol Ther 216:107695
Sun MS, Jin H, Sun X, Huang S, Zhang FL, Guo ZN et al (2018) Free radical damage in ischemia-reperfusion injury: an obstacle in acute ischemic stroke after revascularization therapy. Oxid Med Cell Longev 2018:3804979
Granger DN, Kvietys PR (2015) Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol 6:524–551
Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A (2004) Targets for vascular protection after acute ischemic stroke. Stroke 35:2220–2225
Cui W, Chen S, Chi Z, Guo X, Zhang X, Zhong Y et al (2021) Screening-based identification of xanthone as a novel nlrp3 inflammasome inhibitor via metabolic reprogramming. Clin Transl Med 11:e496
de Bilbao F, Arsenijevic D, Vallet P, Hjelle OP, Ottersen OP, Bouras C et al (2004) Resistance to cerebral ischemic injury in ucp2 knockout mice: evidence for a role of ucp2 as a regulator of mitochondrial glutathione levels. J Neurochem 89:1283–1292
Lu M, Sun XL, Qiao C, Liu Y, Ding JH, Hu G (2014) Uncoupling protein 2 deficiency aggravates astrocytic endoplasmic reticulum stress and nod-like receptor protein 3 inflammasome activation. Neurobiol Aging 35:421–430
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072
Tang D, Chen X, Kang R, Kroemer G (2021) Ferroptosis: molecular mechanisms and health implications. Cell Res 31:107–125
Chen X, Li J, Kang R, Klionsky DJ, Tang D (2021) Ferroptosis: machinery and regulation. Autophagy 17:2054–2081
Fusco R, Scuto M, Cordaro M, D’Amico R, Gugliandolo E, Siracusa R et al (2019) N-palmitoylethanolamide-oxazoline protects against middle cerebral artery occlusion injury in diabetic rats by regulating the sirt1 pathway. Int J Mol Sci 20:19–4845
Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S (2017) Ferroptosis and cell death mechanisms in parkinson’s disease. Neurochem Int 104:34–48
Bao WD, Pang P, Zhou XT, Hu F, Xiong W, Chen K et al (2021) Loss of ferroportin induces memory impairment by promoting ferroptosis in alzheimer’s disease. Cell Death Differ 28:1548–1562
Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E et al (2019) Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177(1262–1279):e1225
Sreedhar A, Lefort J, Petruska P, Gu X, Shi R, Miriyala S et al (2017) Ucp2 upregulation promotes plcgamma-1 signaling during skin cell transformation. Mol Carcinog 56:2290–2300
Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D et al (2014) Ucp2 transports c4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci U S A 111:960–965
Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Li XL et al (2017) Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry 22:1520–1530
Perez-de-Puig I, Miro-Mur F, Ferrer-Ferrer M, Gelpi E, Pedragosa J, Justicia C et al (2015) Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol 129:239–257
McBride DW, Zhang JH (2017) Precision stroke animal models: the permanent mcao model should be the primary model, not transient mcao. Transl Stroke Res 8(5):397–404
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91
Hou K, Li G, Zhao J, Xu B, Zhang Y, Yu J et al (2020) Bone mesenchymal stem cell-derived exosomal microrna-29b-3p prevents hypoxic-ischemic injury in rat brain by activating the pten-mediated akt signaling pathway. J Neuroinflammation 17:46
Li P, Stetler RA, Leak RK, Shi Y, Li Y, Yu W et al (2018) Oxidative stress and DNA damage after cerebral ischemia: potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology 134:208–217
Verma N, Vinik Y, Saroha A, Nair NU, Ruppin E, Mills G, et al 2020 Synthetic lethal combination targeting bet uncovered intrinsic susceptibility of tnbc to ferroptosis. Sci Adv 6. https://doi.org/10.1126/sciadv.aba8968
Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S et al (2003) Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med 9:1062–1068
Normoyle KP, Kim M, Farahvar A, Llano D, Jackson K, Wang H (2015) The emerging neuroprotective role of mitochondrial uncoupling protein-2 in traumatic brain injury. Transl Neurosci 6:179–186
Richard D, Rivest R, Huang Q, Bouillaud F, Sanchis D, Champigny O et al (1998) Distribution of the uncoupling protein 2 mrna in the mouse brain. J Comp Neurol 397:549–560
Hidaka S, Yoshimatsu H, Kakuma T, Sakino H, Kondou S, Hanada R et al (2000) Tissue-specific expression of the uncoupling protein family in streptozotocin-induced diabetic rats. Proc Soc Exp Biol Med 224:172–177
Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D et al (2001) Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105:745–755
Blanc J, Alves-Guerra MC, Esposito B, Rousset S, Gourdy P, Ricquier D et al (2003) Protective role of uncoupling protein 2 in atherosclerosis. Circulation 107:388–390
Deierborg T, Wieloch T, Diano S, Warden CH, Horvath TL, Mattiasson G (2008) Overexpression of ucp2 protects thalamic neurons following global ischemia in the mouse. J Cereb Blood Flow Metab 28:1186–1195
Diano S, Matthews RT, Patrylo P, Yang L, Beal MF, Barnstable CJ et al (2003) Uncoupling protein 2 prevents neuronal death including that occurring during seizures: a mechanism for preconditioning. Endocrinology 144:5014–5021
Sullivan PG, Dube C, Dorenbos K, Steward O, Baram TZ (2003) Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann Neurol 53:711–717
Derdak Z, Mark NM, Beldi G, Robson SC, Wands JR, Baffy G (2008) The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res 68:2813–2819
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X et al (2016) Ferroptosis: process and function. Cell Death Differ 23:369–379
Yu H, Guo P, Xie X, Wang Y, Chen G (2017) Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med 21:648–657
Magtanong L, Ko PJ, Dixon SJ (2016) Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ 23:1099–1109
Liu Q, Liu Y, Li Y, Hong Z, Li S, Liu C (2023) PUM2 aggravates the neuroinflammation and brain damage induced by ischemia-reperfusion through the SLC7A11-dependent inhibition of ferroptosis via suppressing the SIRT1. Mol Cell Biochem 478(3):609–620
Zhu L, Feng Z, Zhang J, Du L, Meng A (2023) MicroRNA-27a regulates ferroptosis through SLC7A11 to aggravate cerebral ischemia-reperfusion injury. Neurochem Res 48(5):1370–1381
Bi Y, Liu S, Qin X, Abudureyimu M, Wang L, Zou R, Ajoolabady A, Zhang W et al (2024) FUNDC1 interacts with GPx4 to govern hepatic ferroptosis and fibrotic injury through a mitophagy-dependent manner. J Adv Res 55:45–60
Kim JY, Park J, Chang JY, Kim SH, Lee JE (2016) Inflammation after ischemic stroke: the role of leukocytes and glial cells. Exp Neurobiol 25:241–251
Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22:391–397
Liu Y, Ai K, Ji X, Askhatova D, Du R, Lu L et al (2017) Comprehensive insights into the multi-antioxidative mechanisms of melanin nanoparticles and their application to protect brain from injury in ischemic stroke. J Am Chem Soc 139:856–862
Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS et al (2015) The role of inflammation in perinatal brain injury. Nat Rev Neurol 11:192–208
Kim JD, Yoon NA, Jin S, Diano S (2019) Microglial ucp2 mediates inflammation and obesity induced by high-fat feeding. Cell Metab 30(952–962):e955
Acknowledgements
We thank Rachel James, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn), for editing the English text of a draft of this manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (82001119), National Natural Science Foundation of Hubei (2024AFD149, 2020CFB224), Fundamental Research Funds for the Central Universities (2042020kf0079, 2042019kf0061), and the Hubei Province Key Laboratory Open Project (2021KFY044).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval
The study was approved by the Ethics Committee of Renmin Hospital of Wuhan University and performed in compliance with the ARRIVE guidelines.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Lei Wang and Xiaona Li contributed equally to this work as first authors.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, L., Li, X., Chen, L. et al. Mitochondrial Uncoupling Protein-2 Ameliorates Ischemic Stroke by Inhibiting Ferroptosis-Induced Brain Injury and Neuroinflammation. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04288-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04288-0