
Abstract
Metastatic progression is a major burden for breast cancer patients and is associated with the ability of cancer cells to overcome stressful conditions, such as nutrients deprivation and hypoxia, and to gain invasive properties. Autophagy and epithelial-to-mesenchymal transition are critical contributors to these processes. Here, we show that the P2X4 purinergic receptor is upregulated in breast cancer biopsies from patients and it is primarily localised in endolysosomes. We demonstrate that P2X4 enhanced invasion in vitro, as well as mammary tumour growth and metastasis in vivo. The pro-malignant role of P2X4 was mediated by the regulation of lysosome acidity, the promotion of autophagy and cell survival. Furthermore, the autophagic activity was associated with epithelial–to-mesenchymal transition (EMT), and this role of P2X4 was even more pronounced under metabolic challenges. Pharmacological and gene silencing of P2X4 inhibited both autophagy and EMT, whereas its rescue in knocked-down cells led to the restoration of the aggressive phenotype. Together, our results demonstrate a previously unappreciated role for P2X4 in regulating lysosomal functions and fate, promoting breast cancer progression and aggressiveness.
Similar content being viewed by others


Introduction
During tumour development and progression, cancer cells acquire new abilities in order to overcome stressful microenvironmental conditions and fulfil energy demands required by their high metabolism [1,2,3]. Cancer cells can use autophagy as a powerful strategy to survive under metabolic challenges such as nutrient deprivation and reduced oxygen supply within the tumour microenvironment [4]. This stringent microenvironment also drives the expression of transcription factors responsible for the epithelial-to-mesenchymal transition (EMT) program [5, 6], another major process for tumour progression. EMT has been correlated with cancer aggressiveness and metastatic potential, and is generally associated with autophagy [7, 8], while the link between the two processes is still elusive.
Lysosomes are intracellular acidic compartments that are essential for the catabolic clearance and recycling of defective macromolecules or organelles, during autophagy and also following endocytosis of extracellular materials [9, 10]. Lysosome properties are dramatically altered in cancer cells [11, 12] and these induce cellular functions that are involved in cancer progression. These include EMT [13], the release of extracellular matrix degrading enzymes such as lysosomal cathepsins [14], cancer cell invasion and metastases [15, 16] and also the acquisition of resistance to anticancer treatments [17]. Nevertheless, relatively little is known about the key molecular determinants that modulate lysosome function and fate in cancers, and their dependence on microenvironmental conditions.
The P2X4 receptor belongs to the family of transmembrane ionotropic P2X receptors. While it acts as an ATP-gated non-selective cation channel, like other members of the family, P2X4 differs from the other subtypes by its predominant localization in endosomal and lysosomal organelles of immune cells, rather than to the plasma membrane [18]. In this study, we show that the P2X4 receptor is significantly overexpressed in human breast cancer samples compared to normal tissues. P2X4 is targeted to acidic compartments of highly invasive mammary cancer cells and sustained invasion in vitro as well as mammary tumour growth and metastatic progression in a syngeneic model of mammary cancer in immunocompetent mice. We further investigated whether lysosomal P2X4 regulates breast cancer cell properties to support aggressiveness. Importantly, P2X4 promoted the autophagic flux and associated maintenance of a mesenchymal phenotype, primarily under metabolic challenges. Collectively, our results provide compelling evidence supporting endolysosomal P2X4 as a critical protagonist driving cancer progression.
Results
Overexpression of P2X4 is associated with mammary tumour growth and metastatic progression
Purinergic receptors have recently emerged as central players in tumour development [19, 20]. Among them, the ATP-gated P2X7 receptor has attracted specific attention [21, 22], however not much is known about the involvement of other P2X receptors. Therefore, we performed bioinformatics gene expression analyses in breast cancer from The Cancer Genome Atlas (TCGA), using the UCSC Xena browser (https://xenabrowser.net). Five out of the seven P2RX genes were deregulated at the transcript level in primary tumours compared to adjacent non-tumoral breast tissues (Fig. 1A). P2RX1, P2RX6 and P2RX7 were significantly down-regulated whereas P2RX4 and P2RX5 were up-regulated in breast tumours (Fig. 1A). For the latter two genes, P2RX4 was more noticeably upregulated in breast cancer (median was 9.562) compared to adjacent non-tumoral tissue (median was 8.955), and this was independent of tumoral grade (Supplementary Fig. S1A). P2RX4 upregulation in primary breast tumours was confirmed using bc-GenExMiner mining tool (Supplementary Fig. S1B). Moreover, P2RX4 was upregulated in multiple types of cancers (15/24 tested types of cancers; Supplementary Fig. S1C). It was also overexpressed in metastases, as compared to both normal and adjacent non-tumoral tissues, but with a similar expression level to that of primary tumours (Fig. 1B). The expression of P2X4 proteins was assessed by immunohistochemistry on normal (10 tissues) and primary breast cancer tissues (108 tissues). No or weak staining was found in normal tissues, whereas 51% of breast cancer biopsies exhibited P2X4 staining, which was mainly cytoplasmic with a granular aspect (Fig. 1C). The staining was moderate in 32% and strong in 19% of breast tumour tissues (Fig. 1D). Overexpression of the P2X4 protein in breast tumours was confirmed using external data from Clinical Proteomic Tumor Analysis Consortium (CPTAC) and UALCAN portal (Supplementary Fig. S1D).

A Box plots showing P2RX1-7 expression in adjacent non-tumoral tissue (blue; n = 114) versus primary tumours (red; n = 1097) in TGCA-BRCA dataset. Expression values were estimated using IlluminaHiSeq and downloaded from UCSC Xena Browser. B Box plots showing P2RX4 gene expression in normal tissue (n = 179), adjacent non-tumoral tissue (n = 113), primary tumours (n = 1205) and metastases (n = 7), using the Genotype-Tissue EXpression (GTEx) normal breast and The Cancer Genome Atlas (TCGA) datasets. Expression values were estimated using RSEM norm_count and downloaded from UCSC Xena Browser. C Representative immunohistochemistry staining for P2X4 in normal (10 tissues) and primary breast cancer tissues (108 tissues). Scale bars, 20 μm. D Percentage of tissues with non-detected (ND)/low, medium or high P2X4 staining in breast tissues as in (C). E P2rx4 mRNA levels, assessed by qPCR, in 4T1 cells transfected with either control CRISPR/Cas9 plasmid (Crctl) or P2X4 CRISPR/Cas9 plasmid (Cr4#1 and Cr4#2). n = 4 independent experiments. F Representative P2X4 protein expression in 4T1 cells transfected as in (E) selected from 4 to 5 independent experiments. G MTT assay showing Cr4#2 cell viability as a function of time. Data are expressed as fold of Crctl. n = 4–7 independent experiments. H Representative images of bioluminescence in mice at day 28 post-injection. Crctl, Cr4#1 and 2 4T1-luc cells were injected into the mammary fat pad of BALB/c mice. Tumour growth was quantified by bioluminescence imaging. n = 7 mice for the Crctl group and n = 8 for the Cr4 groups. I Tumour volume (in mm3) was measured with a calliper in the three groups of mice, and is represented as a function of time after cell inoculation. J Bioluminescence imaging (BLI) of metastases was measured in living animals and calculated as the difference between the total BLI and primary tumour BLI in the three groups of mice. K The number and organ distribution of metastases in the three groups of mice. A, B, E, F, G Box plots are shown. Comparisons were performed using the two-tailed Mann–Whitney test. I, J Graphs show the mean ± s.e.m., and the P values were calculated using a Two-way Anova with Tukey’s multiple comparison test.
In order to assess the potential role of P2X4 in mammary tumour growth, we developed an orthotopic mammary cancer model in immunocompetent BALB/cJ mice implanted with 4T1 syngeneic murine mammary cancer cells. From the parental 4T1 cell line stably expressing the luciferase gene (4T1-Luc), we developed two cell lines with a permanent knockdown of P2rx4, named Cr4#1 and Cr4#2, and a control cell line (Crctl). The efficiency of P2rx4 knockdown was verified by RT-qPCR (Fig. 1E) and western blotting (Fig. 1F). P2X4 silencing reduced cell proliferation/survival, as assessed by MTT reduction after 4 and 5 days of culture, compared to Crctl cells (Fig. 1G). EdU incorporation was also reduced in Cr4#2 cells after 5 days of culture (Supplementary Fig. S1E).
Compared to Crctl, P2X4 knockdown in mammary cancer (Cr4) cells was associated with a significant reduction of tumour growth, as assessed by bioluminescence imaging (BLI) (Fig. 1H and Supplementary Fig. S1F) and calliper (Fig. 1I). Of note, the two Cr4 clones expressed higher levels of luciferase as compared to Crctl cells (relative values were 1.00 for Crctl, 1.28 for Cr4#1 and 1.33 for Cr4#2), as measured in vitro, suggesting that in vivo BLI results may slightly overestimate Cr4 cells colonization. Nevertheless, mice injected with Cr4 cells showed a very weak BLI signal for metastases (no statistical difference between Cr4#1 and Cr4#2), which did not increase over time (Fig. 1J). Ex vivo analyses confirmed these results and revealed that mice bearing Cr4 cells did not develop any metastasis, while Crctl bearing counterparts developed metastases, mainly in bones and lungs (Fig. 1K). Ex vivo immunohistochemical analyses of primary tumours and metastases from Crctl injected mice showed a significantly higher expression of P2X4 in metastases than in the primary tumour (Supplementary Fig. S1G, H).
P2X4 receptor is partially localised in endolysosomal acidic compartments and controls autophagic flux
P2X4 has been found to be localized intracellularly in different cell types, predominantly within endolysosomal compartments [23, 24]. Consistent with previous studies assessing P2X4 subcellular distribution, P2X4 was predominantly expressed within intracellular puncta and demonstrated partial co-localization with lysotracker, a marker of acidic compartments (Pearson’s correlation coefficient (PCC) = 0.49) in 4T1 cells (Fig. 2A). A similar distribution of P2X4 was observed in two highly invasive human cancer cell lines, MDA-MB-231 and MDA-MB-435s (Supplementary Fig. S2A). Interestingly, the knockdown of P2X4 in 4T1 cells was associated with an increase in the number of LAMP-1 positive vesicles suggesting a perturbation in lysosome biogenesis (Fig. 2B, C). While there was no change in lysosomes distance to the nucleus (Supplementary Fig. 2B), the lysosome distance to the cell surface was higher in Cr4#2 cells (medians were 3.130 for Crctl and 4.128 µm for Cr4#2, Fig. 2D). No difference was observed in lysosome average size (Fig. 2E). As lysosome accumulation may be associated with defective autophagic lysosomal degradation [25, 26], the expression of LC3-II and p62 proteins was measured under basal conditions and following treatment with chloroquine (CQ) to inhibit autophagic degradation. The expression levels of both LC3-II (lower band) and p62 proteins were increased in Cr4 cells compared with Crctl cells (Fig. 2F, left; Supplementary Fig. S2C). Autophagic flux (ratio of LC3-II level w/o vs. with CQ) was significantly reduced in Cr4 cells (Fig. 2F, right). Flow cytometry analyses of LC3 levels were in agreement with western blot results (Fig. 2G, left). Under basal conditions, the fluorescence intensity of LC3-FITC in Cr4 cells was higher than in Crctl cells, whereas the intensities were matched following CQ treatment. The median ratios of LC3-II MFI were 4.77, 3.44 and 3.30 in Crctl, Cr4#1 and Cr4#2, respectively (Fig. 2G, right). Similar results were obtained using autophagic reagents from EMD Millipore’s Autophagy LC3-antibody-based kit (Supplementary Fig. S2D). Immunolabelling of p62 showed a higher number of p62 puncta in Cr4 cells (medians were 10.30 for Crctl and 18.83 for Cr4#2, Fig. 2H). Furthermore, the pharmacological inhibition of P2X4 in the Crctl cells using the membrane-permeable antagonist 5-BDBD, significantly increased the p62 expression (Supplementary Fig. S2E). Similarly, the measure of GFP/RFP colocalization using RFP-GFP tandem fluorescent-tagged LC3 confirmed that the knock-down of P2X4 led to autophagosome accumulation (Fig. 2I). In addition, following transient silencing of P2RX4 gene with siRNA (Supplementary Fig. S2F), the autophagic flux was reduced in human MDA-MB-435s cancer cells, although this did not reach significance in the MDA-MB-231 cells (Supplementary Fig. S2G, H). Rescue experiments confirmed that transfection of the wild-type mouse P2rx4 gene in Cr4#2 cells (Supplementary Fig. S2I) significantly reduced p62 level caused by P2X4 knock-down (Fig. 2J and K). The medians were 1.04 and 0.78 in empty vector- and P2X4-expressing Cr4#2 cells, respectively (Fig. 2K, left). However, no significant differences were observed on the autophagic flux in P2X4 rescued Cr4#2 cells. The medians were 2.73 and 3.07 in empty vector- and P2X4-expressing Cr4#2 cells, respectively (Fig. 2K, right). Using RFP-GFP-LC3 reporter, we found that P2X4 overexpression decreased the number of yellow puncta, suggesting a restored autophagic process (Fig. 2L and Supplementary Fig. 2J).

A Representative image of P2X4 immunostaining and co-localization with lysotracker red (LT, top) and WGA (bottom) (Scale bar, 20 µm; n = 3 independent experiments). B Representative confocal images of Lamp-1 immunostaining in Crctl and Cr4#2 4T1 cells (scale bar, 15 µm; from 3 independent experiments). C Quantification of the number of Lamp-1 positive vesicles in 4T1 cells either expressing P2X4 (Crctl) or not (Cr4#2) cultured as in (B) (n = 30–32 images from 3 independent experiments). D Quantification of lysosome distance to plasma membrane in 4T1 cells) cultured as in (B) (n = 24 and 21 cells from 3 independent experiments for Crctl and Cr4#2, respectively.). E Quantification of lysosome size in 4T1 cells) cultured as in (B) (n = 32 images from 3 independent experiments). F LC3-I, LC3-II and p62 expressions analysed by western blot in Crctl, Cr4#1 and Cr4#2 cells either untreated or treated with chloroquine (100 µM) for 6 h. Autophagic flux was calculated as the ratio of LC3-II expression with and without chloroquine (n = 4–6 independent experiments). G Representative histogram of LC3-II mean fluorescence intensities in Crctl (blue) and Cr4#2 (Red) cells, untreated or treated with chloroquine, evaluated by flow cytometry (n = 6 independent experiments). H Representative confocal images of p62 immunostaining Crctl and Cr4#2 cells (scale bar, 15 µm; selected from 3 independent experiments) and quantification of the number of p62 positive vesicles in control Crctl and Cr4#2 cells. n = 36 for Crctl and n = 47 for Cr4#2 images from 3 independent experiments. I The proportion of autophagosomes and autolysosomes in 4T1 cells was measured 24 h after transfection with RFP–GFP–LC3. Cells were treated or not with 100 µM CQ for 4 h before imaging (scale bar, 15 µm; n = 4). J LC3-I, LC3-II and p62 expressions analysed by western blot in Cr4#2 cells transfected with either empty or P2X4 vector and treated or not with chloroquine (100 µM) for 6 h. K Quantification of autophagic flux (n = 6 independent experiments) and p62 protein expressions (n = 7 independent experiments). L Quantification of autophagosomes and autolysosomes in Cr4#2 cells transfected with either empty or P2X4 vector and cultured as in (I). C–I, K Box plots are shown. J, L Median with interquartile range are shown. Comparisons were performed using a two-tailed Mann–Whitney test.
P2X4 receptor regulates autophagy and promotes cancer cell survival under metabolic challenges
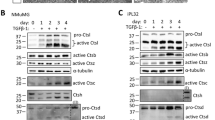
During tumour development, cancer cells are subjected to considerable metabolic stresses such as nutrient deprivation and hypoxia. Given the important role of autophagy in cancer cell survival within a stressful microenvironment, we studied the effects of P2X4 on p62 abundance under hypoxic conditions and with serum deprivation (Fig. 3A). The p62 protein level was upregulated in Cr4#2 cells under serum starvation compared to control cells (Fig. 3B), suggesting an impairment of autophagic flux. The same tendency was observed for LC3-II expression, which was non-significantly increased in Cr4#2 cells (Supplementary Fig. S3). To further investigate whether P2X4 regulates the fusion of lysosomes with autophagosomes under conditions of metabolic challenge, confocal images were taken of Crctl and Cr4#2 cells, co-stained with anti-LC3 and anti-Lamp-1 antibodies (Fig. 3C). Serum deprivation under both normoxic and hypoxic conditions, significantly increased the co-localization of these two proteins in the Crctl cells (median increase, 1.38-fold in 21% O2 and 1.82-fold in 1% O2) but not in the Cr4#2 cells (Fig. 3D). Together, these results suggest that formation of autolysosomes is impaired by knockdown of P2X4. The autophagy defect was correlated with reduced number of autolysosomes in the Cr4#2 cells observed by electron microscopy (Fig. 3E) and was associated with increased apoptosis, as assessed by monitoring caspase-3 activity (Fig. 3F).

A Representative p62 protein expression in Crctl and Cr4#2 cells cultured for 24 h in either 21% or 1% O2 with or without serum, selected from 5 independent experiments. B Quantification of p62 protein expressions, relative to β-actin (n = 5 independent experiments). C Representative confocal images of Lamp1/LC3 co-staining. Cells were cultured as in (A) and were co-immunostained for Lamp-1 (green) and LC3B (red). D Autolysosome formation was evaluated by calculation of the Pearson correlation coefficient for Lamp-1 and LC3 immunostaining; n represents the number of images analysed. Crctl 21% O2/+serum, n = 15; Cr4#2 21% O2/+serum, n = 15; Crctl 1% O2/+serum, n = 15; Cr4#2 1% O2/+serum, n = 16; Crctl 21% O2/−serum, n = 25; Cr4#2 21% O2/−serum, n = 18; Crctl 1% O2/−serum, n = 19; Cr4#2 1% O2/−serum, n = 18, from 3 independent experiments. E Electron microscopy micrographs of autophagic vacuoles in 4T1 cells cultured either in 21% or 1% oxygen for 24 h. Red, blue and white arrows indicate phagophores, autophagosomes and autolysosomes, respectively (scale bar, 2 µm; selected from 3 independent experiments). F Caspase-3 activity was evaluated in Crctl and Cr4#2 cells cultured for 24 h in either 21% or 1% O2 with or without serum. The results are normalized to the control condition (Crctl cells cultured under 21% O2). n = 5 independent experiments. B, Graphs show the mean ± s.e.m., and results were compared using a two-way Anova with Sidak’s multiple comparison test. D, F Box plots are shown. Comparisons were performed using Kruskal-Wallis with Dunn’s multiple comparisons test and two-tailed Mann–Whitney test, respectively.
P2X4 receptor regulates lysosomal exocytosis and invasive capacities
Lysosomal cathepsins have been shown to be related to cancer malignancy and poor patient prognosis [14]. Especially, the aspartyl protease cathepsin D, encoded by the CTSD gene, is correlated with breast cancer aggressiveness [27]. In 4T1 cells, cathepsin D partially colocalizes with P2X4 (PCC = 0.58) (Fig. 4A). At the transcript level, there is a significant correlation between P2RX4 and CTSD expression in human breast cancer tissues (p < 0.001, PCC = 0.334, Fig. 4B). Consistently with reported evidence, CTSD is upregulated in breast primary tumour as well as in metastases compared to adjacent non-tumoral tissues (Fig. 4C). The addition of the aspartate protease inhibitor pepstatin A decreased the ability of the 4T1 cells to invade the extracellular matrix (Supplementary Fig. S4A). The functional activity of extracellular cathepsins was measured using a fluorescent probe in the model of murine breast cancer, and this demonstrated important activity in primary tumours (Fig. 4D), both in vivo and ex vivo. To investigate whether P2X4 regulates lysosomal exocytosis, we studied the release of cathepsin D in cancer cell supernatants. We found that the total amount of cathepsin D (both pro- and active forms) in the extracellular space was significantly decreased in Cr4#2 cells compared to Crctl counterparts (Fig. 4E, F). No significant differences were observed in the intracellular contents (Fig. 4E and Supplementary Fig. S4B), suggesting that maturation of lysosomal cathepsin D is not altered in Cr4#2 compared to Crctl cells. Consistently, the lysosomal pH of Cr4#2 cells was more acidic, as measured by the shift in the ratio of fluorescence of the LysoSensor Yellow/Blue probe (Fig. 4G, H), supporting that Cr4#2 cells are able to induce maturation of lysosomal enzymes. These data might suggest a reduced ability of Cr4#2 cells to fuse lysosomes to the plasma membrane and to secrete intralysosomal components. Silencing P2X4 in human cancer cells also reduced the levels of extracellular cathepsin D (Supplementary Fig. S4C and D). Accordingly, the lack of P2rx4 expression caused a reduction of the invasive potential of 4T1 cancer cells (Fig. 4I). The rescue of P2X4 in Cr4#2 cells led to a marked and significant increase of cell invasiveness (2.98-fold, Fig. 4J). Consistent with these results, inhibiting P2X4 using 5-BDBD reduced invasion of Crctl cells (Supplementary Fig. S4E). Silencing P2X4 also significantly reduced invasiveness of MDA-MB-435s cells, while a tendency was observed in MDA-MB-231 cells (Supplementary Fig. S4F, G). These data provide compelling support for the role of P2X4 in lysosomal exocytosis and cancer cell invasiveness.

A Representative image of P2X4 immunostaining and co-localization with cathepsin D (CTSD) (Scale bar, 20 µm; n = 3 independent experiments). B Positive correlation between P2RX4 and CTSD genes expression in TGCA target GETEx dataset. Expression values were estimated using RNASeq and analysed as log2(norm_count+1). Shown is the Pearson correlation coefficient (1391 samples). C Box plots showing CTSD gene expression in normal tissue (n = 179), adjacent non-tumoral tissue (n = 113), primary tumours (n = 1205) and metastases (n = 7). D Cathepsin activity in vivo and ex vivo. Crctl cells were injected into the mammary fat pad of Balb/cJ mice and cathepsin activity was assessed when tumours reached 100 mm3 threshold, using ProSense-750 fast probe (n = 4 mice). Mice were imaged 24 h following intravenous probe injection and ex vivo after necropsy. E Cathepsin D protein expression was investigated by western blot intracellularly and extracellularly in 4T1 cells expressing (Crctl) or not (Cr4#2) P2X4. F Quantification of cathepsin D release after 24 h of culture (n = 14 independent experiments). G Flow cytometry analysis of lysosomal pH in Crctl and Cr4#2 4T1 cells, using the ratiometric pH Yellow/Blue LysoSensor dye. Chloroquine (CQ; 100 µM) was used as control for lysosome alkalinisation. H Quantification of the ratio of median fluorescence intensities (Blue/Yellow) in Crctl, and Cr4#2 cells as in (F). n = 10 for Crctl and Cr4#2 conditions. n = 4 for Crctl + CQ condition. I Crctl, Cr4#1 and Cr4#2 cell invasiveness was measured over 24 h. Pictures were taken using x10 objective (n = 4–5 independent experiments performed in triplicates). J Invasiveness analysis in Cr4#2 cells transfected with either empty vector or mouse P2X4 vector (n = 8 independent experiments performed in triplicates). Box plots are shown. Comparisons were performed using two-tailed Mann–Whitney test (C, H, I, J) or Wilcoxon matched-pairs signed-rank test (F).
P2X4 receptor drives mammary cancer cells towards a mesenchymal phenotype while improving autophagy
Epithelial-to-mesenchymal transition (EMT) is typically associated with cell aggressiveness and metastasis formation, involving extracellular matrix degradation by released proteases. We found that P2X4 inhibition led to a transcriptional increase of Cdh1 (epithelial marker) and a decrease of Vim (mesenchymal marker), especially in hypoxia, as assessed by RT-qPCR (Fig. 5A). The respective protein expression of E-cadherin and vimentin was assessed by western blot (Fig. 5B). Hypoxia did not significantly modify the level of E-cadherin but this was strongly upregulated when P2X4 was silenced (1.88-fold in 21% O2 and 2.44-fold in 1% O2; Fig. 5C). Hypoxia increased the amount of vimentin in Crctl cells (by 1.47). Silencing P2X4 led to a reduction of vimentin expression (by 62.4% in 21% O2 and 56.6% in 1% O2; Fig. 5C). The rescue of P2X4 in the knocked-down cells restored the mesenchymal phenotype, as assessed by RT-qPCR (Fig. 5D) and western blot (Fig. 5E). While no modification of E-cadherin expression was observed, P2X4 overexpression led to a significant increase of vimentin (1.33-fold and 2.43-fold at mRNA and protein level, respectively; Fig. 5F). Studying the architecture of vimentin intermediate filaments, we found that the organization of its network was notably modified in Cr4#2 cells in which a weaker signal was observed as well as reduction of the quantity of elongated fibres and its subcellular restriction to a perinuclear localization (Fig. 5G). The analysis of another epithelial marker (Zo1) and mesenchymal transcription factors (Twist, Zeb1, Snai1) by RT-qPCR confirmed the role of P2X4 in favouring mesenchymal phenotype (Fig. 5H). Silencing P2X4 caused an increase of Zo1 and a decrease of EMT-related transcription factors Zeb1 and Twist, in both normoxia and hypoxia conditions. Consistent with previous reports, hypoxia triggered upregulation of mesenchymal genes in Crctl cells. Twist and Zeb1 upregulations were even stronger after five days of culture in hypoxia (Supplementary Fig. S5A). Similar results were obtained in human MDA-MB-435s (increase of CDH1 and ZO1, decrease of VIM and ZEB1) and MDA-MB-231 (increase of CDH1, decrease of ZEB1 and SNAI1) cancer cells using siRNA targeting P2RX4 gene expression (Supplementary Fig. S5B, C). Interestingly, the autophagy inhibitor bafilomycin A1 led to the significant reduction of Vim and Twist expressions without modifying the expression of Cdh1 (Fig. 5I) or the other markers (Supplementary Fig. S5D), suggesting that blocking the autophagy process impairs EMT, and that P2X4 has a critical role in controlling these two related functions.

A mRNA level of epithelial marker Cdh1 (E-cadherin) and mesenchymal marker Vim (vimentin) in Crctl and Cr4#2 cells cultured in 21% or 1% O2. Tata Box Protein (Tbp) was used as house-keeping gene. Shown are values and mean from 7 independent experiments performed in triplicates. B E-cadherin and vimentin protein expressions in Crctl and Cr4#2 cells cultured for 24 h in either 21% or 1% O2. C Quantification of E-cadherin (n = 5) and vimentin (n = 6) protein expressions, relative to β-actin, in 4T1 cells as in (B). D mRNA levels of Cdh1 and Vim in Cr4#2 cells transfected with the control vector or P2X4 vector (n = 7 independent experiments performed in triplicates). E E-cadherin and vimentin protein expressions in vector and P2X4 Cr4#2 cells. F Quantification of E-cadherin (n = 7) and vimentin (n = 7) protein expressions, relative to β-actin, in 4T1 cells as in (E). G Confocal imaging of vimentin in Crctl and Cr4#2 4T1 cells. Representative pictures from 3 independent experiments. H mRNA levels of epithelial marker Zo1, and mesenchymal transcription factors Twist, Snai1 and Zeb1, in Crctl and Cr4#2 cells cultured in 21% or 1% O2. Shown are values and mean from 7–9 independent experiments performed in triplicates. I mRNA levels of Cdh1, Vim and Twist in Crctl and Cr4#2 4T1 cells cultured during 24 h with or without 100 nM of bafilomycin A1 (Baf). Shown are values and mean from 3 independent experiments performed in triplicates. Graphs show the individual values and the mean ± s.e.m., and results were compared using a one-way Anova with Tukey’s multiple comparison test (A, C, H, I) or Student’s t-test with Welch’s correction (D, F).
Discussion
A growing body of evidence indicates that the purinergic signalling plays multiple functions during cancer development and progression. Here, we identified the elevated expression of P2X4 in human breast cancer tissues compared to normal tissues, at both mRNA and protein levels. Our results show that P2X4 is mainly distributed in the cytosolic compartment and partially localizes in acidic organelles of breast cancer cells, as previously reported in different non-cancer cell types [24, 28]. The changes that we have observed in autophagic flux and in lysosomes number, location and exocytosis suggest that there is a reduction in lysosome fusion associated with the knockdown of P2X4 in 4T1 cancer cells. These results are consistent with previous studies showing that endolysosomal P2X4 receptors can mediate Ca2+ release from lysosomes to trigger lysosome fusion, P2X4 is orientated with its extracellular domain facing the lumen and levels of ATP within the lumen are sufficiently high, that lysosome alkalinization is sufficient to trigger P2X4 activation and lysosome fusion [29]. The identification of factors that promote lysosome alkalinization in cancer cells remains to be established. It has been shown, however, that endolysosomes are heterogeneous and differ in their pH, and that their anterograde trafficking to the cell surface is associated with alkalinization. Additionally, P2X7 has been shown to be a regulator of lysosome pH, both in microglia and in retinal epithelial cells, and in normal rat kidney (NRK) cells, activation of P2X7 receptors enhanced lysosomal P2X4 receptor activation [30].
We demonstrated that the inhibition of P2X4 in breast cancer cells led to a reduction of both autophagy and lysosomal exocytosis, suggesting that P2X4 promotes fusion of lysosomes to either autophagosomes or plasma membrane. Consistent with this hypothesis, it was previously observed that lysosomal P2X4 interacts with calmodulin for fusion of lysosomes with other compartments [18, 29]. Fusion with autophagosomes is required for autophagy and, here, we found that P2X4 downregulation led to autophagosome accumulation and an increase in caspase-3 activity. In particular, we observed the accumulation of LC3-II in three highly invasive cancer cell lines when P2X4 was knocked-down, by using either Crispr/Cas9 technology or siRNA. The role of P2X4 in autophagy was even more pronounced under serum deprivation than in normal growth medium, as indicated by p62 expression. In turn, overexpression of P2X4 in Cr4#2 cells resulted in a robust decrease of p62 and therefore the restitution of autophagic activity. Supporting these results, a very recent study identifies P2X4 as a target for indophagolin, an autophagy inhibitor, using thermal proteome profiling technology [31]. In this study, the reduced autophagic flux in Cr4#2 cells might partly be responsible for the inhibition of tumour progression observed in our in vivo mammary cancer model. To overcome stressful conditions and fulfil energy demands necessary for their metabolic functions, cancer cells may increase autophagy [3, 32]. In this context, autophagy inhibition has been proposed as a treatment in a variety of cancers, including breast cancer [33, 34].
Depending on tumour stage, autophagy can trigger EMT. Typically, autophagy generates resistance to cell death and several studies have shown that autophagy deregulation inhibits tumour growth and metastatic spreading [35, 36]. By decreasing apoptosis and regulating the tumour microenvironment, autophagy can support EMT, which is considered as a major process for cell invasiveness and metastasis formation [8]. One key finding of this study is to reveal that P2X4 has an important role in modulating EMT. In all three cell lines, P2X4 inhibits the expression of epithelial markers E-cadherin and Zo-1, while increasing the expression of mesenchymal genes. In 4T1 cells, the use of bafilomycin A1 led to an inhibition of vimentin and Twist expressions, suggesting that autophagy inhibition may reduce the expression of some EMT markers. Furthermore, P2X4 may also strengthen EMT by increasing lysosome exocytosis and thereby influencing the tumour microenvironment [37, 38], or by promoting lysosome interaction with cytoskeleton-associated proteins, as previously observed [39, 40]. Indeed, lysosomes interact with vimentin intermediate filaments and vimentin inhibition leads to juxtanuclear lysosome accumulation and subsequent modulation of autophagy [41]. In turn, lysosomal proteins can modify the architecture of vimentin networks [42]. Consistent with these observations, our results indicate that P2X4 downregulation leads to an inhibition of vimentin expression and alteration of its network. Conversely, the rescue of P2X4 restored high vimentin expression in P2X4 knocked-down cells.
A range of studies have reported that vimentin regulates EMT through upregulation of EMT-associated genes [43, 44] and modifications of cell shape, motility, and adhesion [45]. EMT is currently associated with metastatic spreading, which require extracellular matrix degradation by extracellular proteases. Here, we show a positive correlation between expression of P2RX4 and CTSD in breast cancer. Cathepsin D is reported to mediate tumour aggressiveness and expression of EMT genes and is associated with poor patient prognosis [27, 46]. Consistent with studies showing the role of P2X4 in lysosome fusion to the plasma membrane [18, 47], our results suggest that P2X4 regulates the release of the lysosomal cathepsin D in the extracellular compartment. Accordingly, P2X4 downregulation compromises cell invasiveness, whilst its rescue restores this capacity.
In conclusion, our data reveal an important role of P2X4 in breast cancer cell functions, promoting cancer cell invasiveness and resistance to metabolic challenges by regulating autophagy and lysosomal exocytosis. As such, P2X4 may mediate hallmarks of cancer cell malignancy.
Materials and methods
Agonists, antagonists and reagents
Chloroquine and bafilomycin A1 were purchased from Sigma-Aldrich (France). Caspase 3 Assay kit and staurosporine were from Abcam (France). 5-BDBD was purchased from Tocris (Bio-Techne, France) and pepstatin A was from Enzo Life Sciences. LysoSensor Yellow/blue-DND 160, Lysotracker Red DND-99 and Click-iT Plus EdU Flow Cytometry Assay Kit were purchased from Invitrogen (France). ProSense 680 Fluorescent Imaging Agent was from PerkinElmer. FlowCellect™ Autophagy LC3 Antibody-based Assay Kit was purchased from Merck Millipore (France).
Cells and cell culture
Murine mammary cancer cell line 4T1 from the Balb/cJ strain was purchased from LGC Standards (France), and a stable 4T1-luc cell line expressing the luciferase gene (thereafter called “4T1 cells”) was obtained by transduction of cells with lentiviral vectors containing the luciferase gene and blasticidin resistance gene for selection (GIGA Viral Vectors, Belgium). Stable 4T1 cell lines not expressing the P2rx4 gene were obtained using the CRISPR/Cas9 technique by transfection with the P2RX4 Double Nickase Plasmid (Santa Cruz, France). Clonal selection was performed by FACS sorting. Two clones have been kept for this study, called “Cr4#1” and “Cr4#2”. A null-target Double Nickase Plasmid was also used to transfect 4T1 cells and this led to the selection of a control cell line, thereafter called “Crctl” cell line. Efficiency of the CRISPR-mediated knock-down was assessed by RT-qPCR and western blotting, and stability of clones was followed for a minimal duration of 6 weeks. All the 4T1 cell lines were cultured in RPMI supplemented with 10% foetal calf serum (FSC) and regularly tested for mycoplasma contamination (MycoAlertTM Mycoplasma Detection Kit, Lonza, Thermo Fisher Scientific, Villebon-sur-Yvette, France). Cells were grown at 37 °C in a humidified 5% CO2 incubator. For hypoxic experiments, cells were cultured in a hypoxic chamber (Invivo 200, Ruskinn Technology; 1% O2, 5% CO2, 94% N2).
Plasmid transfection
Plasmid pcDNA3.1 encoding wild-type mouse P2rx4 was used for the rescue experiments. Empty plasmid (pcDNA3.1) was used as a control. GFP-RFP-LC3 vector (addgene #21074) was used to distinguish autophagosomes (yellow puncta) from autolysosomes (red puncta). Plasmid transfection was carried out using Opti-MEM (Gibco) and TransIT-X2 (Mirus), according to the manufacturer’s protocol.
RNA extraction, reverse transcription and quantitative polymerase chain reaction (qPCR)
Total RNA was extracted using NucleoSpin® RNA kit (Macherey Nagel EURL, Germany), and reverse-transcribed with the PrimeScript RT Reagent Kit (Ozyme, France). Quantitative PCR were performed using SYBR qPCR Premix Ex Taq (Ozyme, France) and CFX CONNECT (Bio-rad, France). The housekeeping gene was Tata-binding Protein (TBP).
The primers used were as follows: 5’-CCTGGCTTACGTCATTGGGT-3’ (P2rx4 forward), 5’-AAGTGTTGGTCACAGCCACA-3’ (P2rx4 reverse), 5’-AAGGGAGAATCATGGACCAGAAC-3’ (Tbp forward), 5’-GGTGTTCTGAATAGGCTGTGGAG-3’ (Tbp reverse), 5’-CAGTTCCGAGGTCTACACCTT-3’ (Cdh1 forward), 5’-TGAATCGGGAGTCTTCCGAAAA-3’ (Cdh1 reverse), 5’-GCCTCAGAAATCCAGCTTCTCGAA-3’ (Zo1 forward), 5’-GCAGCTAGCCAGTGTACAGTATAC-3’ (Zo1 reverse), 5’-CACACGCTGCCTTGTGTCT-3’ (Snai1 forward), 5’-GGTCAGCAAAAGCACGGTT-3’ (Snai1 reverse), 5’-TTCTCGGTCTGGAGGATGGA-3’ (Twist forward), 5’-TCTCTGGAAACAATGACATCTAGG-3’ (Twist reverse), 5’-ACCGCCGTCATTTATCCTGAG-3’ (Zeb1 forward), 5’-CATCTGGTGTTCCGTTTTCATCA-3’ (Zeb1 reverse), 5’-ACCTCACTGCTGCCCTGCGT-3’ (Vim forward), 5’-CTCATCCTGCAGGCGGCCAA-3’ (Vim reverse).
Western blotting
Cells were washed twice with ice-cold PBS (Gibco), and then lysed with RIPA Buffer including Protease Inhibitors (Roche Lifesciences). Protein concentration was measured using Pierce® BCA Protein Assay Kit (Thermofisher Scientific, France) and 25 µg of total protein were resolved on SDS-PAGE. Proteins were transferred onto nitrocellulose membrane, followed by western blot analysis using indicated antibodies. Quantification of signal intensity was performed using ImageJ software. Antibodies used for immunoblotting were as follows: P2X4 (Alomone, #APR-002, 1:300), LC3B (Abcam, #ab51520, 1:3,000), p62 (Abcam, #ab109012, 1:30,000), E-cadherin (Santa Cruz, #sc-7870, 1:1,000), vimentin (Cell signalling, #5741, 1:5,000), cathepsin D (Abcam, #ab6313, 1:1,000), actin (Santa Cruz, #sc-47778, 1:1,000), and HSC-70 (Santa Cruz, #sc-7298, 1:5,000). For the analysis of autophagic flux, densitometric analyses of the LC3-II bands were normalized to actin (LC3-II/actin). Autophagic flux was then determined by division of the normalized value obtained in presence of chloroquine (CQ + conditions) by the normalized value obtained in absence of chloroquine (CQ- conditions). Unprocessed original immunoblot scans are provided in Supplementary File.
Lysosomal pH measurement
Changes in lysosomal pH were detected using the LysoSensor Yellow/Blue DND-160. 4T1 cells were treated with 3 μM LysoSensor for 10 min at 37 °C. Cells were then washed twice, detached and resuspended in PBS. Before the first experiment, a spectrum scan was performed using Hitachi F2700 spectrofluorimeter (Hitachi, Tokyo, Japan). Then, emission fluorescences were measured by flow cytometry (BD FACS Melody Cell Sorter) with emission wavelengths at 448 nm (BP 448/45 nm) and 500 nm (LP 500/50) with excitation at 405 nm. Lower 448/500 ratio indicates more acidic lysosomal pH. A total of 10,000 cell-gate events were acquired and analysed per sample.
Cell viability / proliferation
Tetrazolium salt (MTT) assay was performed as previously described [48]. Briefly, cells were cultured for 1 to 5 days and cell viability was measured after incubation with MTT during 60 min at 37 °C. The resulting formazan crystals were dissolved in DMSO and the absorbance was measured at 560 nm using a plate reader (Biotek EL800).
Measurement of autophagy by flow cytometry
Autophagic fluxes were carried out by flow cytometry using the FlowCellect™ Autophagy LC3 Antibody-based Assay Kit (Merck Millipore, CF200097) according to the manufacturer’s instructions. Briefly, cells (4 x 104/well) were seeded in 96 well plate 24 h before experiment. The day after, medium was replaced by fresh medium and cells were either left untreated or treated for 6 h with chloroquine (100 µM) or the lysosome inhibitor Autophagy Reagent A (Merck Millipore, CS208212). Cells were subsequently permeabilized and stained with anti-LC3/FITC. The median fluorescence intensity was measured by flow cytometry (BD FACSCanto™ I flow cytometer (BD Biosciences).
Caspase-3 activity
4T1 cells were cultured for 24 h either in normoxic (CO2 incubator, 21% O2) or hypoxic (hypoxic chamber, 1% O2) conditions and either with or without staurosporine (1 µM) which was added 6 h before the end of the culture. Caspase-3 activity was subsequently assessed according to the manufacturer’s protocol (abcam, #ab39401). Absorbances were measured at 405 nm expressed relative to the staurosporine condition, and normalized to the control (Crctl, 21% O2). All experiments were performed in duplicate.
Invasion assays
Cell invasiveness was measured as previously described [48] using 8 µm pore size polyethylene terephthalate membrane inserts covered with Matrigel® matrix (Becton Dickinson, France). Cells in the lower surface of the insert were stained with DAPI and nuclei were counted after imaging with an Evos M7000 microscope (Thermofisher, France).
Immunocytochemistry experiments
Cells were grown for 24 h on 18 mm coverslips in 21% or 1% O2, with or without serum. For experiments using LysoTracker-Red DND-99 (Invitrogen, L7528), cells were incubated with 100 nM LysoTracker for 1 h. Cells were fixed in 4% paraformaldehyde and then permeabilized with 0.1% Triton-X-100 for 15 min. Unspecific antibody binding was blocked with 3% BSA for 20 min. Primary antibodies for P2X4 (Alomone, APR-002, 1:200), Lamp-1 (Abcam, #ab25245, 1:800), LC3B (Abcam, #ab51520, 1:2000), p62 (Abcam, #ab109012, 1:400), vimentin (Cell signalling, #5741, 1:100) or cathepsin D (Abcam, #ab6313, 1:200) were applied for 1.5 h at room temperature or overnight at 4 °C. Cells were then extensively washed with PBS and incubated with diluted Alexa Fluor 488- and/or Alexa Fluor 647-conjugated secondary antibodies for 1 h. Coverslips were mounted on slides using ProLong® Gold Antifade Mountant with DAPI (Invitrogen, France). Images were obtained using either a confocal fluorescence microscope (Leica SP8, 63x objective) or an epifluorescence microscope (EVOS M7000, 40x objective) and were analysed using ImageJ software. LAMP-1 staining was used as a marker to determine lysosome abundance, size and distribution using the particle measurement tool of the ImageJ software. Lysosome distance to the plasma membrane was measured as the shortest distance to cell contour (manually drawn region of interest in WGA-stained cells). The Pearson correlation coefficient for LC3/LAMP-1 co-localization was determined with JACoP [49] for ImageJ software 1.53c.
Clinical samples and immunohistochemistry
The P2X4 protein expression in normal and tumoral mammary tissues was analysed by immunohistochemistry. Normal breast and cancer biopsies from the University-Hospital of Tours were from the tumour collection declared to the French Ministry of Research (No. DC2008-308) and were prepared at the anatomopathology department of the University-Hospital of Tours, France. Briefly, after deparaffinization and rehydration, sections were treated with a high-pH (Tris buffer/EDTA, pH 9.0) target retrieval procedure (Dako PT-link; Dako, USA). Endogenous peroxidase was then blocked by a commercial solution (Dako REAL), and incubated overnight with a 1/200 dilution of the primary polyclonal rabbit anti-P2X4 antibody (APR-002; Alomone) at 4 °C. Sections were then incubated with a commercial anti-rabbit-labelled polymer (Dako EnVision FLEX; Dako) for 30 min at RT. Immunoreaction was finally revealed with 3-3′ diaminobenzidine solution (Dako) for 5 min. A positive reaction was identified by a cytoplasmic dark-brown precipitate. To determine the protein expression in tissues, a qualitative scale was used, for negative or low, medium and high expression.
Metastases and primary tumours from in vivo mouse experiments were fixed in formalin, included in paraffin, and cut in 5 μm tissue sections. Slides were deparaffinized, rehydrated and heated in citrate buffer pH6 for antigenic retrieval, prior to labelling with the anti-P2X4 antibody. Immunohistochemistry was performed using the streptavidin-biotin-peroxidase method with diaminobenzidine as the chromogen (Kit LSAB, Dakocytomation). Slides were finally counterstained with haematoxylin. Negative controls were obtained after omission of the primary antibody or incubation with an irrelevant antibody.
Electron microscopy
4T1 cells were cultured for 24 h in either 21% or 1% O2 and then fixed for 24 h in 4% paraformaldehyde, 1% glutaraldehyde (Sigma, St-Louis, MO) in 0.1 M phosphate buffer (pH 7.2). Samples were washed in PBS and post-fixed by incubation with 2% osmium tetroxide (Agar Scientific, Stansted, UK) for 1 h. Cells were then fully dehydrated in a graded series of ethanol solutions and propylene oxide. The impregnation step was performed with a mixture of (1∶1) propylene oxide/Epon resin (Sigma) and then left overnight in pure resin. Samples were then embedded in Epon resin (Sigma), which was allowed to polymerize for 48 h at 60 °C. Ultra-thin sections (90 nm) of these blocks were obtained with a Leica EM UC7 ultramicrotome (Wetzlar, Germany). Sections were stained with 2% uranyl acetate (Agar Scientific), 5% lead citrate (Sigma) and observations were made with a JEOL 1400 plus transmission electron microscope (JEOL, Tokyo, Japan).
In vivo mammary cancer model
All experiments have been approved by the Comité d’éthique du Centre-Val de Loire and have been performed in accordance with the European Ethics rules (Ref 005377.01 Apafis #12960). All animals were bred and housed at the CNRS UPS44 – TAAM- CIPA (CNRS Campus, Orléans, France), in controlled conditions with a 12 h light/dark cycle at 22 °C, and free to food and water ad libitum. We developed a syngeneic and orthotopic mouse mammary cancer model in female BALB/cJ immunocompetent mice. To do so, 4T1-luciferase-expressing mouse mammary cancer cells were injected into the fifth mammary fat pad of 6 weeks-old mice. The luciferase activity was used to follow tumour appearance and growth in vivo, following D-luciferin (150 mg/kg) intraperitoneal injection and bioluminescent imaging (IVIS Lumina II, Perkin Elmer). No randomization was used to select animals for each group and studies were performed blinded to the experimenter. Primary tumour volume (mm3) and growth over time were most effectively measured with a calliper, twice a week, and calculated as (L x l²)/2 (in mm). Metastases were counted macroscopically at the completion of studies, during autopsies. Animal weight was measured once a week. In the experiments, 1 x 104 Crctl, Cr4#1 or Cr4#2 4T1-derived mammary cancer cells (see section “Cells and cell culture”) in 100 µL of PBS solution were injected in the mammary fat pad, under isoflurane inhalation, of wild-type BALB/cJ mice (Janvier Labs, Saint Berthevin, France).
To evaluate cathepsin activity, ProSense 750 Fast (Perkin Elmer), which becomes fluorescently activated when cleaved by cathepsins, was used. Mice received the fluorescent dyes within a single intravenous injection (4 nmol in 100 μL PBS) once individual tumours reached 100 mm3 size threshold. Mice were imaged 24 and 48 h post-injection using Ivis Lumina II imaging system (Perkin Elmer, USA) and analysed with Living Image (4.4) software. Ex vivo imaging of tumours was also performed after necropsy.
Bioinformatic analyses
Gene expression data were obtained from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) databases using the UCSC Xena Browser (https://xenabrowser.net) [50]. The IlluminaHiSeq (log2-normalized_count+1) files were downloaded from the “TCGA Breast Cancer (TCGA-BRCA)” cohort, in order to compare expressions between adjacent non-tumoral tissues and primary tumour. From the “TCGA TARGET GTEx” cohort, the RSEM_norm_count (log2-normalized_count+1) files were downloaded, in order to compare expressions between normal tissues, adjacent non-tumoral tissues, primary tumour and metastases. In addition, Breast Cancer Gene-Expression Miner v4.5 (bc-GenExMiner v4.5) incorporating TCGA and GTEX data (n = 1234) was used to confirm P2RX4 expression in normal and tumoral tissues (http://bcgenex.ico.unicancer.fr). P2X4 protein expression data were obtained from Clinical Proteomic Tumor Analysis Consortium (CPTAC) and UALCAN interface (http://ualcan.path.uab.edu/analysis.html) [51]. For P2RX4/CTSD correlation, Pearson’s rank rho coefficient was calculated, and a two-dimensional correlation scatter plot was obtained using the UCSC Cancer Browser Interface.
Statistics and reproducibility
Data were displayed as mean ± s.e.m. or as median ± range where indicated in the figure legends. For box plots, the upper and lower edges of the box indicate the first and third quartiles (25th and 75th percentiles) of the data and the middle line indicates the median. Mann-Whitney rank-sum test, Wilcoxon matched-pairs signed-rank test and two-tailed Student’s t-tests were used for comparisons between two groups. Kruskal-Wallis, One- or two-way ANOVAs were used for comparisons of more than two groups, using GraphPad Prism v.9.0.0 (GraphPad Software). Confidence intervals of 95% were used and significance was considered when the P-value was less than 0.05. The number of times an experiment was repeated is indicated in the figure legends.
Data availability
All data and materials used in this study are available upon request.
References
Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7.
Gouirand V, Guillaumond F, Vasseur S. Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front Oncol. 2018;8. Available from: https://www.frontiersin.org/articles/10.3389/fonc.2018.00117/full
Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25:1037–43.
Levine B. Autophagy and cancer. Nature. 2007;446:745–7.
Martin FT, Dwyer RM, Kelly J, Khan S, Murphy JM, Curran C, et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: Stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res Treat. 2010;124:317–26.
Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18:128–34.
Gugnoni M, Sancisi V, Manzotti G, Gandolfi G, Ciarrocchi A. Autophagy and epithelial–mesenchymal transition: an intricate interplay in cancer. Cell Death Dis. 2016;7:e2520–e2520.
Chen H-T, Liu H, Mao M-J, Tan Y, Mo X-Q, Meng X-J, et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol Cancer. 2019;18:101.
Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–35.
Lahiri V, Hawkins WD, Klionsky DJ. Watch what you (Self-) Eat: Autophagic mechanisms that modulate metabolism. Cell Metab. 2019;29:803–26.
Hämälistö S, Jäättelä M. Lysosomes in cancer—living on the edge (of the cell). Curr Opin Cell Biol. 2016;39:69–76.
Kallunki T, Olsen OD, Jäättelä M. Cancer-associated lysosomal changes: Friends or foes? Oncogene. 2013;32:1995–2004.
Janda E, Nevolo M, Lehmann K, Downward J, Beug H, Grieco M. Raf plus TGF β -dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene. 2006;25:7117–30.
Olson OC, Joyce JA. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat Rev Cancer. 2015;15:712–29.
Brix DM, Tvingsholm SA, Hansen MB, Clemmensen KB, Ohman T, Siino V, et al. Release of transcriptional repression via ErbB2-induced, SUMO-directed phosphorylation of myeloid zinc finger-1 serine 27 activates lysosome redistribution and invasion. Oncogene. 2019;38:3170–84.
Morgan MJ, Fitzwalter BE, Owens CR, Powers RK, Sottnik JL, Gamez G, et al. Metastatic cells are preferentially vulnerable to lysosomal inhibition. Proc Natl Acad Sci. 2018;115:E8479–88.
Li S, Song Y, Quach C, Guo H, Jang G-B, Maazi H, et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat Commun. 2019;10:1693.
Murrell-Lagnado RD, Frick M. P2X4 and lysosome fusion. Curr Opin Pharm. 2019;47:126–32.
Burnstock G, Di Virgilio F. Purinergic signalling and cancer. Purinergic Signal. 2013;9:491–540.
Adinolfi E, Capece M, Amoroso F, De Marchi E, Franceschini A. Emerging Roles of P2X Receptors in Cancer. Curr Med Chem. 2015;22:878–90.
Di Virgilio F, Ferrari D, Adinolfi E. P2X7: A growth-promoting receptor—implications for cancer. Purinergic Signal. 2009;5:251–6.
Roger S, Jelassi B, Couillin I, Pelegrin P, Besson P, Jiang L-H. Understanding the roles of the P2X7 receptor in solid tumour progression and therapeutic perspectives. Biochim Biophys Acta. 2015;1848:2584–602.
Huang P, Zou Y, Zhong XZ, Cao Q, Zhao K, Zhu MX, et al. P2X4 Forms Functional ATP-activated Cation Channels on Lysosomal Membranes Regulated by Luminal pH. J Biol Chem. 2014;289(Jun):17658–67.
Murrell-Lagnado RD. A role for P2X4 receptors in lysosome function. J Gen Physiol. 2018;150:185–7.
Brisson L, Bański P, Sboarina M, Dethier C, Danhier P, Fontenille M-J, et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell. 2016;30:418–31.
Majora M, Sondenheimer K, Knechten M, Uthe I, Esser C, Schiavi A, et al. HDAC inhibition improves autophagic and lysosomal function to prevent loss of subcutaneous fat in a mouse model of Cockayne syndrome. Sci Transl Med. 2018;10. Available from: https://stm.sciencemag.org/content/10/456/eaam7510
Westley BR, May FEB. Cathepsin D and breast cancer. Eur J Cancer. 1996;32:15–24.
Toulme E, Garcia A, Samways D, Egan TM, Carson MJ, Khakh BS. P2X4 receptors in activated C8-B4 cells of cerebellar microglial origin. J Gen Physiol. 2010;135:333–53.
Cao Q, Zhong XZ, Zou Y, Murrell-Lagnado R, Zhu MX, Dong X-P. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J Cell Biol. 2015;209:879–94.
Tan S-L, Barri M, Atakpa-Adaji P, Taylor CW, St. John Smith E, Murrell-Lagnado RD. P2X4 receptors mediate Ca2+ release from lysosomes in response to stimulation of P2X7 and H1 histamine receptors. Int J Mol Sci. 2021;22:10492.
Carnero Corrales MA, Zinken S, Konstantinidis G, Rafehi M, Abdelrahman A, Wu Y-W, et al. Thermal proteome profiling identifies the membrane-bound purinergic receptor P2X4 as a target of the autophagy inhibitor indophagolin. Cell Chem Biol. 2021; Available from: https://www.sciencedirect.com/science/article/pii/S2451945621001021
Izuishi K, Kato K, Ogura T, Kinoshita T, Esumi H. Remarkable tolerance of tumor cells to nutrient deprivation: Possible new biochemical target for cancer therapy. Cancer Res. 2000;60:6201–7.
Chittaranjan S, Bortnik S, Dragowska WH, Xu J, Abeysundara N, Leung A, et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin Cancer Res. 2014;20:3159–73.
Liang DH, Choi DS, Ensor JE, Kaipparettu BA, Bass BL, Chang JC. The autophagy inhibitor chloroquine targets cancer stem cells in triple-negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016;376:249–58.
Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18:370–9.
Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF, et al. VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell. 2012;21:532–46.
Zhitomirsky B, Assaraf YG. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget. 2017;8:45117–32.
Kundu ST, Grzeskowiak CL, Fradette JJ, Gibson LA, Rodriguez LB, Creighton CJ, et al. TMEM106B drives lung cancer metastasis by inducing TFEB -dependent lysosome synthesis and secretion of cathepsins. Nat Commun. 2018;9:2731.
Groth-Pedersen L, Aits S, Corcelle-Termeau E, Petersen NHT, Nylandsted J, Jäättelä M. Identification of cytoskeleton-associated proteins essential for lysosomal stability and survival of human cancer cells. PLOS ONE. 2012;7:e45381.
Caviston JP, Zajac AL, Tokito M, Holzbaur ELF. Huntingtin coordinates the dynein-mediated dynamic positioning of endosomes and lysosomes. Mol Biol Cell. 2010;22:478–92.
Biskou O, Casanova V, Hooper KM, Kemp S, Wright GP, Satsangi J, et al. The type III intermediate filament vimentin regulates organelle distribution and modulates autophagy. PLOS ONE. 2019;14:e0209665.
Cogli L, Progida C, Bramato R, Bucci C. Vimentin phosphorylation and assembly are regulated by the small GTPase Rab7a. Biochim Biophys Acta BBA - Mol Cell Res. 2013;1833:1283–93.
Ivaska J, Vimentin. Small GTPases. 2011;2:51–3.
Vuoriluoto K, Haugen H, Kiviluoto S, Mpindi J-P, Nevo J, Gjerdrum C, et al. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30:1436–48.
Mendez MG, Kojima S-I, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838–51.
Ketterer S, Mitschke J, Ketscher A, Schlimpert M, Reichardt W, Baeuerle N, et al. Cathepsin D deficiency in mammary epithelium transiently stalls breast cancer by interference with mTORC1 signaling. Nat Commun. 2020;11:5133.
Miklavc P, Mair N, Wittekindt OH, Haller T, Dietl P, Felder E, et al. Fusion-activated Ca2+ entry via vesicular P2X4 receptors promotes fusion pore opening and exocytotic content release in pneumocytes. Proc Natl Acad Sci. 2011;108:14503–8.
Jelassi B, Chantôme A, Alcaraz-Pérez F, Baroja-Mazo A, Cayuela ML, Pelegrin P, et al. P2X(7) receptor activation enhances SK3 channels- and cystein cathepsin-dependent cancer cells invasiveness. Oncogene. 2011;30:2108–22.
Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–32.
Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38:675–8.
Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK, et al. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649–58.
Acknowledgements
This work was supported by the “Ministère de la Recherche et des Technologies”, the “Ligue Nationale Contre le Cancer – Interrégion Grand-Ouest” to SR, the Région Centre-Val de Loire (grant “CanalEx” to SR), the Institut National du Cancer (grant INCA_16110 “PURIN4EXO” to SR). SR was recipient of a prize “Prix Ruban Rose Avenir 2017” from the Charity “le Cancer du sein: parlons-en!”. SC was recipient of a post-doctoral grant from the School of Life Science, University of Sussex (UK) and from the AURCIL Association in Tours (France). We thank Mrs Carole Desplanches for secretary and administrative assistance. We thank Mrs Stéphanie Rétif, Mrs Marilyne Le Mée and M. Julien Sobilo for the assistance with in vivo experiments performed at CNRS UPS44 CIPA, Orléans. Our microscopy data were obtained with the assistance of the IBiSA Electron Microscopy Facility of the University of Tours, and we are grateful to M. Julien Burlaud-Gaillard for his help with electronic and confocal microscopy. We thank Dr Vinh Ta Phuoc for the insightful discussions on lysosomal distribution analyses. This work was supported by the “Ministère de la Recherche et des Technologies”, the “Ligue Nationale Contre le Cancer – Interrégion Grand-Ouest” to SR, the Région Centre-Val de Loire (grant “CanalEx” to SR), the Institut National du Cancer (grant INCA_16110 “PURIN4EXO” to SR). SR was recipient of a prize “Prix Ruban Rose Avenir 2017” from the Charity “le Cancer du sein: parlons-en!”. SC was recipient of a post-doctoral grant from the School of Life Science, University of Sussex (UK) and from the AURCIL Association in Tours (France).
Author information
Authors and Affiliations
Contributions
All authors contributed extensively to the work presented in this study. SC performed cell culture, molecular and cellular biology experiments, assessed cell viability, apoptosis and invasion, immunofluorescence/confocal imaging, flow cytometry, pH imaging, bioinformatics, and statistical analyses. JA performed invasion and molecular biology experiments. LB and RL participated in cell culture, flow cytometry and scientific input regarding autophagy analysis. AH and JA participated in cell culture. OL-C, JA, and L-HJ participated to rescue experiments. RG and GF performed IHC analyses. SC, SL, and ALP performed in vivo mouse experiments. SC, RM-L, and SR analysed in vivo data. SR and RM-L obtained research grants. DA and L-HJ participated in critical reading of the paper. SC, SR, and RM-L directed the research, designed the study, analysed the data, and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Chadet, S., Allard, J., Brisson, L. et al. P2x4 receptor promotes mammary cancer progression by sustaining autophagy and associated mesenchymal transition. Oncogene 41, 2920–2931 (2022). https://doi.org/10.1038/s41388-022-02297-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-022-02297-8
- Springer Nature Limited
This article is cited by
-
LncRNA PRBC induces autophagy to promote breast cancer progression through modulating PABPC1-mediated mRNA stabilization
Oncogene (2024)
-
Preparation and preliminary evaluation of a tritium-labeled allosteric P2X4 receptor antagonist
Purinergic Signalling (2024)
-
ATP released from dying cancer cells stimulates P2X4 receptors and mTOR in their neighbors
Purinergic Signalling (2024)
-
Lactate in breast cancer cells is associated with evasion of hypoxia-induced cell cycle arrest and adverse patient outcome
Human Cell (2024)
-
Therapeutic targeting of P2X4 receptor and mitochondrial metabolism in clear cell renal carcinoma models
Journal of Experimental & Clinical Cancer Research (2023)