
Abstract
Autism spectrum disorder (ASD) is a neurodevelopmental disorder whose pathophysiological mechanisms are still unclear. Hypotheses suggest a role for glutamate dysfunctions in ASD development, but clinical studies investigating brain and peripheral glutamate levels showed heterogenous results leading to hypo- and hyper-glutamatergic hypotheses of ASD. Recently, studies proposed the implication of elevated mGluR5 densities in brain areas in the pathophysiology of ASD. Thus, our objective was to characterize glutamate dysfunctions in adult subjects with ASD by quantifying (1) glutamate levels in the cingulate cortex and periphery using proton magnetic resonance spectroscopy and metabolomics, and (2) mGluR5 brain density in this population and in a validated animal model of ASD (prenatal exposure to valproate) at developmental stages corresponding to childhood and adolescence in humans using positron emission tomography. No modifications in cingulate Glu levels were observed between individuals with ASD and controls further supporting the difficulty to evaluate modifications in excitatory transmission using spectroscopy in this population, and the complexity of its glutamate-related changes. Our imaging results showed an overall increased density in mGluR5 in adults with ASD, that was only observed mostly subcortically in adolescent male rats prenatally exposed to valproic acid, and not detected in the stage corresponding to childhood in the same animals. This suggest that clinical changes in mGluR5 density could reflect the adaptation of the glutamatergic dysfunctions occurring earlier rather than being key to the pathophysiology of ASD.
Similar content being viewed by others

Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder that begins in utero, exhibiting very varying evolutionary trajectories and degrees of intensity [1]. All subtypes of ASD are characterized by impaired social communication and repetitive and restricted behaviors and interests [2]. In the last decades, a large increase in the prevalence of ASD has been observed making ASD as a major public health problem [3]. As the etiology and pathophysiology of ASD remain unclear, the definition of targets of interest for the development of pharmaceutical ASD therapies has been difficult. Distinct hypotheses describe an imbalance between excitation and inhibition as a common pathophysiological mechanism suggesting a crucial role of glutamate and GABA dysfunctions in ASD [4,5,6].
Research related to glutamate dysfunctions in ASD include many genetic studies depicting the association between ASD and polymorphisms in genes coding subunits of glutamate receptors and transporters [7], along with structural proteins of the excitatory synapse [8, 9]. Post-mortem and in vivo studies reported either increased or decreased glutamate levels in several brain regions and peripheral compartments of subjects with ASD leading to the propositions of hyper- and hypo-glutamate theories of autism followed by the excitatory/inhibitory imbalance theory [10, 11]. Studies related to glutamate receptor densities are scarce, and the involvement of glutamatergic neurotransmission in autism remains unresolved.
Recently, studies highlighted a potential role of the metabotropic glutamate receptors subtype 5 (mGluR5) in the pathophysiology of ASD. This receptor has been shown to play crucial roles in synaptic plasticity and neuronal development [12, 13]. It is also a target of the fragile X mental retardation protein whose expression is compromised in X-fragile syndrome (FXS) frequently associated with ASD [14]. Thus, preclinical arguments supported the development of negative allosteric modulators of mGluR5 as treatments for FXS [15]. Two mGluR5 negative allosteric modulators (basimglurant, mavoglurant) have been evaluated in subjects with FXS [16,17,18], but no therapeutic benefits have been reported for unclear reasons yet. The interest for targeting mGluR5 in ASD has been highlighted by a post-mortem study showing increased cerebellar mGluR5 densities in ASD vs. controls [19], and recently extended to the postcentral gyrus in vivo using positron emission tomography (PET) in few men with ASD [20, 21].
To further understand the role of the glutamatergic transmission and the mGluR5 in the pathophysiology of ASD, we conducted a multimodal clinical study combining proton magnetic resonance spectroscopy (1H-MRS), metabolomics and Positron Emission Tomography (PET) to explore the cerebral and peripheral glutamate levels along with the brain mGluR5 density in adults with ASD and controls. In addition, we performed a longitudinal preclinical PET imaging study in a well characterized animal model of ASD (male rats prenatally exposed to valproic acid) to describe the changes in mGluR5 density at two developmental stages corresponding to childhood and adolescence in human [22].
Materials/subject and methods
Clinical and behavioral evaluations
Participants
Two groups consisting in 12 adult males with ASD (ASD group) and 14 healthy adult males (CTRL group) were recruited through the University Child Psychiatry Center of Tours (France) and matched on age (mean age (years) of 29.4 ± 7.2 and 29.8 ± 8.3 ranging from 24 to 45 and 24 to 47 for ASD and CTRL groups, respectively) (Table 1). Healthy volunteers with history of neurological disorders (including epilepsy and Attention Deficit/Hyperactivity Disorder) were excluded, as those with current or history of neuropsychiatric disorders according to the Mini DSM-IV [23].
ASD subjects were diagnosed by an experienced team of clinicians according to DSM-5 criteria [24], complemented by Autism Diagnostic Interview-Revised assessment (ADI-R) [25]. Autism Diagnostic Observation Schedule (ADOS-2) [26] was also available for 10 participants.
The Wechsler Adult Intelligence Scale (WAIS-III, WAIS-IV) [27, 28] had been administered to assess verbal and performance IQ scores for 11 subjects with ASD, and the PEP-3 (Psychoeducational profile) [29] had been used to assess verbal and non-verbal developmental ages for one individual. The Revised Behavior Summarized Evaluation Scale [30] (BSE-R, 29 items rated on a 5-point scale) and The Repetitive and Restricted Behavior scale [31] (RRB, 35 items rated on a 5-point scale) complemented the quantitative assessment of behavioral difficulties of ASD subjects. In the CTRL group, an estimation of verbal and performance IQ scores was performed using four subtests (vocabulary, similarities, block design, and matrix) of the WAIS. Self-report questionnaires were completed in both groups: the Autism-Spectrum Quotient (AQ) to quantify autistic-like traits [32] and the Empathy Quotient (EQ) [33] to quantify the cognitive and affective domains of empathy.
Informed written consent was obtained from all participants or from their legal guardian. The protocol (clinical trial 2015-001897-16) was approved by the Ethics Committee of the University Hospital of Tours (Comité de Protection des Personnes (CPP) Région Centre Ouest I). The study was conducted according to the ethical principles of the Declaration of Helsinki.
In vivo 1H-MRS acquisitions
Before PET scan, each subject underwent a Magnetic Resonance Imaging (MRI) study for brain anatomical imaging to define Regions Of Interest (ROIs), and to allow co-registration with PET images. MRI was performed using a 3-Tesla whole-body scanner (Verio®, Siemens, Germany), and a 12 channels head coil. The same protocol as previously described [34] was also used to measure the levels of glutamate and glutamine in a voxel of interest (5.94 ml) including parts of the anterior and mid-cingulate cortices. We focused on this brain region based on its implication on a broad range of behaviors and cognitive processes that are modified in ASD individuals [35,36,37], and on the heterogenous results previously reported by other studies. The echo time of 35 ms was chosen to improved SNR (signal to noise ratio) due to reduced T2 relaxation, reduced dependence on assumed metabolite T2 values, and improved SNR for J‐coupled metabolite signals due to reduced dephasing [38].
All spectra were analyzed in Osprey (Open-Source Processing, Reconstruction & Estimation of Magnetic Resonance Spectroscopy Data; committed version 89f5f31) [39] with all possible metabolites in the basis set. The strength of this all-in-one toolbox for processing of magnetic resonance spectroscopy data is to allow tissue correction and quantification into a coherent ecosystem [40,41,42]. Spectra were fitted between 0.2 and 4.0 ppm using baseline correction. Metabolite levels were calculated relative to the unsuppressed voxel water and were expressed in institutional units (I.U., approximately millimolar) [40,41,42], where the fraction of CSF, gray and white matter were calculated by segmentation of the anatomical T1-weighted images using SPM 12 (https://www.fil.ion.ucl.ac.uk/spm/software/spm12/). Metabolite levels were included in further statistical analyses only when the SNR was superior to 3, the full width at half maximum (FWHM) < 0.1 ppm [38], and after visual inspection of each spectrum. Five spectra were excluded from the analyses based on those criteria. To represent the voxel locations, we used a basic visualization approach that give information about the MRS localization and its robustness according to an in-between participants view [43]. Glutamate (Glu) and Glu + Gln (Glx) were compared between ASD and HC groups using Student’s t test or Mann–Whitney U test. Because we analyzed two metabolites, Bonferroni correction was used with a final statistical significance of p < 0.025.
[18F]FPEB radiosynthesis and mGluR5 PET Imaging
The expression of mGluR5 was assessed using [18F]FPEB (3-[(18)F]fluoro-5-(2-pyridinylethynyl)benzonitrile) [44]. No-carrier-added [18F]FPEB was radiolabeled as previously described [34]. [18F]FPEB was demonstrated to meet all quality control criteria for a PET radiopharmaceutical. Molar activity at the end of synthesis was 114.9 ± 58.3 GBq/μmol for the PET study involving both experimental groups.
After a CT acquisition (40 mA, 80 kv) for attenuation correction, healthy volunteers were intravenously injected with [18F]FPEB as a bolus, and underwent a 90 min brain dynamic acquisition using an Ingenuity Philips TF64 tomograph® (Philips, USA). List mode data were reconstructed into 31 frames (6 × 10 s, 8 × 30 s, 4 × 1 min, 5 × 2 min, 8 × 5 min). To shorten the acquisition duration for the ASD group, PET data were collected between 30 and 50 min post [18F]FPEB injection (257 ± 44 and 261 ± 32 MBq injected to ASD and control groups, respectively). Thus, the same time window of 20-min images from 30 to 50 min post [18F]FPEB injection was used for comparison between the two groups (please see the supplementary information for more details regarding the PET protocol). Data were reconstructed with the standard package included with the system (PET view software-Philips Medical Systems). PET sinograms were corrected for attenuation, decay, scatter, and randoms, and reconstructed using a 3D iterative RAMLA algorithm (voxels of 2 × 2 × 2 mm3). PET images were coregistered to subject’s T1 3D MRI, and into maximal standard uptake values (SUVmax) with the brain stem as the reference region. Brain differences in [18F]FPEB SUVmax between ASD and CTRL groups were measured via a voxel-based analysis using unpaired Student’s two-tailed t-test with p values corrected for multiple comparisons using the Benjamini-Hochberg control of false discovery rate [45]. Z-score maps with a threshold of p = 0.05 were generated, along with d value maps to evaluate the effect size (moderate, large or very large effect size for d values inferior to 0.80, between 0.80 and 1.20, or superior to 1.20, respectively) [46, 47]. The Hammer’s template from PMOD v3.2 software (PMOD Technologies Ltd, Switzerland) was applied to z-score and d value maps to obtain cerebral z and d values.
Metabolomics analysis
Liquid Chromatography-High Resolution Mass Spectrometry (LC-HRMS) analysis was performed from 50 µl of serum or 20 µl of urines as previously described [48]. The analysis of metabolites related to glutamate metabolism (Glu, Gln, aspartate, alanine, succinate, fumarate, urocanate in both serum and urines, along with GABA, valine, malate specifically in the serum and isoleucine, leucine specifically in urines) was performed based on our library of standard metabolites (IROA Technologies). The identification criteria were identical to those previously described [48]. The signal value was calculated using Xcalibur software (Thermo Fisher Scientific, San Jose, CA) providing the identity of each metabolite and its corresponding area after validation by a mass spectrometry specialist. Data were normalized to the sum, and the univariate analysis of metabolite levels between groups was based on non-parametric Mann-Whitney tests.
Preclinical evaluations
Model of valproate prenatal exposure
Wistar pregnant females (CERJ, Le Genest) arrived in the animal facility at gestational day 6 (GD6) and were randomly assigned to control or VPA groups (n = 11 per group). Pregnant dams were housed in individual cages (lights on between 20:00 and 8:00; 21 ± 1 °C; 55 ± 5% humidity; ad libitum access to food and water) and received an intraperitoneal injection of saline or valproate (VPA; 600 mg/kg; Sigma®) at GD12.5. After birth, each mother stayed with her litter until postnatal day 21 (PND21). Experiments were performed on the male progeny using the litter as statistical unit. The animals were treated in accordance with the European Community Council Directive 2010/63/EU for laboratory animal care with Regional Ethical Committee authorizations (no. 00620.02).
Social behaviors in VPA vs. control rats
Social interactions and play behaviors were evaluated in VPA and control rats at PND31 and PND33, respectively (n = 6 control vs. 5 VPA rats, one animal/litter). Social preference was assessed as previously described [49] using a 3-chamber apparatus with rats habituated to the device (5 min), and placed back with the possibility to interact with a rat from the same or another litter (5 min) in the opposite areas. The time spent sniffing the familiar or unfamiliar peer was quantified as an index of social preference. Play behavior was quantified by placing two VPA or control rats matched for weight from the same litter in a circular open field (Diameter 40 cm) whose floor was covered with sawdust (2 cm) (PND33). The time spent doing play behaviors (boxing, spinning, jumping, pursuits, attacks) was quantified for 15 min. Differences between VPA and control rats were analyzed using a Mann-Whitney test.
PET imaging and data analysis
[18F]FPEB PET imaging was performed on VPA and control male progenies at PND21 and PND35 as previously described [34]. Each rat was i.v. injected with a bolus of [18F]FPEB (18.83 ± 0.56 MBq and 23.47 ± 0.57 MBq on PND21 and PND35, respectively). PET acquisitions (50 min) were performed using a microPET-CT SuperArgus system (Sedecal, Madrid, Spain). The PET list-mode scans were rebinned into 45 frames (15x20s, 15x40s, 10x60s, 5x300s). Images were corrected for randoms, scatter, attenuation, and reconstructed using a 2-dimensional OSEM algorithm (GE Healthcare, France; voxels of 0.388 × 0.388 × 0.775 mm3). Partial volume effect corrections were applied on all images. A voxel-based analysis was performed with a threshold of p = 0.01 for the z-score maps to minimize the type I error rate [50]. See Supplementary Information for details concerning the MRI template for early adolescent male rats used in this study.
Results
Clinical study
Demographics and clinical characteristics are detailed in Table 1.
Anterior/mid cingulate glutamate levels were not different in adults with ASD and healthy controls
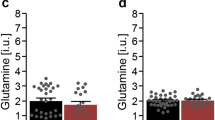
The left and right cingulate Glu and Glx levels in our participants at rest were not significantly different (left Glu levels, 19.29 ± 7.91 and 13.51 ± 4.65 in ASD and HC respectively, U(20) = 2.13, p = 0.04; left Glx levels, 35.18 ± 13.93 vs. 25.30 ± 9.01, t(20) = 2.01, p = 0.06; right Glu levels, 15.21 ± 6.02 vs. 13.06 ± 4.82, t(22) = 0.97, p = 0.34; right Glx levels, 27.83 ± 10.27 vs. 24.81 ± 8.46, U(22) = 0.79, p = 0.44; Fig. 1A, B).

Proton magnetic resonance spectroscopy of the cingulate cortex (A left hemisphere, B right hemisphere) with the voxel overlap density and the boxplot of Glu and Glx in individuals with ASD (n = 10 and 11 for the left and right hemispheres) vs. CTRL (n = 12 and 13 for the left and right hemispheres), each point corresponding to one participant (mean ± standard deviation). Quantification of the Glu sub-metabolomes in the serum and urines of individuals with ASD and CTRL showing the statistical significance of the differences observed for each quantified metabolite between the two groups (C), and the associated boxplot (median ± quartile for each subject) for the significantly different metabolites (D). Ala alanine, Asp aspartate, ASD autism spectrum disorder group, Cing cingulate cortex, CTRL control group, Ile isoleucine, Leu leucine, Glu glutamate, Gln glutamine, Glx glutamate + glutamine, Val valine.
Differences in the glutamate sub-metabolome were detected in the serum of subjects with ASD vs. controls
In the serum, significantly lower levels of glutamate (1.5 × 10−3 ± 0.6 × 10−3 vs. 2.0 × 10−3 ± 0.6 × 10−3 in ASD vs. HC respectively, U(25) = 32, p = 0.013) and higher levels of glutamine (29.4 × 10−3 ± 2.4 × 10−3 vs. 25.6 × 10−3 ± 3.0 × 10−3 in ASD vs. HC respectively, U(25) = 133, p = 0.001), GABA (2.2 × 10−3 ± 0.6 × 10−3 vs. 1.6 × 10−3 ± 0.4 × 10−3 in ASD vs. HC respectively, U(25) = 124, p = 0.009) and succinate (6.9 × 10−3 ± 0.9 × 10−3 vs. 5.8 × 10−3 ± 0.9 × 10−3 in ASD vs. HC respectively, U(25) = 126, p = 0.006) were detected in subjects with ASD vs. controls (Fig. 1C, D). No significant differences in the glutamate sub-metabolome of ASD vs. controls were observed in urines (Fig. 1C).
mGluR5 binding was significantly increased in the brain of ASD vs. controls groups
The z-score maps comparing the [18F]FPEB SUVr obtained in individuals with ASD and controls revealed significant differences in [18F]FPEB binding between the two populations in almost all the studied brain areas at the exception of the pallidum and hippocampus (Fig. 2A, B and Table 2). The d values for all the statistical differences observed ranged between 1.06 and 1.17 showing that these differences are not dependent on the effective of each group. Most of these differences corresponded to significantly higher values in [18F]FPEB SUVr observed in the ASD vs. control groups (Fig. 2A, B and Table 2).

[18F]FPEB brain imaging in individuals with ASD vs. controls (A, B), and in an animal model of ASD (C, D). A Examples of brain [18F]FPEB uptake images obtained in one healthy control (male, 32 years-old) and one individual with ASD (male, 45 years-old) on two axial planes with their corresponding T1 weighted MRI template images. B Fusion images between an MRI template and the z-score map showing the significant differences in [18F]FPEB binding between ASD and CTRL groups (red vs. blue = higher vs. lower [18F]FPEB binding in ASD vs. CTRL groups; n = 12 and 14, respectively). C Significant modifications in social interactions and play behavior displayed at PND31 and PND33 by male rats prenatally exposed to VPA vs. saline at GD12.5. The individual values of each quantified index and the mean ± standard deviation was presented for both VPA and control groups (n = 5 and 6 for VPA and controls groups, respectively). D Significant differences in [18F]FPEB binding between male rats prenatally exposed to VPA vs. controls (n = 11 per group) presented on fusion images between an MRI template and z-score maps at PND21 and PND35 (red vs. blue = higher vs. lower [18F]FPEB binding in VPA vs. control rats). ASD autism spectrum disorder group, CTRL control group, PND21 postnatal day 21, PND35 postnatal day 35, VPA group prenatally exposed to valproic acid.
In the cortex, higher values in [18F]FPEB SUVr were detected in the frontal, parietal, temporal, occipital and insular lobes with z-score values ranging from 3.16 ± 0.27 in the anterior and mid-cingulate cortex (p = 0.0010 in both cases; Table 2) to 3.48 ± 0.52 in the medial occipital cortex (p < 0.0001; Table 2). Lower values in [18F]FPEB SUVr in the ASD group vs. controls were only observed in parts of the medial precentral and inferior temporal cortices (z-score values of −3.35 ± 0.36 and −3.39 ± 0.64 with p values of 0.0008 and 0.0002, respectively).
Subcortically, significantly higher values in [18F]FPEB SUVr were observed in the striatum, thalamus, amygdala, and parahippocampal areas with z-scores ranging from 3.17 ± 0.25 in the nucleus accumbens to 3.39 ± 0.37 in the thalamus (p values of 0.0011 and 0.0003 in the nucleus accumbens and thalamus, respectively; Table 2).
Lastly, differences in [18F]FPEB SUVr values were also observed in the cerebellum, with higher values in the vermis of the ASD group vs. controls (z-score of 3.65 ± 0.60; p value < 0.0001) and lower values in its intermediate part (z-score of −3.38 ± 0.51; p value < 0.0001; Table 2).
VPA vs. control rats showed altered social behaviors and modifications in their brain mGluR5 density at PND35 but not at PND21
At PND31, VPA rats spent significantly less time interacting with a familiar peer compared to controls (ratio of time interacting with an un- vs. familiar rat of 0.72 ± 0.24 vs. 6.55 ± 1.95 for control vs. VPA rats; t = 6.7, df = 9, p < 0.0001; Fig. 2C). In addition, the time spent playing at PND33 was significantly lower for VPA vs. control rats (175 ± 76 vs. 35 ± 27 s for control vs. VPA rats; t = 3.9, df = 9, p = 0.0036; Fig. 2C).
In sharp contrast to the clinical data, rats prenatally exposed to VPA compared to controls do not show any reliable differences in [18F]FPEB binding. The VPA rats showed significantly lower [18F]FPEB SUVr values than controls in the somatosensory cortex and hypothalamus (−3.16 ± 0.33, d value = 0.44, p value = 0.0216 for the somatosensory cortex; −3.05 ± 0.57, p value = 0.0029 for the hypothalamus; Fig. 2D), but these differences were considered non reliable as they were associated with d values inferior to 0.80 suggesting these differences were too dependent on the number of subject included in each group (Table 3).
Reliable significant differences in [18F]FPEB binding between the two groups were only observed at PND35 (Fig. 2D). Significantly higher [18F]FPEB SUVr values in rats prenatally exposed to VPA were detected in the retrosplenial cortex (3.35 ± 0.35, d value = 0.82, p value = 0.0007), striatum (3.08 ± 0.3514, d value = 0.95, p value = 0.0038), amygdala (3.28 ± 0.351, d value = 1.03, p value = 0.0007), and hippocampus (3.57 ± 0.54, d value = 0.821.64, p value = 0.0002; Table 3). Higher values were also observed in the hypothalamus but considered non reliable as associated to a d value lower than 0.80 (3.37 ± 0.35, d value = 0.52, p value = 0.0007; Table 3). The VPA rats also showed significantly lower [18F]FPEB SUVr values than control at this developmental stage in the thalamus (−4.15 ± 1.32, d value = 1.92, p value < 0.0001), mesencephalon (−3.19 ± 0.20, d value = 0.94, p value = 0.0004), and cerebellum (−4.55 ± 1.06, d value = 2.08, p value < 0.0001; Table 3).
Discussion
Our translational and longitudinal approach evidenced that glutamatergic synapse abnormalities in ASD are complex and evolves with age.
Focusing on the mGluR5, PET results suggest a continuous developmental trajectory of the glutamatergic synapse dysfunction in ASD. The temporal evolution is evidenced by almost no changes in mGluR5 density between juvenile rats prenatally exposed to VPA and controls evolving to significant modifications in the subcortical areas at adolescence, and the higher densities in the whole brain observed in young adults with ASD. Only two studies suggested higher densities in adult subjects with ASD in the cerebellum and postcentral gyrus using PET but were either post-mortem or limited by a small number of participants [19,20,21]. We showed that adult individuals with ASD exhibit higher mGluR5 densities in almost all the cortex and subcortical areas, and our translational approach evidenced that these modifications are a dynamic process occurring with time. In this hypothesis, the early adulthood widespread mGluR5 alterations may reflect a compensatory process more than a source of the ASD pathophysiology itself.
Previous genetic studies evidenced the role of proteins involved in the cytoarchitecture of the glutamate synapse in ASD from the early stages of brain development. It has thus been proposed that alterations in mGluR5 expression may be governed in part by Homer1b/c, a post-synaptic scaffolding protein [51] and Shank3 protein dysfunctions [52]. The origin of glutamatergic synapse dysfunction in ASD may thus be related to an aberrant spine construction related to disrupted synaptic plasticity. These results may suggest that glutamatergic dysfunction originates with synaptic ultrastructural alterations, that evolves over childhood toward impairment of the glutamatergic neurotransmission.
At the neurotransmitter level the complexity of glutamatergic synapse dysfunction is also reflected by the similar cingulate Glu and Glx levels observed in subjects with ASD and controls, while their serum Glu sub-metabolomes were modified. The cingulate Glu and Glx literature reported highly variable results with studies reporting either lower or higher levels [53,54,55,56,57,58,59,60], or no differences between subjects with ASD and controls as observed in this study [53, 61, 62]. Such variability has been explained by the heterogeneity of the studied groups, different voxel locations, and differences in data analysis. The sample size of our groups is a limitation; however, our results were not dependent on partial volume effects which are rarely corrected in the other studies. Besides, our voxel included parts of the anterior and mid-cingulate cortex, the whole cingulate cortex being related to ASD-associated impairments [35,36,37] whereas others focused on the anterior cingulate cortex. Taken together, this suggest the difficulty of using 1H-MRS to evaluate changes in excitatory transmission in ASD, or that the glutamate-related modifications are more complex.
In the same vein, the lower serum Glu levels observed in our ASD group are in contradiction with the higher levels reported in the only other study performed on blood samples from adults [63]. However, such variability is in line with studies on children samples reporting either lower, unchanged or higher Glu levels in the blood and urine samples of individuals with ASD vs. controls [64,65,66,67,68,69,70,71,72,73]. Comparing metabolomic data from adult and children’s samples seems hazardous as the blood metabolome evolve with age in controls [74]. Interpretating peripheral glutamate results is complex as brain glutamate levels do not depend on peripheral levels, the blood brain barrier being almost impermeable to glutamate [75]. However, changes in Glu, Gln, and GABA levels observed in ASD individuals suggest that the excitation/inhibition imbalance is not restricted to the brain and affected the whole individual.
Interestingly our results could contribute to explain the failure of phase II and III studies evaluating the therapeutic efficacy of negative allosteric modulators (NAMs) of mGluR5 in fragile X syndrome. Whereas results obtained in Fragile X mental retardation 1 (Fmr1) knock-out mice were promising showing that NAMs could fix several disease-related phenotypes (including learning impairment, electrophysiological and synaptic plasticity alterations, as well as growth abnormalities), subsequent clinical trials did not demonstrate therapeutic benefit in FXS subjects treated with either basimglurant or mavoglurant over placebo. The authors of these studies hypothesized that NAM’s therapeutic intervention might be more effective at earlier stages of neurodevelopment when synaptic plasticity may operate more importantly [16]. Our results suggest that changes in mGluR5 density could reflect the adaptation of the glutamatergic synapse to structural damage occurring at earlier stages of neurodevelopment, with the mGluR5 increased density constituting a consequence within the pathophysiological cascade rather than a primary molecular target.
Several limitations are necessary to address to interpret these findings. First, we did not use a PET-MR for the clinical part of our study. It would have facilitated the protocol enabling simultaneous PET, MRI and MRS acquisitions [76]. Second, our longitudinal evaluation of the brain mGluR5 density was performed in a translational way using PET imaging trying to connect findings in an animal model of ASD and in humans. This was dictated to us due to practical, regulatory and ethical difficulties in performing imaging using radioactivity in young autistic children, requiring them to remain motionless for 30 min. Nevertheless, the model of prenatal exposure to VPA has been extensively used to study ASD pathophysiology in rodents [77], and the correspondences of developmental age between the two species strongly validated [22]. Third, we used SUVmax to quantify [18F]FPEB cerebral binding instead of non-displaceable binding potential (BPND). BPND is known as more reliable and efficient in terms of quantification, but its measurement requires dynamic PET acquisitions starting from the injection of the radiotracer, which was difficult to implement in our ASD group. Fourth, the pons was used as reference tissue for PET quantification of [18F]FPEB binding contrary to the previously described cerebellar white matter. Cerebellar white matter cannot be used here due to the small size of the brain and the spillover effect from cerebellar changes in [18F]FPEB binding between VPA and controls rats. Moreover, the [18F]FPEB binding in the pons was low and not significantly dependent on the studied group.
To conclude, our findings suggest that disturbance of glutamatergic neurotransmission in ASD follows a complex trajectory from childhood to adulthood involving different mechanisms of synapse binding and transmission. The delayed overexpression of mGluR5 may be a compensatory mechanism to earlier alterations of the synaptic architecture. These results reinforce the interest to consider personalized intervention using targeted therapeutics according to the specific needs and the developmental trajectory of each individual.
References
Lord C, Elsabbagh M, Baird G, Veenstra-Vanderweele J. Autism spectrum disorder. Lancet. 2018;392:508–20.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-5. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
Chiarotti F, Venerosi A. Epidemiology of autism spectrum disorders: a review of worldwide prevalence estimates since 2014. Brain Sci. 2020;10:274.
Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev. 2012;36:2044–55.
Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems: model of autism. Genes Brain Behav. 2003;2:255–67.
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–8.
Rojas DC. The role of glutamate and its receptors in autism and the use of glutamate receptor antagonists in treatment. J Neural Transm. 2014;121:891–905.
Vyas Y, Cheyne JE, Lee K, Jung Y, Cheung PY, Montgomery JM. Shankopathies in the developing brain in autism spectrum disorders. Front Neurosci. 2021;15:775431.
Eltokhi A, Santuy A, Merchan-Perez A, Sprengel R. Glutamatergic dysfunction and synaptic ultrastructural alterations in schizophrenia and autism spectrum disorder: evidence from human and rodent studies. IJMS. 2020;22:59.
Carlsson ML. Hypothesis: is infantile autism a hypoglutamatergic disorder? Relevance of glutamate—serotonin interactions for pharmacotherapy. J Neural Transm. 1998;105:525.
Fatemi SH. The hyperglutamatergic hypothesis of autism. Prog Neuro-Psychopharmacol Biol Psychiatry. 2008;32:911.
Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, Mizuno N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci Lett. 1993;163:53–57.
Ferraguti F, Shigemoto R. Metabotropic glutamate receptors. Cell Tissue Res. 2006;326:483–504.
Lee M, Martin GE, Berry-Kravis E, Losh M. A developmental, longitudinal investigation of autism phenotypic profiles in fragile X syndrome. J Neurodev Disord. 2016;8:47.
Berry-Kravis E, Des Portes V, Hagerman R, Jacquemont S, Charles P, Visootsak J, et al. Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci Transl Med. 2016;8:1–11.
Youssef EA, Berry-Kravis E, Czech C, Hagerman RJ, Hessl D, Wong CY, et al. Effect of the mGluR5-NAM Basimglurant on behavior in adolescents and adults with fragile x syndrome in a randomized, double-blind, placebo-controlled trial: FragXis phase 2 results. Neuropsychopharmacology. 2018;43:503–12.
Levenga J, Hayashi S, de Vrij FMS, Koekkoek SK, van der Linde HC, Nieuwenhuizen I, et al. AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol Dis. 2011;42:311–7.
Hagerman R, Jacquemont S, Berry-Kravis E, Des Portes V, Stanfield A, Koumaras B, et al. Mavoglurant in fragile x syndrome: results of two open-label, extension trials in adults and adolescents. Sci Rep. 2018;8:16970.
Fatemi SH, Folsom TD, Kneeland RE, Liesch SB. Metabotropic glutamate receptor 5 upregulation in children with autism is associated with underexpression of both fragile x mental retardation protein and GABAA receptor beta 3 in adults with autism. Anat Rec. 2011;294:1635–45.
Fatemi SH, Wong DF, Brašić JR, Kuwabara H, Mathur A, Folsom TD, et al. Metabotropic glutamate receptor 5 tracer [18F]-FPEB displays increased binding potential in postcentral gyrus and cerebellum of male individuals with autism: a pilot PET study. Cerebellum Ataxias. 2018;5:3.
Brašić JR, Nandi A, Russell DS, Jennings D, Barret O, Martin SD, et al. Cerebral expression of metabotropic glutamate receptor subtype 5 in idiopathic autism spectrum disorder and fragile x syndrome: a pilot study. IJMS. 2021;22:2863.
Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. 2013;106–107:1–16.
Hergueta T, Lecrubier Y, Sheehan D, Weiller E. Mini international neuropsychiatric interview French current DSM-IV. 2015. https://doi.org/10.13140/RG.2.1.2792.9440.
First MB. Diagnostic and statistical manual of mental disorders, 5th edition, and clinical utility. J Nerv Ment Dis. 2013;201:727–9.
Lord C, Rutter M, Le Couteur A. Autism diagnostic interview-revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–85.
Lord C, Risi S, Lambrecht L, Cook EH, Leventhal BL, DiLavore PC, et al. The autism diagnostic observation schedule–generic: a standard measure of social and communication deficits associated with the spectrum of autism. 2000;30:205–23.
Wechsler D. Echelle d’intelligence de Wechsler pour adultes (WAIS-III). 3ème éd. Paris: ECPA; 2000.
Wechsler D. Echelle d’intelligence de Wechsler pour adultes (WAIS-IV). 4ème éd. Paris: ECPA; 2011.
Schopler E, Lansing MD, Reichler RJ, Marcus LM. PEP-3. Profil psycho-educatif. Evaluation fonctionnelle pour enfants autistes. de BoeckBruxelles, Belgique; 2020.
Barfthelemy C, Roux S, Adrien JL, Hameuiy L, Gtieriis P, Garreau B, et al. Validation of the revised behavior summarized evaluation scale. 1997;27:139–53.
Bourreau Y, Roux S, Gomot M, Bonnet-Brilhault F, Barthélémy C. Validation of the repetitive and restricted behaviour scale in autism spectrum disorders. Eur Child Adolesc Psychiatry. 2009;18:675–82.
Baron-Cohen S, Wheelwright S, Skinner R, Martin J, Clubley E. The autism-spectrum quotient (AQ): evidence from Asperger syndrome/high-functioning autism, males and females, scientists and mathematicians. J Autism Dev Disord. 2001;31:5–17.
Baron-Cohen S, Wheelwright S. The empathy quotient: an investigation of adults with Asperger syndrome or high functioning autism, and normal sex differences. J Autism Dev Disord. 2004;34:163–75.
Dupont A-C, Serrière S, Barantin L, Vercouillie J, Tauber C, Gissot V, et al. Study of influence of the glutamatergic concentration of [18F]FPEB binding to metabotropic glutamate receptor subtype 5 with N-acetylcysteine challenge in rats and SRM/PET study in human healthy volunteers. Transl Psychiatry. 2021;11:66.
Apps MAJ, Lockwood PL, Balsters JH. The role of the midcingulate cortex in monitoring others’ decisions. Front Neurosci. 2013;7:1–7.
Apps MAJ, Rushworth MFS, Chang SWC. The anterior cingulate gyrus and social cognition: tracking the motivation of others. Neuron. 2016;90:692–707.
Leech R, Sharp DJ. The role of the posterior cingulate cortex in cognition and disease. Brain. 2014;137:12–32.
Wilson M, Andronesi O, Barker PB, Bartha R, Bizzi A, Bolan PJ, et al. Methodological consensus on clinical proton MRS of the brain: review and recommendations. Magn Reson Med. 2019;82:527–50.
Oeltzschner G, Zöllner HJ, Hui SCN, Mikkelsen M, Saleh MG, Tapper S, et al. Osprey: open-source processing, reconstruction & estimation of magnetic resonance spectroscopy data. J Neurosci Methods. 2020;343:108827.
Gasparovic C, Song T, Devier D, Bockholt HJ, Caprihan A, Mullins PG, et al. Use of tissue water as a concentration reference for proton spectroscopic imaging. Magn Reson Med. 2006;55:1219–26.
Harris AD, Puts NAJ, Edden RAE. Tissue correction for GABA-edited MRS: considerations of voxel composition, tissue segmentation, and tissue relaxations: tissue correction for GABA-Edited MRS. J Magn Reson Imaging. 2015;42:1431–40.
Gussew A, Erdtel M, Hiepe P, Rzanny R, Reichenbach JR. Absolute quantitation of brain metabolites with respect to heterogeneous tissue compositions in 1H-MR spectroscopic volumes. Magn Reson Mater Phys. 2012;25:321–33.
Truong V, Duncan NW. Suggestions for improving the visualization of magnetic resonance spectroscopy voxels and spectra. R Soc Open Sci. 2020;7:200600.
Park E, Sullivan JM, Planeta B, Gallezot J-D, Lim K, Lin S-F, et al. Test–retest reproducibility of the metabotropic glutamate receptor 5 ligand [18F]FPEB with bolus plus constant infusion in humans. Eur J Nucl Med Mol Imaging. 2015;42:1530–41.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc: Ser B (Methodol). 1995;57:289–300.
Cohen J. Statistical power analysis for the behavioral sciences. 2nd ed. Hillsdale, NJ: Lawrence Erlbaum Associates; 1988.
Sawilowsky SS. New effect size rules of thumb. J Mod Appl Stat Meth. 2009;8:597–9.
Blasco H, Bessy C, Plantier L, Lefevre A, Piver E, Bernard L, et al. The specific metabolome profiling of patients infected by SARS-COV-2 supports the key role of tryptophan-nicotinamide pathway and cytosine metabolism. Sci Rep. 2020;10:16824.
Nadler JJ, Moy SS, Dold G, Simmons N, Perez A, Young NB, et al. Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav. 2004;3:303–14.
Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. NeuroImage. 2002;15:870–8.
Kammermeier PJ. Endogenous homer proteins regulate metabotropic glutamate receptor signaling in neurons. J Neurosci. 2008;28:8560–7.
Verpelli C, Dvoretskova E, Vicidomini C, Rossi F, Chiappalone M, Schoen M, et al. Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J Biol Chem. 2011;286:34839–50.
Ford TC, Crewther DP. A comprehensive review of the 1H-MRS metabolite spectrum in autism spectrum disorder. Front Mol Neurosci. 2016;9:1–27.
Bernardi S, Anagnostou E, Shen J, Kolevzon A, Buxbaum JD, Hollander E, et al. In vivo 1H-magnetic resonance spectroscopy study of the attentional networks in autism. Brain Res. 2011;1380:198–205.
Bejjani A, O’Neill J, Kim JA, Frew AJ, Yee VW, Ly R, et al. Elevated glutamatergic compounds in pregenual anterior cingulate in pediatric autism spectrum disorder demonstrated by 1H MRS and 1H MRSI. PLoS ONE. 2012;7:e38786.
Joshi G, Biederman J, Wozniak J, Goldin RL, Crowley D, Furtak S, et al. Magnetic resonance spectroscopy study of the glutamatergic system in adolescent males with high-functioning autistic disorder: a pilot study at 4T. Eur Arch Psychiatry Clin Neurosci. 2013;263:379–84.
Ito H, Mori K, Harada M, Hisaoka S, Toda Y, Mori T, et al. A proton magnetic resonance spectroscopic study in autism spectrum disorder using a 3-Tesla clinical magnetic resonance imaging (MRI) system: the anterior cingulate cortex and the left cerebellum. J Child Neurol. 2017;32:731–9.
Naaijen J, Zwiers MP, Forde NJ, Williams SC, Durston S, Brandeis D, et al. Striatal structure and its association with N-Acetylaspartate and glutamate in autism spectrum disorder and obsessive compulsive disorder. Eur Neuropsychopharmacol. 2018;28:118–29.
Siegel-Ramsay JE, Romaniuk L, Whalley HC, Roberts N, Branigan H, Stanfield AC, et al. Glutamate and functional connectivity—support for the excitatory-inhibitory imbalance hypothesis in autism spectrum disorders. Psychiatry Res: Neuroimaging. 2021;313:111302.
Tebartz van Elst L, Maier S, Fangmeier T, Endres D, Mueller GT, Nickel K, et al. Disturbed cingulate glutamate metabolism in adults with high-functioning autism spectrum disorder: evidence in support of the excitatory/inhibitory imbalance hypothesis. Mol Psychiatry. 2014;19:1314–25.
Jiménez-Espinoza C, Marcano Serrano F, González-Mora JL. N-acetylaspartyl-glutamate metabolism in the cingulated cortices as a biomarker of the etiology in ASD: a 1H-MRS model. Molecules. 2021;26:675.
Libero LE, Reid MA, White DM, Salibi N, Lahti AC, Kana RK. Biochemistry of the cingulate cortex in autism: an MR spectroscopy study: biochemistry of the cingulate cortex in autism. Autism Res. 2016;9:643–57.
Shinohe A, Hashimoto K, Nakamura K, Tsujii M, Iwata Y, Tsuchiya KJ, et al. Increased serum levels of glutamate in adult patients with autism. Prog Neuro-Psychopharmacol Biol Psychiatry. 2006;30:1472–7.
Ghanizadeh A. Increased glutamate and homocysteine and decreased glutamine levels in autism: a review and strategies for future studies of amino acids in autism. Dis Markers. 2013;35:281–6.
Zheng H-F, Wang W-Q, Li X-M, Rauw G, Baker GB. Body fluid levels of neuroactive amino acids in autism spectrum disorders: a review of the literature. Amino Acids. 2017;49:57–65.
Lussu M, Noto A, Masili A, Rinaldi AC, Dessì A, De Angelis M, et al. The urinary 1 H-NMR metabolomics profile of an italian autistic children population and their unaffected siblings: metabolomics profile of autistic children. Autism Res. 2017;10:1058–66.
Zaki MM, Abdel-Al H, Al-Sawi M. Assessment of plasma amino acid profile in autism using cation-exchange chromatography with postcolumn derivatization by ninhydrin. Turk J Med Sci. 2017;47:260–67.
Al-Otaish H, Al-Ayadhi L, Bjørklund G, Chirumbolo S, Urbina MA, El-Ansary A. Relationship between absolute and relative ratios of glutamate, glutamine and GABA and severity of autism spectrum disorder. Metab Brain Dis. 2018;33:843–54.
Bitar T, Mavel S, Emond P, Nadal-Desbarats L, Lefèvre A, Mattar H, et al. Identification of metabolic pathway disturbances using multimodal metabolomics in autistic disorders in a Middle Eastern population. J Pharm Biomed Anal. 2018;152:57–65.
Khalifa D, Shahin O, Salem D, Raafat O. Serum glutamate was elevated in children aged 3-10 years with autism spectrum disorders when they were compared with controls. Acta Paediatr. 2019;108:295–9.
Rangel-Huerta OD, Gomez-Fernández A, de la Torre-Aguilar MJ, Gil A, Perez-Navero JL, Flores-Rojas K, et al. Metabolic profiling in children with autism spectrum disorder with and without mental regression: preliminary results from a cross-sectional case–control study. Metabolomics. 2019;15:99.
Gevi F, Belardo A, Zolla L. A metabolomics approach to investigate urine levels of neurotransmitters and related metabolites in autistic children. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165859.
Yu X, Qian-Qian L, Cong Y, Xiao-Bing Z, Hong-Zhu D. Reduction of essential amino acid levels and sex-specific alterations in serum amino acid concentration profiles in children with autism spectrum disorder. Psychiatry Res. 2021;297:113675.
Chaleckis R, Murakami I, Takada J, Kondoh H, Yanagida M. Individual variability in human blood metabolites identifies age-related differences. Proc Natl Acad Sci USA. 2016;113:4252–9.
Hawkins RA. The blood-brain barrier and glutamate. Am J Clin Nutr. 2009;90:867S–874S.
Gade S, Hjørnevik T, Park JH, Shen B, Gu M, Tseng J, et al. The first awake simultaneous PET-MR study of an adult with fragile X syndrome: a case report. In Review; 2021.
Tartaglione AM, Schiavi S, Calamandrei G, Trezza V. Prenatal valproate in rodents as a tool to understand the neural underpinnings of social dysfunctions in autism spectrum disorder. Neuropharmacology. 2019;159:107477.
Acknowledgements
The clinical study was supported by the French National Agency for Research (“Investissements d’Avenir” no. ANR-11-LABX-0018-01), IRON. This study was funded in part by the “Région centre Val de Loire APR-IA 2014-850”. We thank the staffs at the CERRP, iBrain, CIC 1415, PST-ASB core facility and the staff of the child psychiatry unit and the nuclear medicine department of the university hospital of Tours. We also thank the PRIMEX core facility from the Angers University for the MR-experiments to build the MRI template for male rats at early-adolescence. We thank Jacques Dupont for its technical assistance on preclinical behavioral experiments. We extend our deep gratitude to the participants of this study.
Author information
Authors and Affiliations
Contributions
Conceptualization: FBB, FL, LG, MJSR, PE, and SC; Data acquisition: AL, CB, CR, EHD, FBB, JV, LB, LG, MJSR, SC, SR, and VG; Data analysis: ACD, CT, LB, LG, LN, FB, FBB, MG, MJSR, NA, PE, and SM; Manuscript writing: FB, FBB, LG, MJSR, NA, and PE.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Galineau, L., Arlicot, N., Dupont, AC. et al. Glutamatergic synapse in autism: a complex story for a complex disorder. Mol Psychiatry 28, 801–809 (2023). https://doi.org/10.1038/s41380-022-01860-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-022-01860-9
- Springer Nature Limited
This article is cited by
-
Patterns of infant fecal metabolite concentrations and social behavioral development in toddlers
Pediatric Research (2024)
-
Reduced excitatory activity in the developing mPFC mediates a PVH-to-PVL transition and impaired social cognition in autism spectrum disorders
Translational Psychiatry (2024)
-
Group I and group II metabotropic glutamate receptors are upregulated in the synapses of infant rats prenatally exposed to valproic acid
Psychopharmacology (2023)