
Abstract
Receptors for extracellular nucleotides are widely expressed by mammalian cells. They mediate a large array of responses ranging from growth stimulation to apoptosis, from chemotaxis to cell differentiation and from nociception to cytokine release, as well as neurotransmission. Pharma industry is involved in the development and clinical testing of drugs selectively targeting the different P1 nucleoside and P2 nucleotide receptor subtypes. As described in detail in the present review, P2 receptors are expressed by all tumours, in some cases to a very high level. Activation or inhibition of selected P2 receptor subtypes brings about cancer cell death or growth inhibition. The field has been largely neglected by current research in oncology, yet the evidence presented in this review, most of which is based on in vitro studies, although with a limited amount from in vivo experiments and human studies, warrants further efforts to explore the therapeutic potential of purinoceptor targeting in cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Purinergic signalling, where adenosine 5′-triphosphate (ATP) and adenosine act as extracellular signalling molecules, was first proposed in 1972 [1]. Later, receptors for purines and pyrimidines were cloned and functionally characterised (see [2]). Four subtypes of P1 (adenosine) receptors (A1, A2A, A2B and A3), seven subtypes of P2X ion channel receptors (P2X1-7) and eight subtypes of G protein-coupled receptors (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13 and P2Y14) have been identified (see [3]). These receptors are expressed by most non-neuronal cell types as well as neurons and their physiological roles have been explored (see [4]). In recent years, there have been a number of studies of the pathophysiological roles of purinergic signalling and its therapeutic potential for a variety of diseases (see [5, 6]).
There is growing interest in the therapeutic potential of purinergic signalling for the treatment of cancer (see reviews by [7–17]). The anti-neoplastic activity of ATP was first shown by Rapaport in 1983 [18] (see also [19–21]), who demonstrated that the addition of exogenous ATP to adenocarcinotomous pancreatic and colon cancer cells inhibited cell growth by causing cell cycle arrest in the S phase. In contrast, adenosine has been suggested to promote tumour growth (see [22]). Adenocarcinomas are malignant epithelial tumours arising from glandular structures which are constituent parts of most organs of the body. Subsequent studies have shown an anti-neoplastic action of extracellular nucleotides in colorectal cancer [23], leukaemia [24, 25], oesophageal cancer [26], Ehrlich ascites tumour cells [27], squamous cell skin cancer [28], lung cancer [29], cervical cancer [30], H35 hepatoma cells [31], prostate cancer [32], bladder cancer [33], retinoblastoma [34], neuroblastoma [35], glioma [36] and melanoma [37, 38]. Tumour cells have very high ATP content compared to most healthy cells [39, 40]. ATP-depleting strategies enhance anti-cancer agent activity [41]. Tumour progression was inhibited in ecto-5′-nucleotidase (CD73)-deficient mice [42], while vascular ectonucleoside triphosphate diphosphohydrolase (CD39) directly promoted tumour cell growth [43]. It has been suggested that NTPDase6 may be a tumour suppression gene and a determinant of cisplatin resistance in testicular cancer [44].
While it is generally acknowledged that treatment with ATP or ATP analogues has a strong cytotoxic effect on several tumours, it is also clear that low ATP doses (as occurs, for example, during spontaneous release of this nucleotide from virtually every cell type) have a growth-promoting effect. Depending on the P2 receptor subtypes expressed, tumour cells may be more sensitive to the death inducing or to the trophic effect of ATP. This observation underscores the need for an in-depth characterization of P2 receptors in tumour cells, in order to fully recognise the potential of purinergic signalling in cancer therapy.
Different P2 purinergic receptor subtypes are involved in the growth inhibitory response observed in the different malignant cell types challenged with ATP or other nucleotides. The anti-neoplastic action is either due to an inhibition of cell proliferation, the promotion of cell differentiation (resulting in inhibition of cell proliferation) and cell death or a combination of these three processes. It is likely that the final effect is due to a combination of multiple effects due to stimulation of more than one P2 receptor subtype. To date, five P2 receptor subtypes have primarily been implicated in the growth inhibition of cancer cells, namely P2X5, P2X7, P2Y1, P2Y2 and P2Y11 [10], with differing cell lines responding to receptor stimulation in different ways (see Fig. 1 and Table 1). P2Y1 receptors decrease cell proliferation in melanoma [45] and squamous cell skin cancer [28]. In human oesophageal and colorectal cancer cells, P2Y2 receptor stimulation results in apoptotic cell death [23, 26], while in melanoma, stimulation of the same receptor increases cell proliferation [45]. The explanation for these divergent responses remains unclear at present. Embryonic carcinoma cells are widely used models for studying the mechanisms of proliferation and differentiation occurring during early embryogenesis. A recent investigation has shown that down-regulation of P2X2 and P2X7 receptor expression by RNA interference affects phenotype specification of P19 embryonal carcinoma cells [46].

Schematic diagram illustrating the different mechanisms by which P2 receptor subtypes might alter cancer cell function. P2Y1 and P2Y2 receptors could affect the rate of cell proliferation through altering the intracellular levels of cAMP by modulating adenylyl cyclase (AC) or by increasing intracellular calcium levels through the phospholipase C (PLC) pathway. P2X5 and P2Y11 receptor activation might switch the cell cycle from proliferation into a state of differentiation. The P2X7 receptor activates the apoptotic caspase enzyme system. (Reproduced from [10] with permission.)
In the HL-60 human leukaemic cell line, P2X receptor-mediated events result in growth inhibition [25]. P2X7 receptors induce apoptosis in melanoma [45], squamous cell skin cancer [28], lung cancer [29] and cervical cancer [30] (and see [47]). The P2X7 receptor is most widely accepted as the purinergic receptor mediator of apoptotic or necrotic cell death, as initially suggested by early experiments in mouse tumour cell lines where ATP was shown to trigger cell death via a necrosis or apoptosis, depending on the cell type [48, 49]. Whether this is due to preferential expression by different mouse tumour cells of different truncated P2X7 splice variants is not currently known. Analysis of the effect of the P2X7 receptor on tumour growth is made more complex by the observation that tonic, as opposed to pharmacological, stimulation may have a trophic, growth-promoting, rather than cytotoxic effect [50]. This intriguing effect of P2X7 receptors has been recently shown to be present also in mouse embryonic stem cells [51] and the intracellular signalling pathways have been identified [14, 52]. Besides cell growth, there is evidence from in vitro and in vivo studies that P2X7 might also participate in metastatic dissemination [53, 54]. In epithelia originating from the ectoderm, urogenital sinus and the distal paramesonephric duct, decreased expression of P2X7 receptors precedes or coincides with neoplastic development [55]. An endogenously expressed truncated P2X7 receptor lacking the C-terminus was shown to be preferentially upregulated in epithelial cancer cells, but fails to mediate pore formation and apoptosis [56]. The cell differentiating effects of P2Y11 receptors in leukaemia cells [57] and P2X5 receptors in skeletal muscle cells [18] and keratinocytes [58] may induce alterations to normal cell cycle progression and promote cell death.
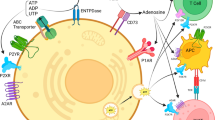
Microarray analysis of lung, breast, prostate and gastric cancers as well as melanoma revealed a significantly higher expression of A2B and P2Y receptors [59]. A3 receptors have also been shown to be highly expressed in tumour compared to normal cells [60]. Surprisingly, proliferation of most tumour cells is inhibited by adenosine, although it promotes cell proliferation via A2 receptors in human epidermoid carcinoma cells. NMR structure and functional characterisation of a human nucleoside triphosphatase involved in human tumorigenesis have been described [61]. Neuroendocrine tumours predominantly express A2A and A2B receptors and their activation leads to increased proliferation and secretion of chromogranin A [62]. One of the crucial issues to understand host–tumour interactions is the biochemical composition of the tumour microenvironment. In vivo studies show that the extracellular milieu of solid tumours has high adenosine content [63]. Due to the well-known immunosuppressive activity of adenosine, this finding gives a crucial hint for the understanding of immunoescape strategies of cancer. The possibility was raised that adenosine may act as an inhibitor of killer T cell activation in the microenvironment of solid tumours [64]. More recently, chimeric plasma membrane-targeted luciferase revealed high extracellular ATP concentrations (in the hundreds micromolar range) in tumours but not tumour-free tissues [65]. Therefore, it seems that the tumour microenvironment is a site of active extracellular ATP release/generation and conversion to adenosine, thus producing a milieu rich in growth-promoting and immunomodulatory factors. Not surprisingly, the inflammatory microenvironment is also very rich in extracellular ATP [66].
It was suggested early that adenosine may regulate the vascular supply to neoplastic tissue and thereby influence the growth of tumours [67]. The major blood vessels that supply tumours are innervated by sympathetic nerves (that release ATP as a cotransmitter with noradrenaline (NA)), but the newly formed blood vessels within tumours are not innervated [68–71]. It has been suggested that P2 purinoceptor antagonists may inhibit neovascularisation in tumour growth and metastases [72]. Inhibition of tumour angiogenesis by targeting endothelial surface ATP synthase with sangivamycin, an anti-tumour agent, was reported [73]. It has been speculated that cancer cells affect endothelial cells during metastasis, perhaps involving P2Y receptor-mediated increases in [Ca2+]i [74].
There is compelling evidence that tumour cells of various kinds release substantial amounts of ATP in response to mechanical deformation, hypoxia and some agents, as well as following necrosis and ischaemia [75, 76]. There is a correlation between levels of ATP in tumour cells and the development of cancer: ATP-depleting agents can markedly enhance cancer therapy (see [77, 78]). Cancer therapy by endogenous or transferred anti-tumour T cells has been used complementary to conventional cancer treatment by surgery, radiotherapy or chemotherapy. However, this approach is limited because tumours can create a hostile immunosuppressive microenvironment that prevents their destruction by anti-tumour T cells (see [79]). However, genetic deletion of immunosuppressive A2A receptors or the use of A2A antagonists can prevent the inhibition of anti-tumour T cells by the tumours, thus opening up a novel therapeutic approach to cancer immunotherapy [63, 80]. Chemotherapy induces ATP release from tumour cells, which leads to apoptotic cell death [81], probably via P2X7 receptors. Studies have shown that certain chemotherapeutic drugs, such as anthracyclines, are potent inducers of immunogenic cancer cell death, thereby triggering anti-tumour immune responses [82]. It was hypothesised that the inflammasome may contribute a third level regulation of immunogenic chemotherapy and that the release of ATP from dying tumour cells is involved in the activation of the inflammasome. Chemotherapeutic drugs such as cadmium, etoposide, mitomycin C, oxaliplatin, cisplatin, staurosporine, thapsigargin, mitoxanthrane and doxorubin may trigger release of ATP from tumour cells before and during apoptosis [81] and also from dendritic cells (DCs) [83]. Mice deficient in P2X7 receptor-expressing tumours failed to respond to oxaliplatin treatment and failed to mount tumour-specific CD8-T cell responses. In this process, the increase of ATP concentration within the tumour microenvironment is crucial, as ATP stimulates the P2X7 receptor of DCs to drive secretion of the key pro-inflammatory cytokine interleukin (IL)-1β. This cytokine potentiates antigen presentation to CD4+ lymphocytes, thus enhancing the anti-tumour immune response. As emphasised in this ‘Introduction’, the ATP level is a crucial determinant for the final outcome, since while high ATP doses will potentiate anti-tumour immunity, low ATP levels are likely to be immunosuppressive, as shown by the finding that human DCs stimulated by low ATP concentrations produce less pro-inflammatory cytokines, more IL-10 and synergize with interferon to upregulate indoleamine 2,3-dioxygenase levels [84]. More recently, in vivo experiments have shown that release of ATP from cancer cells is associated with autophagy, a protective mechanism in cancer, and that the increase in the pericellular environment is essential for a proper anti-cancer immunoresponse and for the efficacy of chemotherapy [85].
The possibility has been raised that ATP may be used for the treatment of both a primary tumour and the systemic side effects of the tumour in patients with advanced disease, as demonstrated in murine in vivo models. This could potentially have a considerable impact on the management of patients with advanced malignancy.
The ultimate goal of any laboratory-based medical research is to see translation of this work to treatment in patients with disease. Intravenous ATP has already been safely trialled in patients with lung cancer. A phase I trial for extracellular ATP in patients with advanced cancer was carried out in 1996 with promising results and acceptable toxicity with a dose rate of 50 μg/kg/min [86]. A phase II trial was later carried out on patients with non-small cell lung cancer [87]. Agteresch et al. [88] investigated the pharmacokinetics of intravenous ATP in 28 patients. Treatment was well tolerated with no side effects in two thirds of the group. Side effects included chest tightness (15 %) or dyspnoea (10 %), which was mild (level 1 or 2 by U.S. National Cancer Institute Criteria) and transient, resolving within minutes of decreasing the infusion rate or stopping the infusion. Other minor side effects included flushing and nausea in 5 %, light headedness in 3 %, headache and sweating in 2 % and palpitations in 1 %. In a later trial by this group, beneficial effects of ATP on nutritional status in advanced lung cancer patients were reported [89]. A recent review discusses the use of kinase inhibitors, which interact with ATP binding sites, in anti-cancer therapeutics [90].
In keeping with murine models, ATP treatment has been shown to maintain body weight and decrease cancer cachexia in human studies [91]. In the murine cancer models, intraperitoneal ATP inhibited weight loss in the animals with advanced tumour growth independent of its primary anti-neoplastic action. This anti-cachectic effect was thought to occur primarily via the ATP breakdown product, adenosine, which had little anti-neoplastic activity, but was effective at reducing weight loss. However, the anti-cachectic effect of ATP was greater than that seen with adenosine alone, implying that some other mechanism must be involved, at least in part [92]. In their trial, Agteresch et al. [91] found intravenous ATP infusions maintained body weight, muscle strength, serum albumin concentrations and quality of life in cachectic patients with advanced lung cancer over the 6-month period of the investigation. In 2003, Agteresch et al. [93] also showed, in a randomized controlled trial, that ATP infusions in patients with advanced non-small cell lung cancer significantly increased overall survival (9.3 months ATP-treated vs. 3.5 months for control), supporting the theory that ATP may treat the underlying malignancy as well as its systemic effects, although larger trials are needed to confirm this. A further trial is currently underway by the same group, investigating the effects of ATP treatment in combination with radiotherapy for non-small cell lung carcinoma. This multi-centre, double-blind, randomized control trial will focus on the effects of ATP on survival, tumour response, nutritional status and quality of life [94]. It has been claimed that intravenous ATP infusions can be safely administered to preterminal cancer patients in the home setting [95–97].
Protective effects of ATP against radiation-induced injury in human blood were reported [98]. Ionizing γ-irradiation is a well-known carcinogen capable of inducing tumours, especially in children, even though radiation is commonly used in cancer treatment protocols. Recent papers suggest that γ-irradiation leads to release of ATP, probably via connexin 43 hemichannels and/or P2X7 receptors, which then acts via activation of P2Y6 and P2Y12 receptors to mediate repair of DNA damage [99–102].
Breast cancer
Breast cancer is the most common malignant tumour in women and a major health problem worldwide. There is a great emphasis on early diagnosis, but more efficacious therapies are in urgent demand. Growth inhibition of human breast cancer cells by exogenous ATP was first shown in 1993, and it was claimed that the growth arrest was mainly due to elongation of the S-phase of the cell cycle [103]. Chemotherapeutic release of ATP from murine breast tumour cells enhanced tumour regression via apoptosis [104]. The agonist potencies of nucleotides on MCF-7 BCC were shown to be uridine 5′-triphosphate (UTP) ≥ ATP > adenosine 5′-diphosphate (ADP) [105], suggesting that P2Y2 and/or P2Y4 receptors were involved. This was later demonstrated with RT-PCR in MCF-7 breast tumour cells and ATP-activated P2Y2 receptor-linked Ca2+ signalling was shown to induce a proliferative response [106]. Extracellular nucleotides co-operate with growth factor to activate c-fos gene expression linked to the proliferative response of MCF-7 cells through activation of P2Y2 receptors [107]. It has been suggested that oestrogen, via ERα receptors, promotes proliferation of breast cancer cells by down-regulating P2Y2 receptor expression and attenuating P2Y2 receptor-induced increase in [Ca2+]i [108]. Expression of CD73 is negatively regulated by oestrogen acting via the ERα receptor and its generation of adenosine may relate to breast cancer progression [109]. Tamoxifen is used for adjuvant treatment of breast cancer because it prevents growth of cancer cells due to a range of effects in addition to blocking oestrogen actions. Hydrolysis of adenine nucleotides is modified in platelets from breast cancer patients taking tamoxifen [110]. ATP depletion due to hypoxia enhances tamoxifen anti-proliferative effects in T47D breast carcinoma cells [111]. Radioiodide therapy has been used against breast cancer and the iodide symporter gene is expressed in breast tumours. ATP and UTP, probably via P2Y2 receptors, stimulate sodium/iodide symporter-mediated iodide transport in breast cancer cells [112].
There is much interest in K+ transport in human breast cancer cells, with the strong possibility that alterations in K+ ion transport may regulate tumour cell proliferation and apoptosis (see [113]). ATP has been shown to increase K+ efflux from cultured human breast cancer cells [114]. Thus, it would not be surprising that apoptosis was activated given the profound caspase-3 stimulatory activity of K+ depletion [115].
Bioluminescence assay of ATP levels in breast tumours has been proposed to detect levels of cell proliferation and hence can be used as a marker for the biological aggressiveness and metastatic potential of breast carcinoma [116]. It has been suggested that over-expression of ATP synthase α-subunit may be involved in the progression of metastasis of breast cancer, representing a potential biomarker for diagnosis, prognosis and therapeutic target for breast cancer [117]. An ATP-based chemotherapy response assay was developed for predicting cell viability [40] and for predicting responses to chemotherapy [118–120].
The Walker 256 rat tumour cell line, which initially arose spontaneously in the mammary gland of a pregnant albino rat, has been used for studies of cancer pathophysiology. Ecto-NTPDases 2 and 5 and CD73 have been identified in Walker 256 tumour cells and are likely to be important in reducing the ratio of ATP/adenosine involved in tumour growth [121, 122]. In a later paper, nucleotide pyrophosphatase/phosphodiesterase (NPP3) was also identified in Walker 256 tumours [123].
MDA-MB-4355 human breast cancer cells secrete nucleoside diphosphate kinase (NDPK) that supports metastases and evidence has been presented to support the notion that secreted NDPK mediates angiogenesis via P2Y1 receptors and suggests that inhibitors of NDPK may be useful as therapeutics [124, 125]. Mitogen-activated protein kinase (MAPK) signalling pathways have been implicated in the regulation of cell proliferation and differentiation. ATP, acting via P2Y2 and/or P2Y4 receptors, activates MAPKs and the P13K/Akt signalling pathway in breast cancer MCF-7 cells [126, 127]. A study of human breast adenocarcinoma, MDA-MB-231 cells, suggests that cell surface ATPase plays important roles in tumour cell migration, drug resistance and the anti-tumour immunoresponse [128, 129]. CD73 facilitates the adhesion, migration and invasion of human breast carcinoma T-47D and MB-MDA-231 cell lines via generation of adenosine [130]. Anti-CD73 antibody therapy inhibits breast tumour growth and metastasis [129]. Bisphosphonates are effective inhibitors of breast cancer as well as for the treatment of metastatic bone disease in women with bone cancer and myeloma [131]. A3 receptor agonists inhibited the growth of breast tumour-derived bone metastasis, raising the possibility of a therapeutic approach to bone-residing breast cancer [132]. The bisphosphonate, zoledronic acid, had a strong anti-tumour effect, measured by the ATP tumour chemosensitivity assay, on primary breast cancer cells in vitro, which was claimed to be equal or superior to commonly used chemotherapeutic regimens for treating breast cancer [133] as did another bisphosphonate, 5-FdU-alendronate [134].
Proteomic analysis of human breast carcinoma showed that ATP synthase was upregulated in tumours and aurovertin B, an ATP synthase inhibitor, was shown to inhibit proliferation of several breast cancer cell lines [135]. It has been suggested that malignant breast carcinoma cells release ATP that makes a pre-metastatic environment suitable for micro-metastasis in lymph nodes and its nearest afferent lymph vessels [136]. Evidence has been presented that blockade of the action of nucleotides in the context of newly diagnosed breast cancer may provide a useful adjunct to current anti-angiogenesis treatment [137].
ATP enhances epidermal growth factor (EGF) activation of c-fos in Hs578T and T47D breast cancer cell lines, c-fos being an immediate early gene and proto-oncogene that plays a role in cell proliferation, differentiation and apoptosis; the combination of ATP and EGF was anti-proliferative and had strong effects on apoptosis and therefore survival of breast cancer cells [138]. Modulation of ATP-induced calcium signalling by progesterone in T47D-Y breast cancer cells has been reported [139]. ATP induced increase in [Ca2+]i and actin cytoskeleton disaggregation via P2X receptors in the rat mammary tumour cell line, WRK-1 [140]. ATP increased [Ca2+]i in breast tumour cells and high concentrations produced apoptosis [141], in retrospect probably via P2X7 receptors. P2X7 receptor-mediated activation of the human breast cancer cell line, MDA-MB-435s, resulted in neurite-like cellular prolongations, an increase in cell migration and the development of metastases, suggesting a potential therapeutic role for P2X7 receptor antagonists [54].
The role of hypoxia in regulating tumour progression is controversial. However, in MCF-7 and MDA-MB-231 breast carcinoma cell lines (as well as the HeLa cervical cancer cell line), the expression of P2X7 receptors is increased by hypoxia and they respond to the P2X7 receptor agonist, 2′(3′)-O-(4-benzoylbenzoyl) adenosine 5′-triphosphate (BzATP), by activating extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), to cause nuclear translocation of nuclear factor-κB [142]. The authors showed further that hypoxia-driven increase in P2X7 receptors enhances invasion and migration of tumour cells. Changes in purinergic signalling during EGF-induced epithelial mesenchymal transition in MDA-MB-468 breast cancer cells have been reported [143]. There was an alteration in the calcium signalling response to ATP, an increase in expression of P2X5 receptor mRNA and a decrease in P2Y13 receptor mRNA. Further, it was shown that silencing of P2X5 receptors, which inhibited cell proliferation, led to a significant reduction in EGF-induced vimentin protein expression and it was suggested that this may represent a novel mechanism for targeting cancer metastasis. Elevated release of ATP in cystic fibrosis is associated with inhibition of breast cancer growth [144].
A1, A2B and A3 receptor mRNA has been identified in MCF-7 cells with A1 receptor agonists leading to MAPK activation [145]. A1 and A3 receptor mRNA was shown to be expressed by human breast tumours [146]. Adenosine promotes tumour cell migration and proliferation of MCF-7 and T-47D breast carcinoma cell lines [147]. However, the A3 receptor-selective agonist, N6-(3-iodobenzyl) adenosine-5′-N-methyluronamide (IB-MECA), down-regulates oestrogen receptor α and suppresses human breast cancer cell proliferation [148, 149]. Adenosine reduces apoptosis in oestrogen receptor-positive (MCF-7 cells) and oestrogen receptor-negative (MDA-MB-468 cells) human breast cancer cells [150]. RNA interference targeting of A1 receptors, which are upregulated in breast cancer (MDA-MB-468) cells, leads to diminished rates of cell proliferation and induction of apoptosis [151]. The human breast cancer cell line, MDA-MB-231, expresses A2B receptors, which probably mediate cell proliferation [152] and A2B receptor blockade has been shown to slow growth of breast tumours [153]. Tenascin C is expressed in invasive solid tumours, although its role is obscure. Tenascin C has been shown to interact with CD73 to regulate adenosine generation in MDA-MB-231 breast cancer cells [154]. Assessment of adenosine deaminase (ADA) and its isoenzymes ADA1 and ADA2 has been proposed as a reliable test for differential diagnosis of benign and malignant breast disease [155].
Ehrlich ascites tumour cells appeared originally as a spontaneous breast carcinoma in mice and have been widely studied. An early paper showed that these tumour cells did not show a deficit of ATP during growth and concluded that there was no clear relationship between ATP supply and tumour growth [156]. However, later it was shown that extracellular ATP increased [Ca2+]i [27, 157] and had a growth inhibitory effect on Ehrlich tumour cells [158, 159]. It was shown that ATP elicits changes in phosphoinositide metabolism in Ehrlich ascites tumour cells similar to those produced by a wide variety of Ca2+-mobilizing hormones and growth factors [160]. ATP-induced tumour growth inhibition in Ehrlich ascites tumour-bearing mice was accompanied by a selective decrease in the content of the tripeptide glutathione within the cancer cells in vivo [161]. UTP as well as ATP activated release of Ca2+ from inositol triphosphate (InsP3)-sensitive stores in Ehrlich cells [162], suggesting that P2Y2 and/or P2Y4 receptors were involved. Mechanical stress results in the release of ATP from Ehrlich ascites tumour cells, which in turn stimulates both P2Y1 and P2Y2 receptors [163]. A calmodulin inhibitor induced short-term Ca2+ entry and a pulse-like secretion of ATP in Ehrlich ascites tumour cells [164]. It was suggested that the increased sensitivity of Ehrlich ascites tumour cells to ATP during the course of tumour growth may be associated with a decrease in ecto-ATPase activity [165].
Prostate cancer
Prostate cancer is the second most common cancer in males and the third leading cause of cancer death. Surgery is the treatment of choice, but post-surgery medical treatment is routinely given. Prostate cancer cells are sensitive to extracellular ATP. Fang et al. [166] first demonstrated that ATP could inhibit the growth of commercially available human hormone-refractory (androgen-independent) prostate cancer PC-3 cells and suggested that this effect was likely to be mediated by P2 receptors. Later, this was shown in DU145 as well as PC-3 prostate cancer cell lines [167, 168]. The potency order of UTP ≥ ATP > adenosine-5′-(γ-thio)-triphosphate (ATPγS) > inosine 5′-triphosphate > uridine diphosphate (UDP) ≥ ADP on human prostate PC-3 cancer cells [169] suggests the presence of P2Y2 and/or P2Y4 receptors. However, P2Y1 receptors were later identified on PC3 cells [170] and a more recent paper claims that activation of P2Y1 receptors, identified by RT-PCR, Western blots and pharmacology, induced apoptosis and inhibited proliferation of these cells [171]. ATP is a potent growth inhibitor of tumours and it was suggested that P2X7 receptors mediate cell death in prostate cancer [172]. Northern blotting showed that both PC-3 and DU145 prostate tumour cells expressed P2Y2, P2Y6 and P2Y11 receptors, and after breakdown of ATP to adenosine, there was A2 receptor activation [173]. It was also shown in this study using RT-PCR that these tumour cells also expressed P2X4 and P2X5 receptors in the DU145 cells and P2X4, P2X5 and P2X7 in the PC-3 cells. ATP inhibited the growth of the tumour cells, but this effect was not mimicked by UTP or adenosine, but BzATP caused an increase in apoptosis in PC-3 cells, probably via P2X7 receptors. Multicellular prostate tumour spheroids prepared from the DU145 prostate cancer cell line were exposed to direct current electrical fields, resulting in ATP release, which activated purinergic receptors to elicit a Ca2+ wave leading to stimulation of tumour growth [174]. ATP-induced inhibition of growth of prostate cancer DU145 cells (as well as lung cancer (A549) and pancreatic cancer (Panc-1) cells) via P2X7 receptors was dependent on the P13 kinase pathway that regulates apoptosis and cell growth [175]. P2Y2 receptors mediated resistance to ursolic acid-induced apoptosis in DU145 cells [176]. P2Y receptor agonists stimulated PC-3 prostate cancer cell invasion, via their down-stream ERK1/2 and p38 protein kinases [177]. Both mechanical and hypotonic stress leads to ATP release from DU145 prostate cancer cells [178]. Calcium waves were elicited by mechanical strain releasing ATP from DU145 cancer cells and purinergic receptor activation [179]. ATP enhances the motility and invasion of prostate cancer cells by activating Rho GTPases Racl and Cdc42 and upregulates the expression of matrix metalloproteinases [180]. A recent study has shown that CD73-deficient mice are resistant to prostate carcinogenesis and concluded that CD73 promotes de novo prostate tumorigenesis and further that anti-CD73 monoclonal antibodies can significantly reduce prostate tumour growth and metastasis [181].
Studies from our own laboratories compared hormone refractory prostate cancer cell lines (PC-3 and DU145) with commercially available normal prostate cells (PNT-2) [182]. Despite the similar mRNA expression, the normal prostate and HRPC cells differed considerably in their response to cell growth. PNT-2 cells were significantly less sensitive to the cytotoxic effect of ATP (19 ± 3.2 vs. 45 ± 2.3 % inhibition of cell growth, after ATP 0.1 mM) and more responsive to the mitogenic effects of UTP. The order of agonist potency also differed from HRPC cells, raising the possibility that the control of growth in normal and cancerous prostate cells is different. This may be due either to the functional involvement of a different receptor complement or an altered downstream response to the stimulation of the same receptor subtypes. Pharmacological characterization suggested that the anti-neoplastic action of ATP was likely to be mediated by P2X5 and/or P2Y11 receptors in DU145 cells. The absence of P2Y11 receptor mRNA in PC-3 cells made the P2X5 receptor the most likely receptor involved in this cell line.
The discovery of P2X7 receptor mRNA in PC-3 cells raised hopes of a pivotal role for this pro-apoptotic receptor in the observed cell death. However, functional studies using the selective antagonist KN-62 and assessment of P2X7 receptor-mediated cell membrane pore formation (using lucifer yellow stain) failed to demonstrate a functional role for these receptors. Coupled with the presence of P2X7 receptor mRNA in the normal PNT-2 cells and its absence in DU145 cells, which despite the absence of this receptor had a similar cytotoxic response to ATP as PC-3 cells, this left the explanation of the exact functional role of this receptor subtype uncertain.
The lack of effect of KN-62 at a human P2X7 receptor has been reported previously, where it failed to block permeability lesions to Ca2+ and Ba2+ and subsequent cytotoxic pore formation [183]. There are known to be many polymorphisms of the P2X7 receptor [184, 185], which, in addition to conferring a loss of function, may alter the activity of the receptor. Another possibility could be the activation of alternative downstream events.
There is a differential expression of P2X7 receptors in patients with normal prostates compared to those with prostate cancer. Slater et al. [186] found expression of P2X7 cytolytic purinergic receptors in all 116 pathology specimens of prostate cancer, irrespective of Gleason grade or patient age. P2X7 receptors were also found in normal epithelial cells adjacent to tumour margins, but not in normal tissues from patients with no evidence of cancer, raising the possibility of the appearance of such receptors as an early marker of prostate cancer. What functional role this may play in the development or treatment of prostate cancer is unclear, and the exact underlying mechanism of action of the P2X7 receptor remains largely unknown. In a later paper, it was shown that P2X7 receptor expression in the glandular epithelium is a marker for early prostate cancer and correlates with increasing levels of prostate-specific serum antigen [187].
The exact control of growth in HRPC is unclear. In hormone-sensitive normal prostate and prostate cancer cells, androgen ablation leads to apoptotic cell death. In these cells, androgen ablation leads to a sustained rise in intracellular calcium ion concentration ([Ca2+]i), leading ultimately to programmed cell death [188]. This response to androgen ablation is lost in hormone refractory cells. However, studies by Martikainen et al. [189] showed that modest elevations in [Ca2+]i for sufficient time, achieved using calcium ionophores such as ionomycin, induced apoptotic cell death in HRPC cells, raising the possibility that alterations in calcium homeostasis could still be the key to apoptosis induction in HRPC.
ATP has been shown to increase [Ca2+]i in various human cancer cell lines in vitro, including prostate cancer [166, 182], and this has been proposed as a possible mechanism for ATP-induced cell death. ATP, acting at P2Y receptors, induces a biphasic increase in [Ca2+]i, with an immediate release of endoplasmic reticulum (ER)-stored Ca2+, and secondary activation of store-operated channels, with resultant capacitative calcium entry of Ca2+ after depletion of ER Ca2+ stores. ATP and UTP were equipotent at increasing [Ca2+]i in HRPC cells, while both were shown to have markedly opposite effects on cell growth (UTP increases viable cell numbers, whereas ATP induces cell death [182]). Complete blockade of [Ca2+]i increase was observed after use of the phospholipase C inhibitor U73122, confirming the role of a G protein-coupled receptor (i.e. P2Y2) in this response, contrary to the cytotoxic effects of ATP in HRPC cell growth. Studies by Vanoverberghe et al. [168] also confirmed this poor correlation between [Ca2+]i and control of HRPC cell growth. They found that varying the concentrations of extracellular Ca2+ in culture media had no significant effect on ATP-induced growth inhibition, thereby denoting either an alternative mechanism, or secondary messenger, in ATP-induced apoptotic cell death. ATP release from erythrocytes is increased in blood samples from prostate cancer patients receiving radiation therapy, which would contribute to its beneficial effects, since ATP is a potent inhibitor of tumour growth [190]. A more recent paper claims that activation of P2Y1 receptors induced cell death and inhibited growth of prostate cancer PC-3 cells and it was suggested that P2Y1 receptor agonists may be a promising therapeutic strategy for prostate cancer [171].
Vanoverberghe et al. [168] hypothesized that decreases in the intracellular Ca2+ pool were more relevant to the observed cell death, backed up by experiments showing pretreatment with thapsigargin, at a level where it had no apoptotic effect itself (1 nM), prevented ATP-induced growth inhibition (100 μM) by decreasing the Ca2+ pool content. Interestingly, while both 1 nM thapsigargin and 100 μM ATP reduced the Ca2+ pool content to a similar extent, only thapsigargin alone had no growth inhibitory effects [168]. As thapsigargin and ATP work on the ER in different ways to lower the Ca2+ pool (sarcoplasmic reticulum/ER Ca2+ ATPase pump inhibitor vs. InsP3 receptor activation), they concluded that the secondary mechanisms involved may be more important than the level of reduction in the intracellular Ca2+ pool alone. One possibility could be potential alterations to the intracellular production and levels of Bcl-2 proteins by extracellular ATP. Overexpression of Bcl-2 proteins is associated with prevention of apoptosis and is a common finding in cancer cells. Miyake et al. [191] previously showed that the treatment of HT1376 bladder cancer cells with ionomycin induced apoptosis and decreased the mRNA and receptor expression of the anti-apoptotic Bcl-2 proteins, while increasing the expression of pro-apoptotic Bax proteins. At present, no studies have explored the effect of ATP on Bcl-2 expression in prostate cancer, or any other malignancy, and this would be an interesting avenue for future research.
The in vitro cytotoxic effects of extracellular ATP have also been confirmed in vivo. We found that daily intraperitoneal injections of ATP significantly reduced the growth of subcutaneously implanted DU145 and PC-3 cells in male nude athymic mice (57–69 % reduction in the growth of freshly implanted or established DU145 and PC-3 cells, respectively) (Fig. 2a, b) [32]. No side effects or complications related to ATP treatment were seen throughout the experiment. Light and electron microscopy were used to confirm that the inoculated tumour cells retained their original phenotype and cellular characteristics. The endothelium has an important role in the regulation of malignant tumour growth [192]. It has been shown recently that secretion of soluble vascular endothelial growth factor (VEGF) receptor-2 by microvascular endothelial cells from human benign prostate cancer is increased by ATP [193]. Although these experiments validated the relevance of the in vitro experiments on the primary growth of HRPC, they gave no information about the effect of ATP on tumour metastases. An orthotopic model of prostate cancer would add to our understanding of this process and the potential effect of ATP (see section on ‘Bone Cancer’).

a Effect of ATP (1 ml of 25 mM i.p.) on the fractional growth of HRPC DU145 tumour cells in vivo after 60 days initial growth and b effect of ATP (1 ml of 25 mM i.p.) on the growth of implanted DU145 tumour cells in vivo after 60 days of initial growth; the lower mouse received ATP treatment vs. no treatment in the upper mouse. (Reproduced from [32] with permission.)
Proliferation of prostate tumour cells is inhibited by adenosine, whereas normal cells are stimulated by adenosine. 2-Chloroadenosine (2-ClAdo) has cytotoxic effects on PC-3 prostate tumour cells, probably by entry into the S-phase of the cell cycle and the induction of DNA strand breaks [194]. It has been claimed that 2-ClAdo induces apoptosis of PC-3 prostate cancer cells [195]. A3 receptor activity by IB-MECA inhibited prostate cancer cell proliferation and induced cell cycle arrest and apoptosis [196, 197]. Activation of A3 receptors also suppressed prostate cancer metastasis by inhibiting nicotinamide adenine dinucleotide phosphate oxidase activity [198].
Colorectal, gastric, oesophageal and neuroendocrine cancer
Colorectal cancer is widespread, but cancer of the oesophagus and gastric and neuroendocrine tumours also occur. Exposure of two colonic adenocarcinoma cell lines, HT-29 and SW-620, to ATP resulted in substantial inhibition of cell growth [199].
Colorectal tumours
ATP and ADP increased [Ca2+]i in the HT-29 human colonic adenoma cell line [200]. HT-29 cells were depolarised by UTP > ATP > ADP > adenosine [201]. There is a report that cultured human tumour cells derived from the colon (LoVo) are resistant to ATP cytotoxicity, but exposure to verapamil increases sensitivity to ATP [202]. HT-29 cells express P2U (i.e. P2Y2 and/or P2Y4) receptors [203]. Both P2 and neurotensin receptors are expressed by HT-29 cells and both increase extracellular acidification, but there was no obvious interaction between the actions of these receptors [204]. HT-29-C116E is a highly differentiated sub-clone of the HT-29 colonic cancer cell line and ATP transiently increased Cl− conductance in these cells [205, 206]. ATP activation of Cl− conductance was also shown in the T84 human colonic adenocarcinoma cell line [207]. In a later paper, HT-29 cells showed a decrease of intracellular Cl− and Na+ and an increase in Ca2+ in response to both ATP and UTP via P2U (P2Y2 and/or P2Y4) receptors [208]. RT-PCR studies confirmed the presence of P2U mRNA in both primary cultures of human colorectal carcinoma cells and HT-29 cells and it was suggested that they play a role in the regulation of cell proliferation and apoptosis [209]. P2Y2 receptors mediated resistance to ursolic acid-induced apoptosis in HT-29 cells [176].
Extracellular ATP induced apoptosis and inhibited growth of primary cultures of colorectal carcinomas [209], perhaps via P2Y2 receptors [210] or by an unidentified ATP receptor, mediating actions on the S-phase of cell cycle by inhibiting protein kinase C [211]. Purinergic responses of HT-29 cells are mediated by G protein α-subunits after activation of P2U receptors [212]. mRNA for P2Y receptor subtypes P2Y2, P2Y4 and P2Y6, acting through MAPK cascades, was located on the apical membranes of human colonic Caco-2 adenocarcinoma cells [213, 214]. Caveolin-1 facilitates the hypotonicity-induced release of ATP from basolateral, but not apical, membranes of Caco-2 cells [215]. Using microphysiometry, to measure extracellular acidification rate, P2Y2 receptors were identified on HT-29 colonic carcinoma cells [216], and in a later study, this group showed that both P2Y2 and P2Y4 receptors were upregulated in human colon cancer [217]. Regulation of increase in [Ca2+]i during P2Y2 receptor activation is mediated by Gβγ-subunits [218]. It has been claimed that P2Y2 receptors have oncogenic potential mediating transformation of colorectal RKO cancer cells [219]. ATP induces proliferation of Caco-2 cells via P2Y receptors [220]. Tissue samples from patients with colorectal cancers showed increased expression of an ATP-binding cassette superfamily transporter, multidrug resistance protein-2 [221].
Modulatory effects of the ectonucleotidase CD39 (NTPDase1) on colorectal tumour growth and liver metastasis, and on the expression of both P2Y2 and P2X7 receptors, indicated the involvement of purinergic signalling in these effects [222]. The activity of both CD73 and ADA was markedly higher in primary human colorectal tumours; the ADA level could be correlated with lymph node metastases and histological type, while CD73 activity could be correlated with tumour location and grade [223]. A 40 % increase in ADA activity in human colorectal adenocarcinomas was reported [224]. Dipeptidyl peptidase is a multifunctional cell surface protein which is a binding protein for ADA. Adenosine that is present in increased levels in the hypoxic tumour microenvironment down-regulates the surface expression of this protein in HT-29 cells [225]. RT-PCR showed that gene expression of adenosine kinase is significantly increased in human colorectal cancer [226].
Heterogeneity of chemosensitivity of colorectal adenocarcinoma was determined by a modified ex vivo ATP-tumour chemosensitivity assay, and it was suggested that this could be used to identify patients who might benefit from specific chemotherapeutic agents alone or in combination [227–229]. For years, surgeons have washed the abdominal cavity with distilled water to lyse colorectal cancer cells left after surgery. A study has shown that water induces autocrine release of ATP from epithelial cells, which then causes cell death of tumour cells via P2X7 receptors [230].
Adenosine accumulates in solid tumours and stimulates tumour growth and angiogenesis, while imparting tumour resistance to the immune system, thereby facilitating tumour survival [22, 231]. Adenosine promotes cell proliferation in poorly differentiated HT-29 cells via A1 receptors; cell growth inhibition was observed in the presence of ADA and A1 receptor antagonists [232]. In contrast, adenosine had less effect on more differentiated cells with lower proliferation rates (e.g. Caco-2, DLD-1 and SW 403 cell lines) [233], but was still found to stimulate proliferation of such cells (including the colorectal carcinoma human cell lines T84, HRT-18, Caco-2, Colo 320 HSR and MCA-38, the murine liver-derived colon carcinoma cell line) at concentrations present within the tumour extracellular environment; RT-PCR showed that all four P1 (adenosine) receptor subtypes were expressed in all the human carcinoma cell lines studied, but it was speculated that A2B receptors might make a major contribution [234]. A more recent paper claims that adenosine suppresses growth of CW2 human colonic cancer cells by inducing apoptosis via A1 receptors [235]. There is enhanced A2B receptor expression in proliferating colorectal cancer cells, suggesting that A2B receptor antagonists may be a promising target for colorectal cancer therapy [236].
A single low level intravenous dose of [32P]ATP significantly inhibited the growth of established xenografted subcutaneous human colon adenocarcinoma cell line, HCT116, in nude mice [237]. 8-ClAdo inhibited growth of colorectal cancer cell lines HCT116 and 80514 in vitro and in vivo [238]. Inhibition of primary colon carcinoma growth was elicited by A3 receptor agonists [239, 240]. However, subsequent papers claimed that A3 receptors mediate a tonic proliferative effect on Caco-2, DLD1 and HT-29 colorectal tumour cell lines [241]. A phase II, multi-centre study showed that an A3 receptor agonist (CF101) stabilized the tumour in 35 % of the patients with refractory metastatic colorectal cancer [242]. Elevated expression of A3 receptors was shown in human colorectal cancer and it was suggested that this could be used as a diagnostic marker and a therapeutic target for colon cancer [243]. 2′-Deoxyadenosine caused apoptotic cell death in the human colon carcinoma cell line, LoVo [244]. Adenosine has also been claimed to induce apoptosis in Caco-2 colonic cancer cell [245].
The chemokine receptor, CXCR4, plays a crucial role in determining the ability of cancer cells to metastasize from the primary tumour. Adenosine upregulates CXCR4 and enhances the proliferative and migratory responses of HT-29 cells [246]. Adenosine down-regulates the cell surface protein CD26, which binds to ADA, on HT-29 colorectal carcinoma cells, thereby facilitating tumour survival [247]. Evidence has been presented that adenosine can stimulate migration of colon cancer cells and that caffeine significantly inhibits this effect [248].
Gastric cancer
The human gastric signet ring cell carcinoma cell line (JR-1) responds to ATP with hyperpolarisation, probably mediated by P2Y receptors [249]. ATP and adenosine reduced proliferation and induced apoptosis in the human gastric carcinoma cell line (HGC-27) [250, 251]. An ATP-based chemotherapy response assay has been used to predict and enhance the benefits of chemotherapeutic drugs in patients with gastric cancer [252, 253]. Helicobacter pylori infection of the gastric body contributes to the progression of gastric carcinoma, perhaps by regulation of H,K-ATPase [254]. RT-PCR showed that gastric cancer cells showed a loss of A3 receptors [255].
Oesophageal cancer
The human oesophageal squamous carcinoma cell line, Kyse-140, and primary cancer cell cultures from patients expressed P2Y2 receptors, which mediated inhibition of growth [26]. A marked heterogeneity of chemosensitivity in oesophageal cancer has been described using the ATP-tumour chemosensitivity assay [256].
Neuroendocrine tumours of the gastrointestinal tract
Neuroendocrine tumours are a heterogeneous group of neoplasms originating from enteric chromaffin cells. RT-PCR showed that these tumours express A2A and A2B receptors and their activation leads to increased proliferation [257], suggesting that they are potential targets for therapy [62].
Biliary cancer
P2Y2 receptors have been identified in human biliary epithelial cancer cells (Mz-Cha-1) [258].
Lung cancer
Lung cancer is the most common cancer in terms of incidence and mortality in the developed world. In males, it is undisputedly the most frequent malignant tumour, but the incidence in females is also rising rapidly. A549 human lung epithelial-like adenocarcinoma cells express P2U (i.e. P2Y2 and/or P2Y4) receptors, which when occupied lead to an increase in [Ca2+]i [259] which does not inhibit forskolin-evoked cyclic adenosine monophosphate (cAMP) accumulation in these cells [260]. Calcium-dependent release of ATP and UTP (with subsequent increase in adenosine levels) from A549 cells has been reported [261].
A phase II study of intravenous ATP in patients with previously untreated non-small cell lung cancer led to the authors concluding that ATP, at least at the dose and administrative schedule employed, was an inactive agent in patients with advanced non-small cell lung cancer [87].
Erythromycin is widely used in the treatment of respiratory tract infections. It has also been shown to selectively inhibit Ca2+ influx induced through P2X4 receptor activation of A549 human lung tumour cells [262]. In this study, it was also shown with RT-PCR that A549 cells express P2Y2, P2Y4 and P2Y6, as well as P2X4 receptors. Transforming growth factor β1 augments ATP-induced Ca2+ mobilization, which leads to an acceleration of migration of A549 cells, but it markedly reduces endogenous ATP release [263].
Cachexia is a common feature of lung cancer patients and is associated with metabolic alterations, including elevated lipolysis, proteolysis and gluconeogenesis. An increase in glucose turnover during high-dose ATP infusion in patients with advanced non-small cell lung cancer occurs, perhaps contributing to the reported beneficial effects of ATP on body weight in patients with advance lung cancer [88]. Later randomized clinical trials led to the conclusion that ATP has beneficial effects on weight, muscle strength and quality of life in patients with advanced non-small cell lung cancer as well as enhancing median survival from 3.5 to 9.3 months [89, 97, 264, 265]. ATP infusion restores hepatic energy levels in patients with advanced lung cancer, especially in weight-losing patients [266]. ATP has been claimed to reduce radiation-induced damage [98] and clinical trials are underway to assess the effect of concurrent ATP and radiotherapy treatment on outcome in non-small cell lung cancer patients [94].
ATP induced a significant dose-dependent growth inhibition of five different cell lines: human large cell lung carcinoma (H460), human papillary lung adenocarcinoma (H441), human squamous cell lung carcinoma (H520), human small cell lung carcinoma (GLC4) and human mesothelioma (MERO82) [93]. ATP also had cytotoxic effects on the PC14 lung adenocarcinoma cell line and further enhanced the anti-tumour effect of etoposide (VP16) in both PC14 and the A549 cell line, a human alveolar epithelial cell carcinoma [267]. ATPγS regulated the production of cyclooxygenase-2 and synthesis of prostaglandin E2 in A549 cells [268].
It has been claimed that extracellular ATP, UTP and UDP stimulate proliferation of A549 lung tumour cells via P2Y2 and P2Y6 receptors as well as an ADP-sensitive receptor that was not the P2Y1 subtype [29]. ATP and ADP strongly inhibited proliferation of the human lung adenocarcinoma cell line, LXF-289, via P2Y receptors [269]. ATP-based chemotherapy response assay has been used to guide the outcomes of platinum-based drug chemotherapy for un-resectable non-small cell lung cancer [270, 271]. Cisplatin, a platinum complex, is a widely used anti-cancer agent for the treatment of lung cancer. ATP increases the cytotoxicity of cisplatin in a human large cell lung carcinoma cell line (H460) [272, 273]. Blockade of ATP synthase, located on the plasma membrane, suppresses adenocarcinoma growth [274].
It has been suggested that tumour-infiltrating immune cells can benefit the tumour by producing factors that promote angiogenesis and suppress immunity and because adenosine levels are high in tumours. It has been proposed that A2B receptors on host immune cells may participate in these effects and confirmed when A2B receptor knock-out mice exhibited significantly attenuated growth in a Lewis lung carcinoma (LLC) isograft model [275]. Exposure of human lung cancer cell lines A549 and H1299 to 8-ClAdo induced cell arrest at the G2/M phase and mitotic catastrophe followed by apoptosis [276–278]. 3-Deoxyadenosine (cordycepin) exerted inhibitory effects on the growth of the mouse LLC cell line by stimulating A3 receptors [279]. The A3 receptor agonist, thio-Cl-IB-MECA, inhibited cell proliferation through cell cycle arrest and apoptosis of A549 human lung carcinoma cells [280]. Adenosine induced apoptosis via A3 receptors in A549 cells [281], SBC-3 [282] and Lu-65 [283] human lung cancer cells. Stanniocalcin-1, a secreted pleiotrophic protein, regulates extracellular ATP-induced calcium waves in monolayers of A549 cancer cells by stimulating ATP release [284]. Lung cancer has been reported to alter the hydrolysis of nucleotides and nucleosides by ecto-nucleotidases in platelets [285].
Cisplatin is widely used for the treatment of cancer, including non-small cell lung cancer. Expression of copper-transporting P-type adenosine triphosphatase, which is associated with platinum drug resistance in tumours, is claimed to be a useful chemoresistance marker for cisplatin actions [286].
Nasopharyngeal cancer
Micromolar concentrations of ATP activated a chloride current that led to shrinking of human nasopharyngeal carcinoma cells [287]. In a later study of human nasopharyngeal carcinoma CNE-2Z cells, it was suggested that the volume-sensitive chloride current is activated via P2Y receptors after autocrine/paracrine release of ATP [288].
Liver cancer
Primary liver malignant tumours are almost always carcinomas and can be further subdivided in hepatocarcinoma, bile duct carcinoma (cholangiocarcinoma) and hepatocholangiocarcinoma. Hepatoma cells have been extensively used to investigate ATP effects. ATP increases calcium uptake by rat hepatoma cells [289]. Nucleotide receptors activate cation, potassium and chloride currents in HTC cells from a rat liver tumour line [290]. CD39 knock-down mice show an increased incidence of spontaneous and induced hepatocellular carcinoma [291]. The hepatoma cell line N1S1-67 has been used to study the signal transduction system activated by ATP, probably P2Y2 or P2Y4 subtypes [292]. An increase in intracellular calcium is followed by the opening of Ca2+-activated K+ channels leading to membrane hyperpolarisation. Direct intra-arterial injection of a potent inhibitor of ATP production has been proposed as a novel therapy for liver cancer [293]. Vesicular exocytosis plays an important role in release of ATP from HTC cells and a Cl− channel inhibitor can be used to specifically stimulate ATP release through exocytotic mechanisms [294]. Tumour necrosis factor-α (TNFα) was the first cytokine used for cancer therapy. It has been shown that healthy liver cells are transiently protected from TNFα-mediated cell death by fructose-induced ATP depletion, while malignant cells are selectively eliminated through TNFα-induced apoptosis [295, 296]. Chrysophanol, a member of the anthraquinone family that is one of the components of a Chinese herb including rhubarb recommended for the treatment of cancer, induces necrosis of J5 human liver cancer cells via reduction in ATP levels [297]. Curcumin, a herbal extract, has been reported to inhibit the growth of a variety of cancer cells, and a recent paper suggests that it acts by inhibiting ecto-ATPase activity leading to increased extracellular ATP in hepatocellular carcinoma HepG2 cells [298]. Further, ATP induces ATP release from HepG2 cells [299]. The in vivo effects of ATP infusions on rat hepatocarcinomas have been investigated [300].
Inhibition of hepatoma cell growth by adenosine was reported [301]. In vivo experiments show that the A3 receptor agonist, CF101, causes inhibition of liver metastasis (following colon carcinoma) [302]. Human hepatocellular carcinoma HepG2 cells express high affinity A1 receptors, which, when occupied, result in decreased adenosine monophosphate (AMP) and erythropoietin production [303]. ATP and adenosine induce cell apoptosis of the human hepatoma cell line Li-7A via the A3 adenosine receptor [302, 304]. CF102, a selective A3 receptor agonist, was claimed to have anti-tumour and anti-inflammatory effects on the liver [305] and has been investigated in a clinical trial for patients with hepatocellular carcinoma [306]. A2B receptors are highly expressed in human hepatoma cellular carcinoma [307].
NTPDase1 (CD39) expression on regulatory T cells inhibits the activity of natural killer cells and promotes hepatic metastatic tumour growth in mice [308]. CD39 deletion, resulting in higher concentrations of extracellular nucleotides, promotes the development of both induced and spontaneous autochthonous liver cancer in mice [309]. Liver metastasis from colorectal cancer is a leading cause of cancer-related morbidity. It is claimed that tailored-chemotherapy, based on ATP-chemotherapy response assay, could be effective for the treatment of initially un-resectable colorectal liver metastasis [310]. The upregulation of ATP-binding cassette transporter genes in hepatocellular carcinoma is mediated by cellular microRNAs [311].
Pancreatic cancer
There are only a few reports about purinergic signalling in pancreatic cancer. Adenocarcinoma arising from pancreatic ducts is responsible for more than 90 % of pancreatic cancers and survival is less than 5 % over a 5-year period. Insulinomas are relatively rare and have a much better prognosis. What we know about purinergic signalling in these cancer cells is mostly from cultured cancer cell lines, which are often used as model systems. An early paper by Rapaport et al. [199] showed growth inhibition of two human pancreatic adenocarcinoma cell lines (CAPAN-1 and PANC-1) in soft agar cultures by treatment with low levels of ATP. A later paper showed that dipyridamole, that prevents uptake of adenosine leading to increased extracellular levels, prevented human pancreatic cancer cell-induced hepatic metastasis in nude mice [312]. Insulinoma cell lines are often compared to isolated islets or β-cells in the same studies and similar conclusions have been reached. For example, ATP at low concentrations promotes insulin secretion from the INS-1 insulinoma cell line and rat islets via P2Y receptors, but inhibits insulin release at high concentrations after being metabolised to adenosine [313]. Also in the CAPAN-1 cell line, derived from human pancreatic adenocarcinoma of ductal origin, ATP and UTP applied to the apical membranes decreased cellular pH indicating HCO3 − secretion, but were inhibitory when applied to the basolateral membranes [314]. CD39 and P2X7, P2Y2 and P2Y6 receptors are significantly increased in biopsies of pancreatic cancer [315]. High levels of mRNA for CD39 significantly correlated with better, long-term survival after tumour resection in patients with pancreatic cancer. It was claimed that extracellular ATP is cytotoxic for pancreatic cancer cells because of its induction of cell cycle arrest at S-phase and cell death by apoptosis [316]. P2Y2 receptors are functionally expressed on human pancreatic cancer cells mediating cell proliferation [317]. Solid pseudopapillary tumours of the pancreas are rare, comprising only 0.3 % of all pancreatic tumours. The in vitro ATP-based chemotherapy response assay has been used effectively for assessing the chemotherapy for these tumours [318].
Bone cancer, osteosarcoma, myeloma and fibrosarcoma
Interest in bone tumours is motivated not only for the treatment of primary tumours, which are relatively rare, but also for the treatment of metastases. Secondary bone metastases arising from prostate and breast cancer are common, but some primary osteosarcomas occur in children. In addition, there is malignant disease of bone marrow (myeloma) and fibrosarcoma, which can arise from bone.
Bone cancer
Bone metastases are radiographically classified as osteoblastic or osteolytic, resulting from imbalances between osteoblast-mediated bone formation and osteoclast-mediated bone resorption. Osteoblastic lesions, characteristic of prostate cancer, are caused by an excess of osteoblast activity leading to abnormal bone formation. In breast cancer, osteolytic lesions are found in 80 % of patients with stage IV metastatic disease [319] and are characterized by increased osteoclast activity and net bone destruction [320]. Breast cancer bone lesions span a spectrum; most are osteolytic, but up to 15 % are osteoblastic or mixed. Bone metastasis can result in significant bone loss, fractures, pain and hypercalcaemia and spinal cord compression. ATP has been reported to inhibit the growth of bone tumour cells (see [10]).
Significant inhibition of bone tumours by an ADPase, APT102, in combination with aspirin has been demonstrated in two experimental models of bone metastases [321]. APT102 is not directly cytotoxic on the tumour cells, but rather acts via platelets, which are known to contribute to the development of metastasis, since cancer cells travel from a primary site to a distant metastatic site co-existing with platelets in thrombi located in organs and the circulatory system [322]. Prostate cancer primarily metastasizes to bones in the axial skeleton. Bisphosphonates, such as zolendrenic acid, licensed for use in the treatment of bone metastases in patients with HRPC, have previously been shown to inhibit prostate carcinoma cell adhesion to bone [323]. Bisphosphonates inhibit growth, attachment and invasion of cancer cells in culture and promote apoptosis. A recent study has shown that this is, in part, due to the formation of a novel ATP analogue (ApppI) which is able to induce apoptosis [324]. Further assessment of this phenomenon and its possible interaction with functional P2X7 receptors found on osteoclasts [325] may help further our understanding of ATP treatment and purinergic receptor pathways in prostate cancer and metastases. Expression of cathepsin L, a cysteine protease associated with cancer metastasis, which activates heparanase, is predominately enhanced in primary bone tumours, such as osteosarcoma, chrondrosarcoma and multiple myeloma, and tumours which preferentially metastasise to bone (i.e. breast and prostate cancer) and in bone metastases [326]. ATP, ADP and adenosine were most effective in stimulating secretion of active heparanase by tumour cells [327]. Further, heparanase secretion was inhibited by antagonists to P2Y receptors, probably the P2Y1 subtype.
Osteosarcoma
This is the most common primary tumour of bone in children and adolescents. It is characterised by poor differentiation and dysregulation of the genes involved in differentiation. P2X5 receptors, which mediate tumour cell differentiation [328], may be involved in this mechanism. Purinergic regulation of cytosolic Ca2+ and phosphoinositide metabolism was reported in rat osteosarcoma cells [329, 330] and human osteoblast-like tumour cells [331]. P2U (i.e. P2Y2 and/or P2Y4) receptors have been implicated in this effect [332]. Modulation of [Ca2+]i and activation of ERK1/2 and P38 MAPK by ATP, acting via P2Y2 receptors, have been described in osteoblast-like osteosarcoma ROS-A 17/2.8 cells [333]. Butyl benzyl phthalate suppresses the ATP-induced cell proliferation in human osteosarcoma HOS cells, perhaps via P2X receptors [334]. Osteosarcoma cell lines SaOs2 and MG63 express P2X7 receptors; however, another osteosarcoma cell, Te85, did not express P2X7 receptors, but rather P2X5 > P2X4 > P2X6 receptor mRNA, showing that the anti-proliferative effect of ATP on these cells was not via P2X7 receptors [335].
Myeloma
Myelomas are a malignancy of plasma cells, e.g. antibody-producing, differentiated B lymphocytes in bone marrow. 8-Aminoadenosine is an effective cytotoxic agent against multiple myelomas [336]. RPMI 8226 multiple myeloma cells express P2X7 receptor mRNA and protein, as well as P2X1, P2X4 and P2X5 mRNA [337]. A2A adenosine and β-2 adrenergic receptors have synergistic anti-proliferative activity in multiple myeloma models [338]. Heat shock protein 90 (HSP90) is over-expressed in multiple myeloma and 8-chloro-adenosine is currently in clinical trials as an enhancer of inhibition HSP90 to treat multiple myeloma [339].
Fibrosarcoma
Fibrosarcoma is a malignant tumour derived from fibrous connective tissue of the bone. In vivo data show that intraperitoneal ATP slows the growth of spontaneous murine fibrosarcomas without adversely affecting bone marrow radiation tolerance [340]. When fibrosarcoma NCTC 2472 cells were co-cultured with nodose neurons, the sensitivity of P2X2/3 and P2X2 receptors to opioid inhibitory control was decreased and it was suggested that this may contribute to the decreased sensitivity of cancer pain to opioids [341]. Cl− channels play an important role in ATP release from human fibrosarcoma HT-1080 cells; release does not appear to involve hemichannels [342]. However, in recent papers, it was claimed that maxi-anion channels and pannexin 1 hemichannels are separate pathways for swelling-induced ATP release from murine L929 fibrosarcoma cells [343, 344]. Adenosine A3 receptor activation elicited inhibition of fibrosarcoma G:5:113 cells [345].
The presence of bone metastases is a major cause of pain [346, 347]. The most common primary sites for bone metastases are breast and prostate with incidence rates for either at 70 % followed by lung at 35 % [348]. Up to 83 % of bone cancer pain patients reported pain that is significantly worse on movement [349]. Despite the availability of bisphosphonates to treat bone cancer pain specifically by preventing bone resorption in addition to NSAIDs and opioids, no new pharmacotherapy has merged in over a decade and patients continue to have bone cancer pain undermanaged [349].
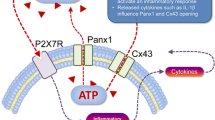
A unifying purinergic hypothesis for the initiation of pain was proposed by Burnstock [350]. One component of the hypothesis was that high concentrations of ATP can be released upon damage of the expanding tumours by bone and connective tissue (see also [351]). It would then stimulate P2X3 receptor-expressing nociceptors present in the afferent nerve endings and result in cancer pain. P2X3 and P2X2/3 receptors have been the most studied purinergic receptors for their role in ATP-mediated nociception since they are highly expressed in a selective subpopulation of nonpeptidergic isolectin B4-positive primary afferents on peripheral and central terminals [352, 353]. Tumour cells contain an abnormally elevated amount of ATP. Spontaneous and evoked release of ATP from cancer cells by mechanical, hypotonic, electrical stimulation and cell swelling has also been demonstrated (see [342]). Upregulation of P2X3 receptors is found on epidermal nerve fibres in models of bone cancer pain [354]. Minodronic acid, which is a third generation of bisphosphonates, was found to exert antagonistic properties on P2X2/3 receptors and showed analgesic effects in non-cancer pain models [355]. Complementary to its inhibition of bone resorption, the compound is proposed to be effective in relieving bone cancer pain. Radiotherapy is effective in relieving bone cancer pain and P2X6 receptors have been implicated in the underlying mechanism [356].
Thyroid cancer
Thyroid cancers are relatively rare and often only found following a post-mortem examination. However, incidence may vary in different geographical areas, and a steady increase in the incidence has occurred since World War II. P2Y receptors were shown to be expressed on thyroid cancer cells, but the ATP-induced Ca2+-phosphatidylinositol signalling cascade was found to be impaired [357]. ATP released from human thyroid ARO tumour cells controls the intracellular levels of apurinic apyrimidinic endonuclease redox effector factor-1, a protein involved in repair of DNA lesions [358], thereby controlling HSP90 expression via P2Y1 and P2Y2 receptors [359]. In addition, extracellular ATP was shown to trigger release of IL-6 from human thyrocytes [360]. This observation is of particular relevance as IL-6 is a well-known growth factor for thyroid cells. Increased expression and function of P2X7 receptors have been reported in thyroid papillary cancer [361], and a loss of function polymorphism in the P2X7 receptor (1513A>C) was shown to have a strong association with the follicular variant of this thyroid cancer histotype [362]. It has been claimed that the expression of X-linked inhibitor of apoptosis and P2X7 receptors may predict the aggressiveness of papillary thyroid cancer [363]. The tumour suppressor gene PTEN plays an important somatic role in both hereditary and sporadic cancer. ATP regulates PTEN subcellular localisation in thyroid as well as breast and colon carcinomas [364]. Clodronate is a bisphosphonate used to improve survival of breast cancer patients and prevent bone metastasis. Clodronate-induced apoptosis in human papillary thyroid carcinoma is mediated via the P2Y receptor signalling pathway [365].
PKA-independent inhibition of proliferation and induction of apoptosis of human thyroid cancer cells by 8-ClAdo have been reported [366]. A3 agonists inhibit thyroid cancer cell proliferation, but apparently independently of receptor activation [367]. Enhanced expression of A1 receptors in human thyroid carcinoma has been reported [368].
Skin cancer
Ultraviolet (UV) light has been implicated in the genesis of several tissues of cutaneous malignancies, including basal cell carcinoma, melanoma and squamous cell carcinoma. The UV-B component has been identified to have the most severe effects and UV-B irradiation was shown to decrease the amount of P2X1 and P2Y2 receptors and destroy P2X7 receptors, possibly contributing to the malignant transformation of keratinocytes [369].
Basal cell and squamous cell carcinomas
Basal cell and squamous cell carcinomas are tumours that usually arise after 50 years of age, squamous cell carcinoma being more frequent and more aggressive than basal cell carcinoma. Local administration of nucleoside analogs inhibited growth of basal cell carcinomas [370]. The A431 human cutaneous squamous cell (epidermal) carcinoma cell line expressed P2 receptors [371], which when occupied led to an increase in [Ca2+]i [372, 373]. Stimulation of A431 cells by ATP caused production of InsP3 [374], suggesting that P2Y receptors were involved. A mechanism based on the release of ATP, perhaps acting at P2X receptors, was shown to be involved in human lymphokine-activated killing of human carcinoma and melanoma cells [375].
An investigation of purinergic signalling on the non-melanoma skin cancers, basal cell carcinoma and cutaneous squamous cell carcinoma was carried out [28]. Immunohistochemical analysis of both frozen and paraffin sections of these human skin carcinomas showed expression of P2X5, P2X7, P2Y2, P2Y2 and P2Y4 receptors. P2X5 and P2Y receptors were heavily expressed on both basal cell and squamous cell carcinomas, and P2X7 receptors were expressed in the necrotic centre of nodular basal cell carcinomas and in apoptotic cells in superficial multifocal and infiltrative basal cell carcinomas. P2Y1 receptors were only expressed on the stroma surrounding tumours. P2Y4 receptors were found in basal cell, but not squamous cell carcinomas. Functional studies on the A431 squamous carcinoma cell line supported the view that low concentrations of ATP and UTP caused an increase in cell number, whereas high concentrations caused a significant decrease, while the potent P2X7 receptor agonist, BzATP, also caused a significant decrease.
In addition to ATP causing apoptosis of cultured A431 cells via P2X7 receptors, it was shown that UTP and adenosine (following breakdown of ATP) also induced cell death [376]. In in vivo experiments in mice, skin papillomas followed by squamous spindle cell carcinomas induced by local treatment with 7,12-dimethyl-benz(a)anthracene (DMBA), followed by tumour promotion with 12-O-tetradecanoylphorbol-13-acetate (TPA) were used to show that application of BzATP, a potent P2X7 receptor agonist, inhibited the formation of DMBA/TPA-induced skin papillomas and carcinomas [377]. At the completion of the study at week 28, the proportion of living animals with cancers in the DMBA/TPA group was 100 % compared to 43 % in the DMBA/TPA + BzATP group. γ-Irradiation, which causes growth arrest and death of tumour cells, induces P2X7 receptor-dependent ATP release from B16 melanoma cells [99]. Skin cancer can be induced by drinking water containing arsenic. A recent paper claimed that arsenic may induce malignancies by reducing calcium release from ER by P2Y4-mediated ATP actions in human primary keratinocytes [378].
It is concluded that P2Y2 receptors mediate proliferation, P2X5 receptors mediate differentiation (and are therefore anti-proliferative) and P2X7 receptors mediate cell death. ADA in saliva has been identified as a diagnostic marker of squamous cell carcinoma of the tongue [379].
Melanoma
Malignant melanoma is an aggressive cancer with a high potential for metastasis that originates from melanocytes, the pigment-producing cells of the skin. ATP inhibited the growth of both animal and human melanoma cells in vivo [18, 380, 381]. Amelanotic hamster melanoma A-Mel3 cells, grown subcutaneously in hamsters, have been used to study ATP levels in relation to blood flow [382, 383]. CD39 is over-expressed in differentiated human melanomas compared to normal melanocytes [384]. Extracellular ATP has growth-inhibiting properties in a highly metastatic liver-colonising murine B16 melanoma cell line in vitro [385].
Increased expression of P2X7 receptors in 80 patients with superficial spreading melanomas was reported [386]. Labelling of P2X7 receptors also extended 2 μm from the melanoma into the keratinocyte layer of the adjacent epidermis. Conversely, P2X1-3 and P2Y2 receptors (found on normal, but not neoplastic skin) were fully de-expressed within 2 μm of the melanoma. A later paper from another group confirmed that human melanomas express functional P2X7 receptors, which produce apoptosis, and it was suggested that they may represent a novel target for melanoma therapy [37]. Overexpression of P2X7 receptors was produced when P2X7 receptor cDNA was transfected into B16 murine melanoma; tumour growth was significantly enhanced in vivo, but not in vitro [387]. A low pH environment (mimicking the hypoxia and acidosis commonly seen in solid tumours) was shown to induce ATP release from B16 melanoma cells to act via P2X7 receptor to increase proliferation and the P2X7 receptor antagonist, oxidised ATP, significantly inhibited tumour growth [388]. Expression of P2Y1, P2Y2 and P2Y6 receptor mRNA and protein in human melanomas was reported [45]. This study also showed that incubation of A375 melanoma cells with the P2Y1 agonist, 2-methylthio ADP, caused a dose-dependent decrease in cell number, while the P2Y2 receptor agonist, UTP, caused an increase in cell numbers. Melanoma is characterised by apoptosis resistance connected to irradiation- and chemo-resistance, and it was claimed that the P2X7 receptor has an anti-apoptotic function in melanoma cells, since ATP activation suppresses induced apoptosis, while with knock-down of P2X7 receptor gene expression, ATP-induced apoptosis was enhanced [389].
Athymic mice, injected with A375 human melanoma cells, were treated daily with intraperitoneal injections of ATP. The in vivo tumour volume and animal weight were measured over the course of the experiment and the final tumour nodule weight was measured at the end of the experiment. Tumour volume decreased by nearly 50 % by 7 weeks in treated mice. Weight loss in untreated animals was prevented by ATP. Histological examination of the excised tumour nodules showed necrosis in the ATP-treated tumours only. The presence of P2Y1 and P2X7 receptors, previously proposed as extracellular targets for melanoma treatment with ATP, was demonstrated in the excised specimens by immunohistochemistry. This paper provides further support for the use of ATP as a treatment for melanoma [38].
ATP released by murine B16 melanoma cells up-regulates the expression of CD39 on tumour-resistant regulatory T cells; the upregulated CD39 degrades ATP to adenosine, which then contributes to the immunosuppressive environment of the tumour [390]. It has been claimed that ATP production by B16 melanoma tumour cells may contribute to recruitment and stimulation of regulatory T cells, resulting in an immunosuppressive environment [391]. They showed further that implanting B16 melanomas into CD73 knock-out mice, which are impaired in adenosine production, led to a significant slowing down of the growth of the tumours.
Adenosine has also been investigated in relation to melanomas. For example, administration of adenosine was shown to potentiate the actions of chemotherapeutic agents in vivo. In mice inoculated with B-16 melanoma cells, adenosine (268 mg/kg) was injected 5 days before administration of cyclophosphamide (50 mg/kg). This combined treatment reduced the number of melanoma foci by 60 %, while the chemotherapy alone only reduced them by 45 %. Moreover, a protective effect of adenosine against chemotherapy-induced decrease in leukocyte counts was seen in this study [392]. Cell motility, an essential component of tumour progression and metastasis, is mediated by adenosine in human melanoma cells, probably via the A1 receptor subtype and the possibility of anti-metastatic therapies based on inhibition of A1 receptor activation was raised [393]. A1, A2A, A2B and A3 receptor subtypes were all identified in the human malignant melanoma A375 cell line with RT-PCR and pharmacological evidence [394]. Adenosine, arising from released ATP, acts on adenosine receptors that are mediators of both reduction of cell proliferation (probably via A3 receptors) and promotion of cell death (probably via A2A receptors) of cultured human melanoma A375 cells [395]. A later paper, from another group, confirmed that A3 receptor activation led to growth inhibition of melanoma cells and showed that this occurs both in vitro and in vivo [396]. A3 receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase 1/2 phosphorylation in A375 human melanoma cells [397]. An A3 receptor agonist, 2-chloro-N 6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine, produces an effective anti-tumour immune response in melanoma-bearing mice, involving the activation of natural killer cells and T cells [398]. However, a recent report claims that adenosine, acting via A3 receptors, promoted cell proliferation of human C32 malignant melanoma cells [399]. A review of adenosine receptors and human melanoma was published in 2003 [400]. A2B receptor blockade can impair IL-8 production, which is elevated in patients with malignant melanoma, while blocking A3 receptors decreases VEGF, which promotes angiogenesis and metastasis of human carcinoma cells [401]. Evidence has been presented to suggest that adenosine could be a potent immunoregulatory factor affecting both cytokine production and cytotoxic activity of anti-melanoma-specific T cells [402]. Serum and peritoneal fluid ADA levels were higher in malignant ovarian neoplasms and it was suggested that this may be a useful biomarker in diagnosis and management of ovarian tumours [403].
It has been proposed that extracellular ATP released by dying tumour cells accumulates in high concentrations that not only act as danger signals in the immune system, but can also directly kill adjacent tumour cells via P2X7 receptors [43]. Using a genetically modified melanoma cell line, they showed that the anti-tumour effect of ATP can be amplified by inhibition of the ectonucleotidase CD39. In another recent paper, it was shown that in a melanoma model, tumour growth is impaired in CD73-deficient mice [42].
Cervical, ovarian and uterine cancer
Cervical cancer
Cervical cancer today can be effectively diagnosed in the very early phases of the disease, thus reducing by three to four times the death rate compared to pre-early diagnosis times. However, due to its high incidence, an efficacious medical treatment, besides surgery, is needed. HeLa cells, derived from human cervical cancer cells, have been widely used in studies of the involvement of purinergic signalling in cancer. Extracellular ATP was shown to activate K+ movements in HeLa cells [404] and to elevate [Ca2+]i following interaction with a nucleotide receptor [405]. Activation of P2Y2 receptors with UTP and ATP caused proliferation and inhibited the activity of Na+/K+-ATPase in HeLa cells [406]. It has been claimed that P2Y6 and P2Y4 receptor expression on HeLa cells increases during their proliferation [407]. UDP activation of P2Y6 receptors also induced proliferation of HeLa cells, but via a different second messenger pathway from that produced by the P2Y2 receptor [408]. Oestrogen reverses the apoptotic effects mediated by P2X7 receptors in normal cervix, but not in human cervical epithelial cancer cells [30]. The authors suggest that oestrogen may have a permissive effect for the development and growth of cervical cancer. A truncated P2X7 receptor variant (P2X7-j), endogenously expressed in cervical cancer cells, antagonises the full-length P2X7 receptor through hetero-oligomerization [409]. ATP induced Ca2+ mobilization and cell proliferation of four different human cervical cancer cell lines via the activation of nuclear factor κB, a transcription factor implicated in regulatory genes involved in neoplastic transcription [410]. The ATP cell viability assay has been recommended for the measurement of intrinsic radiosensitivity in cervical cancer, which shows how well a tumour is responding to radiotherapy [411].
Most human cancers derive from epithelia and the proliferation and differentiation of epithelial cells are crucially dependent on EGF receptor function. Stimulation of P2Y1 receptors on HeLa cells, for as little as 15–10 min, triggers EGF receptor mitogen signalling and P2Y1 antagonists reduced basal cell proliferation [412]. OppA, the ecto-ATPase of Mycoplasma hominis, induced ATP release and cell death of HeLa cells [413]. Human connexin 30.2/31.3 mediates enhanced ATP release from HeLa cells [414]. Gentle mechanical stimulation also released ATP from HeLa cells [415].
ADP and AMP hydrolysis, as well as ADA activity, were enhanced in the early (NIC I) stage of cervical intraepithelial neoplasia and uterine invasive cancer [416]. They also showed that the ADA isoform, ADA 1, was present in platelets from neoplastic patients, suggesting platelet participation in tumour development.
Ovarian cancer
Epithelial ovarian tumours represent about 25 % of female organ malignancies and have a higher mortality rate than cervical or uterine cancers. Extracellular ATP raised [Ca2+]i and stimulated growth of human ovarian carcinoma OVCAR-3 [417] and SKOV-3 cells [418]. ATP enhances the penetration into human ovarian cancer cell lines (OC-109, OC-238 and OC-7-Nu) of adriamycin, a drug used as a cytotoxic agent to reduce tumour progression [419]. It has been suggested that ATP may act as an extracellular messenger in controlling the ovarian epithelial cell cycle through P2Y2 receptors on EFO-21 and EFO-27 human ovarian cancer cells [420]. ATP tumour chemosensitivity assays have been recommended to assess the viability of chemotherapy for the treatment of primary recurrent platinum-resistant epithelial ovarian cancer [421–426]. A combination of zoledronic acid and fluvastatin has been claimed to have activity against ovarian (and breast) cancer based on this assay [427]. A2 receptor antagonism inhibits angiogenic activity of human ovarian cancer cells [428]. Variants of the RB1 gene have been implicated as risk factors for invasive ovarian cancer [429]. A recent study has shown that the presence of ATP during the treatment of human ovarian carcinoma with cisplatin leads to additive cytotoxicity [430].
Uterine cancer
P2Y2 receptors were claimed to participate in control of the cell cycle and suppression of proliferation of HEC-1A and Ishikawa human endometrial carcinoma cells [431]. Expression of copper-transporting P-type ATPase has been proposed as a prognostic factor for human endometrial carcinoma [432]. The P2X7 receptor has been claimed to be a novel biomarker for uterine epithelial cancers [433]. Tissue levels of P2X7 mRNA and protein differentiate between normal and hyperplastic from pre-cancerous and cancerous endometrium; there is decreased expression of P2X7 receptors on endometrial epithelium in pre-cancerous and cancer cells [434]. The reduced expression of P2X7 receptors in uterine cancer cells was claimed to be the result of increased expression of micro RNAs that regulate P2X7 expression [435], but the pathophysiological significance of this phenomenon is unclear.
It was shown, using an ATP tumour chemosensitivity assay (see [436]), that topotecan has a significant cytotoxic effect on uterine squamous cancer cell lines A-431, CaSki and C-33, which appeared to be superior to cisplatin [437]. A reduction in ectonucleotide pyrophosphatase/phosphodiesterase and ADA activities in patients with uterine cervix neoplasia have been reported [438].
Leukaemia
Leukaemia consists of a group of malignant diseases that start in the bone marrow and cause overproduction of blood cells that are massively released into the bloodstream. There are four common types of leukaemia: chronic lymphocytic (or lymphoid) leukaemia (CLL), chronic myeloid leukaemia (CML), acute lymphocytic (lymphoblastic) leukaemia and acute myeloid leukaemia (AML). These basic types can further be subdivided into subtypes. In particular, AML can be further subdivided into myeloblastic, promyelocytic, myelo-monocytic, monocytic, megakaryoblastic leukaemia and erythroleukaemia. Uncontrolled proliferation of lymphoid cells can also start in lymphoid organs, with little spill over of neoplastic cells into the blood, until the late stages of the disease. These tumours are classified into Hodgkin’s or non-Hodgkin’s lymphomas.
Lymphocytic leukaemia
The enzymes concerned with purine degradation, CD73, ADA and purine nucleoside phosphorylase, were measured in the bone marrow or blood of patients with both CML and CLL [439]. The levels of these enzymes varied with the type of leukaemia.
ATP and UTP activated superoxide formation in HL-60 promyelocytic leukaemic cells [440] and ATP also increased [Ca2+]i in these cells [441, 442] probably via P2Y2 receptors [443, 444]. Reduced proliferation was produced by ATP and UTP in both HL-60 promyelocytic and U937 promonocytic human cell lines [445], perhaps via A3 receptors [446]. Two different P2Y receptor subtypes were proposed to be responsible for the increase in [Ca2+]i in HL60 cells, a P2Y2 (or P2Y4) receptor and probably a P2Y1 receptor [447]. ATP enhanced the adherence of HL-60 cells to bovine pulmonary artery endothelial cells [448]. ATP increased cAMP production in undifferentiated HL-60 cells [449] and induced differentiation and suppressed cell growth via an unknown receptor (not P2Y2) [450]. Histamine inhibits ATP-induced rise in [Ca2+]i through the activation of PKA in HL-60 cells [451]. Adenosine, after breakdown of ATP, contributes to the inhibitory effect of ATP on proliferation of HL-60 cells [25]. It was claimed that ATP-dependent suppression of proliferation was largely via adenosine receptors, while ATP induction of differentiation was via P2X receptors [452]. It was proposed that P2Y11 receptors mediated ATP-induced differentiation of both myeloblastic HL-60 and promyelocytic NB4 cells into reactive neutrophil-like cells [57, 453]. RT-PCR analysis showed expression of P2X5, P2X7 and P2Y1-11 receptors (except for P2Y6) mRNA in HL-60 cells during the course of differentiation and CD39 and CD73 were upregulated during maturation [454]. P2Y2 and P2Y11 receptors were upregulated during granulocytic differentiation of HL-60 cells [455]. ATP induced apoptotic cell death of both HL-60 and F-36P leukaemia cell lines [456]. Static magnetic field exposure of HL-60 cells increased the increase in [Ca2+]i produced by ATP [457, 458]. A3 receptor agonists were shown to inhibit proliferation and induce apoptosis of HL-60 leukaemia cells [459, 460]. ADP and ATP increased [Ca2+]i in CB1 cells, isolated from a patient with T-acute lymphoblastic leukaemia [461], probably via P2Y1, P2Y12 or P2Y13 receptors. Apoptotic cells release ATP, which is an early signal to recruit phagocytes. Pannexin 1 channels have been identified that mediate ATP release in Jurkat T lymphoblastoma leukaemia cells [462].
P2X7 receptors have been described in human leukaemic lymphocytes [463, 464]. B cell CLL is one of the most common haemopoietic tumours. Evidence has been presented to suggest that expression and function of P2X7 receptors, which can mediate cell death or proliferation depending on the level of activation, may correlate with the severity of B cell CLL [463]. A 1513C polymorphism of the P2X7 receptor gene has been associated with an increased risk of developing CLL [465], but later studies showed that this might only be relevant in the rare familial form of the disease [466]. P2X7 receptor expression was significantly higher (relative to bone marrow mononuclear cells) in cells from patients with lymphoblastic leukaemia (as well as acute and chronic myelogenous leukaemia) [467]. It has been claimed that the P2X7 receptor-mediated cytotoxic effects on KGla and J6-1 leukaemia cell lines may occur independently of the calcium response [468]. In paediatric acute leukaemia, RT-PCR and Western blots showed that P2X1, P2X4, P2X5 and P2X7 receptors were upregulated, while P2X2, P2X3 and P2X6 receptors were absent or marginally expressed and the highest expression of P2X7 receptors was found in relapsed patients [469]. They also showed a significant decrease in the expression of P2X4, P2X5 and P2X7 receptors after complete remission after chemotherapy.
Adenosine was shown to have cytotoxic effects on mouse leukaemia L1210 cells [470]. In the human leukaemia cell line, U-937, ATP-induced cytotoxicity was biphasic, the initial response due to ATP, while the later response was due to adenosine, after ectoenzymic breakdown of ATP [471]. A3 receptors were also identified on Jurkat cells, a human leukaemia cell line [472]. Guanosine and deoxyguanosine are toxic to Jurkat cells through two mechanisms: ATP depletion, causing necrosis, and the accumulation of dGTP, resulting in apoptosis [473]. 2-ClAdo produced cell death of leukaemic B cells [474]. Adenosine was shown to suppress the growth of the human T lymphocyte leukaemic cell line MOLT-4 [475]. A1 and A2-like receptors exert opposite effects on 5-hydroxytryptamine release from a mastocyte tumour cell line, rat basophilic leukaemic RBL cells [476].
Lymphomas
Adenosine, acting via A2 receptors, and CD39 have been suggested as novel targets for augmenting human follicular lymphoma immunotherapy [477]. It has been suggested that increased CD39 expression on CD4+ T lymphocytes has clinical and prognostic value in CLL [478]. CD73-generated extracellular adenosine favoured growth and survival of CLL cells [479]. 8-ClAdo has been evaluated in phase I clinical trials for the treatment of CLL using the mantle cell lymphoma cell lines, Granta 519, JeKo, Mino and SP-53 [480]. 8-ClAdo inhibited the rates of DNA synthesis and depleted ATP resulting in cell death and inhibition of growth. It has been suggested that the ATP-CD39-A2A receptor pathway is one mechanism for T cell hyporesponsiveness in follicular lymphoma [477]. Purine nucleoside analogs, including clofarabine, nelarabine and forodesine, are being explored for the treatment of CLL [481]. A3 receptors were shown to mediate inhibition of lymphoma cell growth [482]. ADA was immunolocalized on human B cell lymphomas [483]. Extracellular ATP increased cation permeability of CLL lymphocytes [484].
Iron complexed by ATP induces lymphomas in mouse organs [485]. Anaplastic large cell lymphomas are a distinct subset of non-Hodgkin’s lymphomas and multikinase inhibitors have been recommended for the treatment of these tumours [486]. Inhibition of the expression and function of P2X7 receptors attenuated the metastatic capability of murine P388D1 lymphoid neoplasm cells [487]. Activation of P2X7 receptors caused depletion of intracellular ATP in T lymphoma cells [488]. Yac lymphoma cells actively secrete ATP in response to P2X7 receptor activation and the ATP amplifies P2X7 receptor signalling or acts on other purinoceptor subtypes to modulate tumour growth and the anti-tumour immune response [489].
Myeloid (myelogenous) leukaemia
Myeloid leukaemias consist of any of the blood cells originating in the blood-forming (myeloid) tissue of the bone marrow and K562 is a leukaemic cell line established from the pleural effusion of a patient with chronic myelogous leukaemia. ADP was shown to be a potent stimulus for calcium mobilization in K562 cells probably acting via P2T (i.e. P2Y12) receptors [490]. ATP, UTP, BzATP and adenosine were cytotoxic on K562 cells [491].
A frameshift polymorphism of the P2X5 receptor elicits an allogeneic cytotoxic T lymphocyte response associated with remission of CML [492]. Mouse myelomonocytic leukaemic M1 cells were established from spontaneous myeloid leukaemia mouse strains and ATP was shown to enhance differentiation in these cells [493]. 4-Aminopyridine, a voltage-gated potassium channel blocker, induced apoptosis of human AML cells via increasing [Ca2+]i through P2X7 receptor pathways [494]. P2X7 receptor activation induces reactive oxygen species formation in murine erythroleukaemia cells and it was suggested that this may be involved in downstream events of P2X7 receptor activation, other than apoptosis, in erythroid cells [495]. P2X7 receptor agonists mediate cation uptake into human myeloid leukaemic KG-1 cells [496]. Adenosine analogues have been proposed as a possible differentiation-inducing agent against AML in B4 cells [497]. ATP depletion triggers AML differentiation (and is therefore anti-proliferative) through an ATR/Chk1 protein-dependent and a p53 protein-independent pathway and therefore is a promising strategy for treatment of AML [498]. AML cells express P2X1, P2X4, P2X5 and P2X7 and all P2Y receptor subtypes [499]. However, in contrast to that observed in normal human leucocytes, P2 receptor stimulation induced a significant inhibition of both proliferation and migration in vitro and engraftment in immuno-deficient mice.
Differences were detected in ATP-binding cassette subfamily B member 1 (ABCB1) mRNA expression in leukocytes, polymorphonuclear and mononuclear cells in patients with de novo CML [500].
Erythroleukaemia
Extracellular ATP inhibited the growth of murine erythroleukaemia MEL cells [24]. P2X7 receptors mediate cell death and microparticle release in MEL cells [501]. ATP and UTP increased [Ca2+]i in the HEL human erythroleukaemia cell line [502]. It was later proposed that HEL cells expressed both P2Y2 (and/or P2Y4) and probably P2Y1 receptors [503]. The P2U receptor engages the heterotrimeric G protein G16 to mobilize Ca2+ in HEL cells [504].
ATP causes lysis of the monocytic leukaemic cell line THP-1, probably via P2Z (i.e. P2X7) receptors [505]. P2X7 receptor-mediated pore formation was described in THP-1 cells [506]. Adenosine, via cAMP, was shown to inhibit differentiation (and therefore increase proliferation) of mouse erythroleukaemic MEL cells [507].
Bladder cancer
Three main types of bladder cancer are described: transitional cell carcinoma (TCC), squamous cell carcinoma and adenocarcinoma. Bladder cancer is more common in industrialized countries; however, it is also frequent where bilharzial (Schistosoma hematobium) infections occur (i.e. Egypt). The effect of ATP has been investigated in high grade 3 (G3) superficial TCC of bladder where the tumour is confined to the mucosa or submucosa [33]. Commercially available HT-1376 cells were found to express the same purinergic receptor mRNA as PC-3 prostate cancer cells (P2X4,5,7 and P2Y1,2,4,6,11). ATP reduced cell growth in a concentration-dependent manner, via the induction of P2 receptor-mediated apoptosis. Pharmacological profiling implicated P2X5 and/or P2Y11 receptors in this anti-neoplastic response, although a possible contributory effect of P2X7 receptors could not be discounted. This functional receptor profile and the order of agonist potency were the same as that seen in HRPC cells, although G3 TCC cells were more sensitive to the cytotoxic effects of ATP (reduction of growth by 88.5 ± 4.4 vs. 45 ± 2.3 % for PC-3 cells at ATP 0.1 mM). These results suggest that the two most common advanced urological malignancies may have a common therapeutic purinergic target despite their differing cellular type and origin (transitional cells in the bladder vs. prostate adenocarcinoma).
Although studies have demonstrated a potential differentiating role for P2X5 [58, 508] and P2Y11 receptors [57], no studies have implicated these receptors in the induction of apoptosis. Apoptosis has classically been linked to the P2X7 receptor, although, despite the presence of P2X7 receptor mRNA, a significant functional role for this receptor subtype could not be elicited. ATP significantly increased apoptosis after 72 h [33]. Ryten et al. [508] demonstrated that the activation of P2X5 receptors mediated the stimulation of cell differentiation markers and thereby inhibited proliferation in skeletal muscle cells. It is therefore possible that activation of P2X5 receptors in bladder cancer leads to cellular differentiation, resulting in cells unable to continue the cell cycle, which subsequently undergo apoptosis. This may explain the delay in apoptosis detection, with no significant increase noted after 24 h incubation with ATP. Assessment of cell differentiation using markers would help define the contribution of this process to the observed growth inhibition and further clarify the anti-neoplastic mechanism of ATP in bladder cancer.
In vivo experiments mirrored the in vitro findings, with a reduction in mean implanted tumour volume by 64.3 % after daily intraperitoneal treatment with ATP (Fig. 3). No obvious side effects relating to treatment were noted in any experimental group. Histological analysis of the neoplasms in control mice using hematoxylin and eosin staining and transmission electron microscopy (TEM) showed tumours maintained the classical characteristics of urinary TCCs. While ATP-treated tumours were significantly smaller, light microscopy revealed no other histological changes. TEM detected an increase in both apoptotic bodies and necrosis in treated tumours [33]. There is high correlation between adenine nucleotide content and bladder tumour progression [509]. There was upregulation of P2Y receptors in T24, a transitional cell carcinoma cell line [510].

Effect of daily intraperitoneal ATP (1 ml of 25 mM i.p.) from day 0 on the growth of freshly implanted human bladder TCC HT-1376 tumour cells in vivo. (Reproduced from [33] with permission.)
Growth of bladder carcinoma J82 cells was inhibited and apoptosis was induced by adenosine [511]. In the human bladder T24 cell line, adenosine increased [Ca2+]i and cAMP production as well as IL-8 secretion, via A2B receptors [512]. It has been reported that A2B receptor blockade slows the growth of bladder tumours [153].
A differential pattern of ectonucleotidases in the more malignant human bladder cancer cells compared with cells derived from an early stage of bladder cancer has been described [513].
The distinct advantage with bladder tumours is that direct instillation of chemotherapeutic agents via a urinary catheter is easily achievable and allows drugs to be given at more concentrated levels locally to induce a sufficient response, while reducing systemic side effects. This may benefit the use of ATP either alone or in combination in future trials. The primary principle of combination chemotherapy is to maximize anti-neoplastic activity while minimizing toxic side effects of treatment. This is best achieved by combining drugs, which have different mechanisms of action with an additive or synergistic effect and with different patterns of resistance to minimize cross-resistance. In bladder cancer, the combination of ATP with the established anti-tumour antibiotic mitomycin C significantly increased its effect on cell death, reducing the chemotherapeutic drug concentration at which 50 % of cells were killed by a factor of 10 [33] (Fig. 4a). The same effect was seen with ATP and mitoxantrone, an anti-tumour antibiotic approved for use in the treatment of HRPC [32] (Fig. 4b). However, the cytotoxic effect of these combinations was additive only and not synergistic. This is probably explained by the respective mechanisms of action. Both anti-tumour antibiotics are cell cycle non-specific, whereas ATP has previously been shown to induce checkpoint defects leading to S-phase arrest, preventing further progression in the cell cycle, and eventual apoptosis [18]. Cell cycle non-specific drugs work effectively on all cancer cells. With ATP thought to work primarily only in the S-phase, the decrease in the surviving fraction of cells after exposure to the chemotherapeutic drug would decrease the number of viable cells for ATP to induce its cytotoxic effect. With this in mind, ATP would be better in combination with a chemotherapeutic drug known to work in a different phase of the cell cycle, to prevent any overlap and to increase the chance of synergism. To this effect, the addition of docetaxol, active in the G2/M phase and on bcl-2 phosphorylation, would theoretically be more advantageous in combination with ATP. Hemorrhagic cystitis is an adverse effect of cancer therapy with cyclophosphamide. Pretreatment of mice with the selective P2X7 receptor antagonist, A438079, reduced the nociceptive behaviour score and virtually abolished the increases in bladder myeloperoxidase activity, an indicator of neutrophil migration, induced by cyclophosphamide [41].

a Dose–response curve of the effect of combining ATP with mitomycin C (MMC) vs. MMC alone on the viability of human bladder TCC HT1376 cells in vitro. b The effect of combining mitoxantrone and ATP on the viability of HRPC PC-3 cells in vitro. All points are the mean (S.E.M.) unless occluded by the symbol. ***P < 0.001. (a Reproduced from [33] and b from [182] with permission.)
Brain tumours
Neuroblastoma/neuroma
Neuroblastomas are malignant tumours comprised of embryonic nerve cells. They may originate in any part of the sympathetic nervous system, most commonly in the medulla of the adrenal gland and secondary tumours are often widespread in other organs and in bone. Neuromas refer to any tumours derived from cells of the nervous system, often categorised more specifically, e.g. neurofibroma, neurilemmona and reactive neuroma.
Early studies showed that ATP and adenosine stimulated the production of cAMP in the cloned line NS20 of mouse neuroblastoma [514–517]. Later, the mouse neuroblastoma N18TG-2 × rat C6BU-1 glioma hybrid cells, NG108-15, were used to study the effect of nucleotides [518–526], probably via P2U (P2Y2 and/or P2Y4) receptors [527–529]. Later P2Z (= P2X7) receptors were identified on NG108-15 cells [530–534] and maybe also P2Y6 receptors [535]. Subsequently, RT-PCR analysis detected transcripts for both P2Y2 and P2Y6 receptors in NG108-15 cells, but not for P2Y1 or P2Y4 [536]. UDP arrests the cell cycle and induces apoptosis in human neuroblastoma SH-5Y5Y cells over-expressing the P2Y6 receptor [537]. Ecto-alkaline phosphatase was shown to be important for the metabolism of nucleotides by NG108-15 cells [538, 539].
ATP applied to mouse neuroblastoma Neuro-2A cells resulted in a selective enhancement of plasma membrane permeability for Na+ relative to K+, but also inward Cl− pumping [540], probably via P2Y receptors [541]. ATP and UTP were shown to increase [Ca2+]i in the murine neuroblastoma cell line NIE-115 [542], perhaps via P2Y2 and/or P2Y4 receptors. Later, P2X7 receptor mRNA and protein were shown to be present on NIE-115 cells, mediating apoptosis, perhaps via breakdown to adenosine [543], but more likely by direct activation [544]. P2U (i.e. P2Y2 and/or P2Y4) receptors, as well as P1(A2) receptors, have been identified on NCB-20 mouse neuroblastoma × Chinese hamster brain explant hybrid cells [545]. ATP was shown to inhibit atrial natriuretic peptide binding to R1-type receptors on human neuroblastoma NB-OK-1 cell membranes [546]. Neuropeptide Y was shown to modulate ATP-induced increase in [Ca2+]i via the adenylate cyclase/PKA system in the CHP-234 cell line derived from a human neuroblastoma [547].
Extracellular ATP evoked two excitatory responses in hippocampal neuroblastoma cells (HN2): one opened receptor-operated, non-selective cation channels, perhaps via P2X7 receptors, and the other caused a leftward (negative) shift in the Na+ channel activation curve, probably via P2Y receptors [548]. The human SH-SY5Y neuroblastoma cell line expresses a functional P2X7 receptor that modulates voltage-dependent Ca2+ channel function [549]. Extracellular guanosine and guanosine triphosphates promote the expression of differentiation markers and induce S-phase cell cycle arrest in SH-SY5Y cells [550]. Agonist-induced stimulation of both A1 and A2A receptor induced neurite outgrowth and differentiation of SH-SY5Y neuroblastoma cells in vitro [551].
mRNAs for P2Y1, P2Y4 and P2Y6 receptors were expressed in SK-N-BE(2)C human neuroblastoma cells, but Northern blot analysis revealed that P2Y6 receptors were the predominant subtype [552]. In an abstract, the presence of functional P2Y1 and P2X4 receptors (in addition to P2Y6, P2Y11, P2X5, P2X6 and P2X7 receptor protein) was claimed in human SK-N-MC neuroblastoma cells [553].
Neuroblastoma is the most common tumour in infancy and early childhood. Neuroblastoma and the sympathetic nervous system share a common embryological origin, the neural crest. C-1300 neuroblastoma arose spontaneously in mouse and resembles human neuroblastoma in many respects. Evidence has been presented that the sympathetic nervous system secretes a mitogenic trophic factor that enhances growth of C-1300 neuroblastoma cells in vivo [554]. It is now well established that ATP is released as a cotransmitter with NA in sympathetic nerves and that it often has powerful trophic actions (see [555]). Uridine induces differentiation of LAN-5 human neuroblastoma cells [556]. P2Y4 receptors were claimed to participate in differentiation and cell death of human neuroblastoma SH-SY5Y cells [557]. Glucocorticoids inhibit P2X receptor-mediated Ca2+ influx via a PKA-dependent pathway in HT4 mouse neuroblastoma cells [558]. P2X7 receptors expressed by primary human neuroblastoma cells are uncoupled from their well-known cytotoxic effect, but rather support cell growth. This paradoxical effect seems to be due to an inability to induce caspase-3 activation [559]. The growth stimulation was partially due to the release of substance P from nucleotide-activated neuroblastoma cells. Adenosine is claimed to induce apoptosis in mouse neuroblastoma NIE-115 cells, but uptake of adenosine and its subsequent phosphorylation is required [560]. ATP can stimulate neurite outgrowth in mouse neuroblastoma neuro2a cells independent of other neurotrophic factors [561].
Mouse neuro-2a cells differentiated into neuronal-like cells after exposure to retinoic acid, which was associated with a decrease in expression of functional P2X7 receptors [562]. It was further shown that P2X7 receptor antagonists induced neurite outgrowth as did P2X7 receptor knock-outs and it was concluded that decreases in the expression of P2X7 receptors are associated with neuronal differentiation and that ATP release-activated P2X7 receptors are important in maintaining cell survival of N2a neuroblastoma cells. A study of neuro-2a cells from another laboratory [35] also showed that P2X7 receptor inhibition led to an increase in neurite formation and that P2X7 receptors are involved in the maintenance of neuroblastoma cells in the non-differentiated state. P2X7 receptors are expressed in human lingual nerve neuromas [563]. It has been suggested that there is a positive feedback mechanism mediated by P2X7 receptor-stimulated exocytotic release of ATP that would act on P2X7 receptors on the same or neighbouring cells to further stimulate its own release and negatively control mouse neuroblastoma Neuro2-a cells [564]. The therapeutic potential of P2X7 receptor antagonists for the treatment of neuroblastoma has been reviewed [565].
Benzodiazepines modulate adenosine A2 receptor binding sites on 108CC15 neuroblastoma × glioma hybrid cells [566]. Prolonged exposure to A2 receptor agonists was associated with a small, but significant degree of differentiation of IMR32 human neuroblastoma cells [567]. A1 receptors have been identified in peritumoural zone around experimental F98 and C6 rat brain tumours [568].
In summary, P2Y2, P2Y6 and P2X7 receptors, which mediate cytotoxic effects, appear to be the dominant purinoceptor subtypes in most neuroblastoma cell lines.
Gliomas
Glioma is a general term for malignant tumours of glial cells and includes astrocytomas, oligodendrogliomas, medulloblastomas, Schwannomas, ependymonas and glioblastomas.
Adenosine triphosphatase was found to be localised on the cell membranes of gliomas over 35 years ago [569]. Later, an ecto-nucleotide pyrophosphatase (ectoNPPase) was identified for ATP metabolism by C6 glioma cells [570, 571] and ecto-5-nucleotidase [572]. Thyroid hormone upregulates ecto-5′-nucleotidase (CD73) in C6 cells [573]. CD73 has also been identified in human U138MG glioma cells [574]. CD73, a producer of extracellular adenosine, modulates U138MG glioma cell adhesion and tumour cell–extracellular matrix interactions [575]. Medulloblastoma is the most common malignant brain tumour in children and occurs mainly in the cerebellum. ATP was secreted from three malignant human cell lines and absence of CD73 in the D283 cell line, a metastatic medulloblastoma phenotype, suggested that high expression of CD73 could be correlated with a poor prognosis in patients with medulloblastomas [576]. Selective expression of NTPDase 2 modulates growth of rat C6 and COS-7 glioma cell lines in vivo [577]. Overexpression of NTPDase2 in C6 glioma cells promotes systemic inflammation and pulmonary injury [578]. Intracarotid, but not intravenous, administration of adenosine and ATP into intracerebrally transplanted RG-C6 tumours in rats selectively increased blood flow in the tumour, suggesting that they may be used to enhance the delivery of anti-cancer agents to malignant brain tumours [579, 580].
Rat glioma C6 cells have been widely used for studies of gliomas. ATP was shown to stimulate phosphoinositide hydrolysis in C6 cells [581], suggesting that P2Y receptors were involved, as was supported by thapsigargin blockade of ATP-mediated increase in [Ca2+]i [582]. It was suggested that the P2 receptor subtype in C6 cells was comparable to the P2T receptor of platelets, i.e. the P2Y12 receptor [583]. UTP and ATP were equipotent in increasing [Ca2+]i in C6 glioma cells, suggesting mediation via P2U (i.e. P2Y2 and/or P2Y4) receptors [584, 585]. There appeared to be two different signal transduction pathways for P2Y receptors in C6 cells, one is involved in inhibition of adenyl cyclase and the other in the induction of phosphoinositide turnover, indicating the involvement of two P2Y receptor subtypes [586, 587]. A later paper identified both P2Y1 and P2Y2 receptors involved in calcium signalling in C6 cells [588]. In addition to P2Y2 receptors on C6 cells, ADP acts via P2Y1 and P2Y12 receptors, the former linked to PLC, while the latter is coupled to adenylate cyclase [589]. Cross-talk between P2Y1 and P2Y12 receptors has been implicated in growth and differentiation of C6 cells [590, 591]. A shift in receptor expression from P2Y1 to P2Y12 in long-term serum-deprived C6 cells appears to be a self-regulatory mechanism that promotes cell growth rather than differentiation and is a defense mechanism against the effects of serum deprivation [592]. Bradykinin increased resensitization of P2Y receptor signalling in glioma cells [593]. siRNA silencing of P2Y1 receptors alters calcium signalling in C6 cells [594]. In recent papers, P2Y14 receptor activity has been described in C6 cells [595].
Rat C6 glioma cells express functional P2X7 receptors [596] and ATP-induced cell death in mouse GL 261 glioma cells was claimed to be mediated by P2X7 receptors [597]. P2X7 receptor agonists produced cell death in the glioma radiosensitive cell line M059J, but the radioresistant glioma cell line, U138-MG, presented resistance to death when treated with either ATP or BzATP [598]. ATP released from glioma tumour cells may act as the regulator, via P2X7 receptor signalling, that increases macrophage inflammatory protein-1α and monocyte chemoattractant protein-1 expression in tumour-infiltrating microglia [599]. It was claimed that BzATP-mediated calcium signalling in C6 cells was mediated by P2Y (perhaps P2Y2), rather than P2X7 receptors [36]. P2X4 receptors were identified in glioma tumour growth areas, but immunostaining showed that they were largely, if not entirely, localized on infiltrating macrophages and activated microglia [600].
The ATP-forming capacity at the surface of glioma cells was several times greater than that of normal cells [601] and evidence for ectopic aerobic ATP production in C6 glioma cell membranes has been presented recently [602]. Adenosine uptake and ATP release from C6 cells were demonstrated [603], probably via pannexin 1 channels in response to mechanical stress [604]. ATP stimulates chemokine production in C6 glioma cells via a store-operated calcium entry pathway, which was suggested to enhance tumour cell mobility and promote recruitment of microglia into developing tumours, thereby supporting tumour growth [605].
ATP induces c-fos expression in C6 cells by activation of P2Y receptors [606]. The P2Y1 receptor agonist, 2-methythio ADP, markedly increased C6 glioma cell migration, while the selective P2Y1 receptor antagonist, MRS2179, significantly inhibited migration [607]. The authors suggested that P2Y1 receptor antagonists could be a novel therapeutic procedure to slow glioma progression. UTP and ATP, mediated by P2Y2 receptors, elicited proliferation of C6 glioma cells via activation of the Ras/Raf/MEK/MAPK pathways [608] (see also [609]). Both adenosine and ATP were claimed to induce proliferation in human glioma cell lines U87MG, U251MG and U138MG [610]. cAMP-dependent differentiation of C6 glioma cells into astrocyte type II is characterised by inhibition of cell growth and induction of glial fibrillary acidic protein synthesis. Activation of the P2Y12 receptor inhibited β-adrenergic receptor-induced differentiation and a P2Y12 receptor antagonist abolished this effect [611]. ERK 1/2 activity was positively correlated with cell proliferation evoked by both P2Y1 and P2Y12 receptor agonists, but in serum-starved cells, the effect of ADP on ERK 1/2 was primarily mediated by P2Y12 receptors [612]. The mechanisms underlying P2Y12 receptor activation of C6 cells have been studied, and it was concluded that PKB activation proceeds through insulin growth factor I receptor cross-talk and requires activation of Src, Pyk2 and Rap1 [613]. A review discussing P2Y receptor-mediated proliferation and differentiation of glioma cells is available [614].
Growth inhibition has been reported for human WF glioma cells by 8-ClAdo [615] and for C6 glioma cells by N6-substituted cAMP analogs [616]. mRNA and protein for A1, A2 and A3 receptors were shown to be expressed by C6 cells [617]. It has been suggested that adenine nucleotides inhibit C6 cell growth via adenosine after breakdown by CD73 [618]. Hypoxia has been claimed to decrease adenosine A1 receptors, but to increase A2A receptors in C6 cells [619]. U138-MG human glioma cells and C6 rat glioma cells showed greater resistance to death induced by ATP when compared to normal hippocampal organotypic cell cultures, indicating that released ATP can induce cell death of the normal tissue surrounding the tumour, potentially opening space to the fast growth and invasion of the tumour [620]. On the other hand, extracellular ATP might also exert a trophic effect on glioblastoma growth, as shown by the observation that in vivo C6 glioblastoma growth is reduced by co-injection of apyrase [621].
Temozolomide (TMZ) is a DNA-damaging agent, which is widely used for treating primary and recurrent high-grade gliomas. It has been shown that TMZ induces an autophagy-associated ATP surge in U251 cells that protect them and may contribute to drug resistance [622]. Carnosine inhibits growth of cell isolates from human malignant glioma, and recently, carnosine has been shown to inhibit ATP production in both cells from freshly resected gliomas and from the T98G human glioma cell line [623].
Glioblastomas are mainly, but not exclusively, undifferentiated anaplastic cells. This is the most aggressive type of brain tumour derived from glial cells and is characterised by having a cancer stem cell subpopulation essential for tumour survival. It includes astrogliomas, which are undifferentiated cells, but also astrocytomas, which are differentiated cells. Their rapid enlargement destroys brain cells and raises intracranial pressure, causing headache, vomiting and drowsiness. ATP, acting via P2Y receptors, increased [Ca2+]i in primary cultures of human glioblastoma cells [624]. It was claimed that ATP induces IL-1β release from T98G glioblastoma cells through a purinoceptor-independent mechanism [625]. Human U87 glioma cultures presented tumour spheres that express the markers of glioma cancer stem cells. Extracellular ATP reduced tumour sphere growth and cancer stem cell population in the glioblastoma cells [626].
An A2 receptor agonist increased the release of IL-8, an angiogenic factor, from the glioblastoma cell line U87MG, and while mRNA transcripts for A1, A2A and A2B were identified in these cells, only A2B receptors appeared to be functional; further, hypoxia increased A2B receptor mRNA and A2B antagonists inhibited tumour angiogenesis [627]. Adenosine attenuates growth of mouse glioblastoma G1361 cells acting via A1 receptors on microglia [628]. Adenosine modulates VEGF expression via hypoxia-inducible factor-1 in human hypoxic U87MG and A172 glioblastoma cells via A3 receptors [629]. Modulation of metalloproteinase-9 in U87MG cells via A3 receptors has been reported [630]. Pulsed electromagnetic field exposure significantly increased the anti-tumour effect mediated by A3 receptors in a human glioblastoma cell line [631]. Data have been presented to suggest that A3 receptor agonists may be potential therapeutic agents for the induction of apoptosis in human glioma cells [632].
ATP, after breakdown to adenosine, increases intracellular cAMP in human 1321NI astrocytoma cells [633, 634]. However, later papers showed that high concentrations of ATP, acting via P2 receptors, stimulate proliferation of SKMG-1 and U373 human astrocytoma cells [635] or inhibition of proliferation of 132NI astrocytoma cell [636]. P2Y1 receptors mediate stimulation of MAPKs and induction of apoptosis in 132NI astrocytoma cells [637]. The P2X7 receptor agonist, BzATP, induced ERK 1/2 phosphorylation in human astrocytoma cells over-expressing the recombinant rat P2X7 receptor [638]. P2Y6 receptors mediate activation of PKC to protect 132N1 astrocytoma cells against tumour necrosis factor-induced apoptosis [639]. P2Y12 receptors were shown to be expressed in 132N1 cells mediating Ca2+ signals, which may be crucial for regulating cell proliferation and differentiation [640]. An enhanced green fluorescent protein-tagged human P2Y2 receptor was expressed in 1321N1 astrocytoma cells [641]. P2Y14 receptors were also identified on 1321N1 cells, leading to the release of UDP-glucose [642]. Extracellular osmolarity modulates G protein-coupled receptor-dependent ATP release from 1321N1 astrocytoma cells [643].
A2B receptors mediate an increase in IL-6 mRNA and protein synthesis in the human astrocytoma cell line U373MG [644]. An A3 receptor mediates cell spreading and reorganisation of the actin cytoskeleton in human ADF astrocytoma cells [645, 646]. A3 receptor agonists mediated desensitization, internalization and down-regulation of the A3 receptors in human astrocytoma ADF cells [647]. Extracellular adenosine, acting via A1 receptors, activates caspase-9 and then caspase-3 via two independent pathways, leading to cell death of RCR-1 rat astrocytoma cells predominately by apoptosis [648]. ADA inhibition induces apoptosis in a human astrocytoma cell line [649].
Phaeochromocytoma
Phaeochromocytoma describes a tumour of adrenal medulla (or sympathetic nervous system), characterised by an excess of NA and hypertension. PC12 cells are a clonal line of rat phaeochromocytoma and have been extensively studied. They secrete NA, dopamine (DA) and acetylcholine (ACh) by a Ca2+-dependent process.
Extracellular ATP stimulated NA secretion from PC12 cells [650, 651], but also uptake of NA [652, 653]. ATP-activated inward current in PC12 cells was demonstrated with an agonist potency order of ATP > ATPγS > ADP, while adenosine and α,β-methylene ATP were inactive [654]. Suramin antagonised the ATP-activated current [655] and catecholamine secretion [656]. ATP stimulated the release of DA from PC12 cells [657, 658], which was suppressed by reactive blue 2 [657]. ATP and nicotine both activate an inward current, but the binding sites and the open states of the channels appear to be different [659].
The next step was to try to identify the purinoceptor subtype(s) located on PC12 cells (see, for example, [651, 660–663]). It was proposed that PC12 cells express at least two P2Y receptor subtypes: a P2Y subtype that leads to depletion of intracellular Ca2+ and NA release and a P2U (i.e. P2Y2 and/or P2Y4) receptor [664, 665]. The presence of P2X receptors on PC12 cells was first suggested in 1996 [666, 667]. Imipramine, a tricyclic antidepressant, inhibited, via P2X2 receptors, the ATP-evoked increase in [Ca2+]i and DA release by PC12 cells [668]. In a later paper, fluoxetine, another antidepressant, was also shown to inhibit ATP-induced increase in [Ca2+]i in PC12 cells [669]. Data have been presented to suggest that in undifferentiated PC12 cells, ATP acts via P2X4 receptors, but after nerve growth factor (NGF) treatment, the differentiated cells respond largely via P2Y2 receptors [670]. Both P2X2 and P2X4 receptor mRNA was shown to be present on PC12 cells and ATP, acting via both P2X and P2Y receptors, elevated [Ca2+]i, thereby facilitating catecholamine secretion [671]. Further, they showed that Na+ entry through P2X2 receptors effectively activated L-type voltage-sensitive Ca2+ channels. Transcriptional regulation of P2X2 receptors on PC12 cells by retinoic acids has been reported [672]. Dehydroepiandrosterone sulphate, the major circulating steroid in humans, suppresses P2X, but not P2Y, receptor-coupled responses of PC12 cells [673]. ATP triggers catecholamine release from PC12 cells via P2 receptors that desensitize; thus, habituation is increased by UTP [674]. It has been suggested that ATP-induced increase in [Ca2+]i is mainly due to the release of mitochondrial Ca2+ through Na+–Ca2+ exchangers in PC12 cells [675]. The membrane localization of PKCα is regulated by Ca2+ influx through P2X channels and phosphatidylinositol 4,5′-biphosphate in NGF-differentiated PC12 cells [676].
An RT-PCR and electrophysiological study of P2X receptors in PC12 cells showed that only functional P2X2 receptors were expressed in undifferentiated cells, but all seven P2X receptor subtypes were expressed in NGF-differentiated cells [677]. Another paper was consistent with these findings showing that NGF-stimulated differentiation of PC12 cells induced changes in P2 receptor expression and nucleotide-stimulated catecholamine release [678]. In particular, P2X receptor-selective agonists caused greater NA release from differentiated compared to undifferentiated cells, and receptor protein expression was increased for P2X1-4 receptors, but not P2Y receptors. P2Y receptors on PC12 cells mediate the actions of ATP and UTP to activate MAPK activity and promote the tyrosine phosphorylation of RAFTK, the epidermal growth factor receptor [679]. High concentrations of free fatty acids increased the expression of P2X7 receptors in PC12 cells via activation of the p38 MAPK signalling pathway, enhancing the release of IL-6 [680].
PC12 cells were claimed to express P2Y1-like receptors that mediate inhibition of voltage-activated Ca2+ currents in PC12 cells [681]. It has been reported that processes of differentiated PC12 cells possess P2Y12 receptors mediating inhibition of stimulation-evoked calcium entry [682]. Attenuation of P2Y receptor-mediated control of Ca2+ channels in PC12 cells by anti-thrombotic drugs has been claimed [683]. ADP-activated P2Y1 and P2Y12 receptors on PC12 cells are activated by spontaneous release of nucleotides, while ATP/UTP-sensitive P2Y2 receptors require an excess of depolarisation-evoked release to become activated [684]. A later paper showed that spontaneous release of nucleotides may occur independently of vesicle exocytosis, whereas depolarization-evoked release of ATP relies predominantly on exocytotic mechanisms [685]. It has been suggested that regulation of differentiation and cell survival of PC12 cells is mediated by the P2Y-like G protein-coupled GPR17 receptor [686]. ATP enhanced differentiation of PC12 cells by activating PKCα interactions with cytoskeletal proteins [687]. Sustained elevation of [Ca2+]i via P2X receptors causes changes in gene expression via activation of the transcription factor nuclear factor of activated T cells in PC12 cells [688].
Adenosine appears to be an endogenous regulator of tyrosine 3-monooxygenase activity in cell suspensions prepared from transplantable rat phaeochromocytoma [689], but this is defective in adenosine kinase-deficient PC12 cells [690]. Adenosine, acting through P1 receptors coupled to stimulation of adenylate cyclase, enhances the release of NA and ACh from PC12 cells [691]. 2-ClAdo increases the specific activity of choline acetyltransferase in PC12 cells [692]. Later, an A2A receptor was identified on PC12 cells [693–695], which inhibited ATP-induced Ca2+ influx [696]. Chronic hypoxia reduced A2A receptor-mediated inhibition of calcium currents in PC12 cells [697] and induced neurite outgrowth [698]. Adenosine increased DA metabolism in PC12 cells, which may have implications in relation to dopaminergic deficit in Parkinson’s disease [699]. A1 receptor activation was reported to inhibit neurite formation in PC12 cells [700]. Induction of neurite outgrowth in PC12 cells by the bacterial nucleoside, N6 methyldeoxyadenosine, was mediated by A2A receptors [701]. A2A receptor ligands and proinflammatory cytokines induce PC12 cell death through apoptosis [702]. Adenosine is an active component of Antrodia cinnamomea, a medicinal fungus in Taiwan, which prevents PC12 cells from serum deprivation-induced apoptosis through the activation of adenosine A2A receptors [703]. AMP N 1-oxide, a unique compound of royal jelly, induces neurite outgrowth of PC12 cells via A2A receptors [704]. Adenosine potentiates ATP-evoked DA release from PC12 cells [705].
Facilitation of the ATP-activated current in PC12 cells by 5-hydroxytryptamine and DA has been reported [667, 706] and enhancement of ATP-evoked DA release by zinc [707] and cadmium [708]. Reduction of ACh-activated current by ATP has also been observed [709]. ATP, acting through P2Y receptors, leads to the release of arachidonic acid from PC12 cells [710]. Diadenosine tetraphosphate, probably acting via a P2Y receptor, increased [Ca2+]i in PC12 cells [711].
Both catecholamines and ATP were released from PC12 cells in response to elevated intracellular concentrations of calcium [712]. There is enhancement of ATP levels in PC12 cells by the actions of extracellular adenosine [713]. Autoinhibition of transmitter release from PC12 cells (and sympathetic neurons) through P2Y12 receptor-mediated inhibition of voltage-gated Ca2+ channels has been reported [714]. Small transient inward currents were caused by quantal release of endogenous ATP by depolarised PC12 cells in close juxtaposition to the recorded cells [715]. There was enhancement of cellular ATP levels in PC12 cells by 2,5-dideoxyadenosine, a P-site inhibitor of adenylate cyclase [716]. Endothelin-1 inhibited the release of ATP from PC12 cells via ETB receptors by attenuation of the influx of extracellular Ca2+ through L-type channels [717]. β-Nicotinamide adenine dinucleotide was released together with ATP and DA from PC12 cells, but probably with different sites of vesicular release [718].
CD73 activity was inhibited in PC12 cells and was stimulated by treatment with NGF [719]. It was reported that CD73 played a crucial role in differentiation and survival of PC12 cells [720]. Extracellular ATP enhanced lipid peroxidation in PC12 cells and it was suggested that ATP may contribute to cell death by an oxidative mechanism involving lipid peroxidation [721]. Guanosine triphosphate and guanosine synergistically enhance NGF-induced neurite outgrowth from PC12 cells [722–724]. A later paper showed that guanosine stimulated neurite outgrowth in PC12 cells via activation of heme oxygenase and cyclic guanosine monophosphate [725]. ATP activates transcription factor AP1, a regulatory protein that converts extracellular signals into changes in gene expression programs and may modulate expression of target genes involved in cell death pathways in PC12 cells [726]. ATP inhibited starvation-induced apoptosis via P2X2 receptors in differentiated PC12 cells [727]. L-type Ca2+ channels and P2X2 receptor cation channels participated in calcium-tyrosine kinase-mediated PC12 growth cone arrest [728]. Uridine enhances neurite outgrowth of NGF-differentiated PC12 cells, perhaps through UTP as an agonist at P2Y2 receptors [729]. Neurite outgrowth in PC12 cells is also enhanced by ATP released into the medium through connexin hemichannels [730].
PC12 cells develop normal characteristics of sympathetic neurons after treatment with NGF and P2 receptor antagonists prevent NGF-dependent neuritogenesis [731]. P2 receptor agonists can behave as neurotrophic factors and interact with NGF signalling in neurite outgrowth and survival of PC12 cells [732, 733]. ATP-induced mitogenesis is inhibited by PLD2 in PC12 cells [734]. 2-ChloroATP exerts anti-tumoural actions (cell cycle arrest or cell death) in PC12 cells, although it was claimed that this was not mediated by P2 receptors [735]. Ca2+ influx through P2X receptors induces actin cytoskeleton reorganisation by the formation of cofilin rods in neurites of PC12 cells [736].
The parkin gene is one of the eight genes responsible for Parkinson’s disease. Parkin has been shown to potentiate ATP-induced currents via activation of P2X receptors in PC12 cells, suggesting that parkin may play a role in synaptic activity [737].
Phthalates are environmental pollutants and buylbenzylphthalate blocks purinoceptor-mediated Ca2+ signalling in PC12 cells [738]. Toluene disocynate, another toxic pollutant, suppresses calcium signalling produced via P2X receptors in PC12 cells [739].
Cancer pain
There are an increasing number of reports implicating the involvement of purinergic signalling in cancer pain. It was suggested that the exceptionally high levels of ATP contained in tumour cells may be released by mechanical rupture to activate P2X3 receptors on nearby sensory nerve fibres [350]. P2X3 receptor antagonists are one of the targets being explored against cancer pain [740]. Increased expression of P2X3 receptors on calcitonin gene-related peptide (CGRP)-immunoreactive epidermal sensory nerve fibres in a bone cancer pain model was reported [354] and in other tumours that are responsive to mechanical stress. In bone tumours, the mechanical strength of the bone is reduced and antagonists that block ATP receptors in the richly innervated periosteum might reduce movement-associated pain. The hyperalgesia associated with tumours appears to be linked to increase in expression of P2X3 receptors in nociceptive sensory neurones expressing CGRP by analogy with that described for increased P2X3 receptor expression in a model of inflammatory colitis. Increased expression of P2X3 receptors was also shown to be associated with thermal and mechanical hyperalgesia in a rat model of squamous cell carcinoma of the lower gingival [741]. Responses mediated by both P2X3 and P2X2/3 receptors on sensory neurones are inhibited by μ-opioid receptor agonists showing that P2X and μ-opioid receptors are functionally coupled on sensory neurones [742]. In a later paper, it was shown that P2X3 receptors on sensory neurones co-cultured with cancer cells exhibit a decrease in opioid sensitivity [341].
Radiotherapy is effective in relieving bone pain and it has been claimed from studies of a hind paw model of cancer pain by transplanting a murine hepatocarcinoma into the periosteal membrane of the foot that P2X6 receptor expression in the spinal cord was increased fivefold in the tumours, but that this was reversed following radiation [356].
Orthotopic inoculation of B16-BL6 melanoma cells into the hind paw of mice produced spontaneous licking of the tumour-bearing paw, an indication of pain [743]. P2X3 receptor antagonists suppressed the spontaneous licking and it was concluded that P2X3 receptors were involved in skin cancer pain, due to the increased release of ATP and increased expression of P2X3 receptors in the sensory neurons. In an elegant study, systemic blockade of P2X3 and P2X2/3 receptors was shown to attenuate bone cancer pain behaviour in rats [744], and in a later paper, the P2X3 and P2X2/3 antagonist, A317491, was shown to transiently attenuate cancer-induced bone pain in mice [745]. A recent review that discusses the role of purinergic receptors in cancer-induced bone pain is available [746]. μ- and δ-Opioid receptors are expressed on isolectin (IB) 4− (that expresses some P2X3 receptors) and IB4+ (that expresses most P2X3 receptors) neurons, respectively, which control thermal and mechanical pain, and it was shown that IB4+ and IB4− neurones were differentially involved in oral squamous cell carcinoma-related pain [747]. Using δ-opioid agonists and P2X3 receptor antagonists, it was shown that IB4+ neurones play a key role in cancer-induced mechanical allodynia, but not in thermal allodynia.
It has been shown that P2X7 receptor knock-out mice were susceptible to bone cancer pain and had an earlier onset of pain-related behaviours compared with cancer-bearing, wild-type mice [748]. They showed further that the P2X7 receptor antagonist, A-438079, failed to alleviate the pain-related behaviours and concluded that P2X7 receptors play a negligible role in bone cancer pain. Evidence has been presented that P2Y1 receptors in the spinal cord and DRG may mediate bone cancer pain through the ERK pathway [749].
Concluding comments
Overwhelming clinical evidence supports the notion that the approach to a cancer cure must be based on targeting multiple receptors and pathways, and that the best results are obtained when physiological mechanisms for cancer cell elimination are seconded (as in the case of immunochemotherapy) rather than being ignored. Purinergic signalling is a ubiquitous, crucial, pathway responsible for cell-to-cell communication in physiology and more so in pathology. Recent developments in the construction of reliable molecular probes for the measurement of extracellular ATP have unequivocally demonstrated that ATP at sites of inflammation or neoplasia can reach hundreds of micromolar concentrations [65, 66, 750]. The different P1 and P2 receptor subtypes expressed to a different extent by different cells and the large change in concentration that adenosine and ATP may undergo in physiological and pathological conditions offers an enormous plasticity to purinergic signalling. We are now starting to harvest the therapeutic potential of this system, but it is clear that only a deep knowledge of the molecular and biochemical details of involved pathways will allow us to exploit it in full to our benefit. In particular, it will be crucial to identify those conditions where trophic effects on tumour cells due to extracellular ATP overweight the well-known cytotoxic activity due to activation of specific P2 receptors such as P2X7. This is a crucial issue since several in vitro data and scattered in vivo evidence show that ATP may stimulate tumour cell growth [50, 751]; in addition, some tumours seem to be refractory to killing via the P2X7 receptor [559]. It will be necessary to thoroughly investigate in pre-clinical settings the effect of the administration of ATP (or P2X7-selective agonists or antagonists) via routes that better mimic the current procedures of anti-cancer drug administration in humans, e.g. intravenous infusion (see [14, 751–753]).
The picture is made even more complex by the growing awareness that P2 receptor activation/inhibition has a profound immunomodulatory function and thus shapes in a crucial fashion the type and number of tumour-infiltrating inflammatory cells (see [17, 63, 82, 83, 85, 754–758]). Therefore, it will be necessary to monitor the possible adverse effects caused by the systemic administration of P2-targeted drugs. Nevertheless, as our understanding of purinergic signalling increases, so does the range of malignancies found to be dependent on this messenger system. The discovery of purinergic receptor-mediated apoptotic pathways in advanced urological malignancies, irrespective of the cellular type or origin, has raised the possibility of possible future therapies for these aggressive malignancies, although significant differences between tumour types must be recognised. Much of the evidence has so far been derived from in vitro studies, with less information from in vivo animal experiments and little from human observational studies and randomized, controlled trials; thus, the need for more such studies to be performed is great, since it is not possible, based on existing in vitro and in vivo studies, to predict clinical effects. Nevertheless, studies have shown functional roles for the P2X5 and/or P2Y11 receptors, while a contributory effect of P2X7 receptors cannot be discounted. Selective targeting of these aberrant pathways would allow for the development of novel therapeutic agents that could not only treat the primary malignancy, but also improve the systemic symptoms associated with advanced malignancy. Both irradiation and chemotherapy appear to induce the release of ATP from tumour cells, which could exert cytotoxic effects by causing cell death via P2X7 receptors. Recent studies have shown that high levels of ATP are released from tumour cells that activate inflammasomes, thereby triggering a pro-inflammatory cascade leading to the activation of immune responses. ATP secretion from tumour cells is claimed to be involved in immunogenic cancer cell death [759].
One of the most important immunosuppressive regulatory pathways is the phosphohydrolysis of extracellular ATP to adenosine by the high levels of ectonucleotidase expressed by tumour cells (e.g. CD73), a possible novel target for cancer therapy. CD73 is a potent suppressor of anti-tumour immune responses [760]. Blockade of A2A receptors has been proposed as a target for tumour immunotherapy that synergizes with other immunomodulatory approaches currently in clinical trials [761]. A2A receptor signalling is required for T cell homeostasis and control of tumour growth [762]. A2B receptor signalling in antigen-presenting cells suppressed anti-tumour adaptive immune responses [763]. CD73-deficient mice have increased anti-tumour immunity and are resistant to experimental metastasis [764] (see also [754, 765]).
The serine-threonine kinase, Akt, plays a central role in propagating growth signals, metabolism and cell survival, making it a potential therapeutic target for cancer [766]. It was shown in this paper that ATP competitive inhibitors induced increased phosphorylation of Akt, suggesting a mechanism for regulating kinase activity through nucleotide binding. Since ATP is a naturally occurring small molecule, its radiolabelled form, [32P]ATP, poses advantages as a potential anti-cancer therapeutic agent and it was shown to inhibit the growth of xenografted tumours in nude mice [767]. Collectively, these evidences highlight the crucial role of purinergic signalling in cancer growth and dissemination and underline the, as yet, largely unexploited therapeutic potential of P2 receptor targeting. A3 receptors have been proposed as a therapeutic approach in cancer [768].
Hematopoietic stem cell transplantation is being developed as a therapeutic option for patients with hematologic malignancies; release of ATP from CD4 cells in whole blood was increased, which contributed to the clinical management of patients with hematologic malignancies [769]. A single cell, enhanced fluorescence ATP biosensor was developed recently to monitor ATP release from heterogeneous cancer populations in real time [770].
References
Burnstock G (1972) Purinergic nerves. Pharmacol Rev 24:509–581
Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50:413–492
Burnstock G (2007) Purine and pyrimidine receptors. Cell and Mol Life Sci 64:1471–1483
Burnstock G, Knight GE (2004) Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 240:31–304
Burnstock G (2006) Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev 58:58–86
Burnstock G (2007) Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev 87:659–797
Fishman P, Bar-Yehuda S, Madi L, Cohn I (2002) A3 adenosine receptor as a target for cancer therapy. Anticancer Drugs 13:437–443
Abraham EH, Salikhova AY, Rapaport E (2003) ATP in the treatment of advanced cancer. Curr Top Membr 54:415–452
Merighi S, Mirandola P, Varani K, Gessi S, Leung E, Baraldi PG, Tabrizi MA, Borea PA (2003) A glance at adenosine receptors: novel target for antitumor therapy. Pharmacol Ther 100:31–48
White N, Burnstock G (2006) P2 receptors and cancer. Trends Pharmacol Sci 27:211–217
Deli T, Csernoch L (2008) Extracellular ATP and cancer: an overview with special reference to P2 purinergic receptors. Pathol Oncol Res 14:219–231
Pathak R, Bhatnagar S, Dubey AK (2008) Mechanisms underlying the opposing effects of P2Y receptors on the cell cycle. J Recept Signal Transduct Res 28:505–529
Shabbir M, Burnstock G (2009) Purinergic receptor-mediated effects of adenosine 5′-triphosphate in urological malignant diseases. Int J Urol 16:143–150
Di Virgilio F, Ferrari D, Adinolfi E (2009) P2X7: a growth-promoting receptor—implications for cancer. Purinergic Signal 5:251–256
Stagg J, Smyth MJ (2010) Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 29:5346–5358
Gessi S, Merighi S, Sacchetto V, Simioni C, Borea PA (2011) Adenosine receptors and cancer. Biochim Biophys Acta 1808:1400–1412
Di Virgilio F (2012) Purines, purinergic receptors, and cancer. Cancer Res 72:5441–5447
Rapaport E (1983) Treatment of human tumor cells with ADP or ATP yields arrest of growth in the S phase of the cell cycle. J Cell Physiol 114:279–283
Rapaport E (1988) Experimental cancer therapy in mice by adenine nucleotides. Eur J Cancer Clin Oncol 24:1491–1497
Rapaport E (1990) Mechanisms of anticancer activities of adenine nucleotides in tumor-bearing hosts. Ann N Y Acad Sci 603:142–149
Rapaport E, Fontaine J (1989) Anticancer activities of adenine nucleotides in mice are mediated through expansion of erythrocyte ATP pools. Proc Natl Acad Sci USA 86:1662–1666
Spychala J (2000) Tumor-promoting functions of adenosine. Pharmacol Ther 87:161–173
Hoepfner M, Kap H, Jansen A, Lemmer K, Hanski C, Riecken EO, Scheruebl H (1999) Extracellular ATP induces apoptosis and inhibits growth of colorectal carcinomas. Gastroenterology 116:A423
Chahwala SB, Cantley LC (1984) Extracellular ATP induces ion fluxes and inhibits growth of Friend erythroleukemia cells. J Biol Chem 259:13717–13722
Seetulsingh-Goorah SP, Stewart BW (1998) Growth inhibition of HL-60 cells by extracellular ATP: concentration-dependent involvement of a P2 receptor and adenosine generation. Biochem Biophys Res Commun 250:390–396
Maaser K, Höpfner M, Kap H, Sutter AP, Barthel B, von Lampe B, Zeitz M, Scherübl H (2002) Extracellular nucleotides inhibit growth of human oesophageal cancer cells via P2Y2-receptors. Br J Cancer 86:636–644
Dubyak GR, De Young MB (1985) Intracellular Ca2+ mobilization activated by extracellular ATP in Ehrlich ascites tumor cells. J Biol Chem 260:10653–10661
Greig AVH, Linge C, Healy V, Lim P, Clayton E, Rustin MH, McGrouther DA, Burnstock G (2003) Expression of purinergic receptors in non-melanoma skin cancers and their functional roles in A431 cells. J Invest Dermatol 121:315–327
Schäfer R, Sedehizade F, Welte T, Reiser G (2003) ATP- and UTP-activated P2Y receptors differently regulate proliferation of human lung epithelial tumor cells. Am J Physiol Lung Cell Mol Physiol 285:L376–L385
Wang Q, Wang L, Feng YH, Li X, Zeng R, Gorodeski GI (2004) P2X7 receptor-mediated apoptosis of human cervical epithelial cells. Am J Physiol Cell Physiol 287:C1349–C1358
Horstman DA, Tennes KA, Putney JW Jr (1986) ATP-induced calcium mobilization and inositol 1,4,5-triphosphate formation in H-35 hepatoma cells. FEBS Lett 204:189–192
Shabbir M, Thompson CS, Jarmulowicz M, Mikhailidis DP, Burnstock G (2008) Effect of extracellular ATP on the growth of hormone refractory prostate cancer in vivo. BJU Int 102:108–112
Shabbir M, Ryten M, Thompson CS, Mikhailidis DP, Burnstock G (2008) Purinergic receptor-mediated effects of ATP in high-grade bladder cancer. BJU Int 101:106–112
Kim NH, Park KS, Sohn JH, Yeh BI, Ko CM, Kong ID (2011) Functional expression of P2Y receptors in WERI-Rb1 retinoblastoma cells. Korean J Physiol Pharmacol 15:61–66
Gómez-Villafuertes R, del Puerto A, Díaz-Hernández M, Bustillo D, Díaz-Hernández JI, Huerta PG, Artalejo AR, Garrido JJ, Miras-Portugal MT (2009) Ca2+/calmodulin-dependent kinase II signalling cascade mediates P2X7 receptor-dependent inhibition of neuritogenesis in neuroblastoma cells. FEBS J 276:5307–5325
Suplat-Wypych D, Dygas A, Barañska J (2010) 2′,3′-O-(4-benzoylbenzoyl)-ATP-mediated calcium signaling in rat glioma C6 cells: role of the P2Y2 nucleotide receptor. Purinergic Signalling 6:317–325
White N, Butler PEM, Burnstock G (2005) Human melanomas express functional P2X7 receptors. Cell Tissue Res 321:411–418
White N, Knight GE, Butler PEM, Burnstock G (2009) An in vivo model of melanoma: treatment with ATP. Purinergic Signalling 5:327–333
Ahmann FR, Garewal HS, Schifman R, Celniker A, Rodney S (1987) Intracellular adenosine triphosphate as a measure of human tumor cell viability and drug modulated growth. In Vitro Cell Dev Biol 23:474–480
Maehara Y, Kusumoto H, Anai H, Kusumoto T, Sugimachi K (1987) Human tumor tissues have higher ATP contents than normal tissues. Clin Chim Acta 169:341–343
Martins JP, Silva RB, Coutinho-Silva R, Takiya CM, Battastini AM, Morrone FB, Campos MM (2012) P2X7 purinergic receptor and its role in inflammatory and nociceptive alterations associated to cyclophosphamide-induced hemorrhagic cystitis in mice. Br J Pharmacol 165:183–196
Yegutkin GG, Marttila-Ichihara F, Karikoski M, Niemela J, Laurila JP, Elima K, Jalkanen S, Salmi M (2011) Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur J Immunol 41:1231–1241
Feng L, Sun X, Csizmadia E, Han L, Bian S, Murakami T, Wang X, Robson SC, Wu Y (2011) Vascular CD39/ENTPD1 directly promotes tumor cell growth by scavenging extracellular adenosine triphosphate. Neoplasia 13:206–216
Tada Y, Yokomizo A, Shiota M, Song Y, Kashiwagi E, Kuroiwa K, Oda Y, Naito S (2011) Ectonucleoside triphosphate diphosphohydrolase 6 expression in testis and testicular cancer and its implication in cisplatin resistance. Oncol Rep 26:161–167
White N, Ryten M, Clayton E, Butler P, Burnstock G (2005) P2Y purinergic receptors regulate the growth of human melanomas. Cancer Lett 224:81–91
Yuahasi KK, Demasi MA, Tamajusuku AS, Lenz G, Sogayar MC, Fornazari M, Lameu C, Nascimento IC, Glaser T, Schwindt TT, Negraes PD, Ulrich H (2012) Regulation of neurogenesis and gliogenesis of retinoic acid-induced P19 embryonal carcinoma cells by P2X2 and P2X7 receptors studied by RNA interference. Int J Dev Neurosci 30:91–97
Gorodeski GI (2009) P2X7-mediated chemoprevention of epithelial cancers. Expert Opin Ther Targets 13:1313–1332
Zanovello P, Bronte V, Rosato A, Pizzo P, Di Virgilio F (1990) Responses of mouse lymphocytes to extracellular ATP. II. Extracellular ATP causes cell type-dependent lysis and DNA fragmentation. J Immunol 145:1545–1550
Pizzo P, Murgia M, Zambon A, Zanovello P, Bronte V, Pietrobon D, Di Virgilio F (1992) Role of P2z purinergic receptors in ATP-mediated killing of tumor necrosis factor (TNF)-sensitive and TNF-resistant L929 fibroblasts. J Immunol 149:3372–3378
Adinolfi E, Callegari MG, Ferrari D, Bolognesi C, Minelli M, Wieckowski MR, Pinton P, Rizzuto R, Di Virgilio F (2005) Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol Biol Cell 16:3260–3272
Thompson BA, Storm MP, Hewinson J, Hogg S, Welham MJ, Mackenzie AB (2012) A novel role for P2X7 receptor signalling in the survival of mouse embryonic stem cells. Cell Signal 24:770–778
Adinolfi E, Callegari MG, Cirillo M, Pinton P, Giorgi C, Cavagna D, Rizzuto R, Di Virgilio F (2009) Expression of the P2X7 receptor increases the Ca2+ content of the endoplasmic reticulum, activates NFATc1, and protects from apoptosis. J Biol Chem 284:10120–10128
Adinolfi E, Cirillo M, Woltersdorf R, Falzoni S, Chiozzi P, Pellegatti P, Callegari MG, Sandona D, Markwardt F, Schmalzing G, Di Virgilio F (2010) Trophic activity of a naturally occurring truncated isoform of the P2X7 receptor. FASEB J 24:3393–3404
Jelassi B, Chantôme A, Alcaraz-Pérez F, Baroja-Mazo A, Cayuela ML, Pelegrin P, Surprenant A, Roger S (2011) P2X7 receptor activation enhances SK3 channels- and cystein cathepsin-dependent cancer cells invasiveness. Oncogene 30:2108–2122
Li X, Qi X, Zhou L, Fu W, Abdul-Karim FW, Maclennan G, Gorodeski GI (2009) P2X7 receptor expression is decreased in epithelial cancer cells of ectodermal, uro-genital sinus, and distal paramesonephric duct origin. Purinergic Signal 5:351–368
Feng YH, Li X, Zeng R, Gorodeski GI (2006) Endogenously expressed truncated P2X7 receptor lacking the C-terminus is preferentially upregulated in epithelial cancer cells and fails to mediate ligand-induced pore formation and apoptosis. Nucleosides Nucleotides Nucleic Acids 25:1271–1276
van der Weyden L, Conigrave AD, Morris MB (2000) Signal transduction and white cell maturation via extracellular ATP and the P2Y11 receptor. Immunol Cell Biol 78:369–374
Greig AVH, Linge C, Terenghi G, McGrouther DA, Burnstock G (2003) Purinergic receptors are part of a functional signalling system for proliferation and differentiation of human epidermal keratinocytes. J Invest Dermatol 120:1007–1015
Li S, Huang S, Peng SB (2005) Overexpression of G protein-coupled receptors in cancer cells: involvement in tumor progression. Int J Oncol 27:1329–1339
Madi L, Ochaion A, Rath-Wolfson L, Bar-Yehuda S, Erlanger A, Ohana G, Harish A, Merimski O, Barer F, Fishman P (2004) The A3 adenosine receptor is highly expressed in tumor versus normal cells: potential target for tumor growth inhibition. Clin Cancer Res 10:4472–4479
Placzek WJ, Almeida MS, Wüuthrich K (2007) NMR structure and functional characterization of a human cancer-related nucleoside triphosphatase. J Mol Biol 367:788–801
Kalhan A, Gharibi B, Vazquez M, Jasani B, Neal J, Kidd M, Modlin IM, Pfragner R, Rees DA, Ham J (2012) Adenosine A2A and A2B receptor expression in neuroendocrine tumours: potential targets for therapy. Purinergic Signal 8:265–274
Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, Chen JF, Jackson EK, Apasov S, Abrams S, Sitkovsky M (2006) A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A 103:13132–13137
Hoskin DW, Reynolds T, Blay J (1994) Adenosine as a possible inhibitor of killer T-cell activation in the microenvironment of solid tumours. Int J Cancer 59:854–855
Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F (2008) Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One 3:e2599
Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, Krempl CD, Sorichter S, Gerlach UV, Juttner E, Zerweck A, Gartner F, Pellegatti P, Di Virgilio F, Ferrari D, Kambham N, Fisch P, Finke J, Idzko M, Zeiser R (2010) Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med 16:1434–1438
Phillis JW, Wu PH (1981) Adenosine may regulate the vascular supply and thus the growth and spread of neoplastic tissues: a proposal. Gen Pharmacol 12:309–310
Mitchell BS, Schumacher U, Stauber VV, Kaiserling E (1994) Are breast tumours innervated? Immunohistological investigations using antibodies against the neuronal marker protein gene product 9.5 (PGP 9.5) in benign and malignant breast lesions. Eur J Cancer 30A:1100–1103
Ashraf S, Crowe R, Loizidou MC, Turmaine M, Taylor I, Burnstock G (1996) The absence of autonomic perivascular nerves in human colorectal liver metastases. Br J Cancer 73:349–359
Ashraf S, Loizidou M, Crowe R, Turmaine M, Taylor I, Burnstock G (1997) Blood vessels in liver metastases from both sarcoma and carcinoma lack perivascular innervation and smooth muscle cells. Clin Exp Metastasis 15:484–498
Chamary VL, Robson T, Loizidou M, Boulos PB, Burnstock G (2000) Progressive loss of perivascular nerves adjacent to colorectal cancer. Eur J Surg Oncol 26:588–593
Gil M, Skopinska-Rózewska E, Radomska D, Demkow U, Skurzak H, Rochowska M, Beuth J, Roszkowski K (1993) Effect of purinergic receptor antagonists suramin and theobromine on tumor-induced angiogenesis in BALB/c mice. Folia Biol (Praha) 39:63–68
Komi Y, Ohno O, Suzuki Y, Shimamura M, Shimokado K, Umezawa K, Kojima S (2007) Inhibition of tumor angiogenesis by targeting endothelial surface ATP synthase with sangivamycin. Jpn J Clin Oncol 37:867–873
Nejime N, Tanaka N, Yoshihara R, Kagota S, Yoshikawa N, Nakamura K, Kunitomo M, Hashimoto M, Shinozuka K (2008) Effect of P2 receptor on the intracellular calcium increase by cancer cells in human umbilical vein endothelial cells. Naunyn Schmiedebergs Arch Pharmacol 377:429–436
Hazama A, Fan HT, Abdullaev I, Maeno E, Tanaka S, Ando-Akatsuka Y, Okada Y (2000) Swelling-activated, cystic fibrosis transmembrane conductance regulator-augmented ATP release and Cl− conductances in murine C127 cells. J Physiol 523 Pt 1:1–11
Boyd Tressler A, Dubyak G (2012) Multiple mechanisms for ATP release from apoptotic tumour cells. J Immunol 188:46.21
Martin DS, Bertino JR, Koutcher JA (2000) ATP depletion + pyrimidine depletion can markedly enhance cancer therapy: fresh insight for a new approach. Cancer Res 60:6776–6783
Martin DS, Spriggs D, Koutcher JA (2001) A concomitant ATP-depleting strategy markedly enhances anticancer agent activity. Apoptosis 6:125–131
Lukashev D, Sitkovsky M, Ohta A (2007) From "Hellstrom Paradox" to anti-adenosinergic cancer immunotherapy. Purinergic Signal 3:129–134
Sitkovsky M, Lukashev D, Deaglio S, Dwyer K, Robson SC, Ohta A (2008) Adenosine A2A receptor antagonists: blockade of adenosinergic effects and T regulatory cells. Br J Pharmacol 153(Suppl 1):S457–S464
Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Séror C, Métivier D, Perfettini JL, Zitvogel L, Kroemer G (2009) Chemotherapy induces ATP release from tumor cells. Cell Cycle 8:3723–3728
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, Andre F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15:1170–1178
Aymeric L, Apetoh L, Ghiringhelli F, Tesniere A, Martins I, Kroemer G, Smyth MJ, Zitvogel L (2010) Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res 70:855–858
Marteau F, Gonzalez NS, Communi D, Goldman M, Boeynaems JM, Communi D (2005) Thrombospondin-1 and indoleamine 2,3-dioxygenase are major targets of extracellular ATP in human dendritic cells. Blood 106:3860–3866
Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, Vacchelli E, Galluzzi L, Ghiringhelli F, Di Virgilio F, Zitvogel L, Kroemer G (2011) Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334:1573–1577
Haskell CM, Wong M, Williams A, Lee LY (1996) Phase I trial of extracellular adenosine 5′-triphosphate in patients with advanced cancer. Med Pediatr Oncol 27:165–173
Haskell CM, Mendoza E, Pisters KM, Fossella FV, Figlin RA (1998) Phase II study of intravenous adenosine 5′-triphosphate in patients with previously untreated stage IIIB and stage IV non-small cell lung cancer. Invest New Drugs 16:81–85
Agteresch HJ, Dagnelie PC, Rietveld T, van den Berg JW, Danser AH, Wilson JH (2000) Pharmacokinetics of intravenous ATP in cancer patients. Eur J Clin Pharmacol 56:49–55
Agteresch HJ, Rietveld T, Kerkhofs LG, van den Berg JW, Wilson JH, Dagnelie PC (2002) Beneficial effects of adenosine triphosphate on nutritional status in advanced lung cancer patients: a randomized clinical trial. J Clin Oncol 20:371–378
Chahrour O, Cairns D, Omran Z (2012) Small molecule kinase inhibitors as anti-cancer therapeutics. Mini Rev Med Chem 12:399–411
Agteresch HJ, Leij-Halfwerk S, van den Berg JW, Hordijk-Luijk CH, Wilson JH, Dagnelie PC (2000) Effects of ATP infusion on glucose turnover and gluconeogenesis in patients with advanced non-small-cell lung cancer. Clin Sci (Lond) 98:689–695
Rapaport E, Fontaine J (1989) Generation of extracellular ATP in blood and its mediated inhibition of host weight loss in tumor-bearing mice. Biochem Pharmacol 38:4261–4266
Agteresch HJ, van Rooijen MHC, van den Berg JWO, Minderman-Voortman GJ, Wilson JHP, Dagnelie PC (2003) Growth inhibition of lung cancer cells by adenosine 5′-triphosphate. Drug Dev Res 60:196–203
Arts ICW, Swennen ELR, Vandeurzen KGA, Dagnelie PC (2008) Effect of ATP on survival, tumour response, nutritional status and quality of life in lung cancer patients: a multicentre, double-blind randomized trial. Purinergic Signalling 4:S194–S195
Beijer S, Gielisse EA, Hupperets PS, van den Borne BE, van den Beuken-van EM, Nijziel MR, van Henten AM, Dagnelie PC (2007) Intravenous ATP infusions can be safely administered in the home setting: a study in pre-terminal cancer patients. Invest New Drugs 25:571–579
Beijer S, Wijckmans NE, van Rossum E, Spreeuwenberg C, Winkens RA, Ars L, Dagnelie PC (2008) Treatment adherence and patients’ acceptance of home infusions with adenosine 5′-triphosphate (ATP) in palliative home care. Support Care Cancer 16:1419–1424
Beijer S, Hupperets PS, van den Borne BE, Eussen SR, van Henten AM, van den Beuken-van EM, de Graeff A, Ambergen TA, van den Brandt PA, Dagnelie PC (2009) Effect of adenosine 5′-triphosphate infusions on the nutritional status and survival of preterminal cancer patients. Anticancer Drugs 20:625–633
Swennen EL, Dagnelie PC, Van den Beucken T, Bast A (2008) Radioprotective effects of ATP in human blood ex vivo. Biochem Biophys Res Commun 367:383–387
Ohshima Y, Tsukimoto M, Takenouchi T, Harada H, Suzuki A, Sato M, Kitani H, Kojima S (2010) γ-Irradiation induces P2X7 receptor-dependent ATP release from B16 melanoma cells. Biochim Biophys Acta 1800:40–46
Ohshima Y, Tsukimoto M, Harada H, Kojima S (2012) Involvement of connexin43 hemichannel in ATP release after γ-irradiation. J Radiat Res 53:551–557
Mancuso M, Pasquali E, Leonardi S, Rebessi S, Tanori M, Giardullo P, Borra F, Pazzaglia S, Naus CC, Di Majo V, Saran A (2011) Role of connexin43 and ATP in long-range bystander radiation damage and oncogenesis in vivo. Oncogene 30:4601–4608
Nishimaki N, Tsukimoto M, Kitami A, Kojima S (2012) Autocrine regulation of γ-irradiation-induced DNA damage response via extracellular nucleotides-mediated activation of P2Y6 and P2Y12 receptors. DNA Repair (Amst) 11:657–665
Spungin B, Friedberg I (1993) Growth inhibition of breast cancer cells induced by exogenous ATP. J Cell Physiol 157:502–508
Colofiore JR, Stolfi RL, Nord LD, Martin DS (1995) On the relationship of ATP depletion to chemotherapeutically-induced tumor regression. Int J Oncol 7:1401–1404
Flezar M, Heisler S (1993) P2-purinergic receptors in human breast tumor cells: coupling of intracellular calcium signaling to anion secretion. J Pharmacol Exp Ther 265:1499–1510
Dixon CJ, Bowler WB, Fleetwood P, Ginty AF, Gallagher JA, Carron JA (1997) Extracellular nucleotides stimulate proliferation in MCF-7 breast cancer cells via P2-purinoceptors. Br J Cancer 75:34–39
Wagstaff SC, Bowler WB, Gallagher JA, Hipskind RA (2000) Extracellular ATP activates multiple signalling pathways and potentiates growth factor-induced c-fos gene expression in MCF-7 breast cancer cells. Carcinogenesis 21:2175–2181
Li HJ, Wang LY, Qu HN, Yu LH, Burnstock G, Ni X, Xu M, Ma B (2011) P2Y2 receptor-mediated modulation of estrogen-induced proliferation of breast cancer cells. Mol Cell Endocrinol 338:28–37
Spychala J, Lazarowski E, Ostapkowicz A, Ayscue LH, Jin A, Mitchell BS (2004) Role of estrogen receptor in the regulation of ecto-5′-nucleotidase and adenosine in breast cancer. Clin Cancer Res 10:708–717
do Carmo Araujo M, Rocha JB, Morsch A, Zanin R, Bauchspiess R, Morsch VM, Schetinger MR (2005) Enzymes that hydrolyze adenine nucleotides in platelets from breast cancer patients. Biochim Biophys Acta 1740:421–426
Seyedabadi M, Ghahremani MH, Ostad SN (2009) ATP depletion as a consequence of hypoxia enhances tamoxifen antiproliferative effects in T47D breast carcinoma cells. Oncol Res 18:221–228
Dohán O, De la Vieja A, Carrasco N (2006) Hydrocortisone and purinergic signaling stimulate sodium/iodide symporter (NIS)-mediated iodide transport in breast cancer cells. Mol Endocrinol 20:1121–1137
Wang Z (2004) Roles of K+ channels in regulating tumour cell proliferation and apoptosis. Pflugers Arch 448:274–286
Gow IF, Thomson J, Davidson J, Shennan DB (2005) The effect of a hyposmotic shock and purinergic agonists on K+(Rb+) efflux from cultured human breast cancer cells. Biochim Biophys Acta 1712:52–61
Yu SP (2003) Regulation and critical role of potassium homeostasis in apoptosis. Prog Neurobiol 70:363–386
Loo WT, Tong JM, Cheung MN, Chow LW (2006) A new predictive and prognostic marker (ATP bioluminescence and positron emission tomography) in vivo and in vitro for delivering adjuvant treatment plan to invasive breast tumor patients. Biomed Pharmacother 60:285–288
Pan J, Sun LC, Tao YF, Zhou Z, Du XL, Peng L, Feng X, Wang J, Li YP, Liu L, Wu SY, Zhang YL, Hu SY, Zhao WL, Zhu XM, Lou GL, Ni J (2011) ATP synthase ecto-α-subunit: a novel therapeutic target for breast cancer. J Transl Med 9:211
Kim HA, Yom CK, Moon BI, Choe KJ, Sung SH, Han WS, Choi HY, Kim HK, Park HK, Choi SH, Yoon EJ, Oh SY (2008) The use of an in vitro adenosine triphosphate-based chemotherapy response assay to predict chemotherapeutic response in breast cancer. Breast 17:19–26
Koo JS, Jung W, Shin E, Lee HD, Jeong J, Kim KH, Jeong H, Hong SW (2009) Impact of grade, hormone receptor, and HER-2 status in women with breast cancer on response to specific chemotherapeutic agents by in vitro adenosine triphosphate-based chemotherapy response assay. J Korean Med Sci 24:1150–1157
Qi CJ, Ning YL, Zhu YL, Min HY, Ye H, Qian KQ (2009) In vitro chemosensitivity in breast cancer using ATP-tumor chemosensitivity assay. Arch Pharm Res 32:1737–1742
Buffon A, Ribeiro VB, Wink MR, Casali EA, Sarkis JJ (2007) Nucleotide metabolizing ecto-enzymes in Walker 256 tumor cells: molecular identification, kinetic characterization and biochemical properties. Life Sci 80:950–958
Buffon A, Wink MR, Ribeiro BV, Casali EA, Libermann TA, Zerbini LF, Robson SC, Sarkis JJ (2007) NTPDase and 5′ ecto-nucleotidase expression profiles and the pattern of extracellular ATP metabolism in the Walker 256 tumor. Biochim Biophys Acta 1770:1259–1265
Buffon A, Casali EA, Cardoso VV, Zerbini LF, Robson SC, Sarkis JJ, Wink MR (2010) Differential expression of nucleotide pyrophosphatase/phosphodiesterases by Walker 256 mammary cancer cells in solid tumors and malignant ascites. Life Sci 86:435–440
Rumjahn SM, Javed MA, Wong N, Law WE, Buxton IL (2007) Purinergic regulation of angiogenesis by human breast carcinoma-secreted nucleoside diphosphate kinase. Br J Cancer 97:1372–1380
Yokdang N, Tellez JD, Tian H, Norvell J, Barsky SH, Valencik M, Buxton IL (2011) A role for nucleotides in support of breast cancer angiogenesis: heterologous receptor signalling. Br J Cancer 104:1628–1640
Scodelaro Bilbao P, Boland R, Russo de Boland A, Santillán G (2007) ATP modulation of mitogen activated protein kinases and intracellular Ca2+ in breast cancer (MCF-7) cells. Arch Biochem Biophys 466:15–23
Scodelaro Bilbao P, Boland R, Santillán G (2010) ATP modulates transcription factors through P2Y2 and P2Y4 receptors via PKC/MAPKs and PKC/Src pathways in MCF-7 cells. Arch Biochem Biophys 494:7–14
Zhang X, Gao F, Yu LL, Peng Y, Liu HH, Liu JY, Yin M, Ni J (2008) Dual functions of a monoclonal antibody against cell surface F1F0 ATP synthase on both HUVEC and tumor cells. Acta Pharmacol Sin 29:942–950
Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, Dwyer KM, Smyth MJ (2010) Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A 107:1547–1552
Wang L, Zhou X, Zhou T, Ma D, Chen S, Zhi X, Yin L, Shao Z, Ou Z, Zhou P (2008) Ecto-5′-nucleotidase promotes invasion, migration and adhesion of human breast cancer cells. J Cancer Res Clin Oncol 134:365–372
Coleman RE (2004) The role of bisphosphonates in breast cancer. Breast 13(Suppl 1):S19–S28
Varani K, Vincenzi F, Targa M, Paradiso B, Parrilli A, Fini M, Lanza G, Borea PA (2013) The stimulation of A3 adenosine receptors reduces bone-residing breast cancer in a rat preclinical model. Eur J Cancer 49:482–491
Fehm T, Zwirner M, Wallwiener D, Seeger H, Neubauer H (2012) Antitumor activity of zoledronic acid in primary breast cancer cells determined by the ATP tumor chemosensitivity assay. BMC Cancer 12:308
Schott S, Wallwiener M, Kootz B, Seeger H, Fehm T, Neubauer H (2012) Cytotoxicity of the new antimetabolite-bisphosphonate (5-FdU-alendronate) in comparison to standard therapeutics on breast and ovarian cancer cell lines in the ATP tumor chemosensitivity assay. Invest New Drugs 30:1750–1755
Huang TC, Chang HY, Hsu CH, Kuo WH, Chang KJ, Juan HF (2008) Targeting therapy for breast carcinoma by ATP synthase inhibitor aurovertin B. J Proteome Res 7:1433–1444
Kawai Y, Kaidoh M, Ohhashi T (2008) MDA-MB-231 produces ATP-mediated ICAM-1-dependent facilitation of the attachment of carcinoma cells to human lymphatic endothelial cells. Am J Physiol Cell Physiol 295:C1123–C1132
Buxton IL, Yokdang N, Matz RM (2010) Purinergic mechanisms in breast cancer support intravasation, extravasation and angiogenesis. Cancer Lett 291:131–141
Patel V, Rumney R, Wang N, Hipskind RH, Gallagher JA, Gartland A (2008) The effect of ATP, alone and in combination with EGF, on breast cancer cell survival. Purinergic Signalling 4:S205
Lee KL, Dai Q, Hansen EL, Saner CN, Price TM (2010) Modulation of ATP-induced calcium signaling by progesterone in T47D-Y breast cancer cells. Mol Cell Endocrinol 319:109–115
Pubill D, Dayanithi G, Siatka C, András M, Dufour MN, Guillon G, Mendre C (2001) ATP induces intracellular calcium increases and actin cytoskeleton disaggregation via P2x receptors. Cell Calcium 29:299–309
Vandewalle B, Hornez L, Revillion F, Lefebvre J (1994) Effect of extracellular ATP on breast tumor cell growth, implication of intracellular calcium. Cancer Lett 85:47–54
Tafani M, Schito L, Pellegrini L, Villanova L, Marfe G, Anwar T, Rosa R, Indelicato M, Fini M, Pucci B, Russo MA (2011) Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-κB. Carcinogenesis 32:1167–1175
Davis FM, Kenny PA, Soo ET, van Denderen BJ, Thompson EW, Cabot PJ, Parat MO, Roberts-Thomson SJ, Monteith GR (2011) Remodeling of purinergic receptor-mediated Ca2+ signaling as a consequence of EGF-induced epithelial-mesenchymal transition in breast cancer cells. PLoS One 6:e23464
Abraham EH, Vos P, Kahn J, Grubman SA, Jefferson DM, Ding I, Okunieff P (1996) Cystic fibrosis hetero- and homozygosity is associated with inhibition of breast cancer growth. Nat Med 2:593–596
Clark JH, Broadley KJ, Hutcheson IR, Nicholson RI, Kidd EJ (2003) Expression of adenosine receptors in MCF-7 human breast cancer cells. Brit J Pharmacol 138:130P
Panjehpour M, Hemati S, Forghani MA (2012) Expression of A1 and A3 adenosine receptors in human breast tumors. Tumori 98:137–141
Mujoomdar M, Bennett A, Hoskin D, Blay J (2004) Adenosine stimulation of proliferation of breast carcinoma cell lines: evaluation of the [3H]thymidine assay system and modulatory effects of the cellular microenvironment in vitro. J Cell Physiol 201:429–438
Lu J, Pierron A, Ravid K (2003) An adenosine analogue, IB-MECA, down-regulates estrogen receptor alpha and suppresses human breast cancer cell proliferation. Cancer Res 63:6413–6423
Panjehpour M, Karami-Tehrani F (2004) An adenosine analog (IB-MECA) inhibits anchorage-dependent cell growth of various human breast cancer cell lines. Int J Biochem Cell Biol 36:1502–1509
Hashemi M, Karami-Tehrani F, Ghavami S, Maddika S, Los M (2005) Adenosine and deoxyadenosine induces apoptosis in oestrogen receptor-positive and -negative human breast cancer cells via the intrinsic pathway. Cell Prolif 38:269–285
Mirza A, Basso A, Black S, Malkowski M, Kwee L, Pachter JA, Lachowicz JE, Wang Y, Liu S (2005) RNA interference targeting of A1 receptor-overexpressing breast carcinoma cells leads to diminished rates of cell proliferation and induction of apoptosis. Cancer Biol Ther 4:1355–1360
Panjehpour M, Castro M, Klotz KN (2005) Human breast cancer cell line MDA-MB-231 expresses endogenous A2B adenosine receptors mediating a Ca2+ signal. Br J Pharmacol 145:211–218
Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J (2012) Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol 188:198–205
Sadej R, Inai K, Rajfur Z, Ostapkowicz A, Kohler J, Skladanowski AC, Mitchell BS, Spychala J (2008) Tenascin C interacts with ecto-5′-nucleotidase (eN) and regulates adenosine generation in cancer cells. Biochim Biophys Acta 1782:35–40
Aghaei M, Karami-Tehrani F, Salami S, Atri M (2010) Diagnostic value of adenosine deaminase activity in benign and malignant breast tumors. Arch of Med Res 41:14–18
Harris JW, Wong YP, Kehe CR, Teng SS (1975) The role of adenosine triphosphate, chalones, and specific proteins in controlling tumor growth fraction. Cancer Res 35:3181–3186
Artalejo AR, Garcia-Sancho J (1988) Mobilization of intracellular calcium by extracellular ATP and by calcium ionophores in the Ehrlich ascites-tumour cell. Biochim Biophys Acta 941:48–54
Ueno H, Tezuka M, Tamemasa O (1984) Effect of adenosine triphosphate on the proliferation of cultured tumor cells. Yakugaku Zasshi 104:1207–1210
Lasso De la Vega M, Terradez P, Obrador E, Navarro J, Pellicer JA, Estrela JM (1994) Inhibition of cancer growth and selective glutathione depletion in Ehrlich tumour cells in vivo by extracellular ATP. Biochem J 298:99–105
Dubyak GR (1986) Extracellular ATP activates polyphosphoinositide breakdown and Ca2+ mobilization in Ehrlich ascites tumor cells. Arch Biochem Biophys 245:84–95
Estrela JM, Obrador E, Navarro J, Lasso De la Vega M, Pellicer JA (1995) Elimination of Ehrlich tumours by ATP-induced growth inhibition, glutathione depletion and X-rays. Nat Med 1:84–88
Pedersen SF, Pedersen S, Lambert IH, Hoffmann EK (1998) P2 receptor-mediated signal transduction in Ehrlich ascites tumor cells. Biochim Biophys Acta 1374:94–106
Pedersen S, Pedersen SF, Nilius B, Lambert IH, Hoffmann EK (1999) Mechanical stress induces release of ATP from Ehrlich ascites tumor cells. Biochim Biophys Acta 1416:271–284
Zinchenko VP, Kasymov VA, Li VV, Kaimachnikov NP (2005) The calmodulin inhibitor R24571 induces a short-term Ca2+ entry and a pulse-like secretion of ATP in Ehrlich ascites tumor cells. Biofizika 50:1055–1069
Zamay TN, Zamay AS (2006) Influence of ATP on Ehrlich ascites carcinoma cell free cytoplasmic calcium concentration in the course of tumor growth. Biochemistry (Mosc) 71:1090–1095
Fang WG, Pirnia F, Bang YJ, Myers CE, Trepel JB (1992) P2-purinergic receptor agonists inhibit the growth of androgen-independent prostate carcinoma cells. J Clin Invest 89:191–196
Wasilenko WJ, Cooper J, Palad AJ, Somers KD, Blackmore PF, Rhim JS, Wright GL Jr, Schellhammer PF (1997) Calcium signaling in prostate cancer cells: evidence for multiple receptors and enhanced sensitivity to bombesin/GRP. Prostate 30:167–173
Vanoverberghe K, Mariot P, Vanden Abeele F, Delcourt P, Parys JB, Prevarskaya N (2003) Mechanisms of ATP-induced calcium signaling and growth arrest in human prostate cancer cells. Cell Calcium 34:75–85
Dainty IA, Franklin M, McKechnie KCW (1995) Classification of P2-purinoceptors on a human prostate cancer cell line (PC3 cells). Brit J Pharmacol 114:106P
Janssens R, Communi D, Pirotton S, Samson M, Parmentier M, Boeynaems JM (1996) Cloning and tissue distribution of the human P2Y1 receptor. Biochem Biophys Res Commun 221:588–593
Wei Q, Costanzi S, Liu QZ, Gao ZG, Jacobson KA (2011) Activation of the P2Y1 receptor induces apoptosis and inhibits proliferation of prostate cancer cells. Biochem Pharmacol 82:418–425
Tapia-Vieyra JV, Mas-Oliva J (2001) Apoptosis and cell death channels in prostate cancer. Arch Med Res 32:175–185
Janssens R, Boeynaems JM (2001) Effects of extracellular nucleotides and nucleosides on prostate carcinoma cells. Br J Pharmacol 132:536–546
Sauer H, Stanelle R, Hescheler J, Wartenberg M (2002) The DC electrical-field-induced Ca2+ response and growth stimulation of multicellular tumor spheroids are mediated by ATP release and purinergic receptor stimulation. J Cell Sci 115:3265–3273
Ye ZW, Ghalali A, Högberg J, Stenius U (2011) Silencing p110β prevents rapid depletion of nuclear pAkt. Biochem Biophys Res Commun 415:613–618
Limami Y, Pinon A, Leger DY, Pinault E, Delage C, Beneytout JL, Simon A, Liagre B (2012) The P2Y2/Src/p38/COX-2 pathway is involved in the resistance to ursolic acid-induced apoptosis in colorectal and prostate cancer cells. Biochimie 94:1754–1763
Chen L, He HY, Li HM, Zheng J, Heng WJ, You JF, Fang WG (2004) ERK1/2 and p38 pathways are required for P2Y receptor-mediated prostate cancer invasion. Cancer Lett 215:239–247
Nandigama R, Padmasekar M, Wartenberg M, Sauer H (2006) Feed forward cycle of hypotonic stress-induced ATP release, purinergic receptor activation, and growth stimulation of prostate cancer cells. J Biol Chem 281:5686–5693
Sauer H, Hescheler J, Wartenberg M (2000) Mechanical strain-induced Ca2+ waves are propagated via ATP release and purinergic receptor activation. Am J Physiol Cell Physiol 279:C295–C307
Zhang Y, Gong LH, Zhang HQ, Du Q, You JF, Tian XX, Fang WG (2010) Extracellular ATP enhances in vitro invasion of prostate cancer cells by activating Rho GTPase and upregulating MMPs expression. Cancer Lett 293:189–197
Stagg J, Beavis PA, Divisekera U, Liu MC, Moller A, Darcy PK, Smyth MJ (2012) CD73-deficient mice are resistant to carcinogenesis. Cancer Res 72:2190–2196
Shabbir M, Ryten M, Thompson CS, Mikhailidis DP, Burnstock G (2008) Characterisation of calcium-independent purinergic receptor-mediated apoptosis in hormone refractory prostate cancer. BJU Int 101:352–359
Fernando KC, Gargett CE, Wiley JS (1999) Activation of the P2Z/P2X7 receptor in human lymphocytes produces a delayed permeability lesion: involvement of phospholipase D. Arch Biochem Biophys 362:197–202
Shemon AN, Sluyter R, Fernando SL, Clarke AL, Dao-Ung LP, SkarRatt KK, Saunders BM, Tan KS, Gu BJ, Fuller SJ, Britton WJ, Petrou S, Wiley JS (2006) A Thr357 to Ser polymorphism in homozygous and compound heterozygous subjects causes absent or reduced P2X7 function and impairs ATP-induced mycobacterial killing by macrophages. J Biol Chem 281:2079–2086
Fuller SJ, Stokes L, SkarRatt KK, Gu BJ, Wiley JS (2009) Genetics of the P2X7 receptor and human disease. Purinergic Signal 5:257–262
Slater M, Danieletto S, Gidley-Baird A, Teh LC, Barden JA (2004) Early prostate cancer detected using expression of non-functional cytolytic P2X7 receptors. Histopathology 44:206–215
Slater M, Danieletto S, Barden JA (2005) Expression of the apoptotic calcium channel P2X7 in the glandular epithelium. J Mol Histol 36:159–165
Kyprianou N, Isaacs JT (1989) "Thymineless" death in androgen-independent prostatic cancer cells. Biochem Biophys Res Commun 165:73–81
Martikainen P, Kyprianou N, Tucker RW, Isaacs JT (1991) Programmed death of nonproliferating androgen-independent prostatic cancer cells. Cancer Res 51:4693–4700
Abraham EH, Salikhova A, Sterling KM, Johnston N (2001) Modulation of ATP release rates from erythrocytes in blood samples from prostate cancer patients receiving radiation therapy: implications to iv ATP therapy. Radiology 266
Miyake H, Hara I, Yamanaka K, Arakawa S, Kamidono S (1999) Calcium ionophore, ionomycin inhibits growth of human bladder cancer cells both in vitro and in vivo with alteration of Bcl-2 and Bax expression levels. J Urol 162:916–921
Lissbrant IF, Lissbrant E, Damber JE, Bergh A (2001) Blood vessels are regulators of growth, diagnostic markers and therapeutic targets in prostate cancer. Scand J Urol Nephrol 35:437–452
Aweimer A, Stachon T, Tannapfel A, Köller M, Truss MC, Stachon A (2012) Regulation of soluble VEGFR-2 secreted by microvascular endothelial cells derived from human BPH. Prostate Cancer Prostatic Dis 15:157–164
Minelli A, Bellezza I, Agostini M, Bracarda S, Culig Z (2006) Mechanism of 2-chloroadenosine toxicity to PC3 cell line. Prostate 66:1425–1436
Minelli A, Bellezza I, Tucci A, Rambotti MG, Conte C, Culig Z (2009) Differential involvement of reactive oxygen species and nucleoside transporters in cytotoxicity induced by two adenosine analogues in human prostate cancer cells. Prostate 69:538–547
Aghaei M, Panjehpour M, Karami-Tehrani F, Salami S (2011) Molecular mechanisms of A3 adenosine receptor-induced G1 cell cycle arrest and apoptosis in androgen-dependent and independent prostate cancer cell lines: involvement of intrinsic pathway. J Cancer Res Clin Oncol 137:1511–1523
Aghaei M, Karami-Tehrani F, Panjehpour M, Salami S, Fallahian F (2012) Adenosine induces cell-cycle arrest and apoptosis in androgen-dependent and -independent prostate cancer cell lines, LNcap-FGC-10, DU-145, and PC3. Prostate 72:361–375
Jajoo S, Mukherjea D, Watabe K, Ramkumar V (2009) Adenosine A3 receptor suppresses prostate cancer metastasis by inhibiting NADPH oxidase activity. Neoplasia 11:1132–1145
Rapaport E, Fishman RF, Gercel C (1983) Growth inhibition of human tumor cells in soft-agar cultures by treatment with low levels of adenosine 5′-triphosphate. Cancer Res 43:4402–4406
Hitchin BW, Dobson PR, Ruprai A, Hardcastle J, Hardcastle PT, Taylor CJ, Brown BL (1991) Purinoceptors and second messenger signalling in the human colonic adenoma cell line. J Physiol 438:80P
Lohrmann E, Cabantchik ZI, Greger R (1992) Transmitter-induced changes of the membrane voltage of HT29 cells. Pflügers Arch 421:224–229
Correale P, Caraglia M, Procopio A, Marinetti MR, Guarrasi R, Fabbrocini A, Bianco AR, Tagliaferri P (1993) Transmembrane ion flux modifiers verapamil and ouabain modulate cytotoxic effects of extracellular ATP on human tumor cells in vitro. Int J Oncol 3:847–851
Parr CE, Sullivan DM, Paradiso AM, Lazarowski ER, Burch LH, Olsen JC, Erb L, Weisman GA, Boucher RC, Turner JT (1994) Cloning and expression of a human P2U nucleotide receptor, a target for cystic fibrosis pharmacotherapy. Proc Natl Acad Sci U S A 91:13067
Richards M, van Giersbergen P, Zimmermann A, Lesur B, Hoflack J (1997) Activation of neurotensin receptors and purinoceptors in human colonic adenocarcinoma cells detected with the microphysiometer. Biochem Pharmacol 54:825–832
Guo X, Merlin D, Harvey RD, Laboisse C, Hopfer U (1995) Stimulation of Cl− secretion by extracellular ATP does not depend on increased cytosolic Ca2+ in HT-29.cl16E. Am J Physiol 269:C1457–C1463
Guo XW, Merlin D, Laboisse C, Hopfer U (1997) Purinergic agonists, but not cAMP, stimulate coupled granule fusion and Cl− conductance in HT29-Cl.16E. Am J Physiol 273:C804–C809
Dho S, Stewart K, Foskett JK (1992) Purinergic receptor activation of Cl− secretion in T84 cells. Am J Physiol 262:C67–C74
Zhang W, Roomans GM (1997) Regulation of ion transport by P2U purinoceptors and α2A adrenoceptors in HT29 cells. Cell Biol Int 4:195–200
Höpfner M, Lemmer K, Jansen A, Hanski C, Riecken EO, Gavish M, Mann B, Buhr H, Glassmeier G, Scherübl H (1998) Expression of functional P2-purinergic receptors in primary cultures of human colorectal carcinoma cells. Biochem Biophys Res Commun 251:811–817
Höpfner M, Maaser K, Barthel B, von Lampe B, Hanski C, Riecken EO, Zeitz M, Scherubl H (2001) Growth inhibition and apoptosis induced by P2Y2 receptors in human colorectal carcinoma cells: involvement of intracellular calcium and cyclic adenosine monophosphate. Int J Colorectal Dis 16:154–166
Yaguchi T, Saito M, Yasuda Y, Kanno T, Nakano T, Nishizaki T (2010) Higher concentrations of extracellular ATP suppress proliferation of Caco-2 human colonic cancer cells via an unknown receptor involving PKC inhibition. Cell Physiol Biochem 26:125–134
Cummins MM, O’Mullane LM, Barden JA, Cook DI, Poronnik P (2000) Purinergic responses in HT29 colonic epithelial cells are mediated by G protein α-subunits. Cell Calcium 27:247–255
McAlroy HL, Ahmed S, Day SM, Baines DL, Wong HY, Yip CY, Ko WH, Wilson SM, Collett A (2000) Multiple P2Y receptor subtypes in the apical membranes of polarized epithelial cells. Br J Pharmacol 131:1651–1658
Buzzi N, Bilbao PS, Boland R, de Boland AR (2009) Extracellular ATP activates MAP kinase cascades through a P2Y purinergic receptor in the human intestinal Caco-2 cell line. Biochim Biophys Acta 1790:1651–1659
Ullrich N, Caplanusi A, Brône B, Hermans D, Larivière E, Nilius B, Van Driessche W, Eggermont J (2006) Stimulation by caveolin-1 of the hypotonicity-induced release of taurine and ATP at basolateral, but not apical, membrane of Caco-2 cells. Am J Physiol Cell Physiol 290:C1287–C1296
Nylund G, Nordgren S, Delbro DS (2004) Expression of P2Y2 purinoceptors in MCG 101 murine sarcoma cells, and HT-29 human colon carcinoma cells. Auton Neurosci 112:69–79
Nylund G, Hultman L, Nordgren S, Delbro DS (2007) P2Y2- and P2Y4 purinergic receptors are over-expressed in human colon cancer. Auton Autacoid Pharmacol 27:79–84
Hu H, O’Mullane LM, Cummins MM, Campbell CR, Hosoda Y, Poronnik P, Dinudom A, Cook DI (2010) Negative regulation of Ca2+ influx during P2Y2 purinergic receptor activation is mediated by Gβγ-subunits. Cell Calcium 47:55–64
Hatanaka H, Takada S, Choi YL, Fujiwara S, Soda M, Enomoto M, Kurashina K, Watanabe H, Yamashita Y, Sugano K, Mano H (2007) Transforming activity of purinergic receptor P2Y, G-protein coupled, 2 revealed by retroviral expression screening. Biochem Biophys Res Commun 356:723–726
Buzzi N, Boland R, Russo de Boland A (2010) Signal transduction pathways associated with ATP-induced proliferation of colon adenocarcinoma cells. Biochim Biophys Acta 1800:946–955
Hinoshita E, Uchiumi T, Taguchi K, Kinukawa N, Tsuneyoshi M, Maehara Y, Sugimachi K, Kuwano M (2000) Increased expression of an ATP-binding cassette superfamily transporter, multidrug resistance protein 2, in human colorectal carcinomas. Clin Cancer Res 6:2401–2407
Künzli BM, Bernlochner MI, Rath S, Käser S, Csizmadia E, Enjyoji K, Cowan P, d’Apice A, Dwyer K, Rosenberg R, Perren A, Friess H, Maurer CA, Robson SC (2011) Impact of CD39 and purinergic signalling on the growth and metastasis of colorectal cancer. Purinergic Signal 7:231–241
Eroglu A, Canbolat O, Demirci S, Kocaoglu H, Eryavuz Y, Akgül H (2000) Activities of adenosine deaminase and 5′-nucleotidase in cancerous and noncancerous human colorectal tissues. Med Oncol 17:319–324
ten Kate J, Wijnen JT, van der Goes RG, Quadt R, Griffioen G, Bosman FT, Khan PM (1984) Quantitative changes in adenosine deaminase isoenzymes in human colorectal adenocarcinomas. Cancer Res 44:4688–4692
Tan EY, Mujoomdar M, Blay J (2004) Adenosine down-regulates the surface expression of dipeptidyl peptidase IV on HT-29 human colorectal carcinoma cells: implications for cancer cell behavior. Am J Pathol 165:319–330
Giglioni S, Leoncini R, Aceto E, Chessa A, Civitelli S, Bernini A, Tanzini G, Carraro F, Pucci A, Vannoni D (2008) Adenosine kinase gene expression in human colorectal cancer. Nucleosides Nucleotides Nucleic Acids 27:750–754
Whitehouse PA, Knight LA, Di NF, Mercer SJ, Sharma S, Cree IA (2003) Heterogeneity of chemosensitivity of colorectal adenocarcinoma determined by a modified ex vivo ATP-tumor chemosensitivity assay (ATP-TCA). Anticancer Drugs 14:369–375
Cho YB, Lee WY, Song SY, Choi SH, Shin HJ, Ahn KD, Lee JM, Kim HC, Yun SH, Chun HK (2009) In vitro chemosensitivity based on depth of invasion in advanced colorectal cancer using ATP-based chemotherapy response assay (ATP-CRA). Eur J Surg Oncol 35:951–956
Huh JW, Park YA, Lee KY, Sohn SK (2009) Heterogeneity of adenosine triphosphate-based chemotherapy response assay in colorectal cancer—secondary publication. Yonsei Med J 50:697–703
Selzner N, Selzner M, Graf R, Ungethuem U, Fitz JG, Clavien PA (2004) Water induces autocrine stimulation of tumor cell killing through ATP release and P2 receptor binding. Cell Death Differ 11(Suppl 2):S172–S180
Linden J (2006) Adenosine metabolism and cancer. Focus on "Adenosine downregulates DPPIV on HT-29 colon cancer cells by stimulating protein tyrosine phosphatases and reducing ERK1/2 activity via a novel pathway". Am J Physiol Cell Physiol 291:C405–C406
Lelièvre V, Muller JM, Falcòn J (1998) Adenosine modulates cell proliferation in human colonic adenocarcinoma. I. Possible involvement of adenosine A1 receptor subtypes in HT29 cells. Eur J Pharmacol 341:289–297
Lelièvre V, Muller JM, Falcòn J (1998) Adenosine modulates cell proliferation in human colonic carcinoma. II. Differential behavior of HT29, DLD-1, Caco-2 and SW403 cell lines. Eur J Pharmacol 341:299–308
Mujoomdar M, Hoskin D, Blay J (2003) Adenosine stimulation of the proliferation of colorectal carcinoma cell lines. Roles of cell density and adenosine metabolism. Biochem Pharmacol 66:1737–1747
Saito M, Yaguchi T, Yasuda Y, Nakano T, Nishizaki T (2010) Adenosine suppresses CW2 human colonic cancer growth by inducing apoptosis via A1 adenosine receptors. Cancer Lett 290:211–215
Ma DF, Kondo T, Nakazawa T, Niu DF, Mochizuki K, Kawasaki T, Yamane T, Katoh R (2010) Hypoxia-inducible adenosine A2B receptor modulates proliferation of colon carcinoma cells. Hum Pathol 41:1550–1557
Cheng Y, Yang J, Agarwal R, Green GM, Mease RC, Pomper MG, Meltzer SJ, Abraham JM (2011) Strong inhibition of xenografted tumor growth by low-level doses of [32P]ATP. Oncotarget 2:461–466
Carlson CC, Chinery R, Burnham LL, Dransfield DT (2000) 8-Cl-adenosine-induced inhibition of colorectal cancer growth in vitro and in vivo. Neoplasia 2:441–448
Ohana G, Bar-Yehuda S, Arich A, Madi L, Dreznick Z, Rath-Wolfson L, Silberman D, Slosman G, Fishman P (2003) Inhibition of primary colon carcinoma growth and liver metastasis by the A3 adenosine receptor agonist CF101. Br J Cancer 89:1552–1558
Fishman P, Bar-Yehuda S, Ohana G, Barer F, Ochaion A, Erlanger A, Madi L (2004) An agonist to the A3 adenosine receptor inhibits colon carcinoma growth in mice via modulation of GSK-3β and NF-κB. Oncogene 23:2465–2471
Gessi S, Merighi S, Varani K, Cattabriga E, Benini A, Mirandola P, Leung E, Mac Lennan S, Feo C, Baraldi S, Borea PA (2007) Adenosine receptors in colon carcinoma tissues and colon tumoral cell lines: focus on the A3 adenosine subtype. J Cell Physiol 211:826–836
Stemmer SM, Shani A, Klein B, Silverman MH, Lorber I, Farbstein M, Shmueli E, Figer A (2004) A phase II, multi-center study of a new non-cytotoxic A3 adenosine receptor agonist CF101, dose-finding (randomized blinded) in patients (pts) with refractory metastatic colorectal cancer. J Clin Oncol 22:232S
Gessi S, Cattabriga E, Avitabile A, Gafa’ R, Lanza G, Cavazzini L, Bianchi N, Gambari R, Feo C, Liboni A, Gullini S, Leung E, Mac-Lennan S, Borea PA (2004) Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clin Cancer Res 10:5895–5901
Giannecchini M, D’Innocenzo B, Pesi R, Sgarrella F, Iorio M, Collecchi P, Tozzi MG, Camici M (2003) 2′-Deoxyadenosine causes apoptotic cell death in a human colon carcinoma cell line. J Biochem Mol Toxicol 17:329–337
Yasuda Y, Saito M, Yamamura T, Yaguchi T, Nishizaki T (2009) Extracellular adenosine induces apoptosis in Caco-2 human colonic cancer cells by activating caspase-9/-3 via A2a adenosine receptors. J Gastroenterol 44:56–65
Richard CL, Tan EY, Blay J (2006) Adenosine upregulates CXCR4 and enhances the proliferative and migratory responses of human carcinoma cells to CXCL12/SDF-1α. Int J Cancer 119:2044–2053
Tan EY, Richard CL, Zhang H, Hoskin DW, Blay J (2006) Adenosine downregulates DPPIV on HT-29 colon cancer cells by stimulating protein tyrosine phosphatase(s) and reducing ERK1/2 activity via a novel pathway. Am J Physiol Cell Physiol 291:C433–C444
Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Simioni C, Leung E, Maclennan S, Baraldi PG, Borea PA (2007) Caffeine inhibits adenosine-induced accumulation of hypoxia-inducible factor-1α, vascular endothelial growth factor, and interleukin-8 expression in hypoxic human colon cancer cells. Mol Pharmacol 72:395–406
Hamada E, Imai Y, Hazama H, Takahashi M, Nakajima T, Ota S, Terano A, Omata M, Kurachi Y (1993) P2-purinergic receptor in human gastric signet ring cell carcinoma cell line: a patch clamp study. Gastroenterology 104:A829
Saitoh M, Nagai K, Nakagawa K, Yamamura T, Yamamoto S, Nishizaki T (2004) Adenosine induces apoptosis in the human gastric cancer cells via an intrinsic pathway relevant to activation of AMP-activated protein kinase. Biochem Pharmacol 67:2005–2011
Wang MX, Ren LM (2006) Growth inhibitory effect and apoptosis induced by extracellular ATP and adenosine on human gastric carcinoma cells: involvement of intracellular uptake of adenosine. Acta Pharmacol Sin 27:1085–1092
Park JY, Kim YS, Bang S, Hyung WJ, Noh SH, Choi SH, Song SY (2007) ATP-based chemotherapy response assay in patients with unresectable gastric cancer. Oncology 73:439–440
Lee JH, Kim MC, Oh SY, Kwon HC, Kim SH, Kwon KA, Lee S, Jeong JS, Choi SR, Kim HJ (2011) Predictive value of in vitro adenosine triphosphate-based chemotherapy response assay in advanced gastric cancer patients who received oral 5-fluorouracil after curative resection. Cancer Res Treat 43:117–123
Saha A, Hammond CE, Gooz M, Smolka AJ (2008) The role of Sp1 in IL-1β and H. pylori-mediated regulation of H,K-ATPase gene transcription. Am J Physiol Gastrointest Liver Physiol 295:G977–G986
Cornberg M, Schoefl C, Jandl O, Potthoff A, Mix H, Goeke M, Beil W, Manns MP, Wagner S (2000) Differential expression of the adenosine receptor subtypes in human gastric mucosa and cancer cells. Gastroenterology 118:A304
Ling ZQ, Qi CJ, Lu XX, Qian LJ, Gu LH, Zheng ZG, Zhao Q, Wang S, Fang XH, Yang ZX, Yin J, Mao WM (2012) Heterogeneity of chemosensitivity in esophageal cancer using ATP-tumor chemosensitivity assay. Acta Pharmacol Sin 33:401–406
Kalhan A, Kidd M, Modlin I, Pfragner R, Rees DA, Ham J (2009) Adenosine A2 receptor signalling mediates chromogranin A secretion from neuroendocrine tumours. Neuroendocrinology 90:119
Elsing C, Georgiev T, Hubner CA, Boger R, Stremmel W, Schlenker T (2012) Extracellular ATP induces cytoplasmic and nuclear Ca2+ transients via P2Y2 receptor in human biliary epithelial cancer cells (Mz-Cha-1). Anticancer Res 32:3759–3767
Clunes MT, Kemp PJ (1996) P2U purinoceptor modulation of intracellular Ca2+ in a human lung adenocarcinoma cell line: down-regulation of Ca2+ influx by protein kinase C. Cell Calcium 20:339–346
Remsbury A, Rakhit S, Wilson SM (1996) P2U receptor agonists do not inhibit forskolin-evoked cAMP accumulation in A549 lung adenocarcinoma cells. J Physiol 495:179P
Tatur S, Kreda S, Lazarowski E, Grygorczyk R (2008) Calcium-dependent release of adenosine and uridine nucleotides from A549 cells. Purinergic Signal 4:139–146
Zhao DM, Xue HH, Chida K, Suda T, Oki Y, Kanai M, Uchida C, Ichiyama A, Nakamura H (2000) Effect of erythromycin on ATP-induced intracellular calcium response in A549 cells. Am J Physiol Lung Cell Mol Physiol 278:L726–L736
Miki K, Tanaka H, Nagai Y, Kimura C, Oike M (2010) Transforming growth factor β1 alters calcium mobilizing properties and endogenous ATP release in A549 cells: possible implications for cell migration. J Pharmacol Sci 113:387–394
Agteresch HJ, Dagnelie PC, van der Gaast A, Stijnen T, Wilson JH (2000) Randomized clinical trial of adenosine 5′-triphosphate in patients with advanced non-small-cell lung cancer. J Natl Cancer Inst 92:321–328
Dagnelie PC, Agteresch HJ (2004) Promising effects of adenosine triphosphate infusion on nutritional status and quality of life in advanced non-small-cell lung cancer: a randomized clinical trial. Drug Dev Res 59:146–151
Leij-Halfwerk S, Agteresch HJ, Sijens PE, Dagnelie PC (2002) Adenosine triphosphate infusion increases liver energy status in advanced lung cancer patients: an in vivo 31P magnetic resonance spectroscopy study. Hepatology 35:421–424
Hatta Y, Takahashi M, Enomoto Y, Takahashi N, Sawada U, Horie T (2004) Adenosine triphosphate (ATP) enhances the antitumor effect of etoposide (VP16) in lung cancer cells. Oncol Rep 12:1139–1142
Lin CC, Lee IT, Wu WL, Lin WN, Yang CM (2012) Adenosine triphosphate regulates NADPH oxidase activity leading to hydrogen peroxide production and COX-2/PGE2 expression in A549 cells. Am J Physiol Lung Cell Mol Physiol 303:L401–L412
Schäfer R, Hartig R, Sedehizade F, Welte T, Reiser G (2006) Adenine nucleotides inhibit proliferation of the human lung adenocarcinoma cell line LXF-289 by activation of nuclear factor κB1 and mitogen-activated protein kinase pathways. FEBS J 273:3756–3767
Moon YW, Choi SH, Kim YT, Sohn JH, Chang J, Kim SK, Park MS, Chung KY, Lee HJ, Kim JH (2007) Adenosine triphosphate-based chemotherapy response assay (ATP-CRA)-guided platinum-based 2-drug chemotherapy for unresectable nonsmall-cell lung cancer. Cancer 109:1829–1835
Moon YW, Sohn JH, Kim YT, Chang H, Jeong JH, Lee YJ, Chang J, Kim SK, Jung M, Hong S, Choi SH, Kim JH (2009) Adenosine triphosphate-based chemotherapy response assay (ATP-CRA)-guided versus empirical chemotherapy in unresectable non-small cell lung cancer. Anticancer Res 29:4243–4249
Swennen ELR, Ummels V, Bast A, Dagnelie P (2008) Increased cytotoxicity of cisplatin in a human large cell lung carcinoma cell line by ATP. Purinergic Signalling 4:S207
Swennen EL, Ummels V, Buss I, Jaehde U, Bast A, Dagnelie PC (2010) ATP sensitizes H460 lung carcinoma cells to cisplatin-induced apoptosis. Chem Biol Interact 184:338–345
Chang HY, Huang HC, Huang TC, Yang PC, Wang YC, Juan HF (2012) Ectopic ATP synthase blockade suppresses lung adenocarcinoma growth by activating the unfolded protein response. Cancer Res 72:4696–4706
Ryzhov S, Novitskiy SV, Zaynagetdinov R, Goldstein AE, Carbone DP, Biaggioni I, Dikov MM, Feoktistov I (2008) Host A2B adenosine receptors promote carcinoma growth. Neoplasia 10:987–995
Zhang HY, Gu YY, Li ZG, Jia YH, Yuan L, Li SY, An GS, Ni JH, Jia HT (2004) Exposure of human lung cancer cells to 8-chloro-adenosine induces G2/M arrest and mitotic catastrophe. Neoplasia 6:802–812
Gu YY, Zhang HY, Zhang HJ, Li SY, Ni JH, Jia HT (2006) 8-Chloro-adenosine inhibits growth at least partly by interfering with actin polymerization in cultured human lung cancer cells. Biochem Pharmacol 72:541–550
Li WJ, Gu YY, Zhang HJ, Zhou J, Jia HT (2009) Induction of p14ARF by E2F1 contributes to 8-chloro-adenosine-induced apoptosis in human lung cancer H1299 cells. Chemotherapy 55:335–343
Nakamura K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M (2006) Antitumor effect of cordycepin (3′-deoxyadenosine) on mouse melanoma and lung carcinoma cells involves adenosine A3 receptor stimulation. Anticancer Res 26:43–47
Kim SJ, Min HY, Chung HJ, Park EJ, Hong JY, Kang YJ, Shin DH, Jeong LS, Lee SK (2008) Inhibition of cell proliferation through cell cycle arrest and apoptosis by thio-Cl-IB-MECA, a novel A3 adenosine receptor agonist, in human lung cancer cells. Cancer Lett 264:309–315
Kamiya H, Kanno T, Fujita Y, Gotoh A, Nakano T, Nishizaki T (2012) Apoptosis-related gene transcription in human A549 lung cancer cells via A3 adenosine receptor. Cell Physiol Biochem 29:687–696
Kanno T, Nakano T, Fujita Y, Gotoh A, Nishizaki T (2012) Adenosine induces apoptosis in SBC-3 human lung cancer cells through A3 adenosine receptor-dependent AMID upregulation. Cell Physiol Biochem 30:666–677
Otsuki T, Kanno T, Fujita Y, Tabata C, Fukuoka K, Nakano T, Gotoh A, Nishizaki T (2012) A3 adenosine receptor-mediated p53-dependent apoptosis in Lu-65 human lung cancer cells. Cell Physiol Biochem 30:210–220
Block GJ, DiMattia GD, Prockop DJ (2010) Stanniocalcin-1 regulates extracellular ATP-induced calcium waves in human epithelial cancer cells by stimulating ATP release from bystander cells. PLoS One 5:e10237
Zanini D, Schmatz R, Pimentel VC, Gutierres JM, Maldonado PA, Thomé GR, Cardoso AM, Stefanello N, Oliveira L, Chiesa J, Leal DB, Morsch VM, Schetinger MR (2012) Lung cancer alters the hydrolysis of nucleotides and nucleosides in platelets. Biomed Pharmacother 66:40–45
Inoue Y, Matsumoto H, Yamada S, Kawai K, Suemizu H, Gika M, Takanami I, Nakamura M, Iwazaki M (2010) ATP7B expression is associated with in vitro sensitivity to cisplatin in non-small cell lung cancer. Oncology Letters 1:279–282
He QF, Wang LW, Mao JW, Sun XR, Li P, Zhong P, Nie SH, Jacob T, Chen LX (2004) Activation of chloride current and decrease of cell volume by ATP in nasopharyngeal carcinoma cells. Sheng Li Xue Bao 56:691–696
Yang L, Ye D, Ye W, Jiao C, Zhu L, Mao J, Jacob TJ, Wang L, Chen L (2011) ClC-3 is a main component of background chloride channels activated under isotonic conditions by autocrine ATP in nasopharyngeal carcinoma cells. J Cell Physiol 226:2516–2526
Bear CE, Li CH (1991) Calcium-permeable channels in rat hepatoma cells are activated by extracellular nucleotides. Am J Physiol 261:C1018–C1024
Fitz JG, Sostman AH (1994) Nucleotide receptors activate cation, potassium, and chloride currents in a liver cell line. Am J Physiol 266:G544–G553
Wu Y, Sun X, Imai M, Sultan B, Csizmadia E, Enjyoji K, Jackson S, Usheva A, Robson SC (2005) Modulation of RAS/ERK signaling by CD39/ENTPD1 during liver regeneration. Hepatology 42:Absr. 1138
Peres A, Giovannardi S (1995) Characteristics of the signal transduction system activated by ATP receptors in the hepatoma cell line N1S1-67. Biochim Biophys Acta 1265:33–39
Geschwind JF, Ko YH, Torbenson MS, Magee C, Pedersen PL (2002) Novel therapy for liver cancer: direct intraarterial injection of a potent inhibitor of ATP production. Cancer Res 62:3909–3913
Dolovcak S, Waldrop SL, Fitz JG, Kilic G (2009) 5-Nitro-2-(3-phenylpropylamino)benzoic acid (NPPB) stimulates cellular ATP release through exocytosis of ATP-enriched vesicles. J Biol Chem 284:33894–33903
Speicher T, Foehrenbacher A, Pochic I, Weiland T, Wendel A (2010) Malignant but not naïve hepatocytes of human and rodent origin are killed by TNF after metabolic depletion of ATP by fructose. J Hepatol 53:896–902
Weiland T, Klein K, Zimmermann M, Speicher T, Venturelli S, Berger A, Bantel H, Königsrainer A, Schenk M, Weiss TS, Wendel A, Schwab M, Bitzer M, Lauer UM (2012) Selective protection of human liver tissue in TNF-targeting of cancers of the liver by transient depletion of adenosine triphosphate. PLoS One 7:e52496
Lu CC, Yang JS, Huang AC, Hsia TC, Chou ST, Kuo CL, Lu HF, Lee TH, Wood WG, Chung JG (2010) Chrysophanol induces necrosis through the production of ROS and alteration of ATP levels in J5 human liver cancer cells. Mol Nutr Food Res 54:967–976
Fujii T, Minagawa T, Shimizu T, Takeguchi N, Sakai H (2012) Inhibition of ecto-ATPase activity by curcumin in hepatocellular carcinoma HepG2 cells. J Physiol Sci 62:53–58
Espelt MV, Alberti GS, Chara O, Schwarzbaum PJ (2010) ATP induce ATP release in hepatoma cells. Purinergic Signal 6:S67
Frontini AV, De La Vega Elena CD, Nicolorich MV, Naves A, Schwarzbaum P, Venera GD (2011) In vivo effects of adenosine 5′-triphosphate on rat preneoplastic liver. Medicina (B Aires) 71:139–145
Hargrove JL, Granner DK (1982) Inhibition of hepatoma cell growth by analogs of adenosine and cyclic AMP and the influence of enzymes in mammalian sera. J Cell Physiol 111:232–238
Bar-Yehuda S, Stemmer SM, Madi L, Castel D, Ochaion A, Cohen S, Barer F, Zabutti A, Perez-Liz G, Del Valle L, Fishman P (2008) The A 3 adenosine receptor agonist CF102 induces apoptosis of hepatocellular carcinoma via de-regulation of the Wnt and NF-κB signal transduction pathways. Int J Oncol 33:287–295
Ohigashi T, Brookins J, Fisher JW (1993) Adenosine A 1 receptors and erythropoietin production. Am J Physiol 265:C934–C938
Wen LT, Knowles AF (2003) Extracellular ATP and adenosine induce cell apoptosis of human hepatoma Li-7A cells via the A3 adenosine receptor. Br J Pharmacol 140:1009–1018
Cohen S, Stemmer SM, Zozulya G, Ochaion A, Patoka R, Barer F, Bar-Yehuda S, Rath-Wolfson L, Jacobson KA, Fishman P (2011) CF102 an A 3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J Cell Physiol 226:2438–2447
Stemmer S, Silverman MH, Kerns WD, Bar-Yehuda S, Fishman S, Harpaz Z, Farbstein M, Binyaminov O, Medalia G, Fishman P (2010) Phase 1/2 trial of CF102, a selective A3 adenosine receptor (A3AR) agonist, in patients with hepatocellular carcinoma (HCC). Eur J Cancer 8:122
Xiang HJ, Liu ZC, Wang DS, Chen Y, Yang YL, Dou KF (2006) Adenosine A2b receptor is highly expressed in human hepatocellular carcinoma. Hepatol Res 36:56–60
Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Müller CE, Murakami T, Robson SC (2010) CD39/ENTPD1 expression by CD4+ Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 139:1030–1040
Sun X, Han L, Seth P, Bian S, Li L, Csizmadia E, Junger WG, Schmelzle M, Usheva A, Tapper EB, Baffy G, Sukhatme VP, Wu Y, Robson SC (2013) Disordered purinergic signaling and abnormal cellular metabolism are associated with development of liver cancer in Cd39/ENTPD1 null mice. Hepatology 57:205–216
Hur H, Kim NK, Kim HG, Min BS, Lee KY, Shin SJ, Cheon JH, Choi SH (2012) Adenosine triphosphate-based chemotherapy response assay-guided chemotherapy in unresectable colorectal liver metastasis. Br J Cancer 106:53–60
Borel F, Han R, Visser A, Petry H, van Deventer SJ, Jansen PL, Konstantinova P (2012) Adenosine triphosphate-binding cassette transporter genes up-regulation in untreated hepatocellular carcinoma is mediated by cellular microRNAs. Hepatology 55:821–832
Tzanakakis GN, Agarwal KC, Vezeridis MP (1993) Prevention of human pancreatic cancer cell-induced hepatic metastasis in nude mice by dipyridamole and its analog RA-233. Cancer 71:2466–2471
Verspohl EJ, Johannwille B, Waheed A, Neye H (2002) Effect of purinergic agonists and antagonists on insulin secretion from INS-1 cells (insulinoma cell line) and rat pancreatic islets. Can J Physiol Pharmacol 80:562–568
Szücs A, Demeter I, Burghardt B, Óvári G, Case RM, Steward MC, Varga G (2006) Vectorial bicarbonate transport by Capan-1 cells: a model for human pancreatic ductal secretion. Cell Physiol Biochem 18:253–264
Künzli BM, Berberat PO, Giese T, Csizmadia E, Kaczmarek E, Baker C, Halaceli I, Buchler MW, Friess H, Robson SC (2007) Upregulation of CD39/NTPDases and P2 receptors in human pancreatic disease. Am J Physiol Gastrointest Liver Physiol 292:G223–G230
Yamada T, Okajima F, Akbar M, Tomura H, Narita T, Yamada T, Ohwada S, Morishita Y, Kondo Y (2002) Cell cycle arrest and the induction of apoptosis in pancreatic cancer cells exposed to adenosine triphosphate in vitro. Oncol Rep 9:113–117
Chow JY, Shim KN, Ornelas TA, Carethers JM, Dong H (2009) Purinergic P2y2 receptor and Ca2+-mediated proliferation of human pancreatic epithelial cancer cells. Gastroenterology 136:A312
Kang CM, Kim H, Cho Y, Kim YS, Hwang HK, Choi HJ, Lee WJ (2012) In vitro adenosine triphosphate-based chemotherapy response assay (ATP-CRA) in solid pseudopapillary tumor of the pancreas. Pancreas 41:498–500
Kozlow W, Guise TA (2005) Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. J Mammary Gland Biol Neoplasia 10:169–180
Kakonen SM, Mundy GR (2003) Mechanisms of osteolytic bone metastases in breast carcinoma. Cancer 97:834–839
Uluçkan Ö, Eagleton MC, Floyd DH, Morgan EA, Hirbe AC, Kramer M, Dowland N, Prior JL, Piwnica-Worms D, Jeong SS, Chen R, Weilbaecher K (2008) APT102, a novel adpase, cooperates with aspirin to disrupt bone metastasis in mice. J Cell Biochem 104:1311–1323
Gouin-Thibault I, Achkar A, Samama MM (2001) The thrombophilic state in cancer patients. Acta Haematol 106:33–42
Boissier S, Magnetto S, Frappart L, Cuzin B, Ebetino FH, Delmas PD, Clezardin P (1997) Bisphosphonates inhibit prostate and breast carcinoma cell adhesion to unmineralized and mineralized bone extracellular matrices. Cancer Res 57:3890–3894
Mönkkönen H, Kuokkanen J, Holen I, Evans A, Lefley DV, Jauhiainen M, Auriola S, Mönkkönen J (2008) Bisphosphonate-induced ATP analog formation and its effect on inhibition of cancer cell growth. Anticancer Drugs 19:391–399
Korcok J, Raimundo LN, Ke HZ, Sims SM, Dixon SJ (2004) Extracellular nucleotides act through P2X7 receptors to activate NF-kappaB in osteoclasts. J Bone Miner Res 19:642–651
Leto G, Sepporta MV, Crescimanno M, Flandina C, Tumminello FM (2010) Cathepsin L in metastatic bone disease: therapeutic implications. Biol Chem 391:655–664
Shafat I, Vlodavsky I, Ilan N (2006) Characterization of mechanisms involved in secretion of active heparanase. J Biol Chem 281:23804–23811
Hoebertz A, Arnett TR, Burnstock G (2003) Regulation of bone resorption and formation by purines and pyrimidines. Trends Pharmacol Sci 24:290–297
Kumagai H, Sacktor B, Filburn CR (1991) Purinergic regulation of cytosolic calcium and phosphoinositide metabolism in rat osteoblast-like osteosarcoma cells. J Bone Miner Res 6:697–708
Reimer WJ, Dixon SJ (1992) Extracellular nucleotides elevate [Ca2+]i in rat osteoblastic cells by interaction with two receptor subtypes. Am J Physiol 263:C1040–C1048
Schöfl C, Cuthbertson KS, Walsh CA, Mayne C, Cobbold P, von zur Mühlen A, Hesch RD, Gallagher JA (1992) Evidence for P2-purinoceptors on human osteoblast-like cells. J Bone Miner Res 7:485–491
Bowler WB, Birch MA, Gallagher JA, Bilbe G (1995) Identification and cloning of human P2U purinoceptor present in osteoclastoma, bone, and osteoblasts. J Bone Miner Res 10:1137–1145
Katz S, Bilbao PS, Boland RL, Santillán G (2006) Modulation of intracellular calcium and MAPK activation by ATP in osteoblasts and breat cancer cells. J Bone Mineral Res 21:S390
Liu PS, Chen CY (2010) Butyl benzyl phthalate suppresses the ATP-induced cell proliferation in human osteosarcoma HOS cells. Toxicol Appl Pharmacol 244:308–314
Shah K, Patel V, Wang N, Gartland A (2010) The effect of ATP on cancer cell proliferation in vitro—involvement of multiple P2X receptors. Purinergic Signal 6:140
Krett NL, Davies KM, Ayres M, Ma C, Nabhan C, Gandhi V, Rosen ST (2004) 8-Amino-adenosine is a potential therapeutic agent for multiple myeloma. Mol Cancer Ther 3:1411–1420
Farrell AW, Gadeock S, Pupovac A, Wang B, Jalilian I, Ranson M, Sluyter R (2010) P2X7 receptor activation induces cell death and CD23 shedding in human RPMI 8226 multiple myeloma cells. Biochim Biophys Acta 1800:1173–1182
Rickles RJ, Tam WF, Giordano TP III, Pierce LT, Farwell M, McMillin DW, Necheva A, Crowe D, Chen M, Avery W, Kansra V, Nawrocki ST, Carew JS, Giles FJ, Mitsiades CS, Borisy AA, Anderson KC, Lee MS (2012) Adenosine A2A and beta-2 adrenergic receptor agonists: novel selective and synergistic multiple myeloma targets discovered through systematic combination screening. Mol Cancer Ther 11:1432–1442
Cervantes-Gomez F, Nimmanapalli R, Gandhi V (2011) ATP analog enhances the actions of a heat shock protein 90 inhibitor in multiple myeloma cells. J Pharmacol Exp Ther 339:545–554
Froio J, Abraham EH, Soni R, Epstein A, Okunieff P (1995) Effect of intraperitoneal ATP on tumor growth and bone marrow radiation tolerance. Acta Oncol 34:419–422
Chizhmakov I, Mamenko N, Volkova T, Khasabova I, Simone DA, Krishtal O (2009) P2X receptors in sensory neurons co-cultured with cancer cells exhibit a decrease in opioid sensitivity. Eur J Neurosci 29:76–86
Nejime N, Kagota S, Tada Y, Nakamura K, Hashimoto M, Kunitomo M, Shinozuka K (2009) Possible participation of chloride ion channels in ATP release from cancer cells in suspension. Clin Exp Pharmacol Physiol 36:278–282
Dubyak GR (2012) Function without form: an ongoing search for maxi-anion channel proteins. Focus on “Maxi-anion channel and pannexin 1 hemichannel constitute separate pathways for swelling-induced ATP release in murine L929 fibrosarcoma cells”. Am J Physiol Cell Physiol 303:C913–C915
Islam MR, Uramoto H, Okada T, Sabirov RZ, Okada Y (2012) Maxi-anion channel and pannexin 1 hemichannel constitute separate pathways for swelling-induced ATP release in murine L929 fibrosarcoma cells. Am J Physiol Cell Physiol 303:C924–C935
Hoferová Z, Hofer M, Pospíšil M, Znojil V, Chramostová K (2003) Effects of synthetic adenosine receptor agonosts on growth characteristics of murine G:5:113 fibrosarcoma cells in vitro. Drug Dev Res 60:303–311
Mercadante S (1997) Malignant bone pain: pathophysiology and treatment. Pain 69:1–18
Caraceni A, Portenoy RK (1999) An international survey of cancer pain characteristics and syndromes. IASP Task Force on Cancer Pain. International Association for the Study of Pain. Pain 82:263–274
Coleman R (1997) Management of bone metastases. Cancer Treat Rev 23(Suppl 1):S69–S75
Delaney A, Fleetwood-Walker SM, Colvin LA, Fallon M (2008) Translational medicine: cancer pain mechanisms and management. Br J Anaesth 101:87–94
Burnstock G (1996) A unifying purinergic hypothesis for the initiation of pain. Lancet 347:1604–1605
Wirkner K, Sperlagh B, Illes P (2007) P2X3 receptor involvement in pain states. Mol Neurobiol 36:165–183
Chen CC, Akopian AN, Sivilotti L, Colquhoun D, Burnstock G, Wood JN (1995) A P2X purinoceptor expressed by a subset of sensory neurons. Nature 377:428–431
Bradbury EJ, Burnstock G, McMahon SB (1998) The expression of P2X3 purinoceptors in sensory neurons: effects of axotomy and glial-derived neurotrophic factor. Mol Cell Neurosci 12:256–268
Gilchrist LS, Cain DM, Harding-Rose C, Kov AN, Wendelschafer-Crabb G, Kennedy WR, Simone DA (2005) Re-organization of P2X3 receptor localization on epidermal nerve fibers in a murine model of cancer pain. Brain Res 1044:197–205
Kakimoto S, Nagakura Y, Tamura S, Watabiki T, Shibasaki K, Tanaka S, Mori M, Sasamata M, Okada M (2008) Minodronic acid, a third-generation bisphosphonate, antagonizes purinergic P2X2/3 receptor function and exerts an analgesic effect in pain models. Eur J Pharmacol 589:98–101
Park HC, Seong J, An JH, Kim J, Kim UJ, Lee BW (2005) Alteration of cancer pain-related signals by radiation: proteomic analysis in an animal model with cancer bone invasion. Int J Radiat Oncol Biol Phys 61:1523–1534
Schöfl C, Rössig L, Mader T, Börger J, Pötter E, von zur Mühlen A, Brabant G (1997) Impairment of ATP-induced Ca2+-signalling in human thyroid cancer cells. Mol Cell Endocrinol 133:33–39
Pines A, Perrone L, Bivi N, Romanello M, Damante G, Gulisano M, Kelley MR, Quadrifoglio F, Tell G (2005) Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic Acids Res 33:4379–4394
Pines A, Bivi N, Vascotto C, Romanello M, D’Ambrosio C, Scaloni A, Damante G, Morisi R, Filetti S, Ferretti E, Quadrifoglio F, Tell G (2006) Nucleotide receptors stimulation by extracellular ATP controls Hsp90 expression through APE1/Ref-1 in thyroid cancer cells: a novel tumorigenic pathway. J Cell Physiol 209:44–55
Caraccio N, Monzani F, Santini E, Cuccato S, Ferrari D, Callegari MG, Gulinelli S, Pizzirani C, Di Virgilio F, Ferrannini E, Solini A (2005) Extracellular adenosine 5′-triphosphate modulates interleukin-6 production by human thyrocytes through functional purinergic P2 receptors. Endocrinology 146:3172–3178
Solini A, Cuccato S, Ferrari D, Santini E, Gulinelli S, Callegari MG, Dardano A, Faviana P, Madec S, Di Virgilio F, Monzani F (2008) Increased P2X7 receptor expression and function in thyroid papillary cancer: a new potential marker of the disease? Endocrinology 149:389–396
Dardano A, Falzoni S, Caraccio N, Polini A, Tognini S, Solini A, Berti P, Di Virgilio F, Monzani F (2009) 1513A>C polymorphism in the P2X7 receptor gene in patients with papillary thyroid cancer: correlation with histological variants and clinical parameters. J Clin Endocrinol Metab 94:695–698
Gu LQ, Li FY, Zhao L, Liu Y, Chu Q, Zang XX, Liu JM, Ning G, Zhao YJ (2010) Association of XIAP and P2X7 receptor expression with lymph node metastasis in papillary thyroid carcinoma. Endocrine 38:276–282
Lobo GP, Waite KA, Planchon SM, Romigh T, Houghton JA, Eng C (2008) ATP modulates PTEN subcellular localization in multiple cancer cell lines. Hum Mol Genet 17:2877–2885
Yang DM, Teng HC, Chen KH, Tsai ML, Lee TK, Chou YC, Chi CW, Chiou SH, Lee CH (2009) Clodronate-induced cell apoptosis in human thyroid carcinoma is mediated via the P2 receptor signaling pathway. J Pharmacol Exp Ther 330:613–623
Robinson-White AJ, Hsiao HP, Leitner WW, Greene E, Bauer A, Krett NL, Nesterova M, Stratakis CA (2008) Protein kinase A-independent inhibition of proliferation and induction of apoptosis in human thyroid cancer cells by 8-Cl-adenosine. J Clin Endocrinol Metab 93:1020–1029
Morello S, Petrella A, Festa M, Popolo A, Monaco M, Vuttariello E, Chiappetta G, Parente L, Pinto A (2008) Cl-IB-MECA inhibits human thyroid cancer cell proliferation independently of A3 adenosine receptor activation. Cancer Biol Ther 7:278–284
Kondo T, Nakazawa T, Kawasaki T, Mochizuki K, Niu DF, Yamane T, Katoh R (2011) Expression of adenosine receptors in thyroid carcenoma. Mod Pathol 24:137A
Ruzsnavszky O, Telek A, Gönczi M, Balogh A, Remenyik E, Csernoch L (2011) UV-B induced alteration in purinergic receptors and signaling on HaCaT keratinocytes. J Photochem Photobiol B 105:113–118
Klein E, Burgess GH, Bloch A, Milgrom H, Holtermann OA (1975) The effects of nucleoside analogs on cutaneous neoplasms. Ann N Y Acad Sci 255:216–224
Hosoi K, Edidin M (1989) Exogenous ATP and other nucleoside phosphates modulate epidermal growth factor receptors of A-431 epidermoid carcinoma cells. Proc Natl Acad Sci U S A 86:4510–4514
Gonzalez FA, Alfonzo RG, Toro JR, Heppel LA (1989) Receptor specific for certain nucleotides stimulates inositol phosphate metabolism and Ca2+ fluxes in A431 cells. J Cell Physiol 141:606–617
Hosoi K, Fujishita M, Sugita K, Kurihara K, Atsumi T, Murai T, Ueha T (1992) P2 purinergic receptors and cellular calcium metabolism in A 431 human epidermoid carcinoma cells. Am J Physiol 262:C635–C643
Sugita K, Kurihara K, Hosoi K, Atsumi T, Takahashi T, Kohno M, Ueha T (1994) Effects of pertussis toxin on signal transductions via P2-purinergic receptors in A-431 human epidermoidal carcinoma cells. Enzyme Protein 48:222–228
Correale P, Giuliano M, Tagliaferri P, Guarrasi R, Caraglia M, Marinetti MR, Iezzi T, Bianco AR, Procopio A (1995) Role of adenosine 5′ triphosphate in lymphokine activated (LAK) killing of human tumor cells. Res Commun Mol Pathol Pharmacol 87:67–69
Völkl T, Ogilvie A, Neuhuber W, Ogilvie A (2008) Cell death induced by uridine 5′-triphosphate (UTP) in contrast to adenosine 5′-triphosphate (ATP) in human epidermoid carcinoma cells (A-431). Cell Physiol Biochem 22:441–454
Fu W, McCormick T, Qi X, Luo L, Zhou L, Li X, Wang BC, Gibbons HE, Abdul-Karim FW, Gorodeski GI (2009) Activation of P2X7-mediated apoptosis inhibits DMBA/TPA-induced formation of skin papillomas and cancer in mice. BMC Cancer 9:114
Hsu WL, Tsai MH, Lin MW, Chiu YC, Lu JH, Chang CH, Yu HS, Yoshioka T (2012) Differential effects of arsenic on calcium signaling in primary keratinocytes and malignant (HSC-1) cells. Cell Calcium 52:161–169
Rai B, Kaur J, Jacobs R, Anand SC (2011) Adenosine deaminase in saliva as a diagnostic marker of squamous cell carcinoma of tongue. Clin Oral Investig 15:347–349
Kitagawa T, Amano F, Akamatsu Y (1988) External ATP-induced passive permeability change and cell lysis of cultured transformed cells: action in serum-containing growth media. Biochim Biophys Acta 941:257–263
Mure T, Sano K, Kitagawa T (1992) Modulation of membrane permeability, cell proliferation and cytotoxicity of antitumor agents by external ATP in mouse tumor cells. Jpn J Cancer Res 83:121–126
Kuhnle GE, Dellian M, Walenta S, Mueller-Klieser W, Goetz AE (1992) Simultaneous high-resolution measurement of adenosine triphosphate levels and blood flow in the hamster amelanotic melanoma A-Mel-3. J Natl Cancer Inst 84:1642–1647
Walenta S, Dellian M, Goetz AE, Kuhnle GE, Mueller-Klieser W (1992) Pixel-to-pixel correlation between images of absolute ATP concentrations and blood flow in tumours. Br J Cancer 66:1099–1102
Dzhandzhugazyan KN, Kirkin AF, P t S, Zeuthen J (1998) Ecto-ATP diphosphohydrolase/CD39 is overexpressed in differentiated human melanomas. FEBS Lett 430:227–230
Palomares T, Bilbao P, del Olmo M, Castro B, Calle Y, Alonso-Varona A (1999) In vitro and in vivo comparison between the effects of treatment with adenosine triphosphate and treatment with buthionine sulfoximine on chemosensitization and tumour growth of B16 melanoma. Melanoma Res 9:233–242
Slater M, Scolyer RA, Gidley-Baird A, Thompson JF, Barden JA (2003) Increased expression of apoptotic markers in melanoma. Melanoma Res 13:137–145
Tsukimoto M, Ohshima Y, Harada H, Takenouchi T, Sato M, Suzuki A, Kitani H, Kojima S (2008) Over-expression of P2X7 receptor enhances tumor growth in vivo, but not in vitro. Purinergic Signalling 4:S150–S151
Hattori F, Ohshima Y, Seki S, Tsukimoto M, Sato M, Takenouchi T, Suzuki A, Takai E, Kitani H, Harada H, Kojima S (2012) Feasibility study of B16 melanoma therapy using oxidized ATP to target purinergic receptor P2X7. Eur J Pharmacol 695:20–26
Deli T, Varga N, Ádám A, Kenessey I, Ráasó E, Puskás LG, Tóovári J, Fodor J, Fehér M, Szigeti GP, Csernoch L, Tímár J (2007) Functional genomics of calcium channels in human melanoma cells. Int J Cancer 121:55–65
Kretz M, Ring S, Mahnke K (2009) ATP release by B16 melanomas upregulate the expression of the ectonucleotidase CD39 on Treg. J Invest Dermatol 129:S4
Ring S, Enk A, Mahnke K (2011) A role for adenosine triphosphate in regulating immune responses during melanoma growth. J Invest Dermatol 131:S91
Fishman P, Bar-Yehuda S (1998) Extracellular adenosine acts as a chemoprotective agent. Proc Am Assoc Cancer Res 39:3196
Woodhouse EC, Amanatullah DF, Schetz JA, Liotta LA, Stracke ML, Clair T (1998) Adenosine receptor mediates motility in human melanoma cells. Biochem Biophys Res Commun 246:888–894
Merighi S, Varani K, Gessi S, Cattabriga E, Iannotta V, Ulouglu C, Leung E, Borea PA (2001) Pharmacological and biochemical characterization of adenosine receptors in the human malignant melanoma A375 cell line. Br J Pharmacol 134:1215–1226
Merighi S, Mirandola P, Milani D, Varani K, Gessi S, Klotz KN, Leung E, Baraldi PG, Borea PA (2002) Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J Invest Dermatol 119:923–933
Madi L, Bar-Yehuda S, Barer F, Ardon E, Ochaion A, Fishman P (2003) A3 adenosine receptor activation in melanoma cells: association between receptor fate and tumor growth inhibition. J Biol Chem 278:42121–42130
Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Baraldi PG, Borea PA (2005) A3 adenosine receptors modulate hypoxia-inducible factor-1α expression in human A375 melanoma cells. Neoplasia 7:894–903
Morello S, Sorrentino R, Montinaro A, Luciano A, Maiolino P, Ngkelo A, Arra C, Adcock IM, Pinto A (2011) NK1.1+ cells and CD8+ T cells mediate the antitumor activity of Cl-IB-MECA in a mouse melanoma model. Neoplasia 13:365–373
Soares AF, Diniz C, Fresco P (2012) A3-adenosine receptor effects on malignant melanoma cells. FEBS J 279:547
Merighi S, Baraldi PG, Gessi S, Iannotta V, Klotz JN, Leung E, Mirandola P, Tabrizi MA, Varani K, Borea PA (2003) Adenosine receptors and human melanoma. Drug Dev Res 58:377–385
Merighi S, Simioni C, Gessi S, Varani K, Mirandola P, Tabrizi MA, Baraldi PG, Borea PA (2009) A2B and A3 adenosine receptors modulate vascular endothelial growth factor and interleukin-8 expression in human melanoma cells treated with etoposide and doxorubicin. Neoplasia 11:1064–1073
Raskovalova T, Lokshin A, Huang X, Su Y, Mandic M, Zarour HM, Jackson EK, Gorelik E (2007) Inhibition of cytokine production and cytotoxic activity of human antimelanoma specific CD8+ and CD4+ T lymphocytes by adenosine-protein kinase A type I signaling. Cancer Res 67:5949–5956
Urunsak IF, Gulec UK, Paydas S, Seydaoglu G, Guzel AB, Vardar MA (2012) Adenosine deaminase activity in patients with ovarian neoplasms. Arch Gynecol Obstet 286:155–159
Aiton JF, Lamb JF (1980) The effect of exogenous adenosine triphosphate on potassium movements in HeLa cells. Q J Exp Physiol Cogn Med Sci 65:47–62
Smit MJ, Leurs R, Bloemers SM, Tertoolen LG, Bast A, De Laat SW, Timmerman H (1993) Extracellular ATP elevates cytoplasmatic free Ca2+ in HeLa cells by the interaction with a 5′-nucleotide receptor. Eur J Pharmacol 247:223–226
Muscella A, Elia MG, Greco S, Storelli C, Marsigliante S (2003) Activation of P2Y2 purinoceptor inhibits the activity of the Na+/K+-ATPase in HeLa cells. Cell Signal 15:115–121
Okuda A, Furuya K, Kiyohara T (2003) ATP-induced calcium oscillations and change of P2Y subtypes with culture conditions in HeLa cells. Cell Biochem Funct 21:61–68
Muscella A, Greco S, Elia MG, Storelli C, Marsigliante S (2004) Differential signalling of purinoceptors in HeLa cells through the extracellular signal-regulated kinase and protein kinase C pathways. J Cell Physiol 200:428–439
Feng YH, Li X, Wang L, Zhou L, Gorodeski GI (2006) A truncated P2X7 receptor variant (P2X7-j) endogenously expressed in cervical cancer cells antagonizes the full-length P2X7 receptor through hetero-oligomerization. J Biol Chem 281:17228–17237
Lee SG, Choi JK, Choi BH, Lim Y, Kim YH, Lee KH, Shin JC, Ahn WS (2006) The effect of adenosine 5′-triphosphate on calcium mobilization and cell proliferation in cervical cancer cells. Eur J Obstet Gynecol Reprod Biol 127:110–114
Tam KF, Ng TY, Liu SS, Tsang PC, Kwong PW, Ngan HY (2005) Potential application of the ATP cell viability assay in the measurement of intrinsic radiosensitivity in cervical cancer. Gynecol Oncol 96:765–770
Buvinic S, Bravo-Zehnder M, Boyer JL, Huidobro-Toro JP, González A (2007) Nucleotide P2Y1 receptor regulates EGF receptor mitogenic signaling and expression in epithelial cells. J Cell Sci 120:4289–4301
Hopfe M, Henrich B (2008) OppA, the ecto-ATPase of Mycoplasma hominis induces ATP release and cell death in HeLa cells. BMC Microbiol 8:55
Liang WG, Su CC, Nian JH, Chiang AS, Li SY, Yang JJ (2011) Human connexin30.2/31.3 (GJC3) does not form functional gap junction channels but causes enhanced ATP release in HeLa cells. Cell Biochem Biophys 61:189–197
Huidobro-Toro JP, Lopez J, Hinojosa A, Buvinic S, Gonzalez A (2010) ATP released and autocrine signalling following gentle pipetting of HeLa cell medium. Purinergic Signal 6:S70
Maldonado PA, Pimentel VC, Negrini LA, Morsch VM, Schetinger MR (2012) Role of the purinergic system in patients with cervical intraepithelial neoplasia and uterine cancer. Biomed Pharmacother 66:6–11
Popper LD, Batra S (1993) Calcium mobilization and cell proliferation activated by extracellular ATP in human ovarian tumour cells. Cell Calcium 14:209–218
Batra S, Fadeel I (1994) Release of intracellular calcium and stimulation of cell growth by ATP and histamine in human ovarian cancer cells (SKOV-3). Cancer Lett 77:57–63
Maymon R, Bar-Shira MB, Cohen-Armon M, Holtzinger M, Leibovici J (1994) Enhancing effect of ATP on intracellular adriamycin penetration in human ovarian cancer cell lines. Biochim Biophys Acta 1201:173–178
Schultze-Mosgau A, Katzur AC, Arora KK, Stojilkovic SS, Diedrich K, Ortmann O (2000) Characterization of calcium-mobilizing, purinergic P2Y2 receptors in human ovarian cancer cells. Mol Hum Reprod 6:435–442
Konecny G, Crohns C, Pegram M, Felber M, Lude S, Kurbacher C, Cree IA, Hepp H, Untch M (2000) Correlation of drug response with the ATP tumorchemosensitivity assay in primary FIGO stage III ovarian cancer. Gynecol Oncol 77:258–263
Ng TY, Ngan HY, Cheng DK, Wong LC (2000) Clinical applicability of the ATP cell viability assay as a predictor of chemoresponse in platinum-resistant epithelial ovarian cancer using nonsurgical tumor cell samples. Gynecol Oncol 76:405–408
O’Meara AT, Sevin BU (2001) Predictive value of the ATP chemosensitivity assay in epithelial ovarian cancer. Gynecol Oncol 83:334–342
Cree IA, Kurbacher CM, Lamont A, Hindley AC, Love S (2007) A prospective randomized controlled trial of tumour chemosensitivity assay directed chemotherapy versus physician’s choice in patients with recurrent platinum-resistant ovarian cancer. Anticancer Drugs 18:1093–1101
Neubauer H, Stefanova M, Solomayer E, Meisner C, Zwirner M, Wallwiener D, Fehm T (2008) Predicting resistance to platinum-containing chemotherapy with the ATP tumor chemosensitivity assay in primary ovarian cancer. Anticancer Res 28:949–955
Glaysher S, Gabriel FG, Johnson P, Polak M, Knight LA, Parker K, Poole M, Narayanan A, Cree IA (2010) Molecular basis of chemosensitivity of platinum pre-treated ovarian cancer to chemotherapy. Br J Cancer 103:656–662
Knight LA, Kurbacher CM, Glaysher S, Fernando A, Reichelt R, Dexel S, Reinhold U, Cree IA (2009) Activity of mevalonate pathway inhibitors against breast and ovarian cancers in the ATP-based tumour chemosensitivity assay. BMC Cancer 9:38
Barcz E, Sommer E, Janik P, Marianowski L, Skopinska-Rozewska E (2000) Adenosine receptor antagonism causes inhibition of angiogenic activity of human ovarian cancer cells. Oncol Rep 7:1285–1291
Song H, Ramus SJ, Shadforth D, Quaye L, Kjaer SK, Dicioccio RA, Dunning AM, Hogdall E, Hogdall C, Whittemore AS, McGuire V, Lesueur F, Easton DF, Jacobs IJ, Ponder BA, Gayther SA, Pharoah PD (2006) Common variants in RB1 gene and risk of invasive ovarian cancer. Cancer Res 66:10220–10226
Rotte A, Garmann D, Buss I, Jaehde U (2010) Effect of extracellular ATP on cisplatin-induced cytotoxicity in human ovarian carcinoma cells. Chemotherapy 56:1–8
Katzur AC, Koshimizu T, Tomic M, Schultze-Mosgau A, Ortmann O, Stojilkovic SS (1999) Expression and responsiveness of P2Y2 receptors in human endometrial cancer cell lines. J Clin Endocrinol Metab 84:4085–4091
Aida T, Takebayashi Y, Shimizu T, Okamura C, Higasimoto M, Kanzaki A, Nakayama K, Terada K, Sugiyama T, Miyazaki K, Ito K, Takenoshita S, Yaegashi N (2005) Expression of copper-transporting P-type adenosine triphosphatase (ATP7B) as a prognostic factor in human endometrial carcinoma. Gynecol Oncol 97:41–45
Li X, Zhou L, Feng YH, Abdul-Karim FW, Gorodeski GI (2006) The P2X7 receptor: a novel biomarker of uterine epithelial cancers. Cancer Epidemiol Biomarkers Prev 15:1906–1913
Li X, Qi X, Zhou L, Catera D, Rote NS, Potashkin J, Abdul-Karim FW, Gorodeski GI (2007) Decreased expression of P2X7 in endometrial epithelial pre-cancerous and cancer cells. Gynecol Oncol 106:233–243
Zhou L, Qi X, Potashkin JA, Abdul-Karim FW, Gorodeski GI (2008) MicroRNAs miR-186 and miR-150 down-regulate expression of the pro-apoptotic purinergic P2X7 receptor by activation of instability sites at the 3′-untranslated region of the gene that decrease steady-state levels of the transcript. J Biol Chem 283:28274–28286
Tam KF, Ng TY, Tsang PC, Li CF, Ngan HY (2006) Potential use of the adenosine triphosphate cell viability assay in endometrial cancer. J Soc Gynecol Investig 13:518–522
Boabang P, Kurbacher CM, Kohlhagen H, Waida A, Amo-Takyi BK (2000) Anti-neoplastic activity of topotecan versus cisplatin, etoposide and paclitaxel in four squamous cell cancer cell lines of the female genital tract using an ATP-tumor chemosensitivity assay. Anticancer Drugs 11:843–848
Acosta Maldonado P, de Carvalho CM, Vargas Becker L, Flores C, Beatriz Moretto M, Morsch V, Chitolina Schetinger MR (2008) Ectonucleotide pyrophosphatase/phosphodiesterase (E-NPP) and adenosine deaminase (ADA) activities in patients with uterine cervix neoplasia. Clin Biochem 41:400–406
Sylwestrowicz TA, Ma DD, Murphy PP, Massaia M, Prentice HG, Hoffbrand AV, Greaves MF (1982) 5′nucleotidase, adenosine deaminase and purine nucleoside phosphorylase activities in acute leukaemia. Leuk Res 6:475–482
Seifert R, Burde R, Schultz G (1989) Activation of NADPH oxidase by purine and pyrimidine nucleotides involves G proteins and is potentiated by chemotactic peptides. Biochem J 259:813–819
Nonotte I, Mathieu MN, Chevillard C (1989) P2-purinergic-induced ionised calcium elevation in the human promyelocytic cell line HL60. Brit J Pharmacol 96:238P
Lee H, Suh BC, Kim KT (1997) Feedback regulation of ATP-induced Ca2+ signaling in HL-60 cells is mediated by protein kinase A- and C-mediated changes in capacitative Ca2+ entry. J Biol Chem 272:21831–21838
Cowen DS, Sanders M, Dubyak G (1990) P2-purinergic receptors activate a guanine nucleotide-dependent phospholipase C in membranes from HL-60 cells. Biochim Biophys Acta 1053:195–203
Cowen DS, Baker B, Dubyak GR (1990) Pertussis toxin produces differential inhibitory effects on basal, P2-purinergic, and chemotactic peptide-stimulated inositol phospholipid breakdown in HL-60 cells and HL-60 cell membranes. J Biol Chem 265:16181–16189
Cowen DS, Berger M, Nuttle L, Dubyak GR (1991) Chronic treatment with P2-purinergic receptor agonists induces phenotypic modulation of the HL-60 and U937 human myelogenous leukemia cell lines. J Leukoc Biol 50:109–122
Ohana G, Bar-Yehuda S, Barer F, Fishman P (2001) Differential effect of adenosine on tumor and normal cell growth: focus on the A3 adenosine receptor. J Cell Physiol 186:19–23
Montero M, Garcia-Sancho J, Alvarez J (1995) Biphasic and differential modulation of Ca2+ entry by ATP and UTP in promyelocytic leukaemia HL60 cells. Biochem J 305:879–887
Parker AL, Likar LL, Dawicki DD, Rounds S (1996) Mechanism of ATP-induced leukocyte adherence to cultured pulmonary artery endothelial cells. Am J Physiol 270:L695–L703
Choi SY, Kim KT (1997) Extracellular ATP-stimulated increase of cytosolic cAMP in HL-60 cells. Biochem Pharmacol 53:429–432
Jiang L, Foster FM, Ward P, Tasevski V, Luttrell BM, Conigrave AD (1997) Extracellular ATP triggers cyclic AMP-dependent differentiation of HL-60 cells. Biochem Biophys Res Commun 232:626–630
Song SK, Suh BC, Lee H, Kim KT (1997) Histamine inhibits ATP-induced [Ca2+]i rise through the activation of protein kinase A in HL-60 cells. Eur J Pharmacol 322:265–273
Conigrave AD, van der Weyden L, Holt L, Jiang L, Wilson P, Christopherson RI, Morris MB (2000) Extracellular ATP-dependent suppression of proliferation and induction of differentiation of human HL-60 leukemia cells by distinct mechanisms. Biochem Pharmacol 60:1585–1591
van der Weyden L, Rakyan V, Luttrell BM, Morris MB, Conigrave AD (2000) Extracellular ATP couples to cAMP generation and granulocytic differentiation in human NB4 promyelocytic leukaemia cells. Immunol Cell Biol 78:467–473
Adrian K, Bernhard MK, Breitinger HG, Ogilvie A (2000) Expression of purinergic receptors (ionotropic P2X1-7 and metabotropic P2Y1-11) during myeloid differentiation of HL60 cells. Biochim Biophys Acta 1492:127–138
Communi D, Janssens R, Robaye B, Zeelis N, Boeynaems JM (2000) Rapid up-regulation of P2Y messengers during granulocytic differentiation of HL-60 cells. FEBS Lett 475:39–42
Yoon MJ, Lee HJ, Kim JH, Kim DK (2006) Extracellular ATP induces apoptotic signaling in human monocyte leukemic cells, HL-60 and F-36P. Arch Pharm Res 29:1032–1041
Belton M, Commerford K, Hall J, Prato FS, Carson JJ (2008) Real-time measurement of cytosolic free calcium concentration in HL-60 cells during static magnetic field exposure and activation by ATP. Bioelectromagnetics 29:439–446
Rozanski C, Belton M, Prato FS, Carson JJ (2009) Real-time measurement of cytosolic free calcium concentration in DEM-treated HL-60 cells during static magnetic field exposure and activation by ATP. Bioelectromagnetics 30:213–221
Lee EJ, Min HY, Chung HJ, Park EJ, Shin DH, Jeong LS, Lee SK (2005) A novel adenosine analog, thio-Cl-IB-MECA, induces G0/G1 cell cycle arrest and apoptosis in human promyelocytic leukemia HL-60 cells. Biochem Pharmacol 70:918–924
Notarbartolo M, Lo CS, Meli M, Poma P, Labbozzetta M, Cervello M, D’Alessandro N (2005) Induction of apoptosis by the adenosine derivative IB-MECA in parental or multidrug-resistant HL-60 leukemia cells: possible relationship to the effects on inhibitor of apoptosis protein levels. Chemotherapy 51:272–279
Biffen M, Alexander DR (1994) Mobilization of intracellular Ca2+ by adenine nucleotides in human T-leukaemia cells: evidence for ADP-specific and P2y-purinergic receptors. Biochem J 304:769–774
Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS (2010) Pannexin 1 channels mediate 'find-me' signal release and membrane permeability during apoptosis. Nature 467:863–867
Adinolfi E, Melchiorri L, Falzoni S, Chiozzi P, Morelli A, Tieghi A, Cuneo A, Castoldi G, Di Virgilio F, Baricordi OR (2002) P2X7 receptor expression in evolutive and indolent forms of chronic B lymphocytic leukemia. Blood 99:706–708
Cabrini G, Falzoni S, Forchap SL, Pellegatti P, Balboni A, Agostini P, Cuneo A, Castoldi G, Baricordi OR, Di Virgilio F (2005) A His-155 to Tyr polymorphism confers gain-of-function to the human P2X7 receptor of human leukemic lymphocytes. J Immunol 175:82–89
Wiley JS, Dao-Ung LP, Gu BJ, Sluyter R, Shemon AN, Li C, Taper J, Gallo J, Manoharan A (2002) A loss-of-function polymorphic mutation in the cytolytic P2X7 receptor gene and chronic lymphocytic leukaemia: a molecular study. Lancet 359:1114–1119
Dao-Ung LP, Fuller SJ, Sluyter R, SkarRatt KK, Thunberg U, Tobin G, Byth K, Ban M, Rosenquist R, Stewart GJ, Wiley JS (2004) Association of the 1513C polymorphism in the P2X7 gene with familial forms of chronic lymphocytic leukaemia. Br J Haematol 125:815–817
Zhang XJ, Zheng GG, Ma XT, Yang YH, Li G, Rao Q, Nie K, Wu KF (2004) Expression of P2X7 in human hematopoietic cell lines and leukemia patients. Leuk Res 28:1313–1322
Zhang X, Meng L, He B, Chen J, Liu P, Zhao J, Zhang Y, Li M, An D (2009) The role of P2X7 receptor in ATP-mediated human leukemia cell death: calcium influx-independent. Acta Biochim Biophys Sin (Shanghai) 41:362–369
Chong JH, Zheng GG, Zhu XF, Guo Y, Wang L, Ma CH, Liu SY, Xu LL, Lin YM, Wu KF (2010) Abnormal expression of P2X family receptors in Chinese pediatric acute leukemias. Biochem Biophys Res Commun 391:498–504
Cass CE, Selner M, Tan TH, Muhs WH, Robins MJ (1982) Comparison of the effects on cultured L1210 leukemia cells of the ribosyl, 2′-deoxyribosyl, and xylosyl homologs of tubercidin and adenosine alone or in combination with 2′-deoxycoformycin. Cancer Treat Rep 66:317–326
Schneider C, Wiendl H, Ogilvie A (2001) Biphasic cytotoxic mechanism of extracellular ATP on U-937 human histiocytic leukemia cells: involvement of adenosine generation. Biochim Biophys Acta 1538:190–205
Gessi S, Varani K, Merighi S, Morelli A, Ferrari D, Leung E, Baraldi PG, Spalluto G, Borea PA (2001) Pharmacological and biochemical characterization of A3 adenosine receptors in Jurkat T cells. Br J Pharmacol 134:116–126
Batiuk TD, Schnizlein-Bick C, Plotkin Z, Dagher PC (2001) Guanine nucleosides and Jurkat cell death: roles of ATP depletion and accumulation of deoxyribonucleotides. Am J Physiol Cell Physiol 281:C1776–C1784
Bastin-Coyette L, Smal C, Cardoen S, Saussoy P, Van den Neste E, Bontemps F (2008) Mechanisms of cell death induced by 2-chloroadenosine in leukemic B-cells. Biochem Pharmacol 75:1451–1460
Streitová D, Weiterová L, Hofer M, Holá J, Horváth V, Kozubík A, Znojil V (2007) Effect of adenosine on the growth of human T-lymphocyte leukemia cell line MOLT-4. Cancer Invest 25:419–426
Abbracchio MP, Paoletti AM, Luini A, Cattabeni F, De Matteis MA (1992) Adenosine receptors in rat basophilic leukaemia cells: transductional mechanisms and effects on 5-hydroxytryptamine release. Br J Pharmacol 105:405–411
Hilchey SP, Kobie JJ, Cochran MR, Secor-Socha S, Wang JC, Hyrien O, Burack WR, Mosmann TR, Quataert SA, Bernstein SH (2009) Human follicular lymphoma CD39+-infiltrating T cells contribute to adenosine-mediated T cell hyporesponsiveness. J Immunol 183:6157–6166
Perry C, Hazan-Halevy I, Kay S, Cipok M, Grisaru D, Deutsch V, Polliack A, Naparstek E, Herishanu Y (2012) Increased CD39 expression on CD4+ T lymphocytes has clinical and prognostic significance in chronic lymphocytic leukemia. Ann Hematol 91:1271–1279
Serra S, Horenstein AL, Vaisitti T, Brusa D, Rossi D, Laurenti L, D’Arena G, Coscia M, Tripodo C, Inghirami G, Robson SC, Gaidano G, Malavasi F, Deaglio S (2011) CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood 118:6141–6152
Dennison JB, Balakrishnan K, Gandhi V (2009) Preclinical activity of 8-chloroadenosine with mantle cell lymphoma: roles of energy depletion and inhibition of DNA and RNA synthesis. Br J Haematol 147:297–307
Robak T, Robak P (2012) Purine nucleoside analogs in the treatment of rarer chronic lymphoid leukemias. Curr Pharm Des 18:3373–3388
Fishman P, Bar-Yehuda S, Ohana G, Pathak S, Wasserman L, Barer F, Multani AS (2000) Adenosine acts as an inhibitor of lymphoma cell growth: a major role for the A3 adenosine receptor. Eur J Cancer 36:1452–1458
Chechik BE, Schrader WP, Perets A, Fernandes B (1984) Immunohistochemical localization of adenosine deaminase in human benign extrathymic lymphoid tissues and B-cell lymphomas. Cancer 53:70–78
Wiley JS, Dubyak GR (1989) Extracellular adenosine triphosphate increases cation permeability of chronic lymphocytic leukemic lymphocytes. Blood 73:1316–1323
Anghileri LJ, Plenat F, Thouvenot P (2001) Role of iron in lymphoma-induction by ATP. Oncol Rep 8:1153–1157
Kruczynski A, Mayer P, Marchand A, Vispé S, Fournier E, Annereau JP, Brel V, Barret JM, Delsol G, Imbert T, Fahy J, Bailly C (2009) Antitumor activity of pyridoisoquinoline derivatives F91873 and F91874, novel multikinase inhibitors with activity against the anaplastic lymphoma kinase. Anticancer Drugs 20:364–372
Ren S, Zhang Y, Wang Y, Lui Y, Wei W, Huang X, Mao W, Zuo Y (2010) Targeting P2X7 receptor inhibits the metastasis of murine P388D1 lymphoid neoplasm cells to lymph nodes. Cell Biol Int 34:1205–1211
Nader S, Koch-Nolte F, Haag F (2012) Activation of P2X7 causes depletion of intracellular ATP in T lymphoma cells. Purinergic Signal 8:155
Becher J, Nader S, Nicola J, Danquah W, Koch-Nolte F, Haag F (2012) Regulated release of ATP by Yac lymphoma cells. Immunology 137:393
Murgo AJ, Sistare FD (1992) K562 leukemia cells express P2T (adenosine diphosphate) purinergic receptors. J Pharmacol Exp Ther 261:580–585
Bernardo AA, Pinto-Silva FE, Persechini PM, Coutinho-Silva R, Meyer-Fernandes JR, de Souza AL, Rumjanek VM (2006) Effect of extracellular ATP on the human leukaemic cell line K562 and its multidrug counterpart. Mol Cell Biochem 289:111–124
de Rijke B, van Horssen-Zoetbrood A, Beekman JM, Otterud B, Maas F, Woestenenk R, Kester M, Leppert M, Schattenberg AV, de Witte T, van de Wiel-van KE, Dolstra H (2005) A frameshift polymorphism in P2X5 elicits an allogeneic cytotoxic T lymphocyte response associated with remission of chronic myeloid leukemia. J Clin Invest 115:3506–3516
Yamaguchi M, Hirayoshi K, Okuma M, Nagata K (1994) Enhancement of differentiation induction of mouse myelomonocytic leukemic cells by extracellular ATP. J Cell Physiol 159:441–449
Wang W, Xiao J, Adachi M, Liu Z, Zhou J (2011) 4-aminopyridine induces apoptosis of human acute myeloid leukemia cells via increasing [Ca2+]i through P2X7 receptor pathway. Cell Physiol Biochem 28:199–208
Wang B, Sluyter R (2013) P2X7 receptor activation induces reactive oxygen species formation in erythroid cells. Purinergic Signal 9:101–112
Gadeock S, Pupovac A, Sluyter V, Spildrejorde M, Sluyter R (2012) P2X7 receptor activation mediates organic cation uptake into human myeloid leukaemic KG-1 cells. Purinergic Signal 8:669–676
Niitsu N, Honma Y (1999) Adenosine analogs as possible differentiation-inducing agents against acute myeloid leukemia. Leuk Lymphoma 34:261–271
Chakrabarti A, Gupta K, Sharma JP, Yang J, Agarwal A, Glick A, Zhang Y, Agarwal M, Agarwal MK, Wald DN (2012) ATP depletion triggers acute myeloid leukemia differentiation through an ATR/Chk1 protein-dependent and p53 protein-independent pathway. J Biol Chem 287:23635–23643
Salvestrini V, Zini R, Rossi L, Gulinelli S, Manfredini R, Bianchi E, Piacibello W, Caione L, Migliardi G, Ricciardi MR, Tafuri A, Romano M, Salati S, Di Virgilio F, Ferrari S, Baccarani M, Ferrari D, Lemoli RM (2012) Purinergic signaling inhibits human acute myeloblastic leukemia cell proliferation, migration, and engraftment in immunodeficient mice. Blood 119:217–226
Racil Z, Razga F, Polakova KM, Buresova L, Polivkova V, Dvorakova D, Zackova D, Klamova H, Cetkovsky P, Mayer J (2011) Assessment of adenosine triphosphate-binding cassette subfamily B member 1 (ABCB1) mRNA expression in patients with de novo chronic myelogenous leukemia: the role of different cell types. Leuk Lymphoma 52:331–334
Constantinescu P, Wang B, Kovacevic K, Jalilian I, Bosman GJ, Wiley JS, Sluyter R (2010) P2X7 receptor activation induces cell death and microparticle release in murine erythroleukemia cells. Biochim Biophys Acta 1798:1797–1804
Schwaner I, Seifert R, Schultz G (1992) Receptor-mediated increases in cytosolic Ca2+ in the human erythroleukaemia cell line involve pertussis toxin-sensitive and -insensitive pathways. Biochem J 281:301–307
Akbar GK, Dasari VR, Sheth SB, Ashby B, Mills DC, Kunapuli SP (1996) Characterization of P2 purinergic receptors on human erythro leukemia cells. J Recept Signal Transduct Res 16:209–224
Baltensperger K, Porzig H (1997) The P2U purinoceptor obligatorily engages the heterotrimeric G protein G16 to mobilize intracellular Ca2+ in human erythroleukemia cells. J Biol Chem 272:10151–10159
Spranzi E, Djeu JY, Hoffman SL, Epling-Burnette PK, Blanchard DK (1993) Lysis of human monocytic leukemia cells by extracellular adenosine triphosphate: mechanism and characterization of the adenosine triphosphate receptor. Blood 82:1578–1585
Theaker J, Fagura MS, Lawson M, Bowers KC (2000) P2X 7-induced pore formation in a population of THP-1 cells. Brit J Pharmacol 131:189P
Sherman ML, Shafman TD, Kufe DW (1988) Modulation of cyclic AMP levels and differentiation by adenosine analogs in mouse erythroleukemia cells. J Cell Physiol 134:429–436
Ryten M, Dunn PM, Neary JT, Burnstock G (2002) ATP regulates the differentiation of mammalian skeletal muscle by activation of a P2X 5 receptor on satellite cells. J Cell Biol 158:345–355
Koiso K, Nemoto R, Ohtani M, Uchida K, Shimazui T, Noguchi R, Hattori K, Miyanaga N, Shiraiwa H, Iwasaki A (1991) Evaluation of the invasive potential of superficial bladder cancer by adenosine triphosphate measurement. Urol Int 46:145–148
Artim DE, Birder LA, de Groat WC (2007) Purinergic mechanisms in human bladder cancer. FASEB J 21:954.4
Nucciarelli F, Mearini E, Minelli A (2003) Effect of adenosine on prostate adenocarcinoma PC-3 and bladder carcinoma J82 cell lines. Drug Dev Res 58:390–395
Phelps PT, Anthes JC, Correll CC (2006) Characterization of adenosine receptors in the human bladder carcinoma T24 cell line. Eur J Pharmacol 536:28–37
Stella J, Bavaresco L, Braganhol E, Rockenbach L, Farias PF, Wink MR, Azambuja AA, Barrios CH, Morrone FB, Oliveira Battastini AM (2010) Differential ectonucleotidase expression in human bladder cancer cell lines. Urol Oncol 28:260–267
Blume AJ, Dalton C, Sheppard H (1973) Adenosine-mediated elevation of cyclic 3′:5′-adenosine monophosphate concentrations in cultured mouse neuroblastoma cells. Proc Natl Acad Sci U S A 70:3099–3102
Blume AJ, Foster CJ (1976) Mouse neuroblastoma cell adenylate cyclase: regulation by 2-chloroadenosine, prostaglandin E1 and the cations Mg2+, Ca2+ and Mn2+. J Neurochem 26:305–311
Pénit J, Cantau B, Huot J, Jard S (1977) Adenylate cyclase from synchronized neuroblastoma cells: responsiveness to prostaglandin E1, adenosine, and dopamine during the cell cycle. Proc Natl Acad Sci U S A 74:1575–1579
Green RD, Stanberry LR (1977) Elevation of cyclic AMP in C-1300 murine neuroblastoma by adenosine and related compounds and the antagonism of this response by methylxanthines. Biochem Pharmacol 26:37–43
Blume AJ (1978) Opiate binding to membrane preparations of neuroblastoma x glioma hybrid cells NG108-15: effects of ions and nucleotides. Life Sci 22:1843–1852
Hamprecht B, Brandt M, Propst F, van Calker D, Löffler F (1981) Regulation by neurohormones of cyclic AMP concentration in cells derived from the nervous system. Adv Cyclic Nucleotide Res 14:637–645
Ehrlich YH, Garfield MG, Davis TB, Kornecki E, Chaffee JE, Lenox RH (1986) Extracellular protein phosphorylation systems in the regulation of neuronal function. Prog Brain Res 69:197–208
Chau LY, Lin TA, Chang WT, Chen CH, Shue MJ, Hsu YS, Hu CY, Tsai WH, Sun GY (1993) Endothelin-mediated calcium response and inositol 1,4,5-trisphosphate release in neuroblastoma-glioma hybrid cells (NG108-15): cross talk with ATP and bradykinin. J Neurochem 60:454–460
Chueh SH, Kao LS (1993) Extracellular ATP stimulates calcium influx in neuroblastoma x glioma hybrid NG108-15 cells. J Neurochem 61:1782–1788
Okajima F, Tomura H, Kondo Y (1993) Enkephalin activates the phospholipase C/Ca2+ system through cross-talk between opioid receptors and P2-purinergic or bradykinin receptors in NG 108–15 cells. A permissive role for pertussis toxin-sensitive G-proteins. Biochem J 290:241–247
Reetz G, Reiser G (1994) Cross-talk of the receptors for bradykinin, serotonin, and ATP shown by single cell Ca2+ responses indicating different modes of Ca2+ activation in a neuroblastoma x glioma hybrid cell line. J Neurochem 62:890–897
Filippov AK, Brown DA (1996) Activation of nucleotide receptors inhibits high-threshold calcium currents in NG108-15 neuronal hybrid cells. Eur J Neurosci 8:1149–1155
Sak K, Kelve M, Uri A, Järv J (1998) Pyrimidinoceptor potentiation by ATP in NG108-15 cells. FEBS Lett 439:107–109
Lin TA, Lustig KD, Sportiello MG, Weisman GA, Sun GY (1993) Signal transduction pathways coupled to a P2U receptor in neuroblastoma x glioma (NG108-15) cells. J Neurochem 60:1115–1125
Filippov AK, Selyanko AA, Robbins J, Brown DA (1994) Activation of nucleotide receptors inhibits M-type K current [I K(M)] in neuroblastoma x glioma hybrid cells. Pflugers Arch 429:223–230
Reiser G (1995) Ca2+- and nitric oxide-dependent stimulation of cyclic GMP synthesis in neuronal cell line induced by P2-purinergic/pyrimidinergic receptor. J Neurochem 64:61–68
Song SL, Chueh SH (1996) Antagonistic effect of Na + and Mg2+ on P2z purinoceptor-associated pores in dibutyryl cyclic AMP-differentiated NG108-15 cells. J Neurochem 67:1694–1701
Kaiho H, Kimura J, Matsuoka I, Kumasaka T, Nakanishi H (1996) ATP-activated nonselective cation current in NG108-15 cells. J Neurochem 67:398–406
Kaiho H, Matsuoka I, Kimura J, Nakanishi H (1998) Identification of P2X7 (P2Z) receptor in N18TG-2 cells and NG108-15 cells. J Neurochem 70:951–957
Bräter M, Li SN, Gorodezkaya IJ, Andreas K, Ravens U (1999) Voltage-sensitive Ca2+ channels, intracellular Ca2+ stores and Ca2+-release-activated Ca2+ channels contribute to the ATP-induced [Ca2+]i increase in differentiated neuroblastoma x glioma NG 108–15 cells. Neurosci Lett 264:97–100
Watano T, Matsuoka I, Kimura J (2002) Characteristics of ATP-induced current through P2X7 receptor in NG108-15 cells: unique antagonist sensitivity and lack of pore formation. Jpn J Pharmacol 88:428–435
Sak K, Webb TE, Samuel K, Kelve M, Järv J (1999) Only pyrimidinoceptors are functionally expressed in mouse neuroblastoma cell lines. Mol Cell Biol Res Commun 1:203–208
Sak K, Samuel K, Kelve M, Webb TE (2001) Pharmacological characterisation of pyrimidinoceptor responses in NG108-15 cells. Eur J Pharmacol 415:127–133
Apolloni S, Finocchi P, D’Agnano I, Alloisio S, Nobile M, D’Ambrosi N, Volonté C (2010) UDP exerts cytostatic and cytotoxic actions in human neuroblastoma SH-SY5Y cells over-expressing P2Y6 receptor. Neurochem Int 56:670–678
Ohkubo S, Kimura J, Matsuoka I (2000) Ecto-alkaline phosphatase in NG108-15 cells: a key enzyme mediating P1 antagonist-sensitive ATP response. Br J Pharmacol 131:1667–1672
Kaulich M, Qurishi R, Müller CE (2003) Extracellular metabolism of nucleotides in neuroblastoma x glioma NG108-15 cells determined by capillary electrophoresis. Cell Mol Neurobiol 23:349–364
Van Zoelen EJ, Tertoolen LG, Boonstra J, Van der Saag PT, De Laat SW (1982) Effect of external ATP on the plasma membrane permeability and (Na+ + K+)-ATPase activity of mouse neuroblastoma cells. Biochim Biophys Acta 720:223–234
Chen CC, Chen WC (1997) P2Y receptor linked to phospholipase C: stimulation of neuro 2A cells by UTP and ATP and possible regulation by protein kinase C subtype ε. J Neurochem 69:1409–1416
Iredale PA, Martin KF, Alexander SP, Hill SJ, Kendall DA (1992) Inositol 1,4,5-trisphosphate generation and calcium mobilisation via activation of an atypical P2 receptor in the neuronal cell line, N1E-115. Br J Pharmacol 107:1083–1087
Schrier SM, Florea BI, Mulder GJ, Nagelkerke JF, Ijzerman AP (2002) Apoptosis induced by extracellular ATP in the mouse neuroblastoma cell line N1E-115: studies on involvement of P2 receptors and adenosine. Biochem Pharmacol 63:1119–1126
Watano T, Matsuoka I, Ogawa K, Kimura J (2002) Effects of anions on ATP-induced [Ca2+]i increase in NG108-15 cells. Jpn J Pharmacol 89:302–308
Garritsen A, Zhang Y, Cooper DM (1992) Purinergic receptor regulation of signal transduction in NCB-20 cells. Mol Pharmacol 41:743–749
Delporte C, Winand J, Poloczek P, Brunko E, Tastenoy M, Waelbroeck M, Christophe J (1992) Inhibitory effects of ATP and other nucleotides on atrial natriuretic peptide (ANP) binding to R1-type ANP receptors in human neuroblastoma NB-OK-1 cell membranes. Biochim Biophys Acta 1135:323–329
Soares Lemos V, Bucher B, Takeda K (1997) Neuropeptide Y modulates ATP-induced increases in internal calcium via the adenylate cyclase/protein kinase A system in a human neuroblastoma cell line. Biochem J 321:439–444
El-Sherif Y, Wieraszko A, Banerjee P, Penington NJ (2001) ATP modulates Na+ channel gating and induces a non-selective cation current in a neuronal hippocampal cell line. Brain Res 904:307–317
Larsson KP, Hansen AJ, Dissing S (2002) The human SH-SY5Y neuroblastoma cell-line expresses a functional P2X7 purinoceptor that modulates voltage-dependent Ca2+ channel function. J Neurochem 83:285–298
Guarnieri S, Pilla R, Morabito C, Sacchetti S, Mancinelli R, Fanò G, Mariggiò MA (2009) Extracellular guanosine and GTP promote expression of differentiation markers and induce S-phase cell-cycle arrest in human SH-SY5Y neuroblastoma cells. Int J Dev Neurosci 27:135–147
Canals M, Angulo E, Casadó V, Canela EI, Mallol J, Viñals F, Staines W, Tinner B, Hillion J, Agnati L, Fuxe K, Ferré S, Lluis C, Franco R (2005) Molecular mechanisms involved in the adenosine A1 and A2A receptor-induced neuronal differentiation in neuroblastoma cells and striatal primary cultures. J Neurochem 92:337–348
Lee H, Choi BH, Suh BC, Lee SK, Kim KT (2003) Attenuation of signal flow from P2Y6 receptor by protein kinase C-alpha in SK-N-BE(2)C human neuroblastoma cells. J Neurochem 85:1043–1053
Gualix J, León-Otegui M, Recuero M, Bullido MJ, Valdivieso F, Miras-Portugal MT (2008) Presence of functional P2Y1 and P2Y1 receptors in human SK-N-MC neuroblastoma cells. Purinergic Signal 4:S210
Chelmicka-Schorr E, Jones KH, Checinski ME, Yu RC, Arnason BG (1985) Influence of the sympathetic nervous system on the growth of neuroblastoma in vivo and in vitro. Cancer Res 45:6213–6215
Burnstock G, Verkhratsky A (2010) Long-term (trophic) purinergic signalling: purinoceptors control cell proliferation, differentiation and death. Cell Death and Disease 1:e9
Silei V, Politi V, Lauro GM (2000) Uridine induces differentiation in human neuroblastoma cells via protein kinase C epsilon. J Neurosci Res 61:206–211
Cavaliere F, Nestola V, Amadio S, D’Ambrosi N, Angelini DF, Sancesario G, Bernardi G, Volonté C (2005) The metabotropic P2Y4 receptor participates in the commitment to differentiation and cell death of human neuroblastoma SH-SY5Y cells. Neurobiol Dis 18:100–109
Han JZ, Lin W, Chen YZ (2005) Inhibition of ATP-induced calcium influx in HT4 cells by glucocorticoids: involvement of protein kinase A. Acta Pharmacol Sin 26:199–204
Raffaghello L, Chiozzi P, Falzoni S, Di VF, Pistoia V (2006) The P2X7 receptor sustains the growth of human neuroblastoma cells through a substance P-dependent mechanism. Cancer Res 66:907–914
Schrier SM, van Tilburg EW, van der Meulen H, Ijzerman AP, Mulder GJ, Nagelkerke JF (2001) Extracellular adenosine-induced apoptosis in mouse neuroblastoma cells: studies on involvement of adenosine receptors and adenosine uptake. Biochem Pharmacol 61:417–425
Lakshmi S, Joshi PG (2006) Activation of Src/kinase/phospholipase C/mitogen-activated protein kinase and induction of neurite expression by ATP, independent of nerve growth factor. Neuroscience 141:179–189
Wu PY, Lin YC, Chang CL, Lu HT, Chin CH, Hsu TT, Chu D, Sun SH (2009) Functional decreases in P2X7 receptors are associated with retinoic acid-induced neuronal differentiation of Neuro-2a neuroblastoma cells. Cell Signal 21:881–891
Morgan CR, Bird EV, Robinson PP, Boissonade FM (2009) Immunohistochemical analysis of the purinoceptor P2X7 in human lingual nerve neuromas. J Orofac Pain 23:65–72
Gutiérrez-Martín Y, Bustillo D, Gómez-Villafuertes R, Sánchez-Nogueiro J, Torregrosa-Hetland C, Binz T, Gutiérrez LM, Miras-Portugal MT, Artalejo AR (2011) P2X7 receptors trigger ATP exocytosis and modify secretory vesicle dynamics in neuroblastoma cells. J Biol Chem 286:11370–11381
Sun SH (2010) Roles of P2X7 receptor in glial and neuroblastoma cells: the therapeutic potential of P2X7 receptor antagonists. Mol Neurobiol 41:351–355
Snell CR, Snell PH (1984) Benzodiazepines modulate the A2 adenosine binding sites on 108CC15 neuroblastoma X glioma hybrid cells. Br J Pharmacol 83:791–798
Abbracchio MP, Cattabeni F, Clementi F, Sher E (1989) Adenosine receptors linked to adenylate cyclase activity in human neuroblastoma cells: modulation during cell differentiation. Neuroscience 30:819–825
Dehnhardt M, Palm C, Vieten A, Bauer A, Pietrzyk U (2007) Quantifying the A1AR distribution in peritumoural zones around experimental F98 and C6 rat brain tumours. J Neurooncol 85:49–63
Lantos PL (1974) The fine structural localisation of thiamine pyrophosphatase and adenosine triphosphatase in neural tumours induced by N-ethyl-N-nitrosourea in rats. Acta Neuropathol 29:199–209
Grobben B, Anciaux K, Roymans D, Stefan C, Bollen M, Esmans EL, Slegers H (1999) An ecto-nucleotide pyrophosphatase is one of the main enzymes involved in the extracellular metabolism of ATP in rat C6 glioma. J Neurochem 72:826–834
Grobben B, Claes P, Roymans D, Esmans EL, Van Onckelen H, Slegers H (2000) Ecto-nucleotide pyrophosphatase modulates the purinoceptor-mediated signal transduction and is inhibited by purinoceptor antagonists. Br J Pharmacol 130:139–145
Wink MR, Lenz G, Braganhol E, Tamajusuku AS, Schwartsmann G, Sarkis JJ, Battastini AM (2003) Altered extracellular ATP, ADP and AMP catabolism in glioma cell lines. Cancer Lett 198:211–218
Wink MR, Tamajusuku AS, Braganhol E, Casali EA, Barreto-Chaves ML, Sarkis JJ, Battastini AM (2003) Thyroid hormone upregulates ecto-5′-nucleotidase/CD73 in C6 rat glioma cells. Mol Cell Endocrinol 205:107–114
Braganhol E, Tamajusuku AS, Bernardi A, Wink MR, Battastini AM (2007) Ecto-5′-nucleotidase/CD73 inhibition by quercetin in the human U138MG glioma cell line. Biochim Biophys Acta 1770:1352–1359
Cappellari AR, Vasques GJ, Bavaresco L, Braganhol E, Battastini AM (2012) Involvement of ecto-5′-nucleotidase/CD73 in U138MG glioma cell adhesion. Mol Cell Biochem 359:315–322
Cappellari AR, Rockenbach L, Dietrich F, Clarimundo V, Glaser T, Braganhol E, Abujamra AL, Roesler R, Ulrich H, Battastini AM (2012) Characterization of ectonucleotidases in human medulloblastoma cell lines: ecto-5′NT/CD73 in metastasis as potential prognostic factor. PLoS One 7:e47468
Braganhol E, Morrone FB, Bernardi A, Huppes D, Meurer L, Edelweiss MI, Lenz G, Wink MR, Robson SC, Battastini AM (2009) Selective NTPDase2 expression modulates in vivo rat glioma growth. Cancer Sci 100:1434–1442
Braganhol E, Zanin RF, Bernardi A, Bergamin LS, Cappellari AR, Campesato LF, Morrone FB, Campos MM, Calixto JB, Edelweiss MI, Wink MR, Sévigny J, Robson SC, Battastini AM (2012) Overexpression of NTPDase2 in gliomas promotes systemic inflammation and pulmonary injury. Purinergic Signal 8:235–243
Baba T, Fukui M, Sakata S, Tashima T, Takeshita I, Nakamura T, Inoue T (1989) Selective enhancement of intratumoural blood flow in malignant gliomas: experimental study in rats by intracarotid administration of adenosine or adenosine triphosphate. Acta Neurochir (Wien) 101:66–74
Natori Y, Baba T, Moriguchi M, Takeshita I, Fukui M (1992) Effects of theophylline on the selective increases in intratumoral blood flow induced by intracarotid infusion of adenosine and adenosine triphosphate in C6 glioma-transplanted rat brains. Surg Neurol 37:8–14
Kitanaka J, Hashimoto H, Gotoh M, Mayumi T, Baba A (1992) Mechanism of extracellular ATP-stimulated phosphoinositide hydrolysis in rat glioma C6 cells. J Pharmacol Exp Ther 263:1248–1252
Sabala P, Amler E, Baranska J (1997) Intracellular Ca2+ signals induced by ATP and thapsigargin in glioma C6 cells. Calcium pools sensitive to inositol 1,4,5-trisphosphate and thapsigargin. Neurochem Int 31:55–64
Valeins H, Merle M, Labouesse J (1992) Pre-steady state study of β-adrenergic and purinergic receptor interaction in C6 cell membranes: undelayed balance between positive and negative coupling to adenylyl cyclase. Mol Pharmacol 42:1033–1041
Munshi R, DeBernardi MA, Brooker G (1993) P2U-purinergic receptors on C6-2B rat glioma cells: modulation of cytosolic Ca2+ and cAMP levels by protein kinase C. Mol Pharmacol 44:1185–1191
Lazarowski ER, Harden TK (1994) Identification of a uridine nucleotide-selective G-protein-linked receptor that activates phospholipase C. J Biol Chem 269:11830–11836
Boyer JL, Zohn IE, Jacobson KA, Harden TK (1994) Differential effects of P2-purinoceptor antagonists on phospholipase C- and adenylyl cyclase-coupled P2Y-purinoceptors. Br J Pharmacol 113:614–620
Lin WW, Chuang DM (1994) Different signal transduction pathways are coupled to the nucleotide receptor and the P2Y receptor in C6 glioma cells. J Pharmacol Exp Ther 269:926–931
Sabala P, Czajkowski R, Przybylek K, Kalita K, Kaczmarek L, Baranska J (2001) Two subtypes of G protein-coupled nucleotide receptors, P2Y1 and P2Y2 are involved in calcium signalling in glioma C6 cells. Br J Pharmacol 132:393–402
Czajkowski R, Lei L, Sabala P, Baranska J (2002) ADP-evoked phospholipase C stimulation and adenylyl cyclase inhibition in glioma C6 cells occur through two distinct nucleotide receptors, P2Y1 and P2Y12. FEBS Lett 513:179–183
Baranska J, Czajkowski R, Sabala P (2004) Cross-talks between nucleotide receptor-induced signaling pathways in serum-deprived and non-starved glioma C6 cells. Adv Enzyme Regul 44:219–232
Suplat D, Krzeminski P, Pomorski P, Baranska J (2007) P2Y1 and P2Y12 receptor cross-talk in calcium signalling: evidence from nonstarved and long-term serum-deprived glioma C6 cells. Purinergic Signal 3:221–230
Krzeminski P, Suplat D, Czajkowski R, Pomorski P, Baranska J (2007) Expression and functional characterization of P2Y1 and P2Y12 nucleotide receptors in long-term serum-deprived glioma C6 cells. FEBS J 274:1970–1982
López-Valdés HE, Beltran-Parrazal L, Brennan KC, Charles AC (2010) Bradykinin increases resensitization of purinergic receptor signaling in glioma cells. Cancer Cell Int 10:35
Wypych D, Pomorski P (2012) P2Y1 nucleotide receptor silencing and its effect on glioma C6 calcium signaling. Acta Biochim Pol 59:711–717
Krzeminski P, Pomorski P, Baranska J (2008) The P2Y14 receptor activity in glioma C6 cells. Eur J Pharmacol 594:49–54
Wei W, Ryu JK, Choi HB, McLarnon JG (2008) Expression and function of the P2X7 receptor in rat C6 glioma cells. Cancer Lett 260:79–87
Tamajusuku AS, Villodre ES, Paulus R, Coutinho-Silva R, Battasstini AM, Wink MR, Lenz G (2010) Characterization of ATP-induced cell death in the GL261 mouse glioma. J Cell Biochem 109:983–991
Gehring MP, Pereira TC, Zanin RF, Borges MC, Braga FA, Battastini AM, Bogo MR, Lenz G, Campos MM, Morrone FB (2012) P2X7 receptor activation leads to increased cell death in a radiosensitive human glioma cell line. Purinergic Signal 8:729–739
Fang KM, Wang YL, Huang MC, Sun SH, Cheng H, Tzeng SF (2011) Expression of macrophage inflammatory protein-1α and monocyte chemoattractant protein-1 in glioma-infiltrating microglia: involvement of ATP and P2X7 receptor. J Neurosci Res 89:199–211
Guo LH, Trautmann K, Schluesener HJ (2004) Expression of P2X4 receptor in rat C6 glioma by tumor-associated macrophages and activated microglia. J Neuroimmunol 152:67–72
Ågren G, Ronquist G (1975) On the availability of certain metabolites at the outer surface of normal and malignant cells for the membranous de novo synthesis of ATP and other nucleotides. Ups J Med Sci 80:1–4
Ravera S, Aluigi MG, Calzia D, Ramoino P, Morelli A, Panfoli I (2011) Evidence for ectopic aerobic ATP production on C6 glioma cell plasma membrane. Cell Mol Neurobiol 31:313–321
Sinclair CJ, LaRivière CG, Young JD, Cass CE, Baldwin SA, Parkinson FE (2000) Purine uptake and release in rat C6 glioma cells: nucleoside transport and purine metabolism under ATP-depleting conditions. J Neurochem 75:1528–1538
Tressler AM, Lai C, Naus C, Dubyak G (2011) Gating of pannexin 1-mediated ATP release channels by mechanical stress stimuli. FASEB J 25:1007.3
Jantaratnotai N, Choi HB, McLarnon JG (2009) ATP stimulates chemokine production via a store-operated calcium entry pathway in C6 glioma cells. BMC Cancer 9:442
Zhang W, Turner DJ, Segura BJ, Cowles R, Mulholland MW (2000) ATP induces c-fos expression in C6 glioma cells by activation of P2Y receptors. J Surg Res 94:49–55
Jantaratnotai N, McLarnon JG (2011) Calcium dependence of purinergic subtype P2Y receptor modulation of C6 glioma cell migration. Neurosci Lett 497:80–84
Tu MT, Luo SF, Wang CC, Chien CS, Chiu CT, Lin CC, Yang CM (2000) P2Y2 receptor-mediated proliferation of C6 glioma cells via activation of Ras/Raf/MEK/MAPK pathway. Br J Pharmacol 129:1481–1489
Claes P, Grobben B, Van Kolen K, Roymans D, Slegers H (2001) P2YAC−-receptor agonists enhance the proliferation of rat C6 glioma cells through activation of the p42/44 mitogen-activated protein kinase. Br J Pharmacol 134:402–408
Morrone FB, Jacques-Silva MC, Horn AP, Bernardi A, Schwartsmann G, Rodnight R, Lenz G (2003) Extracellular nucleotides and nucleosides induce proliferation and increase nucleoside transport in human glioma cell lines. J Neurooncol 64:211–218
Claes P, Van Kolen K, Roymans D, Blero D, Vissenberg K, Erneux C, Verbelen JP, Esmans EL, Slegers H (2004) Reactive blue 2 inhibition of cyclic AMP-dependent differentiation of rat C6 glioma cells by purinergic receptor-independent inactivation of phosphatidylinositol 3-kinase. Biochem Pharmacol 67:1489–1498
Czajkowski R, Banachewicz W, Ilnytska O, Drobot LB, Baranska J (2004) Differential effects of P2Y1 and P2Y12 nucleotide receptors on ERK1/ERK2 and phosphatidylinositol 3-kinase signalling and cell proliferation in serum-deprived and nonstarved glioma C6 cells. Br J Pharmacol 141:497–507
Van Kolen K, Gilany K, Moens L, Esmans EL, Slegers H (2006) P2Y12 receptor signalling towards PKB proceeds through IGF-I receptor cross-talk and requires activation of Src, Pyk2 and Rap1. Cell Signal 18:1169–1181
Claes P, Slegers H (2004) P2Y receptor activation affects the proliferation and differentiation of glial and neuronal cells: a focus on rat C6 glioma cells. Curr Neuropharmacol 2:207–220
Langeveld CH, Jongenelen CA, Heimans JJ, Stoof JC (1992) Growth inhibition of human glioma cells induced by 8-chloroadenosine, an active metabolite of 8-chloro cyclic adenosine 3′:5′-monophosphate. Cancer Res 52:3994–3999
Zorn M, Maronde E, Jastorff B, Richter-Landsberg C (1993) Differential effects of two structurally related N6-substituted cAMP analogues on C6 glioma cells. Eur J Cell Biol 60:351–357
Castillo CA, Albasanz JL, Fernández M, Martín M (2007) Endogenous expression of adenosine A1, A2 and A3 receptors in rat C6 glioma cells. Neurochem Res 32:1056–1070
Ohkubo S, Nagata K, Nakahata N (2007) Adenosine uptake-dependent C6 cell growth inhibition. Eur J Pharmacol 577:35–43
Castillo CA, León D, Ruiz MA, Albasanz JL, Martín M (2008) Modulation of adenosine A1 and A2A receptors in C6 glioma cells during hypoxia: involvement of endogenous adenosine. J Neurochem 105:2315–2329
Morrone FB, Horn AP, Stella J, Spiller F, Sarkis JJ, Salbego CG, Lenz G, Battastini AM (2005) Increased resistance of glioma cell lines to extracellular ATP cytotoxicity. J Neurooncol 71:135–140
Morrone FB, Oliveira DL, Gamermann P, Stella J, Wofchuk S, Wink MR, Meurer L, Edelweiss MI, Lenz G, Battastini AM (2006) In vivo glioblastoma growth is reduced by apyrase activity in a rat glioma model. BMC Cancer 6:226
Katayama M, Kawaguchi T, Berger MS, Pieper RO (2007) DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 14:548–558
Renner C, Asperger A, Seyffarth A, Meixensberger J, Gebhardt R, Gaunitz F (2010) Carnosine inhibits ATP production in cells from malignant glioma. Neurol Res 32:101–105
Hartmann J, Verkhratsky A (1998) Relations between intracellular Ca2+ stores and store-operated Ca 2+ entry in primary cultured human glioblastoma cells. J Physiol 513:411–424
de Joannon AC, Mancini F, Landolfi C, Soldo L, Leta A, Ruggieri A, Mangano G, Polenzani L, Pinza M, Milanese C (2000) Adenosine triphosphate affects interleukin −1β release by T98G glioblastoma cells through a purinoceptor-independent mechanism. Neurosci Lett 285:218–222
Ledur PF, Villodre ES, Paulus R, Cruz LA, Flores DG, Lenz G (2012) Extracellular ATP reduces tumor sphere growth and cancer stem cell population in glioblastoma cells. Purinergic Signal 8:39–48
Zeng D, Maa T, Wang U, Feoktistov I, Biaggioni I, Belardinelli L (2004) Expression and function of A2B adenosine receptors in the U87MG tumor cells. Drug Dev Res 58:405–411
Synowitz M, Glass R, Färber K, Markovic D, Kronenberg G, Herrmann K, Schnermann J, Nolte C, van Rooijen N, Kiwit J, Kettenmann H (2006) A1 adenosine receptors in microglia control glioblastoma-host interaction. Cancer Res 66:8550–8557
Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Borea PA (2006) Adenosine modulates vascular endothelial growth factor expression via hypoxia-inducible factor-1 in human glioblastoma cells. Biochem Pharmacol 72:19–31
Gessi S, Sacchetto V, Fogli E, Merighi S, Varani K, Baraldi PG, Tabrizi MA, Leung E, Maclennan S, Borea PA (2010) Modulation of metalloproteinase-9 in U87MG glioblastoma cells by A3 adenosine receptors. Biochem Pharmacol 79:1483–1495
Vincenzi F, Targa M, Corciulo C, Gessi S, Merighi S, Setti S, Cadossi R, Borea PA, Varani K (2012) The anti-tumor effect of A3 adenosine receptors is potentiated by pulsed electromagnetic fields in cultured neural cancer cells. PLoS One 7:e39317
Kim TH, Kim YK, Woo JS (2012) The adenosine A3 receptor agonist Cl-IB-MECA induces cell death through Ca2+/ROS-dependent down regulation of ERK and Akt in A172 human glioma cells. Neurochem Res 37:2667–2677
Clark RB, Gross R, Su YF, Perkins JP (1974) Regulation of adenosine 3′:5′-monophosphate content in human astrocytoma cells by adenosine and the adenine nucleotides. J Biol Chem 249:5296–5303
Hughes AR, Harden TK (1986) Adenosine and muscarinic cholinergic receptors attenuate cyclic AMP accumulation by different mechanisms in 1321N1 astrocytoma cells. J Pharmacol Exp Ther 237:173–178
Rathbone MP, Middlemiss PJ, Kim JK, Gysbers JW, DeForge SP, Smith RW, Hughes DW (1992) Adenosine and its nucleotides stimulate proliferation of chick astrocytes and human astrocytoma cells. Neurosci Res 13:1–17
Bradley KK, Bradley ME (2001) Purine nucleoside-dependent inhibition of cellular proliferation in 1321N1 human astrocytoma cells. J Pharmacol Exp Ther 299:748–752
Sellers LA, Simon J, Lundahl TS, Cousens DJ, Humphrey PP, Barnard EA (2001) Adenosine nucleotides acting at the human P2Y1 receptor stimulate mitogen-activated protein kinases and induce apoptosis. J Biol Chem 276:16379–16390
Gendron FP, Neary JT, Theiss PM, Sun GY, Gonzalez FA, Weisman GA (2003) Mechanisms of P2X7 receptor-mediated ERK1/2 phosphorylation in human astrocytoma cells. Am J Physiol Cell Physiol 284:C571–C581
Kim SG, Gao ZG, Soltysiak KA, Chang TS, Brodie C, Jacobson KA (2003) P2Y6 nucleotide receptor activates PKC to protect 1321N1 astrocytoma cells against tumor necrosis factor-induced apoptosis. Cell Mol Neurobiol 23:401–418
Ho MKC, Simon J, Barnard EA, Wong YH (2004) Atypical regulation of calcium signals in astrocytoma 1321N1 cells expressing the purinergic P2Y12 receptor. J Neurochem 88:P33–15
Flores RV, Casallas-Bond A, Garrad R, Weisman GA, Gonzalez FA (2004) Signaling of a P2y2 receptor—EGFP fusion protein expressed in 1321N1 astrocytoma cells. Society for Neuroscience Abstract Viewer/Itinerary Planner, Washington, Program No.631.6
Kreda SM, Seminario-Vidal L, Heusden C, Lazarowski ER (2008) Thrombin-promoted release of UDP-glucose from human astrocytoma cells. Br J Pharmacol 153:1528–1537
Blum AE, Walsh BC, Dubyak GR (2010) Extracellular osmolarity modulates G protein-coupled receptor-dependent ATP release from 1321N1 astrocytoma cells. Am J Physiol Cell Physiol 298:C386–C396
Fiebich BL, Biber K, Gyufko K, Berger M, Bauer J, van Calker D (1996) Adenosine A2b receptors mediate an increase in interleukin (IL)-6 mRNA and IL-6 protein synthesis in human astroglioma cells. J Neurochem 66:1426–1431
Abbracchio MP, Rainaldi G, Giammarioli AM, Ceruti S, Brambilla R, Cattabeni F, Barbieri D, Franceschi C, Jacobson KA, Malorni W (1997) The A3 adenosine receptor mediates cell spreading, reorganization of actin cytoskeleton, and distribution of Bcl-XL: studies in human astroglioma cells. Biochem Biophys Res Commun 241:297–304
Abbracchio MP, Camurri A, Ceruti S, Cattabeni F, Falzano L, Giammarioli AM, Jacobson KA, Trincavelli L, Martini C, Malorni W, Fiorentini C (2001) The A3 adenosine receptor induces cytoskeleton rearrangement in human astrocytoma cells via a specific action on Rho proteins. Ann N Y Acad Sci 939:63–73
Trincavelli ML, Tuscano D, Marroni M, Falleni A, Gremigni V, Ceruti S, Abbracchio MP, Jacobson KA, Cattabeni F, Martini C (2002) A3 adenosine receptors in human astrocytoma cells: agonist-mediated desensitization, internalization, and down-regulation. Mol Pharmacol 62:1373–1384
Sai K, Yang D, Yamamoto H, Fujikawa H, Yamamoto S, Nagata T, Saito M, Yamamura T, Nishizaki T (2006) A1 adenosine receptor signal and AMPK involving caspase-9/-3 activation are responsible for adenosine-induced RCR-1 astrocytoma cell death. Neurotoxicology 27:458–467
Garcia-Gil M, Tozzi MG, Allegrini S, Folcarelli S, Della Sala G, Voccoli V, Colombaioni L, Camici M (2012) Novel metabolic aspects related to adenosine deaminase inhibition in a human astrocytoma cell line. Neurochem Int 60:523–532
Inoue K, Nakazawa K, Fujimori K, Takanaka A (1989) Extracellular adenosine 5′-triphosphate-evoked norepinephrine secretion not relating to voltage-gated Ca channels in pheochromocytoma PC12 cells. Neurosci Lett 106:294–299
Majid MA, Okajima F, Kondo Y (1992) Characterization of ATP receptor which mediates norepinephrine release in PC12 cells. Biochim Biophys Acta 1136:283–289
Hardwick JC, Ehrlich YH, Hendley ED (1989) Extracellular ATP stimulates norepinephrine uptake in PC12 cells. J Neurochem 53:1512–1518
Eshleman A, Dunigan C, Shamoo A, Eldefrawi M (1995) ATP enhances catecholamine uptake into PC12 cells. Life Sci 56:1613–1621
Nakazawa K, Fujimori K, Takanaka A, Inoue K (1990) An ATP-activated conductance in pheochromocytoma cells and its suppression by extracellular calcium. J Physiol 428:257–272
Nakazawa K, Fujimori K, Takanaka A, Inoue K (1990) Reversible and selective antagonism by suramin of ATP-activated inward current in PC12 phaeochromocytoma cells. Br J Pharmacol 101:224–226
Inoue K, Nakazawa K, Ohara-Imaizumi M, Obama T, Fujimori K, Takanaka A (1991) Selective and competitive antagonism by suramin of ATP-stimulated catecholamine-secretion from PC12 phaeochromocytoma cells. Br J Pharmacol 102:581–584
Inoue K, Nakazawa K, Ohara-Imaizumi M, Obama T, Fujimori K, Takanaka A (1991) Antagonism by reactive blue 2 but not by brilliant blue G of extracellular ATP-evoked responses in PC12 phaeochromocytoma cells. Br J Pharmacol 102:851–854
Sela D, Ram E, Atlas D (1991) ATP receptor. A putative receptor-operated channel in PC-12 cells. J Biol Chem 266:17990–17994
Nakazawa K, Fujimori K, Takanaka A, Inoue K (1991) Comparison of adenosine triphosphate- and nicotine-activated inward currents in rat phaeochromocytoma cells. J Physiol 434:647–660
Majid MA, Okajima F, Kondo Y (1993) UTP activates phospholipase C-Ca2+ system through a receptor different from the 53-kDa ATP receptor in PC12 cells. Biochem Biophys Res Commun 195:415–421
Murrin RJ, Boarder MR (1992) Neuronal "nucleotide" receptor linked to phospholipase C and phospholipase D? Stimulation of PC12 cells by ATP analogues and UTP. Mol Pharmacol 41:561–568
Raha S, de Souza LR, Reed JK (1993) Intracellular signalling by nucleotide receptors in PC12 pheochromocytoma cells. J Cell Physiol 154:623–630
Barry VA, Cheek TR (1994) Extracellular ATP triggers two functionally distinct calcium signalling pathways in PC12 cells. J Cell Sci 107(Pt 2):451–462
Nikodijevic B, Sei Y, Shin Y, Daly JW (1994) Effects of ATP and UTP in pheochromocytoma PC12 cells: evidence for the presence of three P2 receptors, only one of which subserves stimulation of norepinephrine release. Cell Mol Neurobiol 14:27–47
de Souza LR, Moore H, Raha S, Reed JK (1995) Purine and pyrimidine nucleotides activate distinct signalling pathways in PC12 cells. J Neurosci Res 41:753–763
Michel AD, Grahames CB, Humphrey PP (1996) Functional characterisation of P2 purinoceptors in PC12 cells by measurement of radiolabelled calcium influx. Naunyn Schmiedebergs Arch Pharmacol 354:562–571
Nakazawa K, Ohno Y (1996) Dopamine and 5-hydroxytryptamine selectively potentiate neuronal type ATP receptor channels. Eur J Pharmacol 296:119–122
Koizumi S, Uneyama H, Ikeda M, Ueno S, Inoue K (1998) Inhibition by imipramine of ATP-evoked responses in rat pheochromocytoma cells. Biochem Biophys Res Commun 244:342–346
Kim HJ, Choi JS, Lee YM, Shim EY, Hong SH, Kim MJ, Min DS, Rhie DJ, Kim MS, Jo YH, Hahn SJ, Yoon SH (2005) Fluoxetine inhibits ATP-induced [Ca2+]i increase in PC12 cells by inhibiting both extracellular Ca2+ influx and Ca2+ release from intracellular stores. Neuropharmacology 49:265–274
Arslan G, Filipeanu CM, Irenius E, Kull B, Clementi E, Allgaier C, Erlinge D, Fredholm BB (2000) P2Y receptors contribute to ATP-induced increases in intracellular calcium in differentiated but not undifferentiated PC12 cells. Neuropharmacology 39:482–496
Hur EM, Park TJ, Kim KT (2001) Coupling of L-type voltage-sensitive calcium channels to P2X2 purinoceptors in PC-12 cells. Am J Physiol Cell Physiol 280:C1121–C1129
Tozaki-Saitoh H, Koizumi S, Sato Y, Tsuda M, Nagao T, Inoue K (2006) Retinoic acids increase P2X2 receptor expression through the 5′-flanking region of P2rx2 gene in rat phaeochromocytoma PC-12 cells. Mol Pharmacol 70:319–328
Liu PS, Hsieh HL, Lin CM (2001) Dehydroepiandrosterone sulfate (DHEAS) suppresses P2X purinoceptor-coupled responses in PC12 cells. Neurochem Int 39:193–198
Keath JR, Westhead EW (2004) Factors affecting habituation of PC12 cells to ATP. Eur J Biochem 271:4034–4041
Maruyama K, Ohta T, Ito S (2004) Involvement of mitochondrial Na+-Ca2+ exchange in intracellular Ca2+ increase induced by ATP in PC12 cells. Brain Res 1013:40–50
Marín-Vicente C, Gómez-Fernández JC, Corbalán-García S (2005) The ATP-dependent membrane localization of protein kinase C α is regulated by Ca2+ influx and phosphatidylinositol 4,5-bisphosphate in differentiated PC12 cells. Mol Biol Cell 16:2848–2861
Sun JH, Cai GJ, Xiang ZH (2007) Expression of P2X purinoceptors in PC12 phaeochromocytoma cells. Clin Exp Pharmacol Physiol 34:1282–1286
Arthur DB, Taupenot L, Insel PA (2007) Nerve growth factor-stimulated neuronal differentiation induces changes in P2 receptor expression and nucleotide-stimulated catecholamine release. J Neurochem 100:1257–1264
Soltoff SP (1998) Related adhesion focal tyrosine kinase and the epidermal growth factor receptor mediate the stimulation of mitogen-activated protein kinase by the G-protein-coupled P2Y2 receptor. Phorbol ester or [Ca2+]i elevation can substitute for receptor activation. J Biol Chem 273:23110–23117
Xu H, Wu B, Jiang F, Xiong S, Zhang B, Li G, Liu S, Gao Y, Xu C, Tu G, Peng H, Liang S, Xiong H (2013) High fatty acids modulate P2X 7 expression and IL-6 release via the p38 MAPK pathway in PC12 cells. Brain Res Bull 94:63–70
Vartian N, Boehm S (2001) P2Y receptor-mediated inhibition of voltage-activated Ca2+ currents in PC12 cells. Eur J Neurosci 13:899–908
Kulick MB, von Kügelgen I (2002) P2Y-receptors mediating an inhibition of the evoked entry of calcium through N-type calcium channels at neuronal processes. J Pharmacol Exp Ther 303:520–526
Kubista H, Lechner SG, Wolf AM, Boehm S (2003) Attenuation of the P2Y receptor-mediated control of neuronal Ca2+ channels in PC12 cells by antithrombotic drugs. Br J Pharmacol 138:343–350
Moskvina E, Unterberger U, Boehm S (2003) Activity-dependent autocrine-paracrine activation of neuronal P2Y receptors. J Neurosci 23:7479–7488
Hussl S, Kubista H, Boehm S (2007) Autoregulation in PC12 cells via P2Y receptors: evidence for non-exocytotic nucleotide release from neuroendocrine cells. Purinergic Signal 3:367–375
Daniele S, Lecca D, Trincavelli ML, Ciampi O, Abbracchio MP, Martini C (2010) Regulation of PC12 cell survival and differentiation by the new P2Y-like receptor GPR17. Cell Signal 22:697–706
Marín-Vicente C, Guerrero-Valero M, Nielsen ML, Savitski MM, Gómez-Fernández JC, Zubarev RA, Corbalán-García S (2011) ATP enhances neuronal differentiation of PC12 cells by activating PKCα interactions with cytoskeletal proteins. J Proteome Res 10:529–540
Prasai P, Stefos GC, Becker W (2011) Extracellular ATP activates NFAT-dependent gene expression in neuronal PC12 cells via P2X receptors. BMC Neurosci 12:90
Erny RE, Berezo MW, Perlman RL (1981) Activation of tyrosine 3-monooxygenase in pheochromocytoma cells by adenosine. J Biol Chem 256:1335–1339
Erny R, Wagner JA (1984) Adenosine-dependent activation of tyrosine hydroxylase is defective in adenosine kinase-deficient PC12 cells. Proc Natl Acad Sci U S A 81:4974–4978
Rabe CS, McGee R Jr (1983) Regulation of depolarization-dependent release of neurotransmitters by adenosine: cyclic AMP-dependent enhancement of release from PC12 cells. J Neurochem 41:1623–1634
Wu N, Armstrong I, Wagner J (1984) Genetic evidence that chloroadenosine increases the specific activity of choline acetyltransferase in PC12 cells via modulation of an adenosine-dependent adenylate cyclase. Neuroscience 13:1365–1371
Williams M, Abreu M, Jarvis MF, Noronha-Blob L (1987) Characterization of adenosine receptors in the PC12 pheochromocytoma cell line using radioligand binding: evidence for A-2 selectivity. J Neurochem 48:498–502
Hide I, Padgett WL, Jacobson KA, Daly JW (1992) A2A adenosine receptors from rat striatum and rat pheochromocytoma PC12 cells: characterization with radioligand binding and by activation of adenylate cyclase. Mol Pharmacol 41:352–359
Chern Y, Lai HL, Fong JC, Liang Y (1993) Multiple mechanisms for desensitization of A2a adenosine receptor-mediated cAMP elevation in rat pheochromocytoma PC12 cells. Mol Pharmacol 44:950–958
Park TJ, Song SK, Kim KT (1997) A2A adenosine receptors inhibit ATP-induced Ca2+ influx in PC12 cells by involving protein kinase A. J Neurochem 68:2177–2185
Kobayashi S, Beitner-Johnson D, Conforti L, Millhorn DE (1998) Chronic hypoxia reduces adenosine A2A receptor-mediated inhibition of calcium current in rat PC12 cells via downregulation of protein kinase A. J Physiol 512:351–363
O’Driscoll CM, Gorman AM (2005) Hypoxia induces neurite outgrowth in PC12 cells that is mediated through adenosine A2A receptors. Neuroscience 131:321–329
Loeffler DA, Camp DM, Juneau PL, Harel E, LeWitt PA (2000) Purine-induced alterations of dopamine metabolism in rat pheochromocytoma PC12 cells. Brain Res Bull 52:553–558
Thevananther S, Rivera A, Rivkees SA (2001) A1 adenosine receptor activation inhibits neurite process formation by Rho kinase-mediated pathways. Neuroreport 12:3057–3063
Charles MP, Adamski D, Kholler B, Pelletier L, Berger F, Wion D (2003) Induction of neurite outgrowth in PC12 cells by the bacterial nucleoside N6-methyldeoxyadenosine is mediated through adenosine A2a receptors and via cAMP and MAPK signaling pathways. Biochem Biophys Res Commun 304:795–800
Trincavelli ML, Falleni A, Chelli B, Tuscano D, Costa B, Gremigni V, Lucacchini A, Martini C (2003) A2A adenosine receptor ligands and proinflammatory cytokines induce PC 12 cell death through apoptosis. Biochem Pharmacol 66:1953–1962
Lu MK, Cheng JJ, Lai WL, Lin YR, Huang NK (2006) Adenosine as an active component of Antrodia cinnamomea that prevents rat PC12 cells from serum deprivation-induced apoptosis through the activation of adenosine A2A receptors. Life Sci 79:252–258
Hattori N, Nomoto H, Mishima S, Inagaki S, Goto M, Sako M, Furukawa S (2006) Identification of AMP N 1-oxide in royal jelly as a component neurotrophic toward cultured rat pheochromocytoma PC12 cells. Biosci Biotechnol Biochem 70:897–906
Koizumi S, Watano T, Nakazawa K, Inoue K (1994) Potentiation by adenosine of ATP-evoked dopamine release via a pertussis toxin-sensitive mechanism in rat phaeochromocytoma PC12 cells. Br J Pharmacol 112:992–997
Nakazawa K, Inoue K, Koizumi S, Inoue K (1994) Facilitation by 5-hydroxytryptamine of ATP-activated current in rat pheochromocytoma cells. Pflugers Arch 427:492–499
Koizumi S, Ikeda M, Inoue K, Nakazawa K, Inoue K (1995) Enhancement by zinc of ATP-evoked dopamine release from rat pheochromocytoma PC12 cells. Brain Res 673:75–82
Ikeda M, Koizumi S, Nakazawa K, Inoue K, Ito K, Inoue K (1996) Potentiation by cadmium ion of ATP-evoked dopamine release in rat phaeochromocytoma cells. Br J Pharmacol 117:950–954
Nakazawa K, Ito K, Koizumi S, Ohno Y, Inoue K (1995) Reduction of acetylcholine-activated current by low concentrations of extracellular adenosine 5′-triphosphate. Life Sci 57:L351–L356
Murayama T, Oda H, Watanabe A, Nomura Y (1995) ATP receptor-mediated increase of Ca ionophore-stimulated arachidonic acid release from PC12 pheochromocytoma cells. Jpn J Pharmacol 69:43–51
Nordone AJ, Pivorun EB (1995) Cytosolic calcium responses to extracellular adenosine 5',5" '-P1,P4-tetraphosphate in PC12 cells. Pharmacol Biochem Behav 52:85–91
Shoji-Kasai Y, Yoshida A, Sato K, Hoshino T, Ogura A, Kondo S, Fujimoto Y, Kuwahara R, Kato R, Takahashi M (1992) Neurotransmitter release from synaptotagmin-deficient clonal variants of PC12 cells. Science 256:1821–1823
Fujimori H, Yasuda M, Pan-Hou H (2002) Enhancement of cellular adenosine triphosphate levels in PC12 cells by extracellular adenosine. Biol Pharm Bull 25:307–311
Lechner SG, Dorostkar MM, Mayer M, Edelbauer H, Pankevych H, Boehm S (2004) Autoinhibition of transmitter release from PC12 cells and sympathetic neurons through a P2Y12 receptor-mediated inhibition of voltage-gated Ca2+ channels. Eur J Neurosci 20:2917–2928
Fabbro A, Skorinkin A, Grandolfo M, Nistri A, Giniatullin R (2004) Quantal release of ATP from clusters of PC12 cells. J Physiol 560:505–517
Fujimori H, Pan-Hou H (2005) Enhancement of cellular adenosine triphosphate levels in PC12 cells by 2,5-dideoxyadenosine, a P-site inhibitor of adenylate cyclase. Biol Pharm Bull 28:358–360
Gardner A, Westfall TC, Macarthur H (2005) Endothelin (ET)-1-induced inhibition of ATP release from PC-12 cells is mediated by the ETB receptor: differential response to ET-1 on ATP, neuropeptide Y, and dopamine levels. J Pharmacol Exp Ther 313:1109–1117
Yamboliev IA, Smyth LM, Durnin L, Dai Y, Mutafova-Yambolieva VN (2009) Storage and secretion of β-NAD, ATP and dopamine in NGF-differentiated rat pheochromocytoma PC12 cells. Eur J Neurosci 30:756–768
Richter-Landsberg C, Maronde E, Besser AS (1993) Ecto-5′-nucleotidase activity in PC 12 cells is synergistically modulated by nerve growth factor and 8-bromo-cAMP. Neurosci Res Comm 12:51–54
Heilbronn A, Maienschein V, Carstensen K, Gann W, Zimmermann H (1995) Crucial role of ecto-5′-nucleotidase in differentiation and survival of developing neural cells. Neuroreport 7:257–261
Cheng Y, Chen M, James-Kracke M, Wixom P, Sun AY (1996) Enhanced lipid peroxidation by extracellular ATP in PC12 cells. Neurochem Res 21:27–33
Gysbers JW, Rathbone MP (1996) GTP and guanosine synergistically enhance NGF-induced neurite outgrowth from PC12 cells. Int J Dev Neurosci 14:19–34
Gysbers JW, Guarnieri S, Mariggiò MA, Pietrangelo T, Fanò G, Rathbone MP (2000) Extracellular guanosine 5′ triphosphate enhances nerve growth factor-induced neurite outgrowth via increases in intracellular calcium. Neuroscience 96:817–824
Guarnieri S, Fanò G, Rathbone MP, Mariggiò MA (2004) Cooperation in signal transduction of extracellular guanosine 5′ triphosphate and nerve growth factor in neuronal differentiation of PC12 cells. Neuroscience 128:697–712
Bau C, Middlemiss PJ, Hindley S, Jiang S, Ciccarelli R, Caciagli F, Diiorio P, Werstiuk ES, Rathbone MP (2005) Guanosine stimulates neurite outgrowth in PC12 cells via activation of heme oxygenase and cyclic GMP. Purinergic Signal 1:161–172
Chen Y, Sun AY (1998) Activation of transcription factor AP-1 by extracellular ATP in PC12 cells. Neurochem Res 23:543–550
Fujita N, Kakimi M, Ikeda Y, Hiramoto T, Suzuki K (2000) Extracellular ATP inhibits starvation-induced apoptosis via P2X2 receptors in differentiated rat pheochromocytoma PC12 cells. Life Sci 66:1849–1859
Schindelholz B, Reber BF (2000) L-type Ca2+ channels and purinergic P2X2 cation channels participate in calcium-tyrosine kinase-mediated PC12 growth cone arrest. Eur J Neurosci 12:194–204
Pooler AM, Guez DH, Benedictus R, Wurtman RJ (2005) Uridine enhances neurite outgrowth in nerve growth factor-differentiated PC12 [corrected]. Neuroscience 134:207–214
Belliveau DJ, Bani-Yaghoub M, McGirr B, Naus CC, Rushlow WJ (2006) Enhanced neurite outgrowth in PC12 cells mediated by connexin hemichannels and ATP. J Biol Chem 281:20920–20931
D’Ambrosi N, Cavaliere F, Merlo D, Milazzo L, Mercanti D, Volonté C (2000) Antagonists of P2 receptor prevent NGF-dependent neuritogenesis in PC12 cells. Neuropharmacology 39:1083–1094
D’Ambrosi N, Murra B, Cavaliere F, Amadio S, Bernardi G, Burnstock G, Volonté C (2001) Interaction between ATP and nerve growth factor signalling in the survival and neuritic outgrowth from PC12 cells. Neuroscience 108:527–534
Behrsing HP, Vulliet PR (2004) Mitogen-activated protein kinase mediates purinergic-enhanced nerve growth factor-induced neurite outgrowth in PC12 cells. J Neurosci Res 78:64–74
Lee CS, Bae YS, Lee SD, Suh PG, Ryu SH (2001) ATP-induced mitogenesis is modulated by phospholipase D2 through extracellular signal regulated protein kinase dephosphorylation in rat pheochromocytoma PC12 cells. Neurosci Lett 313:117–120
D’Ambrosi N, Costanzi S, Angelini DF, Volpini R, Sancesario G, Cristalli G, Volonté C (2004) 2-ClATP exerts anti-tumoural actions not mediated by P2 receptors in neuronal and glial cell lines. Biochem Pharmacol 67:621–630
Homma K, Niino Y, Hotta K, Oka K (2008) Ca2+ influx through P2X receptors induces actin cytoskeleton reorganization by the formation of cofilin rods in neurites. Mol Cell Neurosci 37:261–270
Sato A, Arimura Y, Manago Y, Nishikawa K, Aoki K, Wada E, Suzuki Y, Osaka H, Setsuie R, Sakurai M, Amano T, Aoki S, Wada K, Noda M (2006) Parkin potentiates ATP-induced currents due to activation of P2X receptors in PC12 cells. J Cell Physiol 209:172–182
Liu PS, Chen YY (2006) Butyl benzyl phthalate blocks Ca2+ signaling coupled with purinoceptor in rat PC12 cells. Toxicol Appl Pharmacol 210:136–141
Liu PS, Chiung YM, Kao YY, Chen HT (2006) 2,4-Toluene diisocyanate suppressed the calcium signaling of ligand gated ion channel receptors. Toxicology 219:167–174
Mantyh PW, Clohisy DR, Koltzenburg M, Hunt SP (2002) Molecular mechanisms of cancer pain. Nat Rev Cancer 2:201–209
Nagamine K, Ozaki N, Shinoda M, Asai H, Nishiguchi H, Mitsudo K, Tohnai I, Ueda M, Sugiura Y (2006) Mechanical allodynia and thermal hyperalgesia induced by experimental squamous cell carcinoma of the lower gingiva in rats. J Pain 7:659–670
Chizhmakov I, Yudin Y, Mamenko N, Prudnikov I, Tamarova Z, Krishtal O (2005) Opioids inhibit purinergic nociceptors in the sensory neurons and fibres of rat via a G protein-dependent mechanism. Neuropharmacology 48:639–647
Fujita M, Andoh T, Sasaki A, Saiki I, Kuraishi Y (2010) Involvement of peripheral adenosine 5′-triphosphate and P2X purinoceptor in pain-related behavior produced by orthotopic melanoma inoculation in mice. Eur J Neurosci 31:1629–1636
Kaan TK, Yip PK, Patel S, Davies M, Marchand F, Cockayne DA, Nunn PA, Dickenson AH, Ford AP, Zhong Y, Malcangio M, McMahon SB (2010) Systemic blockade of P2X3 and P2X2/3 receptors attenuates bone cancer pain behaviour in rats. Brain 133:2549–2564
Hansen RR, Nasser A, Falk S, Baldvinsson SB, Ohlsson PH, Bahl JM, Jarvis MF, Ding M, Heegaard AM (2012) Chronic administration of the selective P2X3, P2X2/3 receptor antagonist, A-317491, transiently attenuates cancer-induced bone pain in mice. Eur J Pharmacol 688:27–34
Falk S, Uldall M, Heegaard AM (2012) The role of purinergic receptors in cancer-induced bone pain. J Osteoporos 2012:758181
Ye Y, Dang D, Viet CT, Dolan JC, Schmidt BL (2012) Analgesia targeting IB4-positive neurons in cancer-induced mechanical hypersensitivity. J Pain 13:524–531
Hansen RR, Nielsen CK, Nasser A, Thomsen SI, Eghorn LF, Pham Y, Schulenburg C, Syberg S, Ding M, Stojilkovic SS, Jorgensen NR, Heegaard AM (2011) P2X7 receptor-deficient mice are susceptible to bone cancer pain. Pain 152:1766–1776
Chen J, Wang L, Zhang Y, Yang J (2012) P2Y1 purinoceptor inhibition reduces extracellular signal-regulated protein kinase 1/2 phosphorylation in spinal cord and dorsal root ganglia: implications for cancer-induced bone pain. Acta Biochim Biophys Sin (Shanghai) 44:367–372
Pellegatti P, Falzoni S, Pinton P, Rizzuto R, Di Virgilio F (2005) A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol Biol Cell 16:3659–3665
Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, Bianchi G, Kroemer G, Pistoia V, Di Virgilio F (2012) Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res 72:2957–2969
Roger S, Pelegrin P (2011) P2X7 receptor antagonism in the treatment of cancers. Expert Opin Investig Drugs 20:875–880
Gorodeski GI (2012) P2X7 receptors and epithelial cancers. WIREs Membr Transp Signal 1:349–371
Blay J, White TD, Hoskin DW (1997) The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res 57:2602–2605
Ghiringhelli F, Bruchard M, Chalmin F, Rébe C (2012) Production of adenosine by ectonucleotidases: a key factor in tumor immunoescape. J Biomed Biotechnol 2012:473712
Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z (2011) Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol 187:676–683
Mandapathil M, Whiteside TL (2011) Targeting human inducible regulatory T cells (Tr1) in patients with cancer: blocking of adenosine-prostaglandin E2 cooperation. Expert Opin Biol Ther 11:1203–1214
Zhang B (2012) Opportunities and challenges for anti-CD73 cancer therapy. Immunotherapy 4:861–865
Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, Annaert W, Golab J, de Witte P, Vandenabeele P, Agostinis P (2012) A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 31:1062–1079
Beavis PA, Stagg J, Darcy PK, Smyth MJ (2012) CD73: a potent suppressor of antitumor immune responses. Trends Immunol 33:231–237
Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD (2012) Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother 61:917–926
Cekic C, Sag D, Linden J (2012) Cell-intrinsic adenosine A2A receptor signalling is required for T cell homeostasis and control of tumor growth b. J Immunol 188:47.1
Cekic C, Linden J (2012) Adenosine A2B receptor signalling in antigen presenting cells suppress anti-tumor adaptive immune responses. J Immunol 188:127.10
Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, Smyth MJ (2011) CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res 71:2892–2900
MacKenzie WM, Hoskin DW, Blay J (1994) Adenosine inhibits the adhesion of anti-CD3-activated killer lymphocytes to adenocarcinoma cells through an A3 receptor. Cancer Res 54:3521–3526
Lin K, Lin J, Wu WI, Ballard J, Lee BB, Gloor SL, Vigers GP, Morales TH, Friedman LS, Skelton N, Brandhuber BJ (2012) An ATP-site on-off switch that restricts phosphatase accessibility of Akt b. Sci Signal 5:ra37
Cheng Y, Senthamizhchelvan S, Agarwal R, Green GM, Mease RC, Sgouros G, Huso DL, Pomper MG, Meltzer SJ, Abraham JM (2012) [32P]ATP inhibits the growth of xenografted tumors in nude mice. Cell Cycle 11:1878–1882
Gaspar A, Silver T, Borges F (2011) Adenosine A3 receptors: a new therapeutic approach in cancer. Química Nova 34:1417–1424
Manga K, Serban G, Schwartz J, Slotky R, Patel N, Fan J, Bai X, Chari A, Savage D, Suciu-Foca N, Colovai AI (2010) Increased adenosine triphosphate production by peripheral blood CD4+ cells in patients with hematologic malignancies treated with stem cell mobilization agents. Hum Immunol 71:652–658
Zadran S, Sanchez D, Zadran H, Amighi A, Otiniano E, Wong K (2013) Enhanced-acceptor fluorescence-based single cell ATP biosensor monitors ATP in heterogeneous cancer populations in real time. Biotechnol Lett 35:175–180
Coutinho-Silva R, Stahl L, Cheung K-K, Ojcius DC, Burnstock G (2005) P2X and P2Y purinergic receptors on human intestinal epithelial carcinoma cell lines: effects of extracellular nucleotides on apoptosis and cell proliferation. Am J Physiol Gastrointest Liver Physiol 288:G1024–G1035
Di Virgilio F, Borea PA, Illes P (2001) P2 receptors meet the immune system. Trends Pharmacol Sci 22:5–7
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Burnstock, G., Di Virgilio, F. Purinergic signalling and cancer. Purinergic Signalling 9, 491–540 (2013). https://doi.org/10.1007/s11302-013-9372-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-013-9372-5