
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder with a complex pathogenesis. Senile plaques composed of the amyloid-β (Aβ) peptide in the brain are the core hallmarks of AD and a promising target for the development of disease-modifying therapies. However, over the past 20 years, the failures of clinical trials directed at Aβ clearance have fueled a debate as to whether Aβ is the principal pathogenic factor in AD and a valid therapeutic target. The success of the recent phase 3 trials of lecanemab (Clarity AD) and donanemab (Trailblazer Alz2), and lessons from previous Aβ clearance trials provide critical evidence to support the role of Aβ in AD pathogenesis and suggest that targeting Aβ clearance is heading in the right direction for AD treatment. Here, we analyze key questions relating to the efficacy of Aβ targeting therapies, and provide perspectives on early intervention, adequate Aβ removal, sufficient treatment period, and combinatory therapeutics, which may be required to achieve the best cognitive benefits in future trials in the real world.
Similar content being viewed by others
In 1901, Dr. Alois Alzheimer, a German psychiatrist, saw a female patient who presented with early-onset progressive cognitive impairment and abnormal behaviors. After her death, an autopsy of the brain revealed two now ‘classical’ neuropathological lesions: senile plaques and neurofibrillary tangles. Because the pathological changes were different from those found in others with cognitive disorders and mental diseases at that time, he considered it to be an independent disease and Kraepelin subsequently named it Alzheimer’s disease (AD) [1]. In the 1980s, amyloid-β (Aβ), the main component of the senile plaques and cerebral amyloid angiopathy was identified [2, 3].
Aβ is a metabolite of the amyloid precursor protein (APP), a type I transmembrane protein concentrated at neuronal synapses, and the proteolytic Aβ fragment is situated in its transmembrane domain. APP can be degraded by either the “amyloidogenic pathway” or “the non-amyloidogenic pathway” [4]. The non-amyloidogenic pathway of APP is sequentially mediated by α-secretase and γ-secretase, which doesn’t produce Aβ because the cleavage site of α-secretase is located within the Aβ fragment. In the amyloidogenic pathway, APP is first cleaved by β-secretase in the extra-membrane proximal region to release the soluble N-terminal (sAPPβ), and the residual C-terminal (CTF-β) on the membrane is then released by γ-secretase to produce Aβ which is then located into the extracellular compartment. Physiologically, Aβ monomers may regulate excitation/ inhibition balance and synaptic vesicle transport in nerve cells, and are primarily involved in long term potentiation and synaptic plasticity [4]. However, in the pathological process of AD, Aβ is not efficiently cleared, and aggregates to form oligomers, protofibrils, fibrils and, ultimately, plaques and perivascular deposits, which are the neuropathognomonic hallmarks of AD required for definitive diagnosis [5]. With the discovery of pathogenic mutations in familial autosomal dominant AD, which lead to overproduction of Aβ in the brain, Aβ accumulation was identified as the proximal causative pathway of AD, and became the most widely accepted theory for the etiology of AD [6,7,8,9]. Exploration of targeting Aβ for diagnosis and therapy of AD was then initiated.
Development of anti-Aβ therapeutics
Since Aβ is produced by sequential cleavage of APP by β-secretase and γ-secretase, β-secretase inhibitors and γ-secretase inhibitors/modulators have been developed for the treatment of AD in order to reduce the production of Aβ, but were terminated prematurely due to adverse cognitive effects. After due consideration, it was concluded that β-secretase inhibitors and γ-secretase inhibitors (GSI) were not suitable for the treatment of AD because they would inhibit the physiological effects of β-secretase and γ-secretase and bring about a series of adverse effects. However, current debate is considering their re-introduction at lower doses. Similarly, γ-secretase modulators (GSM) and other small molecules targeting Aβ aggregation are still in development.
In 1990, Mönning et al. discovered the presence of anti-Aβ auto-antibodies in humans [10] and subsequent studies found that anti-Aβ antibodies have the ability to inhibit Aβ aggregation and promote Aβ depolymerization [11, 12]. In 1999, the late Dale Schenk pioneered anti-Aβ immunotherapy by administering Aβ42 vaccine to PDAPP transgenic mice and confirmed that active immunotherapy could attenuate AD pathologic change and improve cognitive function [13]. In 2002, AN-1792 entered clinical trials as the first synthetic vaccine against Aβ42, which was unfortunately terminated prematurely because of adverse effects from vasculitis/encephalitis because of T-cell activation [14]. Subsequently, to avoid activation of T cells, scientists proposed passive immunotherapy, i.e., direct infusion of antibodies. 10D5 and pabAβ1-42 were the first antibodies shown to reduce Aβ levels in the mouse brain by 81% and 93%, respectively [15]. In 2005, passive immunotherapy entered clinical trials. Bapineuzumab, the first tested antibody in clinical trials, showed that although it tended to improve cognitive function, the results were not statistically significant [16]. After thorough analysis, it was considered that the lack of benefit may have been due to late intervention or insufficient dosage. Subsequently, more than five Aβ-targeted monoclonal antibodies have been studied in clinical trials, mainly in prodromal and early AD patients, but most of them failed to meet their primary objectives [17,18,19,20].The consecutive failures of clinical trials targeting Aβ raised doubts about whether Aβ is the major pathogenic agent or a valid target for AD [21].
AD is currently defined by pathological hallmarks, but these could be either the causes or the results of the disease. So, whether Aβ is a consequence, or an etiological agent of AD is a critical question to be answered. Previous studies (especially genetic) provided strong evidence to confirm the pivotal role of Aβ in the pathogenesis of AD. However, to test whether Aβ is the causative agent, the most critical evidence needed is the efficacy of disease-modifying therapies targeting Aβ accumulation which improve both the pathologic changes and cognitive decline in AD patients.
What has the success of lecanemab and donanemab clarified?
In 2021, the anti-Aβ auto-antibody aducanumab, which was potent in clearing brain Aβ deposits and effective in delaying cognitive decline in AD patients in a phase 3 trial (EMERGE), was given accelerated approval by the Food and Drug Administration (FDA) for AD [22]. This decision caused intense debate in the scientific community, as another phase 3 trial of aducanumab (ENGAGE), did not show a similar cognitive benefit [19].
Just as the debate was surging, the Clarity AD trial, a phase 3 trial testing the efficacy of anti-Aβ antibody, lecanemab, met all of its expected endpoints [23]. This trial included 1,795 subjects with early AD including mild cognitive impairment (MCI) and mild dementia due to AD, at the highest dose (10 mg/kg intravenously biweekly). After 18 months of treatment, all the primary and secondary outcomes were met. Compared with placebo, lecanemab reduced global cognitive decline measured with the Clinical Dementia Rating-Sum-of-Boxes (CDR-SB) by 27% at 18 months, which represented a treatment difference in the score change of −0.45. Statistically significant improvements were also achieved in all secondary endpoints, with the key secondary endpoint being the change in brain levels of Aβ measured by Aβ positron emission tomography (PET), the AD Assessment Scale-cognitive subscale14 (ADAS-cog14), the AD Composite Score (ADCOMS) and the AD Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS MCI-ADL). Aβ-PET showed that after 18 months of treatment, the intracerebral Aβ load in all patients treated with lecanemab was below 30 centiloids, with an average reduction of 59.1 centiloids. Assessing the patients’ clinical function through different scales, the ADAS-cog14 showed that the patients’ cognitive decline slowed by 26%, and the ADCOMS showed a 24% improvement in patients’ general abilities, and the ADCS MCI-ADL showed a 37% slowing in functional decline, all of which were consistent with the results of the primary outcome measured by the CDR-SB. Last but not least, patients treated with lecanemab had significantly lower plasma and CSF levels of phosphorylated Tau (p-Tau), significantly attenuated Tau- PET signal in the temporal lobe, and lower plasma neurofilament light chain (NfL) levels.
In summary, the Clarity AD trial demonstrates that the clearance of Aβ from the brain can slow the progression of cognitive decline and attenuate the advancement of AD. This finding is further supported by the recent phase 3 trial of donanemab [24], which exhibited a remarkable reduction of intracerebral Aβ load by 88 centiloids after 72 weeks of treatment and showed that donanemab could delay cognitive decline by up to 35% on its primary endpoint (iADRS). Moreover, the study revealed that 52% donanemab subjects achieved complete clearance of Aβ plaques from the brain within 12 months treatment, and 47% of the subjects did not show any clinical progression (defined as no decline in the CDR-SB score) [24].It is worth noting that even though these three antibodies have been successful, it is not appropriate to compare their efficacy solely based on the outcomes of the different trials. Future vigorous studies should be designed for head-to-head comparisons between the different monoclonal antibodies to determine the superiority.
Is Aβ the etiological agent?
To determine whether a substance is a causative factor, there are two major criteria: one is whether the substance can cause the occurrence of the disease; another is whether targeting this substance has a modifying effect on disease progression. A series of studies have now shown that the increased production and/or deficient clearance of Aβ can lead to the occurrence of AD, including: (1) in familial AD, Aβ overproduction due to mutations in the APP and presenilin (PS) genes is highly penetrant for the development of AD [25]; (2) in the elderly population, the APP gene mutation that decreases Aβ production significantly reduce the onset of AD [26]; (3) homozygotes of APOE4 alleles that increase Aβ accumulation also increase the risk of sporadic AD by 10-14 times [27]; (4) brain accumulation of Aβ is the initial event of the AD process even before cognitive impairment in both familial and sporadic AD [28, 29]; (5) experimental studies confirm the neurotoxic effects of Aβ [30]. However as noted above, a series of clinical trials on Aβ production and clearance failed to achieve significant effects on cognition and function, thus raising doubts on Aβ as the etiologic factor.
The Clarity and Trailblazer Alz2 AD trials have shown that after clearing Aβ in the brain, not only is the cognitive and functional decline slowed, but also the progression of AD is attenuated. These trials provide strong support for the idea that Aβ is a pathogenic factor and a viable therapeutic target, as they demonstrate that reducing brain Aβ accumulation through Aβ removal can effectively retard disease progression by slowing cognitive decline. In addition, there is clear evidence that the more the Aβ deposition is cleared, the lower the rate of cognitive decline in the analysis of pooled data from previous immunotherapy trials [31]. These data support the proposition that clearance of Aβ will bring cognitive benefits.
It should be noted that in the Clarity and Trailblazer Alz2 AD trials, the benefits of cognitive function are limited [21], and the patients’ cognition continues to decline, suggesting that other factors, such as Tau hyperphosphorylation, gliosis and oxidative stress, are at play in the development of AD. Therefore, future work is needed to investigate to what extent Aβ contributes to the development of AD, and how Aβ interacts with other pathological processes to drive the progression of AD.
Perspectives on Aβ clearance therapies
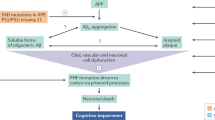
The success of the Clarity and Trailblazer Alz2 AD trials suggests that targeting Aβ is the right direction for future drug development of AD. It is expected that with the success of these trials, Aβ-targeting therapies will become a focal point in drug development. Several key questions need to be answered to validate and enlarge the therapeutic benefits of Aβ-targeting therapies (Fig. 1).

In the development of AD, Aβ firstly accumulates and deposits as plaques and perivascular aggregate and aggregated Tau in neurons. Other processes such as glycosis, oxidative stress, and microvascular dysfunction also participate. The Clarity AD trial and others demonstrate that clearance of Aβ brings cognitive benefits. Early intervention, targeting different Aβ species, robust Aβ clearance, longer intervention time and targeting multiple processes are essential to further validate and improve efficacy of Aβ targeting therapies.
Mechanisms of passive immunotherapy
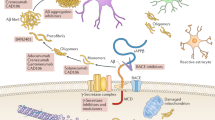
Anti-Aβ antibodies function through multiple mechanisms to eliminate Aβ from both the blood and the brain. In the blood, these antibodies bind specifically to Aβ, preventing its re-entry into the brain across the blood-brain barrier (BBB). The disruption of the balance of free Aβ between the central and peripheral compartments facilitates the efflux of Aβ from the brain. Approximately 0.1–0.3% of the anti-Aβ antibodies penetrate the BBB and directly bind to Aβ aggregates. Some antibodies can solubilize Aβ fibrils and inhibit the aggregation of Aβ into plaques. This promotes clearance by enhancing the accessibility of Aβ to other clearance mechanisms. More importantly, antibodies can opsonize Aβ, marking it for recognition by microglia. Subsequently, microglia engulf and degrade the Aβ through a process of phagocytosis, effectively clearing Aβ from the brain. These mechanisms work synergistically to clear Aβ [32], although further investigation is necessary to ascertain which mechanism plays a dominant role.
In terms of immunotherapy, the decline and dysregulation of immune function are also significant factors contributing to AD [33]. GWAS conducted on AD populations have identified a series of AD risk genes that are highly expressed in microglia and macrophages, indicating the crucial role of the innate immune system in AD [34]. In the aging brain, the phagocytic efficiency of microglia declines, resulting in ineffective clearance of Aβ and continuous accumulation of Aβ. Studies have demonstrated that the impaired ability of microglia to engulf Aβ is a major mechanism underlying the development of AD [35]. Furthermore, microglia release a substantial amount of pro-inflammatory factors and neurotoxic molecules which may contribute to cognitive impairment [36]. Moreover, in the aging brain, the reduced uptake and phagocytic capacity of peripheral monocytes towards Aβ, the dysregulation of T and B cell functions, and the imbalance between pro-inflammatory and anti-inflammatory factors may be crucial mechanisms in the progression of AD [37]. Therefore, targeting immune function represent a promising approach for therapy in AD.
At what stage will Aβ clearance produce maximal benefit?
It is likely that the failure of previous Aβ clearance trials could be attributed to late timing of the intervention, i.e., in the dementia phase of AD [38]. And this leads to the consensus of early intervention. It has been shown that AD begins 15-20 years before the onset of clinical symptoms and progresses through asymptomatic preclinical, and symptomatic prodromal, and dementia stages [39]. In recent trials, intervention has been shifted from the dementia phase to prodromal and preclinical stages [19, 40,41,42]. The pattern of efficacy in cognitive impairment slowing in previous trials in MCI and mild dementia stages suggest that the Aβ clearance treatment in the early clinical phases of AD may be too late to reverse the progression of the disease. It is well recognized that there is a functionally compensated stage of the disease where Aβ accumulation is approaching an advanced stage, and where subjects have no overt cognitive impairment [43]. In recent years, it has been argued that Aβ, as a trigger of AD pathogenesis, may be effective for intervention only in the preclinical stages of AD [44]. The success of the Clarity and Trailblazer Alz2 AD trials demonstrate that targeting Aβ is effective in MCI and mild dementia phases, suggesting that Aβ still plays a substantial role in the biologically advanced stage, i.e. not just a trigger effect, and that clinical efficacy can be achieved with interventions at biologically advanced stages of the disease. These findings encourage the investigation on the efficacy of Aβ clearance in moderate-to-severe dementia.
However, the Clarity and Trailblazer Alz2 AD trials also showed that the clinical benefits were limited when the treatment starts from the prodromal and dementia stages, even if Aβ accumulation is reduced to normal levels. The underlying reason may lie in the complex pathophysiology in the advanced stage of the disease, including Tau aggregation, and synaptic loss. At this stage, even if the effect of Aβ is removed, other events may form vicious cycles to promote cognitive deterioration [45]. From all viewpoints, preclinical intervention seeming to be the best future approach.
Which species of Aβ should be targeted?
Aβ is produced by the proteolytic cleavage of APP and exists in various lengths, including the most abundant forms, Aβ40 and Aβ42. Not all Aβ monomers are toxic [46]. Studies have confirmed that the toxicity of Aβ is independent of its size and depends mainly on its conformation, especially the exposure of the hydrophobic regions of Aβ peptide (residues 16–22 and 30–42) [47].The presence of the two hydrophobic amino acids at the C-terminus makes Aβ42 more aggregable than Aβ40, and thus Aβ42 is more toxic. Under physiological conditions, the production and clearance of Aβ maintain a balance. However, in AD, excessive Aβ in the brain aggregates through hydrophobic interactions, forming soluble Aβ oligomers, protofibrils, fibrils and ultimately, plaques and perivascular deposits. Microinjection of Aβ fibrils into the cerebral cortex of primates has been shown to cause neurodegeneration, reflecting the neurotoxicity of insoluble Aβ aggregates [48]. In addition, studies have shown that neurons exposed to Aβ oligomers fail to form new synapses, thus proving that soluble Aβ oligomers are neurotoxic [49]. Aβ dimers also show significant neurotoxicity [50]. In fact, soluble Aβ species seem to be in a complex equilibrium with the insoluble fibrils and plaques [51]. Thus, immunotherapy targeting Aβ clearance, both for soluble oligomers and insoluble fibrils, should be protective.
In the structure of Aβ fibrils, the N-terminus of the Aβ peptide is exposed on the surface, and the middle and C-terminal domains of Aβ are ‘masked’ inside. Therefore, antibodies targeting the N-terminus of Aβ can depolymerize and remove the aggregates by binding to the free N-terminus exposed on the surface, inducing microglial phagocytosis and enzymatic proteolysis. In contrast, antibodies targeting the middle segment and C-terminus of Aβ could not bind to fibrils and thus fail to remove Aβ plaques. To date, 17 drugs targeting Aβ have been tested in phase 3 clinical trials, among these, seven are monoclonal antibodies targeting Aβ clearance [52] (Table 1). Of these, solanezumab and crenezumab target the hydrophobic region in the middle of the Aβ peptide and bind specifically to Aβ monomers, and have been found to have little effect on global Aβ-PET levels or cognitive decline despite reducing the level of free Aβ in cerebrospinal fluid [40, 53]. The remaining five antibodies, which primarily bind to the immuno-dominant N-terminus of the Aβ and target Aβ aggregates such as oligomers, fibrils or plaques, all presented more or less signals of efficiency in Aβ clearance and cognitive benefit in phase 2 or phase 3 clinical trials [16, 19, 54, 55]. Aducanumab and gantenerumab showed higher affinities for Aβ oligomers and fibrils [56, 57]. The trials of aducanumab and gantenerumab both confirmed that they could effectively clear Aβ and improve cognitive function [58, 59]. Lecanemab primarily targets Aβ soluble oligomers [60]. Phase 3 clinical trials of lecanemab also demonstrated effective reduction of Aβ load in the brain and significant improvement in cognitive function, confirming a dynamic balance between soluble Aβ and Aβ load [23]. Donanemab, primarily targets N-terminal pyroglutamylated forms of Aβ, a post-translational structure that exists only in Aβ fibrils and plaques, and thus donanemab has a high affinity for insoluble Aβ aggregates. Recently published phase 3 clinical trial has shown donanemab to be even more effective, hitting the pause button for nearly half of AD patients [24]. Overall, the toxicity of Aβ species has not been fully elucidated. Developing antibodies targeting the conformation of toxic Aβ and specific post-translational modifications is a direction for future immunotherapy.
How much Aβ removal is effective to achieve cognitive benefits?
To date, seven antibodies targeting Aβ have undergone phase 3 clinical trials. Among them, lecanemab and donanemab trials have achieved most of the expected objectives, while aducanumab demonstrated effectiveness in slowing cognitive decline in one of the two trials. It is worth noting that there is a clear correlation between the clearance of brain Aβ deposition and the deceleration of cognitive decline [19, 52, 61]. Recently in two high-profile phase 3 trials, GRADUATE I and II, gantenerumab also failed to meet a primary objective of attenuating cognitive impairment, probably due to a lower level of Aβ clearance than expected [62]. The Clarity AD trial revealed that after 1.5 years of lecanemab treatment, 60% of patients experienced a decrease in Aβ deposition in the brain to threshold levels. Additionally, the Trailblazer Alz2 AD trial showed that after 18 months of treatment with donanemab, approximately 70% of patients had a reduction in Aβ burden to the threshold level. In contrast, gantenerumab, after 2 years of treatment, only achieved this threshold level in 28% of patients. These findings indicate that gantenerumab has a significantly lower ability to clear Aβ compared to lecanemab and donanemab. Based on analysis of past clinical trials, it is estimated that Aβ should be reduced to below 20–25 centiloid, which is the current conservative Aβ threshold to achieve significant clinical benefit [63,64,65,66]. This sets a criterion for the prediction of efficacy in future clinical trials. It should be noted that there are numerous factors that affect clinical efficacy, such as side effects of passive immunization, the disease stage at intervention, the length of treatment, anti-drug antibodies, dosage, and duration of treatment.
How long should the intervention last to achieve efficacy?
More recently, results from the open label extension trial of gantenerumab showed an increasing gap in cognitive improvement between patients who continued on high-dose gantenerumab and controls after an intervention lasting 104 weeks, suggesting that Aβ-clearing therapy needs sufficient time to show benefits [59]. Considering that the level of Aβ deposition in the brain does not correlate strongly with cognitive impairment and that Aβ may affect cognition through indirect pathways, such as inducing the hyperphosphorylation of Tau protein [67]. Sufficient time may be required for repairing neuronal damage and improving cognitive function after Aβ clearance [52].
Based on an analysis of previous clinical trials, it was proposed that within a given time, the shorter the duration required to reach amyloid negativity (TΔA), the longer the period required to reveal statistically significant clinical efficacy between treatment and placebo groups (TΔE), the greater the difference in cognitive function between the treatment and placebo groups [52]. Of the four anti-Aβ antibodies capable of Aβ clearance, lecanemab and donanemab were able to clear brain Aβ loads below the threshold within 12 months, whereas aducanumab required nearly 18 months to clear Aβ near the positive threshold, which may be a key reason for the success of lecanemab and donanemab [24, 52]. In addition, a “Quantitative ATN” (Q-ATN) model of AD was developed to predict changes in cognition as a function of Aβ removal [68]. Data from numerous observational studies were used to derive mathematical relationships, and the model’s predictions correlated well with actual clinical trial results. This model was applied to forecast the results of five years of treatment with gantenerumab, suggesting that the CDR-SB decline would reach 0.87 points at 27 months and that this difference would expand to 5.2 points after five years [69]. Eisai used an AD Archimedes condition-event (ACE) simulator to predict the long-term benefits of clearing Aβ. Projections based on the phase 2 trial of lecanemab showed that with lifetime administration, each stage of dementia would be delayed by 2.5–3 years in patients with an average age of 72; In patients with a mean age of 65, progression to mild and moderate AD would be expected to be delayed by 3.3 and 3.4 years, respectively [61]. These estimations suggest that the failure of past clinical trials of Aβ clearance may be related to insufficient observation time, which is 18 months in previous and current trials, and longer treatment periods should be considered in future trials.
Is clearing Aβ alone sufficient?
Although the Clarity AD study met all of the expected endpoints, the benefits in cognition were limited, as evidenced by the difference of only −0.45 in CDR-SB score from placebo [23]. This value is statistically significant but may not be clinically meaningful. It is well known that whether changes in scale scores are statistically significant depend largely on the sample size and the magnitude of the differences between groups. Even small differences can yield statistically significant p-values if a sufficiently large sample size is achieved, but this does not mean that they are clinically significant. How much of a change in a scale score is clinically significant depends on whether the change reaches the minimum clinically important difference (MCID). Prior work has suggested that the MICD for the CDR-SB assessment in mild AD is 1.63 [70, 71], which is greater than the 0.45 detected in the Clarity AD study. To date, all the anti-Aβ antibodies that target the reduction of Aβ levels in the brain have shown limited benefits in cognitive function. Also of interest is the fact that while numerous Aβ-lowering therapies have efficacy signals pointing toward clinical benefit, the disease still continues to progress despite Aβ load being normalized. This suggests that there may be additional factors promoting cognitive decline in the brain in addition to Aβ [45].
Aβ deposition levels do not correlate well with cognitive function [72], indicating that Aβ is not a proximal cause of cognitive dysfunction, although previous animal studies have shown that Aβ is neurotoxic and affects synaptic function [73]. Studies have shown that the presence or accumulation of Tau is a better predictor of cognitive impairment and is strongly correlated with the degree of cognitive impairment [67, 74]. There is much evidence that Aβ provokes the accumulation of Tau that ultimately correlates with neuronal loss [75]. In addition, there are other downstream events in the AD brain, such as oxidative stress and energy metabolism disorders, which might form vicious circles to affect the function of neural circuits and promote cognitive decline [76]. There are many ways that Aβ can cause cognitive impairment, including a direct effect on synapses, as well as indirect ways such as through the induction of Tau phosphorylation, gliosis, oxidative stress, energy dysmetabolism and vascular dysfunction, finally causing dysfunction of neurons and neural circuits [45, 77]. This could explain the limited benefits in cognition after Aβ clearance, as assessed by Aβ-PET.
How to improve the effect on cognition is a key issue to be addressed in the future for AD therapies. Emphasis is being placed on multi-targeted combination interventions. In terms of Aβ targeted therapy, reducing Aβ production and aggregation remains the principal direction. For example, specific γ- and β-secretase inhibitors and modulators have been developed to specifically reduce Aβ production without affecting other physiological mechanisms mediated by these secretases. The C-terminal region of apolipoprotein E (APOE-CT) can also selectively inhibit the cleavage of APP by γ-secretase, thus effectively reducing the production of Aβ [78]. However, whether it can be used for clinical treatment needs to be confirmed by further clinical trials. Current intervention strategies for Tau include inhibition of Tau hyperphosphorylation and promotion of aggregated Tau clearance. However, initial clinical trials have not been successful [79, 80]. The formation of neurofibrillary tangles is a downstream event of Aβ in terms of pathogenesis; it is recognized as a secondary change of AD and a result of driving factors that contribute to the development of the disease. The possibility of achieving efficacy by removing hyperphosphorylated Tau alone is limited. Therefore, intervention strategies other than removal of aggregated Tau may be more effective, especially to prevent Tau from aggregating, rather than to remove tangles after they have been formed. The relationship between Aβ and Tau has been extensively studied in the past and blocking Aβ induced Tau aggregation is an important intervention strategy for AD, especially in the early stage of the disease. An important observation in past clinical trials of Aβ clearance is that cognitive function continues to deteriorate after Aβ clearance, implying that processes including Tau aggregation are not exclusively dependent on the role of Aβ after disease initiation and that there are other factors that contribute to the development of AD [81, 82]. Future anti-Aβ therapies are likely to be combined with one or multiple co-therapies against oxidative stress, microglial dysfunction, mitochondrial dysfunction, or disruption of the blood-brain barrier. Importantly, neuronal injury and damage to neuronal circuits serve as the foundation for cognitive decline. Neuroprotection is crucial in preserving the cognition of patients with AD. To this purpose, the Alzheimer’s disease neuroprotection research initiative (ADNRI) has been proposed recently [83]. In general, effective interventions for AD require a comprehensive approach and a tertiary prevention strategy [84].
What do accompanying adverse effects tell us?
Although the fundamental purpose of developing a drug is to make it available to all AD patients, each drug has its own specific efficacy and side effect profile based on its mechanism of action. Aβ monoclonal antibodies are no exception. In the Clarity AD trial, subgroup analyses breaking down participants by age, sex, race, ethnicity, geographic region, disease stage, and use of symptomatic AD medications found treatment benefits of lecanemab across the board. However, results showed that patients who carried two copies of APOE4 appeared to post no treatment effect on the CDR-SB, considering that APOE4 began to cause Aβ deposition earlier than APOE4 noncarriers, resulting in a more severe and complex Aβ burden in the brain [23, 85]. The main side effects of Aβ monoclonal antibody are amyloid-related imaging abnormalities (ARIA) with edema (ARIA-E) or microhemorrhage (ARIA-H), the exact pathogenesis of which are uncertain and are currently presumed to be related to the clearance of aggregated Aβ from brain vessels [86]. All clinical trials to date have shown that ARIA-E and ARIA-H events were related to APOE4 genotype and occurred rarely in the placebo arm [18, 87]. In the Clarity AD trial, the rate of ARIA-E in lecanemab-treated patients was 12.6%, significantly higher than in the placebo group. APOE homozygous patients had the highest incidence of ARIA-E, reaching one-third, and one patient developed severe symptoms. The aducanumab trial also showed consistent results, with up to 66% of APOE4 homozygous patients developing ARIA-E. Meanwhile, lecanemab and aducanumab trials suggested that the incidence of ARIA-E in APOE4 heterozygous patients was 10% and 36%, respectively, which were significantly higher than those in non-APOE4 carriers (5% and 20%). Similarly, ARIA-E occurred in 40.6% of patients treated with donanemab who were APOE4 homozygous, significantly higher than the 22.8% of APOE4 heterozygotes and 15.7% of non-carriers. Two APOE4 heterozygous carriers eventually died as a result of severe ARIA-E [88]. These results suggest that patients with the APOE4 allele, especially APOE4 homozygote patients, have significantly higher risks than benefits when initially receiving Aβ antibodies treatment, and patients and their families should be fully informed of the risks of drug administration in subsequent clinical applications.
Notably, two patients in the Clarity AD trial experienced drug-related adverse events that resulted in severe cerebral hemorrhage or even death [23]. One was a man with atrial fibrillation who was on anticoagulants; another, a woman with cerebral amyloid angiopathy (CAA) who received thrombolytic tissue plasminogen activator (tPA) after a presumed stroke. Twenty years ago, research by Jucker suggested that tPA and anti-Aβ antibodies should not be given to the same AD patient, especially in the presence of CAA [89]; the Clarity AD trial seems to bear this out. Therefore, for patients who are taking anticoagulants for underlying diseases or have clear MRI evidence of CAA and are at higher risk of cerebral hemorrhage. Patients and their families should be fully informed of these risks.
Studies based on the aducanumab trial have also shown that baseline microhemorrhages and APOE4 carrier status are associated with an increase in the incidence of ARIA-E [87]. Caution is required when accepting these patients for treatments. Nevertheless, most ARIA-E events are asymptomatic and only some present as transient headaches which resolve typically within 12 to 16 weeks after initial detection, and without significant sequelae [18, 23, 87].
Another interesting result was that ventricular volume was significantly increased (by 0.5–1.0%) in patients treated with Aβ antibodies and significantly correlated with the frequency of ARIA [90]. However, it is important to clarify that enlarged ventricles do not mean that brain atrophy is aggravated. It is well known that ventricular volume depends on the volume of brain parenchyma and changes in ventricular contents. As the study showed, the Aβ antibody did not cause significant changes in hippocampal and whole brain volume, meaning that the volume of the brain parenchyma did not change significantly, so that enlarged ventricles may be due to an increase in ventricular contents, with changes in CSF at the core. Clearance of Aβ from the cerebral vasculature may lead to an increase in vascular permeability and a decrease in colloidal osmotic pressure, which partially manifests itself in the parenchyma as ARIA, whereas changes of the choroid plexus capillaries may lead to an increase in the production of CSF, and at the same time, interstitial fluid joins the CSF, leading to an increase in the volume of the CSF, which in turn may lead to ventricular enlargement. Significant interstitial edema was also seen on brain images of patients presenting with ARIA, which was also confirmed from an imaging perspective [91]. It is also in line with findings that there is a significant correlation between ventricular volume and the frequency of ARIA. Consequently, the patients’ cognitive function eventually improved rather than suffering once the ARIA is controlled. In addition, as mentioned in our previous study, immunotherapy targeting Aβ clearance might produce a “dust-raising effect “, i.e., the conversion of deposited Aβ into more toxic soluble oligomers, which might cause accelerated neuronal degeneration, leading to a reduction in brain parenchymal volume and enlargement of ventricles [32]. Last but not least, the loss of brain volume does not exclude a secondary alteration of the inflammatory response in the brain attenuated by immunotherapy. In conclusion, the loss of brain volume could be the result of a combination of factors, and whether it is essentially a protective secondary alteration or a result of toxic effects needs to be further verified in future studies.
Conclusions
The success of Aβ targeting humanized antibodies lecanemab and donanemab supports the theory that Aβ is the etiologic agent of AD, and encourages researchers to explore the pathogenesis of AD from the perspective of Aβ. Currently, a number of phase 3 clinical trials of immunotherapy for AD are underway, including an ongoing trial of lecanemab targeting the presymptomatic stage (the AHEAD Study) and the Trailblazer-Alz3 trial of donanemab. The next generation of donanemab (remternetug) is showing exceptional efficacy [92]. While the Clarity and Trailblazer Alz2 AD trials give us confidence in targeting Aβ, there are still a multitude of challenges which need to be addressed to further validate and improve therapeutic benefits for the future. Furthermore, we also advocate for a multi-target approach in the treatment of AD, beyond Aβ clearance, especially focusing on neuroprotective therapies for damaged neuronal circuits.
References
Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66.
Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–5.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–9.
Hampel H, Hardy J, Blennow K, Chen C, Perry G, Kim SH, et al. The Amyloid-beta Pathway in Alzheimer’s Disease. Mol Psychiatry. 2021;26:5481–503.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608.
Beyreuther K, Masters CL. Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1991;1:241–51.
Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–98.
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharm Sci. 1991;12:383–8.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5.
Kumar M, Cohen D, Eisdorfer C. Serum IgG brain reactive antibodies in Alzheimer disease and Down syndrome. Alzheimer Dis Assoc Disord. 1988;2:50–55.
Solomon B, Koppel R, Hanan E, Katzav T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta-amyloid peptide. Proc Natl Acad Sci USA. 1996;93:452–5.
Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci USA. 1997;94:4109–12.
Schenk D. Hopes remain for an Alzheimer’s vaccine. Nature. 2004;431:398.
Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54.
Hartman RE, Izumi Y, Bales KR, Paul SM, Wozniak DF, Holtzman DM. Treatment with an amyloid-beta antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25:6213–20.
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl J Med. 2014;370:322–33.
Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017;9:95.
Ostrowitzki S, Bittner T, Sink KM, Mackey H, Rabe C, Honig LS, et al. Evaluating the Safety and Efficacy of Crenezumab vs Placebo in Adults With Early Alzheimer Disease: Two Phase 3 Randomized Placebo-Controlled Trials. JAMA Neurol. 2022;79:1113–21.
Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s Dement. 2021;17:696–701.
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease. N. Engl J Med. 2014;370:311–21.
Kepp KP, Robakis NK, Høilund-Carlsen PF, Sensi SL, Vissel B. The amyloid cascade hypothesis: an updated critical review. Brain. 2023;146:3969–90.
Alexander GC, Knopman DS, Emerson SS, Ovbiagele B, Kryscio RJ, Perlmutter JS, et al. Revisiting FDA Approval of Aducanumab. N. Engl J Med. 2021;385:769–71.
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in Early Alzheimer’s Disease. N. Engl J Med. 2023;388:9–21.
Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. 2023;330:512–27.
Rosenberg RN, Lambracht-Washington D, Yu G, Xia W. Genomics of Alzheimer Disease: A Review. JAMA Neurol. 2016;73:867–74.
Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive. Nature. 2012;488:96–99.
Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl J Med. 2012;367:795–804.
Wang HF, Shen XN, Li JQ, Suckling J, Tan CC, Wang YJ, et al. Clinical and biomarker trajectories in sporadic Alzheimer’s disease: A longitudinal study. Alzheimers Dement. 2020;12:e12095.
Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–57.
CLINICAL PHARMACOLOGY AND BIOPHARMACEUTICS REVIEW(S). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761178Orig1s000ClinPharm_Redacted.pdf, 2021, Accessed Date Accessed 2021.
Liu YH, Giunta B, Zhou HD, Tan J, Wang YJ. Immunotherapy for Alzheimer disease: the challenge of adverse effects. Nat Rev Neurol. 2012;8:465–9.
Wang X. A Bridge Between the Innate Immunity System and Amyloid-β Production in Alzheimer’s Disease. Neurosci Bull. 2021;37:898–901.
Bertram L, Tanzi RE. Alzheimer disease risk genes: 29 and counting. Nat Rev Neurol. 2019;15:191–2.
Sarlus H, Heneka MT. Microglia in Alzheimer’s disease. J Clin Investig. 2017;127:3240–9.
Franco-Bocanegra DK, McAuley C, Nicoll JAR, Boche D. Molecular Mechanisms of Microglial Motility: Changes in Ageing and Alzheimer’s Disease. Cells. 2019;8:639.
Chen X, Holtzman DM. Emerging roles of innate and adaptive immunity in Alzheimer’s disease. Immunity. 2022;55:2236–54.
Fan DY, Wang YJ. Early Intervention in Alzheimer’s Disease: How Early is Early Enough? Neurosci Bull. 2020;36:195–7.
Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397:1577–90.
Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl J Med. 2018;378:321–30.
Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimer’s Res Ther. 2021;13:80.
Rafii MS, Sperling RA, Donohue MC, Zhou J, Roberts C, Irizarry MC, et al. The AHEAD 3-45 Study: Design of a prevention trial for Alzheimer’s disease. Alzheimer’s Dement. 2023;19:1227–33.
Golde TE, Schneider LS, Koo EH. Anti-abeta therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69:203–13.
De Strooper B, Karran E. The Cellular Phase of Alzheimer’s Disease. Cell. 2016;164:603–15.
Golde TE. Disease-Modifying Therapies for Alzheimer’s Disease: More Questions than Answers. NeuroTherapeutics. 2022;19:209–27.
Izuo N, Murakami K, Sato M, Iwasaki M, Izumi Y, Shimizu T, et al. Non-toxic conformer of amyloid β may suppress amyloid β-induced toxicity in rat primary neurons: implications for a novel therapeutic strategy for Alzheimer’s disease. Biochem Biophys Res Commun. 2013;438:1–5.
Ladiwala AR, Litt J, Kane RS, Aucoin DS, Smith SO, Ranjan S, et al. Conformational differences between two amyloid β oligomers of similar size and dissimilar toxicity. J Biol Chem. 2012;287:24765–73.
Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med. 1998;4:827–31.
Zhao Y, Sivaji S, Chiang MC, Ali H, Zukowski M, Ali S, et al. Amyloid Beta Peptides Block New Synapse Assembly by Nogo Receptor-Mediated Inhibition of T-Type Calcium Channels. Neuron. 2017;96:355–72.e356.
Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, et al. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science. 2019;365:559–65.
Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2019;15:73–88.
Karran E, De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov. 2022;21:306–18.
Meilandt WJ, Maloney JA, Imperio J, Lalehzadeh G, Earr T, Crowell S, et al. Characterization of the selective in vitro and in vivo binding properties of crenezumab to oligomeric Aβ. Alzheimers Res Ther. 2019;11:97.
Salloway S, Farlow M, McDade E, Clifford DB, Wang G, Llibre-Guerra JJ, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat Med. 2021;27:1187–96.
Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res Ther. 2021;13:80.
Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, et al. Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J Alzheimer’s Dis. 2012;28:49–69.
Arndt JW, Qian F, Smith BA, Quan C, Kilambi KP, Bush MW, et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci Rep. 2018;8:6412.
Alexander GC, Emerson S, Kesselheim AS. Evaluation of Aducanumab for Alzheimer Disease: Scientific Evidence and Regulatory Review Involving Efficacy, Safety, and Futility. JAMA. 2021;325:1717–8.
Klein G, Delmar P, Voyle N, Rehal S, Hofmann C, Abi-Saab D, et al. Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res Ther. 2019;11:101.
Söderberg L, Johannesson M, Nygren P, Laudon H, Eriksson F, Osswald G, et al. Lecanemab, Aducanumab, and Gantenerumab - Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics. 2023;20:195–206.
Tahami Monfared AA, Tafazzoli A, Ye W, Chavan A, Zhang Q. Long-Term Health Outcomes of Lecanemab in Patients with Early Alzheimer’s Disease Using Simulation Modeling. Neurol Ther. 2022;11:863–80.
Gantenerumab Falls Short in Phase 3. https://www.alzforum.org/news/research-news/gantenerumab-falls-short-phase-3-0, 2022, Accessed Date Accessed 2022 Accessed.
Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: a prospective cohort study. Lancet Neurol. 2012;11:669–78.
La Joie R, Ayakta N, Seeley WW, Borys E, Boxer AL, DeCarli C, et al. Multisite study of the relationships between antemortem [(11)C]PIB-PET Centiloid values and postmortem measures of Alzheimer’s disease neuropathology. Alzheimer’s Dement. 2019;15:205–16.
Amadoru S, Doré V, McLean CA, Hinton F, Shepherd CE, Halliday GM, et al. Comparison of amyloid PET measured in Centiloid units with neuropathological findings in Alzheimer’s disease. Alzheimers Res Ther. 2020;12:22.
Knopman DS, Lundt ES, Therneau TM, Albertson SM, Gunter JL, Senjem ML, et al. Association of Initial β-Amyloid Levels With Subsequent Flortaucipir Positron Emission Tomography Changes in Persons Without Cognitive Impairment. JAMA Neurol. 2021;78:217–28.
Long JM, Holtzman DM. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell. 2019;179:312–39.
Delor I, Charoin JE, Gieschke R, Retout S, Jacqmin P. Modeling Alzheimer’s Disease Progression Using Disease Onset Time and Disease Trajectory Concepts Applied to CDR-SOB Scores From ADNI. CPT: Pharmacomet Syst Pharmacol. 2013;2:e78.
Could Benefit of Plaque Removal Grow in Time? https://www.alzforum.org/news/conference-coverage/could-benefit-plaque-removal-grow-time, 2022, Accessed Date Accessed 2022 Accessed.
Andrews JS, Desai U, Kirson NY, Zichlin ML, Ball DE, Matthews BR. Disease severity and minimal clinically important differences in clinical outcome assessments for Alzheimer’s disease clinical trials. Alzheimer’s Dement. 2019;5:354–63.
Liu KY, Schneider LS, Howard R. The need to show minimum clinically important differences in Alzheimer’s disease trials. lancet Psychiatry. 2021;8:1013–6.
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–81.
Liu F, Sun J, Wang X, Jin S, Sun F, Wang T, et al. Focal-type, but not Diffuse-type, Amyloid Beta Plaques are Correlated with Alzheimer’s Neuropathology, Cognitive Dysfunction, and Neuroinflammation in the Human Hippocampus. Neurosci Bull. 2022;38:1125–38.
Consortium AB, Jia YJ, Wang J, Ren JR, Chan P, Chen S, et al. A framework of biomarkers for brain aging: a consensus statement by the Aging Biomarker Consortium. Life Med. 2023;2:lnad017.
Sato C, Barthélemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron. 2018;97:1284–98.e1287.
Ying Y, Wang JZ. Illuminating Neural Circuits in Alzheimer’s Disease. Neurosci Bull. 2021;37:1203–17.
Lu MH, Zhao XY, Yao PP, Xu DE, Ma QH. The Mitochondrion: A Potential Therapeutic Target for Alzheimer’s Disease. Neurosci Bull. 2018;34:1127–30.
Hou X, Zhang X, Zou H, Guan M, Fu C, Wang W, et al. Differential and substrate-specific inhibition of γ-secretase by the C-terminal region of ApoE2, ApoE3, and ApoE4. Neuron. 2023;111:1898–913.e1895.
Tsai RM, Miller Z, Koestler M, Rojas JC, Ljubenkov PA, Rosen HJ, et al. Reactions to Multiple Ascending Doses of the Microtubule Stabilizer TPI-287 in Patients With Alzheimer Disease, Progressive Supranuclear Palsy, and Corticobasal Syndrome: A Randomized Clinical Trial. JAMA Neurol. 2020;77:215–24.
Vaz M, Silvestre S. Alzheimer’s disease: Recent treatment strategies. Eur J Pharmacol. 2020;887:173554.
Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2019;15:73–88.
Busche MA, Grienberger C, Keskin AD, Song B, Neumann U, Staufenbiel M, et al. Decreased amyloid-β and increased neuronal hyperactivity by immunotherapy in Alzheimer’s models. Nat Neurosci. 2015;18:1725–7.
Liu J, van Beusekom H, Bu XL, Chen G, Henrique Rosado de Castro P, Chen X, et al. Preserving cognitive function in patients with Alzheimer’s disease: The Alzheimer’s disease neuroprotection research initiative (ADNRI). Neuroprotection. 2023.in press.
Bu XL, Jiao SS, Lian Y, Wang YJ. Perspectives on the Tertiary Prevention Strategy for Alzheimer’s Disease. Curr Alzheimer Res. 2016;13:307–16.
Zhang Q, Xie C. Apolipoprotein E Drives Early Blood-Brain Barrier Damage in Alzheimer’s Disease. Neurosci Bull. 2021;37:281–3.
Tolar M, Abushakra S, Hey JA, Porsteinsson A, Sabbagh M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res Ther. 2020;12:95.
Salloway S, Chalkias S, Barkhof F, Burkett P, Barakos J, Purcell D, et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients With Early Alzheimer Disease. JAMA Neurol. 2022;79:13–21.
Gueorguieva I, Willis BA, Chua L, Chow K, Ernest CS, Shcherbinin S, et al. Donanemab Population Pharmacokinetics, Amyloid Plaque Reduction, and Safety in Participants with Alzheimer’s Disease. Clin Pharmacol Therap. 2023;113:1258–67.
Winkler DT, Biedermann L, Tolnay M, Allegrini PR, Staufenbiel M, Wiessner C, et al. Thrombolysis induces cerebral hemorrhage in a mouse model of cerebral amyloid angiopathy. Ann Neurol. 2002;51:790–3.
Alves F, Kalinowski P, Ayton S. Accelerated Brain Volume Loss Caused by Anti-β-Amyloid Drugs: A Systematic Review and Meta-analysis. Neurology. 2023;100:e2114–24.
Filippi M, Cecchetti G, Spinelli EG, Vezzulli P, Falini A, Agosta F. Amyloid-Related Imaging Abnormalities and β-Amyloid-Targeting Antibodies: A Systematic Review. JAMA Neurol. 2022;79:291–304.
Safety and Amyloid Plaque Reduction Effects of Remternetug in Patients with Alzheimer’s Disease: Interim Analysis from a Phase 1 Study. https://assets.ctfassets.net/mpejy6umgthp/51Sv0wOrFxfiHJNqcyuNYu/0c5df804e8a23256262fcb40489ae181/REMIPT3_ADPD2023_JIN_SAFETY_PLAQUE_REDUCTION_Ph1LAKB.pdf, 2023, Accessed Date Accessed 2023 Accessed.
Vandenberghe R, Rinne JO, Boada M, Katayama S, Scheltens P, Vellas B, et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res Ther. 2016;8:18.
Sperling RA, Donohue MC, Raman R, Rafii MS, Johnson K, Masters CL, et al. Trial of Solanezumab in Preclinical Alzheimer’s Disease. N Engl J Med. 2023;389:1096–107.
Acknowledgements
This study is supported by Natural Science Foundation of China (Grant No. 82120108010 and 81930028).
Author information
Authors and Affiliations
Contributions
All authors contributed to conceptualisation, drafting, review and editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
YJW received research funding from the Natural Science Foundation of China (Grant No. 82120108010 and 81930028). The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lian, Y., Jia, YJ., Wong, J. et al. Clarity on the blazing trail: clearing the way for amyloid-removing therapies for Alzheimer’s disease. Mol Psychiatry 29, 297–305 (2024). https://doi.org/10.1038/s41380-023-02324-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-023-02324-4
- Springer Nature Limited