
Abstract
Pharmacological blockers of the cannabinoid receptor type-1 (CB1) have been considered for a long time as the holy grail of obesity pharmacotherapy. These agents were hastily released in the clinical setting, due to their clear-cut therapeutic efficacy. However, the first generation of these drugs, which were able to target both the brain and peripheral tissues, had serious neuropsychiatric effects, leading authorities to ban their clinical use. New peripherally restricted CB1 blockers, characterized by low brain penetrance, have been developed over the past 10 years. In preclinical studies, these molecules seem to overcome the neuropsychiatric negative effects previously observed with brain-penetrant CB1 inhibitors, while retaining or even outperforming their efficacy. The mechanisms of action of these peripherally restricted compounds are only beginning to emerge, and a balanced discussion of the risk/benefits ratio associated to their possible clinical use is urgently needed, in order to avoid repeating past mistakes. Here, we will critically discuss the advantages and the possible hidden threats associated with the use of peripheral CB1 blockers for the pharmacotherapy of obesity and its associated metabolic complications. We will address whether this novel pharmacological approach might ‘compete’ with current pharmacotherapies for obesity and diabetes, while also conceptualizing future CB1-based pharmacological trends that may significantly lower the risk/benefits ratio associated with the use of these drugs.
Similar content being viewed by others
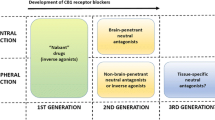

Introduction
Evolution has programmed our metabolism to accumulate energy, allowing survival during periods of famine. Such innate trait may contribute to the current obesity epidemics [1] in a society characterized by constant access to highly palatable/energy-dense food and a sedentary lifestyle. The biological mechanisms underlying energy accumulation are still poorly understood, reflecting the scarce number of pharmacological targets allowing achieving sizable and long-term weight loss. The cloning of the cannabinoid type-1 receptor (CB1) [2], which binds to the psychotropic component of marijuana (Δ9-tetrahydrocannabinol or THC [3]), and the discovery of its endogenous ligands, named endocannabinoids [4,5,6] have shed new light into our comprehension of these mechanisms. Endocannabinoids, CB1 and the closely related type-2 (CB2) receptors, together with the enzymatic machinery directing endocannabinoids synthesis and degradation constitute the endocannabinoid system (ECS). Research efforts over the past three decades have led to recognize that this system has one key purpose under physiological conditions: to maximize the introduction, the accumulation, and the storage of energy (food) in the body, thanks to redundant mechanisms involving CB1 located in the brain and in peripheral tissues [7, 8]. But how does such knowledge help us understand the aetiology of obesity, and to possibly achieve novel solutions against this disease? A link between ECS activity and obesity was established thanks to the observation that hypothalamic levels of the main endocannabinoids arachidonoylethanolamide (anandamide, AEA) and 2-arachidonoylglycerol (2-AG) were elevated in several murine models of obesity [9], causing overfeeding and impaired action of the appetite-suppressant hormone leptin in these animals [9]. Hence, it was hypothesized that a dysregulated/overactive endocannabinoid tone may be causally linked with obesity, a concept confirmed by studies suggesting upregulation of ECS activity in human obesity [8], and by the clear-cut anti-obesity action of pharmacological CB1 blockers [8, 10]. However, the path from basic knowledge to clinical translation is not always straight, and serious neuropsychiatric side-effects were observed with brain-penetrant CB1 inhibitors [8, 11], halting the use of CB1 as an anti-obesity target. Scepticism in the field might remain, despite the generation of peripherally restricted CB1 blockers with similar therapeutic efficacy and safer neuropsychiatric profile relative to the cognate brain-penetrant molecules. In order to comprehend whether these ‘safer’ CB1 blockers may have a future in the pharmacotherapy of obesity and of its most frequent complication, type-2 diabetes (T2D), it is necessary to critically discuss both the bright sides and hidden threats of this novel class of molecules, as further detailed below.
ECS dysregulation in human obesity: marker or mechanism of disease?
Endocannabinoids can be quantified in plasma and serum. While the origin of this circulating pool is still a matter of debate and considered by many ‘spill-over’ from peripheral tissues [11, 12], the levels of these messengers change in relation to food intake. In normal-weight subjects, fasting or presentation of food before its consumption increases plasma AEA levels, while the levels of this endocannabinoid are decreased after food intake [12,13,14], independently of the pleasure associated with specific food components [13]. Differently, 2-AG levels are elevated after the presentation of highly palatable food, but not in response to food deprivation [13]. Hence, AEA and 2-AG may play different roles in the regulation of eating behaviour, with the first favouring the intake of calories, and the second guiding the choice of food by influencing the hedonic aspect of feeding. Intriguingly, obese subjects have high circulating AEA and 2-AG levels in response to the presentation of both rewarding and non-rewarding food [12], indicating that obesity is characterized by an altered endocannabinoid tone in response to a meal, which may ultimately drive overfeeding. Dysregulated ECS activity may contribute to the aetiology of obesity, as in obese subjects plasma endocannabinoid levels positively correlate with body mass index (BMI) and with the chronic negative consequences of the disease, including insulin resistance and dyslipidemia [8]. These association studies, however, are in most part performed in small cohorts of patients and do not allow excluding the potential influencing effects of such as sex, age, pharmacological treatments, and hormonal status [8]. A recent study performed in a large cohort of lean and obese subjects has revealed that changes in plasma 2-AG levels may predict the occurrence of dyslipidemia and insulin resistance in lean males [15], particularly in aged subjects, while the levels of this endocannabinoid correlate with metabolic impairments during menopause in females, independently of BMI [15]. Thus, the progressive reduction of sex hormone action during menopause in women, and the metabolic impairment associated with aging in men, may both lead to a dysregulated ECS tone, which likely precedes obesity. This also implies that a dysregulated ECS tone in obesity may be causally implicated in the disease. Accordingly, genetic variants of the CNR1 gene (coding for CB1) have been linked to metabolic syndrome [16] and dyslipidemia [17], while a missense polymorphism involving the fatty acid amide hydrolase, one of the key enzymes controlling AEA degradation, is associated with high BMI [18].
Pharmacotherapy of obesity using CB1 antagonists
When the first potent and selective CB1 inverse agonist (rimonabant) was developed in 1994 [19], the first idea was to explore its efficacy as a smoking cessation agent, due to some evidence linking brain CB1 activity with nicotine addiction [20]. For this reason, the compound had to have high brain permeability. Intriguingly tobacco users treated with rimonabant not only stopped smoking but also showed a reduced weight gain following smoking cessation [20], which spurred investigation of the anti-obesity effects of this molecule. The first human trials in obese subjects were promptly performed, and rimonabant effectively reduced food intake and body weight (BW), while also improving insulin and leptin resistance, glucose metabolism, dyslipidemia, and hepatic lipid metabolism [21,22,23,24,25]. These favourable outcomes led the European authorities to approve this drug as an anti-obesity agent by mid-2006, albeit it was already clear from every trial by then performed, that serious complications were observed in a small, but significant fraction of patients treated with rimonabant, including anxiety, depression, and/or suicidal ideation [10], which were presumably occurring due to Central Nervous System (CNS) action of the drug. Hence, in 2008, the European medicine association re-considered the risk-benefits ratio of rimonabant and banned it from the market. The development of other CB1 blockers by other pharmaceutical industry was consequently halted, raising serious doubts about the therapeutic potential of the entire class of molecules.
From debacle to renaissance: peripheral CB1 inhibitors
When rimonabant was removed from the market, there had been already published evidence of functional CB1 receptors in peripheral metabolic organs. The role of intestinal endocannabinoids and CB1 on food intake was documented [26], activation of CB1 on adipocytes was shown to increase lipogenesis [27], while activation of CB1 receptors in hepatocytes and skeletal muscle was linked with systemic glucose intolerance and insulin resistance in animal models of obesity [28, 29]. This led to the hypothesis that selective inhibition of peripheral CB1 could represent a strategy to rescue the metabolic benefits of brain-penetrant CB1 antagonists while avoiding their neuropsychiatric negative effects. Thus, a competition started among drug companies and academic institutions, with the final goal of developing novel functional CB1 blockers with a safer pharmacological profile, a quest that has allowed the generation of several entities with a relevant profile (Table 1). The most significant preclinical studies have demonstrated that, depending on chemical optimization, CB1 affinity, CB1 vs. CB2 specificity, and brain penetrance, this second generation of CB1 blockers has significant anti-obesity efficacy, which, in most cases, is not associated with apparent signs of neuropsychiatric side-effects (Table 1). A ‘wonder-molecule’ named JD5037 (from the Jenrin group) seems to combine all the positive effects so far observed with other peripheral CB1 blockers, including good CB1 binding affinity, minimal brain penetration, full CB1 specificity (assessed by lack of efficacy in CB1-KO mice), and the absence of neurobehavioral effects on locomotion, catalepsy, and anxiety [30, 31] (Table 1). JD5037 is as effective as a globally acting CB1 antagonist in reducing BW, fat mass, and several cardiometabolic risk factors in diet-induced obese (DIO) mice, including insulin and leptin resistance, dyslipidemia, and hepatic steatosis [30, 32, 33]. It also shows an interesting anti-diabetic profile in Zucker diabetic fatty (ZDF) rats, as it can almost completely normalize loss of pancreatic β-cells and diabetic nephropathy [34, 35] (Table 1). Thanks to these highly favourable observations, the Food and Drug Administration (FDA) has approved JD5037 towards Phase 1 trials. More recently, a ‘third generation’ hybrid inhibitor (MRI-1867), that blocks the activity of both peripheral CB1 and the enzyme inducible NO synthase (iNOS) has been synthetized [36, 37]. Due to its dual-action, MRI-1867 not only has clear-cut-weight-lowering effects in DIO mice, but it also shows robust anti-fibrotic effects in kidneys and liver (see next section) [36, 37], making it attractive for further clinical development.
The beneficial effects of peripheral CB1 blockade
The key role played by the peripheral ECS in the regulation of energy balance and the main metabolic mechanisms directed by central and peripheral CB1 have been previously discussed [11], but whether these mechanisms are engaged by recently discovered peripheral CB1 blockers remains poorly understood. In the next section, we will review the efficacy and the mechanisms of action of the most characterized and promising peripheral CB1 blockers (Table 1), including the recent hybrid inhibitor MRI-1867.
Hypophagia
Among other alterations, obesity is accompanied by a dysregulated brain leptin action, which contributes to the development and the progression of the disease [38]. The increased circulating leptin levels (hyperleptinemia) found in overweight/obese subjects play a causal role in such altered hormonal action [39], likely because the excess of the hormone provokes an oversaturation of the signalling capacity of the neuronal leptin receptors (LepR), which then creates a vicious circle favouring fat mass accumulation [39, 40]. JD5037 rapidly (within 72 h) reverses hyperleptinemia in DIO mice, and this cannot be explained by treatment-induced weight loss [30], but likely involves two mechanisms: (i) JD5037 might downregulate the local leptin synthesis machinery [30] by blocking adipocyte CB1 signalling, which favours leptin secretion and expression [41]; (ii) the drug might fine-tune the sympathetic nervous system (SNS) activity, which, in turn, suppresses leptin production in adipocytes, thanks to the local release of norepinephrine (NE) and the consequent activation of ß3-receptors [42]. Accordingly, JD5037 treatment in DIO increases NE levels and ß3-receptors expression in the adipose tissue [30], and chemical sympathetic denervation of visceral fat pads attenuates JD5037-mediated reductions in plasma leptin levels and BW in DIO mice [30].
In a recent work, Tam et al. have observed that such JD5037/leptin axis calls into play the activation of hypothalamic pro-opiomelanocortin neurons [43], which control food intake by regulating central melanocortin 4 receptors (MC4R) [38]. JD5037 restores hypothalamic leptin signalling in DIO mice, and the anorectic and weight-lowering effects of this drug are blunted in DIO mice co-treated with a pharmacological MC4R antagonist [43]. Thus, by lowering circulating leptin levels, JD5037 may decrease LepR oversaturation at the level of hypothalamic melanocortin neurons controlling food intake, thereby leading to empowered hypothalamic leptin sensitivity, and ultimately to reduced food intake and BW in DIO mice.
However, other cellular populations located in extra-hypothalamic brain areas or peripheral tissues might also contribute. For instance, LepR activity in vagal afferent neurons innervating the gastrointestinal (GI) tract could play a role, as the activation of these vagal LepR promotes satiety [44], as also observed with several peripheral CB1 blockers (Table 1), while functional removal of the vagus nerve in rats abolishes the hypophagic effects of rimonabant [26]. JD5037-mediated improvements in central leptin action could also originate from an increased leptin transport across the brain, as expression of megalin, a protein involved in the transport of leptin through the blood–brain barrier (BBB) is reduced in DIO mice as compared with chow-fed mice, while the levels of this protein are restored after chronic administration of JD5037 [30]. Moreover, tanycytes, non-neuronal glial-like cells lining the floor of the cerebral third ventricle, can shuttle blood-born leptin into the hypothalamus [45] and express the enzymatic machinery involved in the production of endocannabinoids [46]. Endocannabinoids produced by tanycytes may modulate the function of nearby CB1 expressing hypothalamic neurons [47, 48]. Since these glial cells are in direct contact with the peripheral blood-stream via fenestrations of the BBB [45], they may be targeted by peripheral CB1 inhibitors and may, therefore, contribute to their effects, an intriguing possibility that awaits confirmation.
Augmented energy expenditure and substrate metabolism
Although both JD5037 and compound 1 promote hypophagia in DIO mice, this is not a universal feature of peripheral CB1 blockers (Table 1). Indeed, BPR697 and AJ5012 can significantly lower BW in DIO mice without inducing changes in food consumption [49,50,51]. These compound-specific differences, which may be related to specific pharmacodynamic properties (Table 1), suggest that the weight-lowering efficacy of peripheral CB1 antagonists do not necessarily require a decreased food intake. Food intake-independent mechanisms involve heightened systemic energy dissipation, increased lipolysis, and elevated oxidation of dietary fatty acids, which are concomitantly observed with JD5037, AJ5012, and AJ5018 [43, 51, 52]. Since JD5037 can restore endogenous leptin action, and leptin itself increases energy expenditure and SNS activity [53], these systemic changes in energy handling may also involve the peripheral SNS tone, as observed with rimonabant [54]. Furthermore, CB1 is expressed in the pituitary and the thyroid gland [55, 56], and brain-penetrant CB1 blockers can heighten the production of both thyroid hormone (TH) and thyroid-stimulating hormone [55,56,57]. TH affects energy metabolism by modulating leptin action, adrenergic signalling, energy expenditure, and peripheral substrate metabolism [58], similar to the effects observed with peripheral CB1 inhibitors. Hence, we may speculate that part of the anti-obesity action of these drugs may derive from enhanced TH release and action, a hypothesis that awaits confirmation. CB1 receptors are also expressed and functionally active in other peripheral organs, such as the adipose tissue. Using a transgenic mouse model with selective and inducible CB1 deletion in adipocytes, we have demonstrated that loss of CB1 function in adipocytes leads to a reduction in adipocytes size and lipid content and to the upregulation of genes controlling thermogenesis and mitochondrial biogenesis in adipose tissues [59]. These fat-specific changes make these animals resistant to DIO [59]. Thus, the pharmacological blockade of peripheral CB1 might favour fat mass and BW loss by redirecting fat storage towards lipolysis and thermogenesis. Accordingly, the peripheral antagonists AJ5012 and AJ5018 promote weight loss and increased energy expenditure in DIO mice, while also augmenting the expression of the mitochondrial thermogenic protein uncoupling protein-1 in brown adipose tissue [51, 52]. Functional CB1 receptors can be then found in the plasma membrane and in inner membrane of mitochondria of skeletal muscle, where they can regulate cellular respiration, energy production, and energy substrates oxidation [60, 61]. Modulation of skeletal muscle mitochondria function might contribute to the anti-obesity efficacy of these pharmacological agents, an interesting possibility requiring further scrutiny.
Fat preference
Mammals show an adaptive advantage in seeking fat-rich food, an essential dietary component scarcely available in most natural habitats. Such innate preference can become maladaptive in the presence of unrestricted fatty foods availability and may thus contribute to the current epidemics of obesity. Although the rewarding value of food is classically controlled by neuronal brain circuits, food preference, and palatability can be also ‘sensed’ by the gut or by taste cells localized in the chemosensory organs [38, 62]. The sham-feeding paradigm allows exploring the ‘liking’ aspect of feeding at the level of the oral tract, independently from food processing or absorption. Using this approach [63], oral exposure to corn oil has been shown to trigger the production of 2-AG and AEA in the jejunum of rats [64]. These local changes are essential for fat preference, as intra-intestinal administration of URB447 (Table 1) inhibits sham feeding in these animals [63]. Notably, specific components of dietary fat selectively initiate such endocannabinoid mobilization in the gut [65], since both URB447 and AM6545 reduced the preference for dienoic fatty acids, which cause the accumulation of jejunal endocannabinoids [65]. Thus, local ECS action in the gut controls the intake of dietary fats, based on their orosensory characteristics. Pharmacological curbing of such local endocannabinoid activity by peripheral CB1 blockers selectively lowers the intake of fat-rich food, likely via gut-to-brain feedback mechanisms. These mechanisms are not completely understood, but they may involve the vagus nerve, as its resection blocks the increase in intestinal endocannabinoid signalling occurring during sham feeding of fat [63]. Another possible mechanism could be a lowered production of ghrelin, a GI hormone that favours fat intake [66]. This is supported by the ability of globally acting CB1 antagonists to lower ghrelin production [66]. Thus, peripheral CB1 blockers may not only affect homoeostatic feeding, but could also impact the rewarding value of fat intake, via gut–brain feedback mechanisms controlled by taste receptors located in the oral cavity, and possibly, the vagus nerve and ghrelin.
Improvement of systemic glucose metabolism
Peripheral CB1 antagonists improve systemic glucose metabolism in DIO mice, at least in part independently of their weight-lowering action, as suggested by the following evidence: (i) compound 2p shows no significant effects on food intake and BW in obese rats, but it significantly lowers plasma glucose levels in an oral glucose tolerance test after chronic administration [67]. (ii) In DIO mice, 7-day treatment with JD5037 blunts their BW, but not to the same level of normal-weight, chow-fed animals. Intriguingly, however, this treatment completely normalizes glucose homoeostasis in DIO animals, to a level fully comparable with that of normal-weight mice [33]. (iii) Chronic treatment with AJ5012 in DIO mice is not as efficient in reducing weight as rimonabant, but the glucometabolic improvements achieved with this drug are fully comparable with those obtained with the cognate brain-penetrant molecule [51].
These direct glycemic effects involve an improved systemic insulin action, as chronic treatment with both JD5037 and AJ5012 in DIO mice results in heightened insulin-induced plasma glucose clearance [30, 33, 51], which is partly due to ameliorated hepatic insulin signalling [32, 33]. Such heightened hepatic insulin sensitivity depends upon the activation of the Sirt1/mTORC2/Akt pathway [32], and from the upregulation of hepatic enzymes causing increased degradation of ceramide, a sphingolipid implicated in the aetiology of insulin resistance [33]. These liver-specific effects are possibly directed by hepatic CB1 in a cell-autonomous manner, as transgenic mice with hepatocyte-specific deletion of CB1 are protected from high-fat diet (HFD)-induced insulin resistance, but are obese [68]. CB1 located in skeletal muscle might also contribute to the insulin-sensitizing and glucometabolic effects of these drugs, as JD5037 treatment enhances insulin-induced sensitivity in the skeletal muscle of DIO mice, as assessed by euglycemic-hyperinsulinemic clamp, and also reverses diet-induced accumulation of ceramides in this tissue [33]. The glucoregulatory effects of JD5037 could be also explained by an improved pancreatic function and insulin secretion. Indeed, ex-vivo pre-treatment of human islets of Langerhans with JD5037 stimulates insulin secretion [69], albeit these data should be interpreted with caution for two reasons: (i) the role of ECS in the endocrine pancreas is still controversial, as some studies have reported that CB1 stimulates insulin secretion, while others have shown the contrary [64], (ii) synthetic cannabinoid agonists and antagonists may have negative effects on pancreatic insulin secretion, due to off-target (non-CB1-dependent) actions at the level of a ATP-sensitive potassium (KATP) channel in pancreatic islets [70]. Thus, the in vivo contribution of JD5037 on pancreatic insulin release may be hampered by these possible off-target effects.
Improvement of dyslipidemia and non-alcoholic fatty liver disease (NAFLD)
The obesity epidemic is associated with the rising prevalence and severity of NAFLD, a condition leading to hepatic inflammation and non-alcoholic steatohepatitis (NASH) and liver failure [71]. NAFLD starts as increased lipid accumulation in the liver, which is linked to systemic dyslipidemia, as an imbalance between the production and the use of fatty acids, alters their systemic secretion in the form of very-low-density lipoprotein (VLDL) cholesterol [71], ultimately causing changes in the circulating levels of other lipoproteins interacting with VLDL [71]. In normal conditions, CB1 is expressed in fairly low levels in the liver [54], while CB1 expression and activity are increased in this organ during NAFLD [72]. Transgenic mice with selective deletion of hepatic CB1 are fully protected from liver steatosis and the chronic systemic lipid dysregulation induced by a HFD, independently from changes in BW or fat mass [28]. Likewise, chronic administration of JD5037 in DIO mice increases systemic fatty acid oxidation, and reduces hepatic inflammation, lipogenesis, and plasma triglyceride and LDL levels [30]. JD5037 also attenuates the HFD-induced increase in several ceramide species in the liver [33], again, in a BW-independent manner [33]. Since hepatic ceramide accumulation is implicated in the development of NAFLD and the progression to NASH [73], these changes in hepatic ceramide content highlight a possible therapeutic role for peripheral CB1 blockade in the treatment of NASH. This perspective is supported by the profile observed with the hybrid CB1/iNOS inhibitor MRI-1867, which mitigates liver fibrosis, (a deleterious consequence of NASH) in an animal model of chemically induced liver dysfunction [37, 74], thanks to its ability to concomitantly target both CB1 and iNOS.
Thus, peripherally restricted CB1 blockade show potential for the treatment of the long-term negative consequences of obesity on liver function and systemic dyslipidemia, a scenario supported by the known positive association between circulating and hepatic endocannabinoid levels and altered liver function in patients with NAFLD, regardless of BMI [75, 76].
Improvement in kidney injury
Diabetic nephropathy is a leading cause of kidney disease in patients with obesity and T2D [1]. Treatment with JD5037 completely prevents the development of diabetic nephropathy in ZDF rats, which partly resembles the diabetic nephropathy observed in humans [35]. These positive effects are possibly mediated by the modulation of CB1 located in the renal proximal tubular cells (RPTC) [77], which are responsible for active reabsorption of a large quantity (>80%) of the filtrate. Transgenic mice with selective deletion of CB1 in this renal cell population are protected from HFD-induced kidney injury [78]. RPTC-specific CB1 deletion leads to protective effects in the kidneys by reducing intracellular lipid accumulation, and consequently, inflammation and tissue fibrosis [78]. Likewise, in cultured RPTC, JD5037 reverses lipid-induced changes in the expression of pro-inflammatory cytokines [78], increases mitochondria respiration, and reduces the accumulation of fat droplets [78]. Thus, by blocking CB1 activity in RPTC, JD5037 might empower local fatty acids utilization and β-oxidation, thereby protecting the kidneys from obesity-induced dysfunction and injury, albeit this mechanism still requires in vivo proofs. Of note, RPTC are sensitive to the deleterious effects of chronic hyperglycaemia, typically observed in diabetic patients. In diabetes, the expression of the facilitative glucose transporter 2 (GLUT2) is increased across the proximal tubule [79], causing increased glucose reabsorption, RPTC inflammation, and fibrosis [80]. Diabetic (STZ-treated) mice treated with JD5037 have significant reduction in serum glucose levels, which is accompanied by increased glycosuria and decreased GLUT2 expression in RPTC [81], suggesting that peripheral CB1 blockade might control renal glucose homoeostasis via a decreased renal GLUT2 expression and function [81, 82]. One of the outcome of diabetic nephropathy is kidney fibrosis, which accelerates the functional deterioration of the organ [83]. The dual CB1/iNOS inhibitor MRI-1867 provides superior efficacy in attenuating obesity-induced kidney injury, fibrosis, and dysfunction compared with the peripheral CB1 antagonist (JD5037) or to the iNOS inhibitor (1400 W) alone [36], supporting the potential role of combined inhibition of iNOS and CB1 in the treatment of the tissue-specific fibrotic alterations in obesity and T2D.
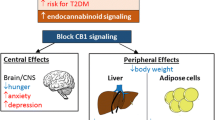
In conclusion, multiple metabolic mechanisms involving peripheral CB1 are engaged in response to treatment with peripheral CB1 blockers, which reflect the pleiotropic ability of this receptor to impact energy balance at both central and peripheral level [8, 11]. The sum of these effects, rather than the activation of one unique metabolic process, likely explains the clear-cut anti-obesity efficacy of peripherally restricted CB1 antagonists, which goes beyond weight loss and hypophagia (Fig. 1).

These drugs can also induce weight loss by enhancing energy dissipation, by increasing the Sympathetic Nervous System (SNS) activity, and by lowering leptin production from adipocytes. Additional beneficial metabolic effect (depicted in the picture) occurs thanks to the targeting of CB1 receptors in peripheral organs such as kidneys, liver, and skeletal muscle.
The hidden threats of peripheral CB1 antagonism
Despite the clear therapeutic potential of peripherally restricted CB1 inhibitors, these drugs are proceeding towards clinical development at a slower rate than other similarly promising anti-obesity and anti-diabetic agents, likely due to still existing concerns about their pharmacokinetic and safety profiles, which we will interrogate in the next section.
The importance of pharmacokinetic
To allow for a low brain penetrance, several peripheral CB1 blockers are endowed of physicochemical optimizations influencing the total polar surface area and/or their ability to interact with efflux transporters, such as P-glycoproteins found in brain endothelial cells. These optimizations, observed for JD5037 [31], compound 38 (from RTI) [84], and compound 4 (NHRI) [85], may compromise drug bioavailability. For instance, increasing polar surface area would affect the compound’s polarity, and therefore its permeability through biological membranes. Although the majority of these agents display a low brain/plasma ratio, which is in most cases <0.1 (Table 1) the majority of the evidence collected so far takes only into account drug concentrations in mice or rats in response to acute dosing, and in lean animals fed with laboratory chow diet. These observations, which are valid for amide D4 [86], TM38837 [87], Compound 1 (Dong pharmaceuticals) [88], compound 2p (Janssen) [67], and AJ5012 [89], do not allow excluding whether these specific molecules may accumulate in the body (adipose tissue) and then leak out to the brain after chronic dosing. This is a relevant question since obesity and diabetes treatment requires long-term pharmacological approaches, albeit we should mention that, in the case of JD5037 and MRI-1867, the brain/plasma ratio of these compounds have been analyzed after both acute and chronic (28 days) dosing, and no signs of drug leakage to the brain over time have been observed [30, 37]. Moreover, obesity is characterized by an altered BBB function [45], and the brain penetrance of these drugs might be altered under obesogenic conditions, a possibility rarely tested in the screenings performed so far. Another aspect underlying the pharmacokinetic profile that requires further investigation is the elimination half-life, which is very long (771 h) for the TM38837 in healthy human subjects [90], an undesirable profile that might explain why this molecule has not been moved forward in clinical studies after completion of a phase 1 clinical trial. MRI-1867 and JD5037 display a shorter elimination half-life in mice (4–6 h) [37, 91], however the elimination half-life and bioavailability of JD5037 in rats and dogs vary significantly between doses, animals species, and sex [91]. Thus, the pharmacokinetic profile of these molecules may change significantly across different animal species, and may be influenced by several factors including dose, duration of treatment, and sex, all variables mostly neglected in the studies performed so far.
Potential neuroendocrine negative effects
The hypothalamic–pituitary–adrenal (HPA) axis is a critical stress system that involves both neuronal and endocrine effectors. The ECS plays a key role in the regulation of HPA axis activity, thanks to redundant neurophysiological mechanisms controlled by CB1 expressed in several brain stress centres, as reviewed in ref. [55]. Peripheral sites of action, such as adrenal glands, could also participate in the endocannabinoid-mediated modulation of the HPA function. Indeed, CB1 agonism decreases adrenocortical steroidogenesis and epinephrine release from human adrenal cells [92, 93]. Our group has shown that peripheral mechanisms involving heightened sympathetic neurotransmission are implicated in the anxiogenic effects of rimonabant, as pharmacological blockade of peripheral SNS activity blunts the effects of this drug on anxiety in mice [94]. Since the pharmacological blockade of peripheral CB1 might similarly lead to elevated SNS activity (see the previous section), we may speculate that peripheral CB1 might influence the stress response thanks to their ability to modulate the SNS. The preclinical characterization of some peripherally restricted CB1 blockers has not evidenced so far any sign of anxiety-like behaviour (Table 1). However, this conclusion seems premature since: (i) most of the studies performed (see refs. [51, 67, 86,87,88,89, 95]) have explored the effects of acute drug injection on the stress response, without investigating the influence of chronic administration and of different dosing, which are crucial aspect in terms of clinical translation. (ii) A long-term (35 days) administration of JD5037 in rats leads to a higher incidence of stereotypic behaviours, which could reflect anxiety [96], including repetitive grooming, excessive/repetitive scratching or shaking of the head [91]. (iii) Although TM38837 showed reduced fear-promoting effects compared with rimonabant in mice, a higher dose of this drug (100 mg/kg) can induce a sustained fear response [97], observed in relation to stressful events [96]. (iv) Finally, behavioural tests aimed at identifying anxiety-like behaviours or other abnormal behaviours in rodents are influenced by environmental variables and by the type of animal model used, including gender, hormonal status, age, strain, day-light cycle, and type of housing [96]. None of these variables have been taken into account or explicitly described during the studies performed so far, raising the issue that the lack of anxiogenic effects observed may have been masked by these possible influencing factors. We should also mention that CB1 is expressed and functional at the level of the hypothalamic–pituitary–gonadal axis [55]. High levels of endocannabinoids may negatively affect reproduction, fertility, and sexual behaviour by acting at different central and peripheral sites [55], suggesting that CB1 blockade should not harm these behaviours [55]. Accordingly, brain-penetrant CB1 blockers improve the sexual activity of male rats [98, 99], albeit the effects of cannabinoids on sexual behaviour and gonadal hormone function may differ between genders [100]. Our understanding of the impact of cannabinoids on female sexual function is still limited [101], and further clarification of this topic will be needed to exclude potential negative effects of peripheral CB1 inhibitors in females.
Are peripheral CB1 antagonists safe at the cardiovascular level?
The ECS has complex and still controversial roles in cardiovascular pathologies. While some studies have shown positive effects of rimonabant on the cardiovascular function in animal models of obesity, this drug can also decrease cardiac contractility, a defect observed in some obese patient [102]. Moreover, rimonabant treatment in humans may induce a subthreshold increase in blood pressure, which may counteract, and thus limit, the blood pressure reduction achieved through weight loss [102]. CB1-KO mice show subtle but significant alterations in sleep-wake cardiorespiratory control in response to chronic feeding with HFD [103], which are associated with a reduced cardiac baroreflex sensitivity (BRS). BRS is severely reduced in obese subjects [104], and is a negative prognostic factor in obesity-associated cardiovascular diseases. Thus, pharmacological CB1 blockade may have negative cardiovascular consequences by worsening BRS control in obese subjects.
None of the studies published so far have addressed the cardiovascular profile of animal models treated with peripheral CB1 antagonists, which, in the light of the current controversy on the topic, deserves further investigation.
GI side-effects
Weight-loss and anti-diabetic medications are generally characterized by poor adherence, mainly due to their associated side-effects. Although the low adherence to rimonabant treatment in humans can be mainly explained by its psychiatric effects, GI side-effects such as nausea, vomiting, and diarrhoea were the most frequent dose-related complications of this drug [10]. Since peripheral CB1 located in the GI tract and the vagus nerve may be implicated in these side-effects [26], it is plausible that peripheral CB1 blockers may have similar unwanted actions. Although JD5037 (3 mg/kg) does not induce nausea-like effects in lean mice, as assessed by conditioned taste aversion [30], little is known on the potential aversive effects of escalating doses of this drug. Of note, JD5037 can reverse CB1-induced decrease in GI motility [30]. Hence, such prokinetic activity may favour diarrhoea and thus undermine drug adherence. In certain pathological conditions of the GI tract, such as Crohn’s disease or irritable bowel syndrome (IBD), the ECS conveys protection to the GI tract, e.g. from inflammation and abnormal gastric and enteric secretion [105]. Moreover, CB1 activation inhibits gastric acid secretion, possibly preventing the formation of gastric ulcers [106]. Thus, chronic peripheral CB1 blockade might lead to increased intestinal motility and intestinal permeability, potentially causing diarrhoea and negative gastric consequences such as IBD and ulcers, all possibilities that still require scrutiny.
In conclusion, although peripheral CB1 blockers may have lower psychiatric side-effects relative to brain-penetrant agents, several open questions on the safety profile of these agents remain (Fig. 2) and warrant further investigation.

By acting at the level of the adrenal gland, these drugs may interfere with the stress axis. Potential negative effects on the cardiovascular system may include impaired baroreflex sensitivity and increased blood pressure. Chronic peripheral CB1 blockade might also lead to increased intestinal motility and intestinal permeability, potentially causing diarrhea and negative gastric consequences such as irritable bowel syndrome and ulcers.
Can peripheral CB1 inhibitors ‘compete’ with current pharmacotherapies or with new drugs on the horizon?
The major FDA-approved anti-obesity medications currently available include phentermine, orlistat, phentermine/topiramate extended release, lorcaserin, naltrexone sustained release (SR)/bupropion SR and the glucagon-like peptide-1 receptor (GLP-1R) agonist liraglutide, which is the only injectable formulation [107]. Recently, the GLP-1R agonist semaglutide has been approved as a once-weekly therapy, due to its promising efficacy in patients with obesity, but without T2D [108]. While these medications offer a solution for the clinical management of those patients who fail to respond to lifestyle modifications, adherence to therapy is still challenging due to side-effects, the modest weight-lowering efficacy (which rarely exceed 10%), and the high number of non-responders [107]. The latter aspect represents a significant obstacle, which occurs due to the high biological heterogeneity of obesity, translating into heterogeneity to the treatment response. Thus, our available pharmacological armamentarium is still limited, and it does not allow reaching a transformative impact against this disease or substituting the large therapeutic effects of bariatric surgery, a procedure with its risks [109]. In this context, pharmacological blockers of peripheral CB1 may have additional therapeutic value for the following reasons: (1) our ability to measure circulating endocannabinoids and to correlate these changes with different profiles of ‘obesities’ has been recently improved [15, 110] (see also “ECS dysregulation in human obesity: marker or mechanism of disease?” section of this review), which may allow predicting whether specific sub-clusters of patients may have a higher benefits/risk ratio in response to peripheral CB1 blockade. (2) Based on preclinical studies, peripheral CB1 inhibition shows potential for the treatment of insulin resistance, dyslipidemia, diabetic nephropathy, and NASH, independently from weight loss. Should these preclinical observations be confirmed in humans, peripheral CB1 inhibition may represent a competitive/complementary pharmacological approach, as none of the currently available pharmacotherapies can concomitantly tackle both fat mass excess and long-term systemic and tissue-specific complications. Interesting new options are on the horizon, such as GLP-1/Glucagon co-agonists, GLP-1/GIP co-agonists, or GLP-1/GIP/Glucagon triagonists. In preclinical models, these molecules allow prominent weight-loss and clear-cut improvements in systemic glycemic control and lipid metabolism [111]. Of note, brain-penetrant CB1 inhibitors can empower the insulinotropic and glycemic effects of a GLP-1 agonist [112], and chronic co-treatment of rimonabant and a GLP-1 agonist in DIO mice allows greater weight-lowering effects than individual monotherapies [113]. Likewise, peripheral CB1 inhibitors may be used to potentiate the glucometabolic and weight-lowering efficacy of these GLP-1 based molecules. However, as previously discussed, several open questions remain concerning the safety profile of peripheral CB1 blockers (Fig. 2). Only a clear answer to these questions will allow predicting whether this class of drugs might compete with current or future available pharmacotherapies or whether it may be used as adjuvant therapy. Nonetheless, none of the drugs available against obesity is deprived of risks [107], and these risks should always be critically assessed taking into account the potentially lethal consequences of obesity [1].
Concluding remarks: will we ever use again pharmacological CB1 blockers for the therapy of obesity?
The whirlwind recent generation of peripherally restricted CB1 blockers and the clear ability of these agents to profoundly ameliorate obesity and its multiple comorbidities in preclinical models might reanimate the use of pharmacological CB1 blockers for the clinical management of this disease. While this second generation of CB1 antagonists may have a safer profile due to their low brain penetrance, decades of research have firmly established that ECS activity is controlled by redundant mechanisms involving both the brain and the periphery [8, 11]. This should warn us on the appearance of the same side-effects previously observed with the brain-penetrant rimonabant, a possibility that still requires detailed scrutiny. Luckily, thanks to the recent determination of the crystal structure of CB1 [114], we are learning that this receptor can plastically respond to a large array of ligands [114], providing an inherent advantage for the rational design of unimolecular compounds with multiple targets. The recent development of the fusion molecule MRI-1867, and the unique ability of this drug to improve the chronic consequences of obesity on kidneys and liver function, represents a concrete example of how such structural plasticity can translate into CB1-based molecules with a larger window of therapeutic efficacy [36, 37]. Should these molecules retain their exceptional preclinical efficacy in humans, this might out-weight the still residual safety concerns associated with this class of molecules.
Obesity is heterogeneous and requires pharmacological approaches targeting multiple mechanisms of action. Unimolecular GLP-1 based multi-agonists able to concomitantly target multiple metabolic pathways demonstrate exceptional preclinical efficacy and are currently under clinical scrutiny [111, 115]. The chemical fusion of peripheral CB1 inhibitors to this variegate molecular portfolio may lead to unimolecular pharmacological entities able to close the still-too-large gap between the efficacy of currently available pharmacological tools relative to the efficacy of bariatric surgery. By closing this gap, we may be able to treat severe obesity and diabetes using pharmacology, while overcoming the risk associated with these surgical procedures [109]. Towards this ultimate goal, we should continue studying the bright side of peripheral CB1 blockade, while investigating also its dark side, in order to avoid the same mistakes of the past.
References
GBD 2015 Obesity Collaborators, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377:13–27.
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–4.
Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International union of pharmacology. XXVII. classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202.
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–9.
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90.
Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, et al. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97.
Piazza PV, Cota D, Marsicano G. The CB1 receptor as the cornerstone of exostasis. Neuron. 2017;93:1252–74.
Quarta C, Mazza R, Obici S, Pasquali R, Pagotto U. Energy balance regulation by endocannabinoids at central and peripheral levels. Trends Mol Med. 2011;17:518–26.
Di Marzo V, Goparaju SK, Wang L, Liu J, Bátkai S, Járai Z, et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature. 2001;410:822–5.
Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. The Lancet. 2007;370:1706–13.
Mazier W, Saucisse N, Gatta-Cherifi B, Cota D. The endocannabinoid system: pivotal orchestrator of obesity and metabolic disease. Trends Endocrinol Metab. 2015;26:524–37.
Gatta-Cherifi B, Matias I, Vallée M, Tabarin A, Marsicano G, Piazza PV, et al. Simultaneous postprandial deregulation of the orexigenic endocannabinoid anandamide and the anorexigenic peptide YY in obesity. Int J Obes. 2012;36:880–5.
Monteleone P, Piscitelli F, Scognamiglio P, Monteleone AM, Canestrelli B, Di Marzo V, et al. Hedonic eating is associated with increased peripheral levels of ghrelin and the endocannabinoid 2-arachidonoyl-glycerol in healthy humans: a pilot study. J. Clin. Endocrinol Metab. 2012;97:E917–924.
Monteleone AM, Di Marzo V, Aveta T, Piscitelli F, Dalle Grave R, Scognamiglio P, et al. Deranged endocannabinoid responses to hedonic eating in underweight and recently weight-restored patients with anorexia nervosa. Am J Clin Nutr. 2015;101:262–9.
Fanelli F, Mezzullo M, Belluomo I, Di Lallo VD, Baccini M, Ibarra Gasparini D, et al. Plasma 2-arachidonoylglycerol is a biomarker of age and menopause related insulin resistance and dyslipidemia in lean but not in obese men and women. Mol Metab. 2017;6:406–15.
Bordicchia M, Battistoni I, Mancinelli L, Giannini E, Refi G, Minardi D, et al. Cannabinoid CB1 receptor expression in relation to visceral adipose depots, endocannabinoid levels, microvascular damage, and the presence of the Cnr1 A3813G variant in humans. Metab Clin Exp. 2010;59:734–41.
Baye TM, Zhang Y, Smith E, Hillard CJ, Gunnell J, Myklebust J, et al. Genetic variation in cannabinoid receptor 1 (CNR1) is associated with derangements in lipid homeostasis, independent of body mass index. Pharmacogenomics. 2008;9:1647–56.
Sipe JC, Waalen J, Gerber A, Beutler E. Overweight and obesity associated with a missense polymorphism in fatty acid amide hydrolase (FAAH). Int J Obes. 2005;29:755–9.
Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–4.
Foll BL, Forget B, Aubin H-J, Goldberg SR. Blocking cannabinoid CB1 receptors for the treatment of nicotine dependence: insights from pre-clinical and clinical studies. Addiction Biology. 2008;13:239–52.
Després J-P, Golay A, Sjöström L, Rimonabant in Obesity-Lipids Study Group. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005;353:2121–34.
Després JP, Ross R, Boka G, Alméras N, Lemieux I. ADAGIO-lipids investigators. effect of rimonabant on the high-triglyceride/ low-HDL-cholesterol dyslipidemia, intraabdominal adiposity, and liver fat: the ADAGIO-Lipids trial. Arterioscler Thromb Vasc Biol. 2009;29:416–23.
Hollander PA, Amod A, Litwak LE, Chaudhari U, ARPEGGIO Study Group.Effect of rimonabant on glycemic control in insulin-treated type 2 diabetes: the ARPEGGIO trial. Diabetes Care. 2010;33:605–7.
Pi-Sunyer FX, Aronne LJ, Heshmati HM, Devin J, Rosenstock J, RIO-North America Study Group. Effect of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: RIO-north america: a randomized controlled trial. JAMA. 2006;295:761–75.
Scheen AJ, Finer N, Hollander P, Jensen MD, Van Gaal LF. Efficacy and tolerability of rimonabant in overweight or obese patients with type 2 diabetes: a randomised controlled study. The Lancet. 2006;368:1660–72.
Gómez R, Navarro M, Ferrer B, Trigo JM, Bilbao A, Del Arco I, et al. A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J Neurosci. 2002;22:9612–7.
Cota D, Marsicano G, Tschöp M, Grübler Y, Flachskamm C, Schubert M, et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Investig. 2003;112:423–31.
Osei-Hyiaman D, Liu J, Zhou L, Godlewski G, Harvey-White J, Jeong W, et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Investig. 2008;118:3160–9.
Liu YL, Connoley IP, Wilson CA, Stock MJ. Effects of the cannabinoid CB1 receptor antagonist SR141716 on oxygen consumption and soleus muscle glucose uptake in Lep(ob)/Lep(ob) mice. Int J Obes. 2005;29:183–7.
Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T, et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16:167–79.
Chorvat RJ, Berbaum J, Seriacki K, McElroy JF. JD-5006 and JD-5037: peripherally restricted (PR) cannabinoid-1 receptor blockers related to SLV-319 (Ibipinabant) as metabolic disorder therapeutics devoid of CNS liabilities. Bioorg Med Chem Lett. 2012;22:6173–80.
Liu J, Godlewski G, Jourdan T, Liu Z, Cinar R, Xiong K, et al. Cannabinoid-1 receptor antagonism improves glycemic control and increases energy expenditure through sirtuin-1/mechanistic target of RAPAMYCIN complex 2 and 5’adenosine monophosphate-activated protein kinase signaling. Hepatology. 2019;69:1535–48.
Cinar R, Godlewski G, Liu J, Tam J, Jourdan T, Mukhopadhyay B, et al. Hepatic cannabinoid-1 receptors mediate diet-induced insulin resistance by increasing de novo synthesis of long-chain ceramides. Hepatology. 2014;59:143–53.
Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med. 2013;19:1132–40.
Jourdan T, Szanda G, Rosenberg AZ, Tam J, Earley BJ, Godlewski G, et al. Overactive cannabinoid 1 receptor in podocytes drives type 2 diabetic nephropathy. Proc Natl Acad Sci USA. 2014;111:E5420–5428.
Udi S, Hinden L, Ahmad M, Drori A, Iyer MR, Cinar R, et al. Dual inhibition of cannabinoid-1 receptor and iNOS attenuates obesity-induced chronic kidney disease. Br J Pharmacol. 2019. https://doi.org/10.1111/bph.14849.
Cinar R, Iyer MR, Liu Z, Cao Z, Jourdan T, Erdelyi K, et al. Hybrid inhibitor of peripheral cannabinoid-1 receptors and inducible nitric oxide synthase mitigates liver fibrosis. JCI Insight. 2016;1:e87336.
Morton GJ, Meek TH, Schwartz MW. Neurobiology of food intake in health and disease. Nat Rev Neurosci. 2014;15:367–78.
Myers MG. Leptin keeps working, even in obesity. Cell Metab. 2015;21:791–2.
Zhao S, Zhu Y, Schultz RD, Li N, He Z, Zhang Z, et al. Partial leptin reduction as an insulin sensitization and weight loss strategy. Cell Metab. 2019;30:706–19.e6.
Bensaid M, Gary-Bobo M, Esclangon A, Maffrand JP, Le Fur G, Oury-Donat F, et al. The cannabinoid CB1 receptor antagonist SR141716 increases Acrp30 mRNA expression in adipose tissue of obese fa/fa rats and in cultured adipocyte cells. Mol Pharmacol. 2003;63:908–14.
Mantzoros CS, Qu D, Frederich RC, Susulic VS, Lowell BB, Maratos-Flier E, et al. Activation of beta(3) adrenergic receptors suppresses leptin expression and mediates a leptin-independent inhibition of food intake in mice. Diabetes. 1996;45:909–14.
Tam J, Szanda G, Drori A, Liu Z, Cinar R, Kashiwaya Y, et al. Peripheral cannabinoid-1 receptor blockade restores hypothalamic leptin signaling. Mol Metab. 2017;6:1113–25.
Peters JH, Ritter RC, Simasko SM. Leptin and CCK selectively activate vagal afferent neurons innervating the stomach and duodenum. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1544–1549.
Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab. 2014;19:293–301.
Suárez J, Romero-Zerbo SY, Rivera P, Bermúdez-Silva FJ, Pérez J, De Fonseca FR, et al. Endocannabinoid system in the adult rat circumventricular areas: an immunohistochemical study. J Comp Neurol. 2010;518:3065–85.
Palma-Chavez A, Konar-Nié M, Órdenes P, Maurelia F, Elizondo-Vega R, Oyarce K, et al. Glucose increase DAGLα levels in tanycytes and Its inhibition alters orexigenic and anorexigenic neuropeptides expression in response to glucose. Front Endocrinol. 2019;10.
Morozov YM, Koch M, Rakic P, Horvath TL. Cannabinoid type 1 receptor-containing axons innervate NPY/AgRP neurons in the mouse arcuate nucleus. Mol Metab. 2017;6:374–81.
Hsiao WC, Shia KS, Wang YT, Yeh YN, Chang CP, Lin Y, et al. A novel peripheral cannabinoid receptor 1 antagonist, BPR0912, reduces weight independently of food intake and modulates thermogenesis. Diabetes Obes Metab. 2015;17:495–504.
Vijayakumar RS, Lin Y, Shia KS, Yeh YN, Hsieh WP, Hsiao WC, et al. Induction of fatty acid oxidation resists weight gain, ameliorates hepatic steatosis and reduces cardiometabolic risk factors. Int J Obes. 2012;36:999–1006.
Han JH, Shin H, Park JY, Rho JG, Son DH, Kim KW, et al. A novel peripheral cannabinoid 1 receptor antagonist, AJ5012, improves metabolic outcomes and suppresses adipose tissue inflammation in obese mice. FASEB J. 2019;33:4314–26.
Han JH, Shin H, Rho JG, Kim JE, Son DH, Yoon J, et al. Peripheral cannabinoid 1 receptor blockade mitigates adipose tissue inflammation via NLRP3 inflammasome in mouse models of obesity. Diabetes Obes Metab. 2018;20:2179–89.
Buettner C, Muse ED, Cheng A, Chen L, Scherer T, Pocai A, et al. Leptin controls adipose tissue lipogenesis via central, STAT3–independent mechanisms. Nat Med. 2008;14:667–75.
Quarta C, Bellocchio L, Mancini G, Mazza R, Cervino C, Braulke LJ, et al. CB(1) signaling in forebrain and sympathetic neurons is a key determinant of endocannabinoid actions on energy balance. Cell Metab. 2010;11:273–85.
Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev. 2006;27:73–100.
Porcella A, Marchese G, Casu MA, Rocchitta A, Lai ML, Gessa GL, et al. Evidence for functional CB1 cannabinoid receptor expressed in the rat thyroid. Eur J Endocrinol. 2002;147:255–61.
da Veiga MA, Fonseca Bloise F, Costa-E-Sousa RH, Souza LL, Almeida NA, Oliveira KJ, et al. Acute effects of endocannabinoid anandamide and CB1 receptor antagonist, AM251 in the regulation of thyrotropin secretion. J Endocrinol. 2008;199:235–42.
Mullur R, Liu Y-Y, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94:355–82.
Ruiz de Azua I, Mancini G, Srivastava RK, Rey AA, Cardinal P, Tedesco L, et al. Adipocyte cannabinoid receptor CB1 regulates energy homeostasis and alternatively activated macrophages. J Clin Investig. 2017;127:4148–62.
Mendizabal-Zubiaga J, Melser S, Bénard G, Ramos A, Reguero L, Arrabal S, et al. Cannabinoid CB1 receptors are localized in striated muscle mitochondria and regulate mitochondrial respiration. Front Physiol. 2016;7:476.
Cavuoto P, McAinch AJ, Hatzinikolas G, Cameron-Smith D, Wittert GA. Effects of cannabinoid receptors on skeletal muscle oxidative pathways. Mol Cell Endocrinol. 2007;267:63–69.
DiPatrizio NV. Is fat taste ready for primetime? Physiol Behav. 2014;136:145–54.
DiPatrizio NV, Astarita G, Schwartz G, Li X, Piomelli D. Endocannabinoid signal in the gut controls dietary fat intake. Proc Natl Acad Sci USA. 2011;108:12904–8.
González-Mariscal I, Egan JM. Endocannabinoids in the Islets of Langerhans: the ugly, the bad, and the good facts. Am J Physiol Endocrinol Metab. 2018;315:E174–E179.
DiPatrizio NV, Joslin A, Jung K-M, Piomelli D. Endocannabinoid signaling in the gut mediates preference for dietary unsaturated fats. FASEB J. 2013;27:2513–20.
Müller TD, Nogueiras R, Andermann ML, Andrews ZB, Anker SD, Argente J, et al. Ghrelin. Mol Metab. 2015;4:437–60.
Matthews JM, McNally JJ, Connolly PJ, Xia M, Zhu B, Black S, et al. Tetrahydroindazole derivatives as potent and peripherally selective cannabinoid-1 (CB1) receptor inverse agonists. Bioorg Med Chem Lett. 2016;26:5346–9.
Liu J, Zhou L, Xiong K, Godlewski G, Mukhopadhyay B, Tam J, et al. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology. 2012;142:1218–28.e1.
González-Mariscal I, Krzysik-Walker SM, Doyle ME, Liu QR, Cimbro R, Santa-Cruz Calvo S, et al. Human CB1 receptor isoforms, present in hepatocytes and β-cells, are involved in regulating metabolism. Sci Rep. 2016;6:33302.
Lynch CJ, Zhou Q, Shyng SL, Heal DJ, Cheetham SC, Dickinson K, et al. Some cannabinoid receptor ligands and their distomers are direct-acting openers of SUR1 KATP channels. Am J Physiol Endocrinol Metab. 2011;302:E540–E551.
Amor AJ, Perea V. Dyslipidemia in nonalcoholic fatty liver disease. Curr Opin Endocrinol Diabetes Obes. 2019;26:103–8.
Osei-Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Bátkai S, et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Investig. 2005;115:1298–305.
Pagadala M, Kasumov T, McCullough AJ, Zein NN, Kirwan JP. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol Metab. 2012;23:365–71.
Iyer MR, Cinar R, Katz A, Gao M, Erdelyi K, Jourdan T, et al. Design, synthesis, and biological evaluation of novel, non-brain-penetrant, hybrid cannabinoid CB1R inverse agonist/inducible nitric oxide synthase (iNOS) inhibitors for the treatment of liver fibrosis. J Med Chem. 2017;60:1126–41.
Zelber-Sagi S, Azar S, Nemirovski A, Webb M, Halpern Z, Shibolet O, et al. Serum levels of endocannabinoids are independently associated with nonalcoholic fatty liver disease. Obesity. 2017;25:94–101.
Westerbacka J, Kotronen A, Fielding BA, Wahren J, Hodson L,Perttilä, et al. Splanchnic balance of free fatty acids, endocannabinoids, and lipids in subjects with nonalcoholic fatty liver disease. Gastroenterology. 2010;139:1961–71.e1.
Tam J. The emerging role of the endocannabinoid system in the pathogenesis and treatment of kidney diseases. J Basic Clin Physiol Pharmacol. 2016;27:267–76.
Udi S, Hinden L, Earley B, Drori A, Reuveni N, Hadar R, et al. Proximal tubular cannabinoid-1 receptor regulates obesity-induced CKD. J Am Soc Nephrol. 2017;28:3518–32.
Marks J, Carvou NJC, Debnam ES, Srai SK, Unwin RJ. Diabetes increases facilitative glucose uptake and GLUT2 expression at the rat proximal tubule brush border membrane. J Physiol. 2003;553:137–45.
Larkins RG, Dunlop ME. The link between hyperglycaemia and diabetic nephropathy. Diabetologia. 1992;35:499–504.
Hinden L, Udi S, Drori A, Gammal A, Nemirovski A, Hadar R, et al. Modulation of renal GLUT2 by the cannabinoid-1 receptor: implications for the treatment of diabetic nephropathy. JASN. 2018;29:434–48.
Tam J, Hinden L, Drori A, Udi S, Azar S, Baraghithy S. The therapeutic potential of targeting the peripheral endocannabinoid/CB1 receptor system. Eur J Intern Med. 2018;49:23–29.
Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 2005;28:164–76.
Amato G, Manke A, Wiethe R, Vasukuttan V, Snyder R, Yueh YL, et al. Functionalized 6-(Piperidin-1-yl)-8,9-diphenyl purines as peripherally restricted inverse agonists of the CB1 receptor. J Med Chem. 2019;62:6330–45.
Chang CP, Wu CH, Song JS, Chou MC, Wong YC, Lin Y, et al. Discovery of 1-(2,4-dichlorophenyl)-N-(piperidin-1-yl)-4-((pyrrolidine-1-sulfonamido)methyl)-5-(5-((4-(trifluoromethyl)phenyl)ethynyl)thiophene-2-yl)-1H-pyrazole-3-carboxamide as a novel peripherally restricted cannabinoid-1 receptor antagonist with significant weight-loss efficacy in diet-induced obese mice. J Med Chem. 2013;56:9920–33.
Receveur JM, Murray A, Linget JM, Nørregaard PK, Cooper M, Bjurling E, et al. Conversion of 4-cyanomethyl-pyrazole-3-carboxamides into CB1 antagonists with lowered propensity to pass the blood–brain-barrier. Bioorg Med Chem Lett. 2010;20:453–7.
Takano A, Gulyás B, Varnäs K, Little PB, Noerregaard PK, Jensen NO, et al. Low brain CB1 receptor occupancy by a second generation CB1 receptor antagonist TM38837 in comparison with rimonabant in nonhuman primates: a PET study. Synapse. 2014;68:89–97.
Son MH, Kim HD, Chae YN, Kim MK, Shin CY, Ahn GJ, et al. Peripherally acting CB1-receptor antagonist: the relative importance of central and peripheral CB1 receptors in adiposity control. Int J Obes. 2010;34:547–56.
Han JH, Shin H, Park JY, Rho JG, Son DH, Kim KW, et al. A novel peripheral cannabinoid 1 receptor antagonist, AJ5012, improves metabolic outcomes and suppresses adipose tissue inflammation in obese mice. FASEB J. 2018;33:4314–26.
Klumpers LE, Fridberg M, de Kam ML, Little PB, Jensen NO, Kleinloog HD, et al. Peripheral selectivity of the novel cannabinoid receptor antagonist TM38837 in healthy subjects. Br J Clin Pharmacol. 2013;76:846–57.
Kale VP, Gibbs S, Taylor JA, Zmarowski A, Novak J, Patton K, et al. Preclinical toxicity evaluation of JD5037, a peripherally restricted CB1 receptor inverse agonist, in rats and dogs for treatment of nonalcoholic steatohepatitis. Regul Toxicolo Pharmacol. 2019;109:104483.
Ziegler CG, Mohn C, Lamounier-Zepter V, Rettori V, Bornstein SR, Krug AW, et al. Expression and function of endocannabinoid receptors in the human adrenal cortex. Horm Metab Res. 2010;42:88–92.
Niederhoffer N, Hansen HH, Fernandez-Ruiz JJ, Szabo B. Effects of cannabinoids on adrenaline release from adrenal medullary cells. Br J Pharmacol. 2001;134:1319–27.
Bellocchio L, Soria-Gómez E, Quarta C, Metna-Laurent M, Cardinal P, Binder E, et al. Activation of the sympathetic nervous system mediates hypophagic and anxiety-like effects of CB1 receptor blockade. PNAS. 2013;110:4786–91.
LoVerme J, Duranti A, Tontini A, Spadoni G, Mor M, Rivara S, et al. Synthesis and characterization of a peripherally restricted CB1 cannabinoid antagonist, URB447, that reduces feeding and body-weight gain in mice. Bioorg Med Chem Lett. 2009;19:639–43.
Hånell A, Marklund N. Structured evaluation of rodent behavioral tests used in drug discovery research. Front Behav Neurosci. 2014;8:252.
Micale V, Drago F, Noerregaard PK, Elling CE, Wotjak CT. The Cannabinoid CB1 Antagonist TM38837 with limited penetrance to the brain shows reduced fear-promoting effects in mice. Front Pharmacol. 2019;10:207.
Gorzalka BB, Morrish AC, Hill MN. Endocannabinoid modulation of male rat sexual behavior. Psychopharmacology. 2008;198:479–86.
Succu S, Mascia MS, Sanna F, Melis T, Argiolas A, Melis MR. The cannabinoid CB1 receptor antagonist SR 141716A induces penile erection by increasing extra-cellular glutamic acid in the paraventricular nucleus of male rats. Behav Brain Res. 2006;169:274–81.
Gorzalka BB, Hill MN, Chang SCH. Male–female differences in the effects of cannabinoids on sexual behavior and gonadal hormone function. Horm Behav. 2010;58:91–99.
Lynn B, Gee A, Zhang L, Pfaus JG. Effects of cannabinoids on female sexual function. Sex Med Rev. 2020;8:18–27.
Pacher P, Mukhopadhyay P, Mohanraj R, Godlewski G, Bátkai S, Kunos G. Modulation of the endocannabinoid system in cardiovascular disease. Hypertension. 2008;52:601–7.
Silvani A, Berteotti C, Bastianini S, Cohen G, Lo Martire V, Mazza R, et al. Cardiorespiratory anomalies in mice lacking CB1 cannabinoid receptors. PLoS ONE. 2014;9:e100536.
Skrapari I, Tentolouris N, Perrea D, Bakoyiannis C, Papazafiropoulou A, Katsilambros N. Baroreflex sensitivity in obesity: relationship with cardiac autonomic nervous system activity. Obesity. 2007;15:1685–93.
Ruiz de Azua I, Lutz B. Multiple endocannabinoid-mediated mechanisms in the regulation of energy homeostasis in brain and peripheral tissues. Cell Mol Life Sci. 2019;76:1341–63.
Massa F, Monory K. Endocannabinoids and the gastrointestinal tract. J Endocrinol Investig. 2006;29:47–57.
Srivastava G, Apovian CM. Current pharmacotherapy for obesity. Nat Rev Endocrinol. 2018;14:12–24.
Christou GA, Katsiki N, Blundell J, Fruhbeck G, Kiortsis DN. Semaglutide as a promising antiobesity drug. Obes Rev. 2019;20:805–15.
Marcotte E, Chand B. Management and prevention of surgical and nutritional complications after bariatric surgery. Surg Clin North Am. 2016;96:843–56.
Fanelli F, Di Lallo VD, Belluomo I, De Iasio R, Baccini M, Casadio E, et al. Estimation of reference intervals of five endocannabinoids and endocannabinoid related compounds in human plasma by two dimensional-LC/MS/MS. J Lipid Res. 2012;53:481–93.
Müller TD, Clemmensen C, Finan B, DiMarchi RD, Tschöp MH. Anti-obesity therapy: from rainbow pills to polyagonists. Pharmacol Rev. 2018;70:712–46.
González-Mariscal I, Krzysik-Walker SM, Kim W, Rouse M, Egan JM. Blockade of cannabinoid 1 receptor improves GLP-1R mediated insulin secretion in mice. Mol Cell Endocrinol. 2016;423:1–10.
Patel KN, Joharapurkar AA, Patel V, Kshirsagar SG, Bahekar R, Srivastava BK, et al. Cannabinoid receptor 1 antagonist treatment induces glucagon release and shows an additive therapeutic effect with GLP-1 agonist in diet-induced obese mice. Can J Physiol Pharmacol. 2014;92:975–83.
Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature. 2017;547:468–71.
Quarta C, Clemmensen C, Zhu Z, Yang B, Joseph SS, Lutter D, et al. Molecular integration of incretin and glucocorticoid action reverses immunometabolic dysfunction and obesity. Cell Metab. 2017;26:620–32.e6.
Cluny NL, Vemuri VK, Chambers AP, Limebeer CL, Bedard H, Wood JT, et al. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br J Pharmacol. 2010;161:629–42.
Amato GS, Manke A, Harris DL, Wiethe RW, Vasukuttan V, Snyder RW, et al. Blocking alcoholic steatosis in mice with a peripherally restricted purine antagonist of the Type 1 cannabinoid receptor. J Med Chem. 2018;61:4370–85.
Zhang YM, Greco MN, Macielag MJ, Teleha CA, DesJarlais RL, Tang Y, et al. 6-Benzhydryl-4-amino-quinolin-2-ones as potent cannabinoid Type 1 (CB1) receptor inverse agonists and chemical modifications for peripheral selectivity. J Med Chem. 2018;61:10276–98.
Acknowledgements
CQ is supported by INSERM, the French Society of Diabetes (SFD), the French Society of Endocrinology (SFE), the French Society of Nutrition (SFN). DC is supported by INSERM, Nouvelle Aquitaine Region, Labex BRAIN ANR-10-LABX-43, ANR-10-EQX-008-1 OPTOPATH, ANR-17-CE14-0007 BABrain, and ANR-18-CE14-0029 Mitobesity.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Quarta, C., Cota, D. Anti-obesity therapy with peripheral CB1 blockers: from promise to safe(?) practice. Int J Obes 44, 2179–2193 (2020). https://doi.org/10.1038/s41366-020-0577-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-020-0577-8
- Springer Nature Limited
This article is cited by
-
Poly-Agonist Pharmacotherapies for Metabolic Diseases: Hopes and New Challenges
Drugs (2024)
-
The CB1 cannabinoid receptor regulates autophagy in the tibialis anterior skeletal muscle in mice
Biological Research (2023)
-
Enhancing the antidiabetic and antidyslipidemic activity of a 1,5-diarylpyrazole by solid dispersion pre-formulation
Chemical Papers (2022)
-
Cannabinoid receptors distribution in mouse cortical plasma membrane compartments
Molecular Brain (2021)
-
Hypothalamic endocannabinoids in obesity: an old story with new challenges
Cellular and Molecular Life Sciences (2021)