
Abstract
The endocannabinoid (eCB) system is widely expressed in many central and peripheral tissues, and is involved in a plethora of physiological processes. Among these, activity of the eCB system promotes energy intake and storage, which, however, under pathophysiological conditions, can favour the development of obesity and obesity-related disorders. It is proposed that eCB signalling is evolutionary beneficial for survival under periods of scarce food resources. Remarkably, eCB signalling is increased both in hunger and in overnutrition conditions, such as obesity and type-2 diabetes. This apparent paradox suggests a role of the eCB system both at initiation and at clinical endpoint of obesity. This review will focus on recent findings about the role of the eCB system controlling whole-body metabolism in mice that are genetically modified selectively in different cell types. The current data in fact support the notion that eCB signalling is not only engaged in the development but also in the maintenance of obesity, whereby specific cell types in central and peripheral tissues are key sites in regulating the entire body’s energy homeostasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Obesity is defined as a chronic low-grade inflammatory condition contributing to many comorbidities known as metabolic syndrome. The aetiology of obesity is the result of an imbalance between food intake and metabolic rate, resulting in an abnormal expansion of white adipose tissue (WAT). Obesity and obesity-related-disorders, such as type-2 diabetes and cardiovascular diseases among others, have become a global epidemic, causing a serious burden to public health with tremendous economic and social consequences. Solely dietary restriction and exercise appear to fail to maintain reduced body weight over the time [201], and, therefore, there is a demand of new long-term therapeutic approaches to tackle obesity, with reduced side effects and improved clinical efficacy. Unfortunately, the current lack of any efficacious therapeutic options predicts that the prevalence of obesity will steadily increase. One strategy is to target the central nervous system (CNS) to reduce appetite or to increase the feeling of satiety. Another strategy is to modulate peripheral mechanisms, such as to reduce fat storage, or to restore the sensitivity to weight-regulating hormones in adipose tissue.
In this context, the endocannabinoid (eCB) system has been identified, among others, as an important endogenous orexigenic signal. It is involved in the regulation of energy metabolism through activation of cannabinoid receptors in central and peripheral tissues. Importantly, obesity has been widely associated with an increase in eCB tone (for a review, see [133, 153]), but the cause of this increase has not yet been well understood, in particular, it would be important to elucidate whether this increased eCB tone is the cause or the consequence of obesity. It was reported that cannabinoid type 1 receptor (CB1)-deficient mice (called CB1–KO) showed a decreased body weight, reduced fat mass, hypophagia, and resistance to develop obesity [36, 174]. Accordingly, chronic treatment with rimonabant, a CB1 inverse agonist, also leads to a decrease in body weight, improvement in the metabolic profile, insulin sensitivity, and cardiovascular risk profile in obese mice [74] and humans [213].
Rimonabant (Acomplia, Zimulti) was approved as anti-obesity drug in Europe in 2006. Four large clinical trials on rimonabant were published [42, 168, 184, 213], reporting a weight loss of 4–6 kg in a period of 6–12 months compared to placebo [33]. Rimonabant was not only successful to reduce body weight and fat mass, but also numerous metabolic impairments associated with obesity. However, after chronic treatment, in some patients, serious neuropsychiatric side effects, such as anxiety, depression, and even suicidal ideation, were reported [33, 146], raising the question about the benefits of the treatment as an anti-obesity drug. Finally, rimonabant was withdrawn from the European market in 2008, leading also to the stop of this research line in the other pharmaceutical companies. In the light of the serious psychiatric side effects observed, it is important to understand the underlying peripheral and central mechanisms of the eCB-mediated regulation of energy homeostasis. During the last 2 decades, the role of the eCB system in the regulation of energy metabolism has been extensively studied. Several reviews on this theme have recently been published [47, 66, 160, 166, 189, 191]. In this review, we will mainly focus on the role of eCBs in metabolism at central versus peripheral tissues, as investigated by the use of cell type-specific mutant mice, leading to possible implications regarding new and improved therapeutic strategies to tackle obesity.
General features of the endocannabinoid system
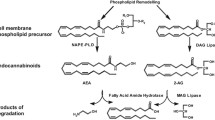
The eCB system consists of cannabinoid receptors, ligands, and enzymes involved in ligand synthesis and degradation (Fig. 1). There are two main cannabinoid receptors, CB1 [137] and CB2 [151], both belonging to the family of G protein-coupled receptors (GPCRs). In addition, two major endogenous cannabinoids have been identified, arachidonoyl ethanolamide (anandamide, AEA) [43] and 2-arachidonoyl glycerol (2-AG) [142], together with their synthesizing and degrading enzymes.

Pathways involved in the formation and degradation of endocannabinoids (in blue) and other bioactive lipids (orange). Endocannabinoids are synthesized from plasma membrane phospholipids. A major pathway of AEA synthesis requires N-acyltransferase (NAT) and NAPE-phospholipase D (NAPE-PLD). 2-AG involves phospholipase C (PLC) and diacylglycerol lipase (DAGL) enzymes. The biosynthesis pathway of AEA is the same as for the anti-inflammatory palmitoylethanolamide (PEA) and the anorexigenic oleoyl ethanolamide (OEA). The degradation of AEA and 2-AG by fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively, results in arachidonic acid (AA), which can be converted by cyclooxygenase into eicosanoids, such as prostaglandins, prostacyclins, and thromboxanes
In the neurons, the prevalent view is that eCBs are synthesized on demand at the postsynaptic site after synaptic activation, where they are released and modulate presynaptic CB1 activity to inhibit neurotransmitter release. This is considered as a retrograde negative feedback mechanism. In non-neuronal cells, it has been described that eCBs can act as in a paracrine or autocrine manner, being synthesize in a different or in the same cell type expressing CB1, as for instance in the liver and in the endocrine pancreas [90, 120, 121, 159].
CB1 and CB2 expression
CB1 is one of the most abundant GPCRs in the CNS [81]. However, it is also expressed in many non-neuronal tissues, including glial cells and peripheral non-neuronal cell types, though mostly at low to very low levels. However, the relative expression of CB1 in a cell type does not necessarily correlate with its functional relevance [72, 180]. In the brain, CB1 is widely distributed in GABA and glutamatergic neurons [145], but also in other neuronal subtypes, such as serotonergic [72], noradrenergic [24], and cholinergic neurons [156], as well as in glial cells (reviewed in [144]). Neuronal CB1 is preferentially expressed at presynaptic sites, although the latest findings have also described a postsynaptic, i.e., somatic and dendritic localization of CB1 [6, 122,123,124, 155]. Moreover, CB1 was also found in the inner membrane of mitochondria in neurons and skeletal muscle, where it regulates cellular respiration and energy production [13, 70, 101, 143]. Regarding the periphery, CB1 was found in adipose tissue, liver, skeletal muscle, endocrine pancreas, kidney, and gastrointestinal tract (reviewed in [166, 191]).
CB2 is mainly, although not exclusively, expressed in immune and blood cells [50, 151], but also in adipocytes, liver, neurons, and astrocytes [86, 108, 144, 176, 214]. Only a few publications reported on the role of CB2 in regulating energy balance [2, 216], and therefore, the role of CB2 in energy homeostasis is still a matter of debate, which needs, e.g., the genetic dissection of CB2 functions using conditional mutagenesis in mice.
It is worth noting that AEA is a promiscuous ligand which can target other receptors than cannabinoid receptors, such as transient receptor potential vanilloid 1 (TRPV1), peroxisome proliferator-activated receptors (PPARs), and GPR55 [20, 45, 83]. Therefore, genetic or pharmacological approaches targeting synthesis or degradation pathways, which modify AEA levels, could elucidate cellular responses which are not directly associated with eCB-related responses.
Synthesis and degradation of eCBs
eCBs are lipids synthesized from plasma membrane phospholipids, which are derived from arachidonic acid (AA). Several pathways have been proposed to lead to AEA and 2-AG formation (reviewed in [147]). The major pathway of AEA synthesis is the hydrolysis of the precursor N-arachidonoyl phosphatidyl ethanolamine (NAPE) by the NAPE-selective phospholipase D (NAPE–PLD) (Fig. 1). However, the generation of total NAPE–PLD knockout mice revealed the presence of additional pathways [107]. Presently, it is accepted that there are at least two additional pathways different from NAPE–PLD. One pathway involves the serine hydrolase ABDH4 (α/β hydroxylase 4). Another pathway engages the protein tyrosine phosphatase PTPN22, and the latest has been characterized in macrophage RAW264.7 cells [115]. The 2-AG biosynthesis occurs in two steps involving phospholipase C (PLC) and diacylglycerol lipase (DAGL) (Fig. 1). A second pathway leading to 2-AG has been proposed, but, since the physiological relevance of this alternative pathway is not clear yet, we will not include it in this review (reviewed in [147]. Degradation pathways of AEA and 2-AG are mediated by fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively (Fig. 1). It has been proposed that FAAH-2 in humans and N-acylethanolamide-hydrolyzing acid amidase (NAAA) could be alternative ways for AEA degradation, although their contribution to AEA hydrolysis is still on debate (see [147]. Finally, using a functional proteomic approach, two additional enzymes were identified as 2-AG hydrolases, ABHD6, and ABDH12 [17]. Therefore, synthesis and degradation of AEA and 2-AG involve redundant pathways, as it will be discussed below in further details.
Bioactive lipids of eCB synthesis and degradation pathways
It is of note that bioactive lipids other than eCBs are also generated from the above-mentioned synthesis and degradation pathways, contributing to the regulation of whole-body metabolism (see Fig. 1). In particular, this crosstalk is important to be considered when genetic or pharmacological strategies are used to modulate the endogenous levels of eCBs, thereby complicating the interpretation of some findings that we will refer below.
AEA is a member of the N-acyl ethanolamide (NAE) family, which also includes the anti-inflammatory palmitoylethanolamide (PEA) [55] and the anorexigenic oleoyl ethanolamide (OEA) [177] and, therefore, share similar synthesis and degradation pathways (Fig. 1). Both AEA congeners regulate energy homeostasis through different mechanisms. Administration of PEA to mildly obese ovariectomized rats decreased body weight, food intake, and fat mass [138]. OEA is an anorectic lipid that reduces food intake and body weight gain [177]. In addition, OEA has recently been implicated in the enhancement of β-adrenergic-mediated thermogenesis in rats [199].
Furthermore, the degradation of AEA and 2-AG results in AA, which is, in turn, a pro-inflammatory molecule. AA is converted by cyclooxygenases, finally leading to prostaglandins, prostacyclins, and thromboxanes [102] (Fig. 1). The effect of AA, prostacyclins, prostaglandins, and thromboxanes in metabolism remains to be further detailed.
Cytosolic endocannabinoid binding proteins
Recent studies have identified cytosolic lipid-binding proteins involved in cellular uptake and intracellular trafficking of the hydrophobic eCBs, AEA, and 2-AG, as well as other fatty acids [82, 96, 98]. There are at least three cytosolic chaperone proteins that can bind to endocannabinoids: fatty acid-binding proteins (FABPs); sterol carrier protein 2 (SCP-2); heat shock 70 kDa protein (HSP70) [96, 109, 157]. These lipid carriers were reported to be involved in the cellular uptake of AEA [109, 126]. In addition, FABPs facilitates the transport of AEA and 2-AG to intracellular degradation pathways [96, 140]. Accordingly, FABP inhibitors increased brain eCB levels and evoked analgesia [15, 98]. Finally, FABPs also mediate the nuclear translocation of NAEs to activate PPARα [97].
Diet-induced obesity (DIO), as model of western society feeding habits, regulates eCB tone
The unlimited availability of fat- and calorie-rich diets in the western modern society is a major contributor to the obesity pandemic, by promoting overfeeding due to high palatability. The diet-induced obesity (DIO) model, where rodents have ad libitum access to high fat diet (HFD), mimics the overconsumption of calorie-enriched diets and the development of obesity as observed in humans. In obese humans and DIO mice, there is a positive correlation between body weight and CB1 expression together with circulating eCB levels [18, 37, 65, 132, 133, 153, 182], as well as with circulating fatty acids, such as AA and linoleic acid, being both precursors of AA-derived AEA and 2-AG (reviewed in [133, 153]). It has been showed that dietary linoleic acid, which is present in high contents in western diets, facilitates eCB synthesis [4, 181]. Therefore, it is generalized that obesity is correlated with an overactivation of eCB system [133, 153].
Another interesting model to test the impact of dietary fat content without the contribution of increased energy intake is the pair-fed HFD model [128], where HFD-fed mice consumed the same amount of calories as control mice. Using this model, the authors observed that there was an increase in body weight, no further alterations of liver function, but hepatic AEA, 2-AG, and CB1 protein levels were oppositely affected in male and female mice [128].
It is interesting to note that highly caloric palatable diet during progestational and gestational stages can also lead to alterations in eCB levels, where pups from palatable diet-fed dams displayed lower levels of AEA and PEA in the hippocampus, finally leading to increased anxiety in the adulthood [171]. In the hypothalamus, AEA, 2-AG, and AA were decreased at birth. However, in the adulthood, mice displayed adiposity. Interestingly and surprisingly, diet restriction during pregnancy similarly decreased eCB levels and, finally, led to adiposity in adulthood [172]. This phenomenon is not understood and should be studied in detail.
Sexual dimorphism of eCB system
Only a few studies have investigated sex-dependent changes in the eCB system. Sexual dimorphism in the peak of CB1 expression at postnatal development in rats was reported (reviewed in [39]). In addition, in humans, females seem to be more sensitive than males to the effects of cannabinoids [39], which may be related to different pharmacokinetic responses. In rodents, female mice have higher brain AEA and 2-AG levels than males [126, 127], although these differences seem to be brain region-specific (reviewed in [179]). However, this situation is different in the liver, where there are lower AEA, but higher 2-AG and CB1 protein levels in female mice than in male mice [128]. Moreover, DIO model also seems to affect differently to male and female mice, so ad libitum HFD showed a profound impact in brain AEA levels in males but no in females [129]. Further characterization of sexual differences in the eCB system concomitantly with a better knowledge of the endocrinological mechanism underlying sexual dimorphism in the eCB system is mandatory in the next years. Therefore, we would like to stress the need to include females in such studies.
Multiple mechanisms of eCB-mediated regulation of energy homeostasis
The eCB system is strongly involved in energy storage promoting fat accumulation and caloric intake, which has been evolved to conserve energy under the conditions of scarce food [47, 166]. Thus, paradoxically, eCBs also facilitates the overconsumption of high-energy enriched palatable foods and fat accumulation as a maladaptive process, such as present in the western society or in animal models with ad libitum access to HFD.
It is well documented that cannabinoids increase appetite [12], even in a satiated state [101], and the consumption of highly palatable food such as sweets and fat-enriched food (reviewed in [161]). Indeed, Δ9-tetrahydrocannabinol (THC), the main active compound of Cannabis sativa, is prescribed as the treatment of certain diseases associated with a lack of appetite, such as chemotherapy-induced nausea and in AIDS patients (for a review, see [10]). The orexigenic effect of THC is dose-dependent, where low THC doses induce hyperphagia, whereas high doses had the opposite effect, indicating a bimodal effect of eCB signalling in feeding behaviour [12].
Accordingly, eCBs are also involved in promoting of feeding behaviour and, besides, in regulating of energy expenditure, both by central and peripheral mechanisms. Here, we will first discuss the contribution of different neuronal subpopulations in various brain regions, as evidenced by experiments using genetically modified mice.
Forebrain glutamatergic neurons
CB1 in cortical glutamatergic neurons was shown to mediate the orexigenic effect of eCBs when mice were in a hunger state [12]. Using the Cre/loxP strategy, the conditional deletion of CB1 in dorsal telencephalic glutamatergic neurons (Glu-CB1–KO mice) was sufficient to reduce food intake after overnight fasting, while the deletion of CB1 in GABAergic neurons (GABA–CB1–KO mice) induced a hyperphagic effect in the same fasted state. Both effects were observed in fasting conditions, but mutant mice also showed similar phenotype, when they were exposed to novel palatable food without the previous fasting.
In wild-type mice, THC can induce a biphasic effect on food intake during fasting-refeeding experiments, whereby a hyperphagic effect at low dose (1 mg/kg) and a hypophagic effect at high dose (2.5 mg/kg) were observed. The THC-induced hyperphagia was completely blunted in Glu-CB1–KO mice. Conversely, GABA–CB1–KO mice only showed the hyperphagic effect of THC. Interestingly, CB1-expressing projections to the ventral striatum, a brain area involved in the hedonic valence of food intake, were absent in GABA–CB1–KO mice. In accordance, bilateral injections of a CB1 antagonist into the ventral striatum were able to fully block THC-induced hypophagia but not THC-induced hyperphagia. Strikingly, both conditional CB1 mutant mice did not change body weight when fed ad libitum with regular chow.
CB1-mediated control of feeding was shown to involve the olfactory system [195]. CB1 in cortical glutamatergic neurons promotes feeding in hungry mice by increasing odour perception, linking the feeling of hunger to stronger odour processing [195]. CB1 protein in the glomerular cell layer (GCL) of olfactory bulb was not present in Glu-CB1–KO mice. GCL mainly contains GABAergic neurons that receive glutamatergic projections from olfactory cortical areas [21, 125], which would explain the lack of the presynaptic CB1 in GCL from these projections in the Glu-CB1–KO mice. In addition, 24 h fasting conditions increased AEA levels in olfactory bulb and hypothalamus. Notably, the selective CB1 deletion in olfactory cortical areas was sufficient to induce a hypophagic response after fasting conditions, similar to Glu-CB1–KO and total CB1–KO mice. To link olfactory processes to the regulation of food intake, investigators measured odour sensitivity in 24 h fasted mice. In wild-type mice, a low dose of THC (1 mg/kg) increased odour sensitivity, and this effect was also correlated with enhanced food intake. Importantly, the same dose of THC did not have any effect on odour sensitivity in Glu-CB1–KO mice. These experiments can explain the role of CB1 in the regulation of food intake in fasted mice, which is mediated by CB1 expression in glutamatergic neurons of the olfactory cortex, controlling the odour detection of food.
Apart from orexigenic function of the eCB system, there are many evidences of the involvement of the central eCB system in the regulation of whole-body metabolism. The deletion of CB1 receptor in forebrain principle projecting (CaMKII-positive) neurons and sympathetic neurons (called CaMKII–CB1–KO mice) revealed the control of energy balance through the regulation of thermogenesis in brown adipose tissue (BAT) [169]. CaMKII–CB1–KO mice were leaner than control littermates, and they did not respond to rimonabant-induced effects on body weight and on basal metabolic rate. In the DIO model, mutant mice were resistant to DIO, which was linked to enhanced lipid oxidation and BAT thermogenesis via an increased sympathetic tone [169], indicating that CB1 located in CaMKII-positive neurons play a critical role in the regulation of peripheral sympathetic activity. According to these results, chemical or surgical sympathetic denervation blunted the increased BAT thermogenesis in the mutant mice. Interestingly, the deletion of CB1 receptor in CaMKII-positive neurons did not alter the food intake, similar to the Glu–CB1–KO mice when they have ad libitum access to food.
As it has been discussed above, mice in the DIO model showed an increased eCB tone by enhanced eCB production. Thus, the impact of reduced 2-AG levels was investigated in mice with overexpressed MAGL in CaMKII-positive neurons (CaMKII–MAGL–Tg mice) [95]. Both MAGL protein expression and activity were increased in cortical areas, hippocampus, and hypothalamus of the transgenic mice, without further alterations in the gene expression of the other components of eCB system. In consequence, 2-AG levels in forebrain regions were reduced by 50% in the mutant mice, without changes in the levels of AEA or other eCB-related lipids. Similar to CaMKII–CB1–KO mice, MAGL transgenic mice on regular chow showed a lean phenotype linked to a reduced fat mass. Surprisingly, decreased 2-AG levels in forebrain neurons induced a hyperphagic phenotype, which may be a compensatory mechanism against reduced body weight. CaMKII–MAGL–Tg mice were also resistant to DIO, which correlated with a hypersensitivity to β3-adrenergic stimulated thermogenesis and enhanced mitochondrial density in isolated BAT. In the DIO model, both wild-type and CaMKII-MAGL-Tg mice consumed equal amount of food, although the decreased 2-AG levels reduced feed efficiency in the transgenic mice. Thus, central 2-AG regulates energy balance by controlling heat dissipation.
In summary, eCB signalling in glutamatergic neurons of forebrain regions is centrally involved in the regulation of energy balance through the modulation of sympathetic activity and, in extension, the regulation of BAT thermogenesis without affecting feeding behaviour. In contrast, the CB1 expression in glutamatergic neurons in the olfactory cortex plays a critical role in the orexigenic properties of the eCB system through the modulation of odour processing, but only in the conditions of immediate energy needs. Therefore, CB1 expression in particular neuronal subpopulations will elicit different metabolic responses, depending on the physiological role of the projecting areas that are affected by the eCB signalling, and these different circuits will be activated to cope with the internal state of the body. However, the exact involvement of eCBs and CB1 in these regulatory circuits is far from being understood, and circuit-specific genetic manipulations are required.
Hypothalamus
The hypothalamus is a hub for homeostatic feeding regulation and energy balance. It comprises several nuclei that regulate many physiological responses, such as food intake, lactation, sexual behaviour, and others. Several evidences have pointed to the role of the hypothalamic eCB system in obesity as well as in the regulation of appetitive behaviour. For example, obese rats with genetic deficiency of leptin signalling (ob/ob and db/db) showed elevated levels of eCBs in the hypothalamus [44], and both ob/ob and db/db rats showed a hyperphagic phenotype. Furthermore, acute anorexigenic leptin treatment in ob/ob or normal rats reduced hypothalamic AEA and 2-AG levels [44]. In the ventromedial nucleus of the hypothalamus (VMH), a local injection of AEA induced a significant increase in caloric intake in presatiated rats; this response was completely blocked by rimonabant, indicating that the hyperphagic effect of AEA in the VMH was CB1-dependent [87]. Likewise, local administration of THC into the paraventricular nucleus of the hypothalamus (PVN) stimulated food intake in satiated rats in a CB1-dependent manner, while local rimonabant administration alone did not affect feeding [215]. According to these data, CB1 is directly involved in the regulation of feeding behaviour in different hypothalamic subregions.
The partial CB1 deletion (about 58%) in the hypothalamus, using an adeno-associated viral (AAV) approach, revealed a reduced body weight linked to an increase in energy expenditure, but no changes in food intake in standard diet-fed mice [25]. The authors reasoned that the lack of cell-type specificity of the viral approach could explain the absence of changes in feeding behaviour; however, the above-described pharmacological injections into the hypothalamus are also devoid of cell-type specificity.
To gain cell-type specificity, the same group investigated the physiological relevance of CB1 in VMH. VMH was described as the satiety centre, and ablation of VMH induced a voracious feeding (reviewed in [32]. Gene expression studies have identified that steroidogenic factor-1 (SF-1, also named NR5A1) is exclusively expressed in the VMH. Therefore, the conditional deletion of CB1 in the VMH, crossing SF1-Cre mice with CB1 floxed mice, showed the importance of CB1 in metabolic flexibility [26]. Metabolic flexibility is the ability of an animal to adjust to different diets. SF1–CB1–KO mice showed decreased adiposity on regular chow, while HFD exposure increased adiposity as compared to wild-type controls. [26]. The mild lean phenotype on regular chow was associated with increased glucose and insulin sensitivity, concomitantly with an increased lipid oxidation and lipolysis in white adipose tissue (WAT) mediated by increased sympathetic nervous system activity. In contrast, the obese phenotype of SF1–CB1–KO mice on HFD was linked to hyperphagia and reduced lipolysis and lipid oxidation in WAT. In accordance, loss-of-function of the 2-AG degrading enzyme ABHD6 in VMH neurons confirmed the importance of the eCB system in metabolic flexibility [59]. Deletion of ABHD6 in VMH neurons increased the fasting-induced elevated 2-AG levels in this area compared to wild-type mice. The enhanced 2-AG levels contributed to reduced fasting-refeeding response, suggesting that ABHD6 activity may be involved in the bimodal modulation of feeding behaviour by eCBs. In fact, VMH-selective ABHD6 deficiency promoted body weight gain and reduced energy expenditure in a DIO model.
PVN is a primary brain region that integrates anorexigenic and orexigenic signals from the arcuate nucleus (ARC) of the hypothalamus. Cota and co-workers have elucidated the role of CB1 in the PVN [27]. CB1 deletion was performed in single-minded 1 (Sim1)-positive neurons, which account for a vast number of PVN neuron, by generating Sim1–CB1–KO mice. Sim1–CB1–KO mice showed a very mild phenotype without metabolic changes on standard diet. In the DIO model, these mutants showed reduced body weight gain by elevating energy expenditure, without affecting food intake. Gene expression studies in BAT displayed enhanced thermogenic and mitochondrial activity, suggesting an increase in sympathetic drive. Indeed, pharmacological sympathetic denervation reverted the decreased body weight in HFD-fed Sim1–CB1–KO mice. Therefore, genetic CB1 blockade in Sim1-positive neurons in the PVN did not affect feeding behaviour, but was able to modulate energy expenditure in a DIO model. Of note, ablation of Sim1-positive neurons caused obesity by increasing food intake and reducing energy expenditure [208], confirming the pathophysiological relevance of these neurons in obesity.
In the hypothalamic arcuate nucleus (ARC), agouti-related peptide-expressing neurons promote food intake, whereas POMC-positive neurons promote satiety. Thus, ablation of POMC neurons caused severe hyperphagia and obesity [35]. Surprisingly, a hyperphagic dose of a CB1 agonist resulted in the activation of a subset of POMC neurons, as assessed by the expression of c-fos protein and ex vivo electrophysiological recordings [101]. In addition, local CB1 agonist injections into the ARC resulted in enhanced feeding to a similar extent as intraperitoneal administration. The POMC gene encodes both the anorexigenic peptide α-melanocyte-stimulating hormone (α-MSH) and the orexigenic opioid peptide β-endorphin. Strikingly, CB1 stimulation triggered an increase of β-endorphin secretion, but not of α-MSH, in PVN, a main site of POMC efferents controlling food intake. As a cellular mechanism, the authors, furthermore, proposed that mitochondrial CB1 stimulation and generation of reactive oxygen species are involved in increased POMC neuronal activity, which then promotes food intake.
The hypothalamus includes several regions that regulate feeding behaviour; however, selective genetic deletion of eCB system components in some of these neurons did not affect caloric intake. Paradoxically, the powerful effect of marijuana driving palatable food consumption seems to be related to unusual activation of a hypothalamic brain area involved in feeding suppression. In summary, hypothalamic CB1 is involved in different metabolic responses, finally favouring energy storage.
Mesolimbic dopamine system
Feeding behaviour is also controlled by the reward system, which promotes overfeeding beyond energy homeostatic requirements. Overeating is a major risk to develop obesity. Concerning the reward system, the eCB system influences the positive-reinforcing hedonic effects of natural rewards, including palatable foods [161]. Thus, both exogenous 2-AG and AEA increase extracellular dopamine levels in the nucleus accumbens (NAc), a brain area part of the mesocorticolimbic dopamine pathway strongly linked to the incentive values of the stimuli, in a CB1-dependent manner [193]. Dopamine signals the sensation of pleasure in the brains, which can also be increased by the conditioning stimulus associated with the reward, promoting the motivation and drive of those behaviours that provide the palatable food. In addition, CB1 is expressed throughout the regions implicated in reward and addiction [161]. Importantly, THC increases the motivation to obtain food in a progressive ratio schedule as well as free food intake [77, 192], suggesting that increased food reinforcement might underlie the hyperphagic effect of THC. Indeed, pharmacological and genetic CB1 blockade resulted in a strong reduction in sweet and fat reinforcement and motivation in rodents [119, 192, 217]. Accordingly, local infusion 2-AG into the NAc shell can induce voracious feeding in satiated animals [100]. Similar data were reported using local injections of AEA in the same brain region [194]. Taken together, CB1 is a crucial neuronal substrate for positive reinforcement and motivational properties of highly palatable food. These data strongly indicate that central CB1 plays a key role in the regulation of the rewarding properties of foods, which could have a pivotal role in the overconsumption of tasty foods in obesity. Here, regarding the underlying mechanism, it would be important to elucidate the role of the eCB system in the conditioning stimulus to control rewarding behaviours, which could lead to loss of control of eating in obesity. Finally, the crosstalk between the eCB and the dopamine system should also be clarified in more detail.
Peripheral tissues
The peripheral eCB system has been identified in adipose tissue, liver, endocrine pancreas, kidney, immune cells, skeletal muscle, gastrointestinal tract, and oral cavity. Here, we will summarize important findings regarding these different peripheral cell types, as obtained by genetic and pharmacological approaches.
Adipocytes
There are two main types of adipose tissues (WAT and BAT), which exert different physiological function in the regulation of energy homeostasis. While WAT is involved in energy storage and adipokine secretion, BAT is specialized in thermogenesis and energy dissipation. In obesity, there is an abnormal increase in adiposity, which is a major risk factor for type-2 diabetes, hepatic steatosis, cardiovascular diseases, and other obesity-related comorbidities.
Several studies have confirmed the expression of CB1 in cultured primary adipocyte as well as in the mouse 3T3 F442A adipocyte cell line [14, 36], where activation of CB1 leads to increased adipogenesis and lipogenesis [11, 36, 132]. CB1 expression is higher in mature adipocytes than in preadipocytes [14, 176]. The expression of CB2 was also reported in adipocytes [176], although its functional relevance needs still to be elucidated in detail. In addition, adipose tissue expresses high levels of PPARγ, a master regulator of adipocyte differentiation [209]. AEA can also act as a PPARγ agonist, thereby increasing adipocyte differentiation and lipid accumulation [20, 99].
The eCB system is largely involved in energy storage in adipose tissue by promoting adipogenesis and lipogenesis [36, 63]. Indeed, obesity is characterized by increased eCB levels and CB1 gene expression in adipose tissue [14, 41, 220], although others reported opposite results [18, 197]. The eCB system in adipose tissue seems to be part of a positive feedback loop, where it is a key regulator of adipogenesis and lipogenesis and, in turn, eCB signalling is also increased during obesity, thereby further promoting fat accumulation. Nisoli and co-workers showed that stimulation of CB1 impaired mitochondrial biogenesis in mouse and human primary white adipocyte cells [205, 206], which was restored by pharmacological or genetic CB1 blockade [205, 206]. Furthermore, CB1 activation regulates adipokine secretion as shown in studies using cell culture models [14, 63, 132]. Altogether, these data strongly support the role of the eCB system in the regulation of adipocyte function using in vitro studies, either in 3T3F442A cells or in primary adipocyte cell cultures from rodents and humans. Below, we will discuss these studies targeting the eCB system selectively in adipocytes in genetically modified mice.
The first study analyzed the conditional deletion of NAPLE–PLD in adipocytes [67]. To reach cell-type specificity, the authors crossed NAPE–PLD floxed mice with Fabp4-Cre (also called aP2-Cre) mice, in which Cre recombinase is expressed under the control of aP2 (fatty acid-binding protein 4) promoter. Other studies, however, described that Cre recombinase activity from aP2-Cre mice also occurred in non-adipocyte cells [88]. Nevertheless, the authors reported that the reduction in NAPE–PLD expression was restricted to adipocytes [67]. Notably, AEA levels in adipose tissue did not change between mutant and wild-type mice, confirming the existence of alternative synthesis pathways for AEA [67, 107, 188]. In contrast, the deletion of NAPE–PLD in adipocytes was able to significantly decrease other members of the NAE family (PEA, OEA, and stearoyl ethanolamide) in the mutant mice fed on standard diet. Interestingly, a similar reduction in these NAE levels was found in HFD-fed NAPE–PLD–WT mice, but not any further decrease in HFD-fed mutant mice. It may be possible that the ceiling effect in the reduction of NAE levels was reached by HFD treatment, and no further reduction could be reached after the deletion of NAPE–PLD. It is remarkable that both adipocyte-specific NAPE–PLD deletion and HFD treatment had the same effect reducing the levels of these bioactive lipids in adipose tissue. Accordingly, the metabolic characterization of adipocyte-specific NAPE–PLD–KO mice correlated well with HFD-fed obese mice. Thus, adipocyte-specific NAPE–PLD knockout mice were prone to obesity and fat mass accumulation, glucose intolerance, and insulin resistance, and they showed an increase in circulating triglycerides and cholesterol levels as well as decreased thermogenesis. Moreover, the deficiency of NAPLE-PLD in adipocytes induced an increased inflammation, which could be explained by decreased levels of the anti-inflammatory lipid PEA. Mutant mice also showed a reduced WAT browning, which might be associated with decreased OEA levels in adipose tissue. Recently, OEA was shown to be involved in β-adrenergic-mediated thermogenesis in rats [199]. In addition, a lipidomic analysis of mutant adipose tissue also showed alterations in other bioactive lipids, such as eicosanoids, and ceramides, which could be linked to metabolic disturbances. Finally, adipose-specific NAPE–PLD deficiency also affected gut microbiota composition through an unknown mechanism. In summary, adipocyte NAPE–PLD control fat mass, glucose homeostasis, browning of WAT, and gut microbiota. Regarding the underlying mechanisms, since adipocyte-specific NAPLE–PLD deletion did not alter AEA levels in adipose tissue, and hence, the metabolic phenotype of these mutant mice might be related to the alterations of other bioactive lipids, such as other NAEs, eicosanoids, or ceramides; thus, further studies are needed to clarify these points.
Our work recently demonstrated that adipocyte-specific deletion of CB1 in mature adipocytes (Ati–CB1–KO mice) was sufficient to protect mice against deleterious effects of diet-induced obesity [180], confirming the crucial role of CB1 in adipocytes in the control of energy metabolism and adipocyte physiology. For the generation of the mutant mice, we used tamoxifen-inducible AdipoqCreERT2 mice [183], expressing Cre recombinase under the regulatory elements of the adiponectin gene. The Cre recombinase expression of adiponectin-Cre mouse lines has been shown in several investigations to be highly specific to adipocytes [88, 105, 150, 180, 183]. Ati–CB1–KO mice showed a reduced body weight, reduced total adiposity, improved glucose homeostasis, and improved plasma metabolic profile in a diet-induced obesity model. Remarkable, adipocyte CB1 deficiency had a profound impact in adipocyte remodelling toward lowered energy storage capacity, in a fat depot-specific manner. Thereby, we found an impaired differentiation state in epididymal WAT, browning of subcutaneous WAT, and an increase in thermogenic gene program in BAT. Accordingly, Ati-CB1–KO mice showed an enhanced energy expenditure and increased sympathetic tone. Caloric intake was reduced in the mutant mice compared to control littermates, although the pair-feeding experiment suggested that the lean phenotype of Ati–CB1–KO mice was primarily due to feeding-independent mechanisms. Strikingly, CB1 deletion in adipocytes caused an increase in alternatively activated M2 macrophages. In adipose tissue, these macrophages are dominant in lean mice, while obesity induces a recruitment and accumulation of classically activated pro-inflammatory M1 macrophages [116, 117]. Recent investigations have suggested that M2 macrophages are a local source of norepinephrine which may contribute to browning of subcutaneous fat and adaptive thermogenesis [154, 170], although latest investigations have raised questions about this process [58, 167]. Regardless whether or not alternatively activated M2 macrophages are able to synthesize norepinephrine, the M2 macrophage polarization in adipose tissue is thought to cause a beneficial effect in whole-body metabolism [84, 173, 200, 218, 219]. Importantly, both adipocyte remodelling and M2 macrophage polarization precede the appearance of body weight differences, i.e., obesity. Finally, the tamoxifen-inducible mouse model allowed evaluating the effect of the deletion in a preexisting obese condition. CB1 deletion selectively in adipocytes in obese mice was sufficient to induce body weight loss, to improve metabolic deleterious effects of obesity, and to reduce obesity-related behavioural alterations. In conclusion, above findings revealed a key role of adipocyte CB1 function in energy balance. The selective deletion of CB1 in adipocytes promotes a profound remodelling not only in adipocytes but also in adipocyte resident cells, including alternatively activated macrophages. These processes finally lead to increased sympathetic innervation, reduced energy storage capacity of adipocytes, and enhanced thermogenic program in subcutaneous WAT and BAT.
In summary, eCB signalling in adipocytes is a key regulator of adipocyte physiology and whole-body energy homeostasis, promoting a profound remodelling of adipocytes and resident adipose tissue cells. However, further investigations are needed to understand the mechanisms underlying the adipocyte-brain and adipocyte-immune cell crosstalk as well as the contribution of each cell type to the reduced body weight and the suppression of the metabolic syndrome.
Liver
Several studies showed the relevance of eCB signalling in liver functions. First, both eCB levels (AEA and 2-AG) in the liver are similar to those found in the brain [90, 158, 187], and they were detected in different cell types of the liver, such as hepatocytes and stellate cells. Importantly, increased serum levels of eCBs correlated to non-alcoholic fatty liver disease (NAFLD) in humans, independently of obesity [223]. Second, HFD increased selectively AEA levels, but not 2-AG levels, in the liver, which was caused by a decrease in hepatic FAAH activity [158]. Third, total CB1–KO mice were protected against diet-induced hepatic lipogenesis and steatosis [158]. Hepatic steatosis is characterized by an ectopic fat accumulation, and is a result of increased hepatic lipogenesis, decreased hepatic free fatty acid oxidation, and an increased free fatty acid uptake into the liver. Hepatic CB1 activation induced the expression of several enzymes involved in de novo lipogenesis, promoting fat accumulation in the liver, which can lead to hepatic steatosis [158]. The role of CB1 in hepatic lipogenesis was also shown by the strong reduction in hepatic steatosis in obese Zucker rat after pharmacological blockade of CB1 [64]. Indeed, the selective deletion of CB1 in hepatocytes was sufficient to protect against diet-induced liver steatosis, hepatic fatty acid oxidation, insulin and leptin resistance, as well as dyslipidemia [159]. However, hepatocyte-specific CB1 knockout mice developed obesity and adiposity to a similar extent as wild-type mice when maintained on HFD [159].
FABP1 is the major cytosolic-binding protein for AEA and 2-AG in mouse and human liver [82, 140]. FABP1 helps trafficking eCBs to degrading enzymes for hydrolysis [82, 140, 141]. Thus, FABP1 ablation markedly increased hepatic and brain AEA and 2-AG levels in male mice, but not in females [82, 126,127,128]. Indeed, hepatic FABP1 expression is sexual dimorphic, showing lower expression levels in females compared to males [127], which suggests that other cytosolic lipid-binding proteins could be involved in trafficking of eCBs in female mice. These FABP1 trafficking proteins chaperone not only eCBs but also other fatty acids. The increased brain eCB levels correlated with elevated free and total arachidonic acid (AA) levels in brain and serum of mutant mice [126]. Therefore, it is plausible that brain eCB levels were enhanced by local synthesis in the liver, thereby increasing plasma availability of their precursor AA, because FABP1 also contributes to hepatic AA clearance. Finally, FABP1 ablation alters hepatic eCB responses to pair-fed HFD treatment [128].
Similar to HFD treatment, chronic alcohol consumption can also lead to fatty liver by increasing hepatic lipogenesis and reducing hepatic lipolysis. The eCB system was also shown to be a master regulator of alcoholic fatty liver [90]. In contrast to diet-induced hepatic steatosis, chronic ethanol intake resulted in a selective increase in 2-AG levels and DAGLβ gene expression in hepatic stellate cells [90]. Importantly, hepatocyte-specific CB1 knockout mice were resistant to alcohol-induced hepatic steatosis, hepatic lipogenesis, and decreased hepatic fatty acid oxidation [90]. The authors concluded that paracrine activation of hepatic CB1 by 2-AG synthesis in stellate cells is responsible for alcoholic fatty liver, by increasing lipogenesis and reducing fatty acid oxidation in hepatocytes.
Glucocorticoids are stress hormones that are relevant players in obesity. Hypercortisolemia shares many characteristics with the metabolic syndrome. One hypothesis is that glucocorticoid-related obesity and metabolic syndrome are mediated by an increase in eCB signalling. Chronic exposure to excess of corticosterone (CORT, 100 µg/ml in drinking water) in mice induced obesity, adiposity, hormonal dysregulation, and hepatic steatosis [22]. Chronic CORT exposure leads to twofold increased hepatic AEA levels, while hepatic 2-AG levels were significantly reduced [22]. Similar to DIO, hepatocyte-specific CB1-KO mice were protected from CORT effects in dyslipidemia and hepatic steatosis, although they still developed CORT-induced obesity [22].
Furthermore, a liver regeneration model by partial hepatectomy showed that FAAH works in a reversal manner and drives AEA synthesis instead of hydrolysis [86, 149]. The reactions require high concentrations of AA and ethanolamine, which also require concomitantly an overactivation of phospholipase A2 and phospholipase D to provide high concentration of both precursors of AEA synthesis. Phospholipase D activity is increased during liver regeneration, and it might be dependent on CB1 stimulation [149]. Finally, CB2 expression levels are only detectable in the liver under pathophysiological conditions (steatosis, cirrhosis, and NAFLD). In summary, partial hepatectomy promotes AEA synthesis by reversal FAAH activity, and AEA production concomitantly with CB1 and CB2 activation is, in turn, involved in liver regeneration.
Above findings are compelling evidence about the critical role of hepatic CB1 in the regulation of hepatic lipid metabolism, insulin resistance, and pathogenesis of hepatic steatosis; otherwise, it has a minor contribution to increased body weight and fat mass in diet-induced or cortisolinemia-related obesity. The selective expression of the eCB chaperone FABP1 is attractive for targeting the eCB hepatic system, although the fact that FABP1 also binds other fatty acids must be kept in mind as off-target side effects. Finally, there are still open questions regarding the contradictory role of the eCB system in liver regeneration and the contribution of CB2 in liver pathophysiological conditions.
Endocrine pancreas
The endocrine pancreas is involved in the regulation of glucose homeostasis by producing and releasing pancreatic hormones. It is composed by five different cell types (α-cells, β-cells, γ-cells, δ-cells, and ε-cells), and every cell type produces and releases different hormones (glucagon, insulin, somatostatin, pancreatic polypeptide, and ghrelin), which all regulate glucose levels by independent mechanisms. For example, insulin is secreted by pancreatic β-cells to regulate glucose uptake in skeletal muscle and adipocytes, and to suppress lipolysis in adipocytes, thereby promoting fat accumulation. Insulin secretion can be regulated via different mechanisms acting in central and peripheral tissues [30, 207]. Below, we will summarize the evidences of the physiological role of the eCB system in the regulation of insulin secretion.
The presence of the eCB system was described in endocrine pancreas [16, 132, 197, 120, 121]. Expression analyses confirmed the presence of enzymes involved in eCB synthesis and degradation, together with CB1, CB2 and TRPV1 receptors, in isolated pancreatic islets of mice [120, 121, 197], of humans [16], in rat insulinoma RIN-m5F pancreatic β-cells [132], and in rat β-cell-derived INS-1E cells [120]. Double immunofluorescence experiments showed CB1 expression mostly restricted to insulin-positive β-cells in foetal and adult endocrine pancreas [120, 121], although other studies found opposite findings with more abundant expression in glucagon-positive α-cells [16, 197]. In contrast, TRPV1 and DAGLα are present in both α- and β-cells [120, 121], while MAGL and ABHD6 were found mainly in non-β-cells, indicating that both autocrine and paracrine 2-AG signals exert CB1-stimulated insulin release in β-cells [120]. Regarding CB2, the expression is quite low in the endocrine pancreas, and it seems to be restricted to the exocrine pancreas [16, 120, 132].
Agonist-induced CB1 activation stimulated basal (low glucose) and glucose-dependent insulin secretion in pancreatic β-cell lines as well as in pancreatic islets isolated from mouse and human [120, 132]. In rat insulinoma cells, both insulin responses were blocked by CB1 antagonists, but not by TRPV1 or CB2 antagonist [120, 132]. Importantly, confirming the latter data, genetic ablation of CB1 suppressed the CB1 agonist-stimulated insulin secretion from isolated pancreatic islets [120]. These observations strongly suggest that, despite the expression of other non-CB1 cannabinoid receptors in the endocrine pancreas, CB1 signalling is involved in eCB-stimulated insulin secretion in β-cells. These findings, together with high glucose-induced AEA and 2-AG synthesis in insulinoma pancreatic β-cells [120, 132], indicate a positive feedback mechanism between glucose and eCBs.
Insulin is released from β-cells in two phases. In the first step, insulin is released from rapid fusion of insulin granules to the plasma membrane, and this requires calcium stimulation. In the second phase, the released vesicles need to be replenished from reserve insulin granules that move to the plasma membrane. The replenishment depends on focal adhesion kinases that induce a cytoskeletal remodelling to facilitate the insulin secretion. As mechanistic insight, in rat INS-1E cells, AEA treatment increased focal adhesion plaque formation enabling trafficking and release of insulin granules [120]. The authors concluded that CB1 signalling in pancreatic β-cells plays a critical role in dynamic cytoskeletal remodelling, suggesting a physiological function of eCBs in the glucose-stimulated insulin release.
Finally, eCBs regulate α- and β-cell organization during the development of pancreatic islet formation [121]. Importantly, colocalization immunofluorescence experiments detected the expression of CB1, TRPV1, and 2-AG metabolic enzymatic pathway in fetal endocrine pancreas (embryonic day 16.5), showing a similar cellular expression pattern as in adulthood, only DAGLα was preferentially expressed in α-cells during the embryonic stage. Thus, 2-AG signalling works in a paracrine manner at prenatal stage, being α-cells the source and β-cells the sensors of 2-AG responses. In addition, morphological analysis of pancreatic islets in adult mice from total MAGL- and CB1-deficient mice suggests that 2-AG signalling impairs cell segregation. Therefore, paracrine 2-AG signalling determines cell segregation in fetal mouse pancreas via CB1 [121].
Overall, the above studies support that chronic CB1 activation in pancreatic β-cells may be involved in the development of insulin resistance in the metabolic syndrome. Nevertheless, conditional deletion of CB1 or 2-AG metabolism targeting endocrine pancreatic cells will be needed to shed light on the functional relevance of the eCB system in insulin and glucagon secretion in physiological and pathological conditions, such as obesity and type-2 diabetes.
Kidney
The eCB system was also shown to regulate renal functions during pathological conditions, such as obesity and type-2 diabetes [78, 92, 211]. Obesity induces several renal dysfunctions even at the early stage, whereas diabetic nephropathy is a serious complication associated to type-1 and type-2 diabetes. In both metabolic disorders, upregulation of CB1 expression was found in kidney [92, 211].
CB1 is expressed in several cell types in the kidney; for example, in glomerular podocytes [7, 92, 152], mesangial cells [111], and in renal proximal tubular cells [89, 92, 110, 211]. Importantly, peripheral CB1 blockade improved albuminuria, renal inflammation, and alteration of the renin–angiotensin system in prediabetic Zucker diabetic fatty (ZDF) rats (6 weeks old), preventing diabetic nephropathy [92]. However, peripheral CB1 antagonist only reversed fully developed diabetic nephropathy without affecting hyperglycemia in diabetic ZDF rats (15 weeks old) [92]. Thus, CB1 blockade was able to prevent and reverse diabetic nephropathy.
Regarding cell-type specific function of CB1, the selective CB1 deletion in renal proximal tubular cells (RPTC–CB1–KO mice) was sufficient to alleviate obesity-induced lipid accumulation, oxidative stress, inflammation, and fibrosis in the kidney as well as obesity-related renal damage [211]. However, RPTC–CB1–KO mice developed obesity and metabolic syndrome on HFD treatment [211]. A follow-up study showed that deletion of CB1 in these renal cells decreased glucose transporter 2 (GLUT2) expression, which reduced glucose reabsorption in the kidney, protecting from diabetic nephropathy after streptozotocin-induced diabetes [78]. One hypothesis is that hyperglycemia affects renal function and induces diabetic nephropathy via increasing GLUT2 expression in RPTC [104, 135]. Nevertheless, the role of CB1 in other renal cells in metabolic-induced kidney damage cannot be excluded.
Taken together, these results indicate an important role of CB1 signalling in renal homeostasis and function, in particular, during metabolic pathological conditions. Similar to the situation in the liver, renal CB1 signalling does not affect the whole-body energy homeostasis.
Immune cells
A plethora of studies have demonstrated the importance of immune cells in the regulation of whole-body energy homeostasis (reviewed in [23, 80]. First strong evidence of a link between obesity and immune cells came from the observation that macrophages secrete inflammatory cytokines, thereby inducing insulin resistance in adipose tissue [162].
In this scenario, CB1 signalling was reported to induce pancreatic β-cell failure by promoting M1 macrophages infiltration in pancreatic islets in ZDF rats [91]. The activation of CB1 in macrophages leads to the activation of Nlrp3-ASC inflammasome, a protein complex involved in β-cell loss in type-2 diabetes [91]. First, they observed that peripheral CB1 blockade reduced macrophage infiltration and promoted M2 macrophage polarization in isolated ZDF pancreatic islets. Furthermore, the authors demonstrated that pharmacologically induced macrophage depletion delayed the onset of insulin resistance in ZDF rats, and decreased AEA content and CB1 mRNA levels in isolated pancreatic islets [91], indicating that macrophages are the main source of eCBs and CB1 expression in pancreatic islets. Importantly, selective knockdown of macrophage CB1 by CB1 siRNA delivery using β-1,3-d-glucagon particles (i.p., for 10 days) normalized the blood glucose and plasma insulin levels, decreased macrophage infiltration and inflammation into the islets, similar to the pharmacological macrophage depletion treatment. In addition, AEA incubation of RAW264.7 macrophages and human macrophages, but not of MIN6 insulinoma cells, increased the secretion of pro-inflammatory cytokines, such as IL-1β, TNFα, and MCP-1 [91].
In a follow-up study [93], the same group generated ZDF rats with global deletion of CB1 (ZDF–CB1–KO rats). These rats showed a reduction in β-cell failure, hyperglycemia, and diabetic-derived nephropathy compared to ZDF rats [93]. They also displayed a reduced food intake, delayed body weight gain, reduced plasma lipid profile, and improved hypoadiponectinemia compared to control ZDF rats. ZDF rats also developed extreme hyperglycemia due to β-cell loss, but ZDF–CB1–KO rats were euglycemic for more than 6 months, which was reflected in an improved glucose homeostasis [93]. Thus, ZDF-CB1-KO rats were protected against β-cell failure, which was associated with a reduced CD68+ macrophage infiltration. Importantly, using an irradiation bone marrow transplantation approach, bone marrow from ZDF–CB1–KO donors to ZDF rats induced a normalization of blood glucose levels and pancreatic islet function, but no changes in body weight, food intake, or plasma lipid profile were observed.
The above findings demonstrate that macrophage CB1 play a prominent role in the progressive loss of β-cell function in ZDF rats through the activation of Nlrp3 inflammasome. However, it is unknown whether macrophage CB1 also play a similar role in other tissues. Liver resident macrophages, called Kupffer cells, are the major source of pro-inflammatory cytokines and play a critical role in hepatic inflammatory responses to different insults as, for example, in NAFLD. Indeed, using GeRP (β-1,3-d-glucagon-encapsulated siRNA particles) technology of intravenous CB1 siRNA delivery, treated mice showed a selective knockdown of CB1 gene expression in Kupffer cells [94]. In DIO mice, CB1 knockdown in liver macrophages improved in vivo glucose tolerance and insulin sensitivity, and decreased hepatic inflammation [94]. Importantly, in isolated CB1 siRNA treated Kupffer cells, there was a shift from pro-inflammatory M1 to anti-inflammatory M2 macrophages together with a decreased gene expression of pro-inflammatory markers, such as TNFα, CCL2, IL-6, and l-1β [94]. Remarkably, hepatocytes incubated with conditioned media (CM) from LPS-stimulated wild-type Kupffer cells showed a significantly reduced insulin signalling compared to hepatocytes incubated with CM from CB1-deficient Kupffer cells, indicating that cytokines from Kupffer cells inhibit hepatic insulin responses [94]. Thus, CB1 signalling in liver macrophages is important in hepatic insulin resistance.
Significant advances in understanding the role of immune cells in obesity have been achieved during the last years. The metabolic syndrome is defined as a low chronic inflammatory disease where M1 macrophages regulate the expression and release of different pro-inflammatory cytokines. CB1 in pancreatic and hepatic macrophages is an important regulator of insulin sensitivity in the metabolic syndrome, suggesting macrophage CB1 as a potential therapeutic target for type-2 diabetes. It is needed to extent the knowledge of the role of CB1 macrophages in other metabolically relevant tissues, for example, in adipose tissues.
Skeletal muscle
Skeletal muscle is a major player of total resting energy expenditure and insulin-induced glucose uptake, thereby being an important player in whole-body energy metabolism and insulin sensitivity. Expression of CB1, CB2, TPRV1, and other components of the eCB system was reported in skeletal muscle [28, 52, 54]. Strikingly, the majority of CB1 in skeletal and myocardial muscle is present in the mitochondria [143].
The eCB system seems to regulate different cellular responses in skeletal muscle. CB1 activation decreased insulin-mediated glucose uptake in primary human skeletal muscle cells [52]. Consistently, pharmacological CB1 blockade increased glucose uptake in L6 myotubes [54] and in rat isolated soleus muscle [112, 114]. CB1 knockdown by siRNA in cultured skeletal muscle cells revealed similar effect in glucose uptake as CB1 antagonists [54]. However, it was also reported that AEA increased 2-deoxy-d-glucose uptake in human skeletal muscle [52], although this opposed effect could also be mediated via non-CB1 cannabinoid receptors. AEA is a promiscuous ligand binding to different receptors, such as CB1, CB2, PPAR, and TRPV. CB1 agonist treatment also inhibits mitochondrial biogenesis and mitochondrial respiration in cultured primary skeletal muscle cells [29, 143, 206]. It was also reported that high dose of AEA increased PGC1-α expression possibly through TRPV1 activation [118]. Finally, in in vitro models, a negative effect of CB1 signalling on muscle oxidative pathways was described [29], which correlated with the increased whole-body oxygen consumption associated with CB1 antagonism [1, 75, 114].
Overall, CB1 signalling in skeletal muscle negatively affects insulin-dependent glucose uptake, mitochondrial biogenesis and respiration, as well as fatty acid oxidation pathways. Therefore, it is tentative to speculate about the role of eCBs in skeletal muscle in the context of obesity and exercise. As mentioned above, obesity is associated with a hyperactive eCB system, while voluntary running also increased plasma AEA levels, but not 2-AG levels [62, 76, 196]. Strikingly, dietary fat intake reduced CB1 expression levels in skeletal muscle in humans [53] and rats [40], possibly as a compensatory mechanism of increased tissue levels of eCBs. Further studies should shed light on the detailed physiological role of the eCB system in skeletal muscle in the context of DIO as well as in the potential beneficial effects of aerobic exercise in metabolic disorders.
Gastrointestinal tract
eCB activity also regulates functions in the gut, such as gastric emptying, gastrointestinal (GI) motility, and gastric acid secretion. The eCB-mediated reduction of gastrointestinal motility seems to depend on the inhibition of smooth muscle contraction through the modulation of vagal (parasympathetic) outflow (reviewed in [164]. Compelling evidence showed that enteric CB1 inhibits acetylcholine release from cholinergic neurons at the enteric synapses to coordinate the gastrointestinal transit [38, 79]. Importantly, the inhibitory effect of THC on gastric emptying can be abolished by bilateral vagotomy at the midcervical level [103]. However, the role of eCBs on gastric acid secretion needs further investigation.
CB1 are mainly involved in the eCB regulation of the gastrointestinal motility, whereas the role of CB2 remains to be further detailed. CB1 is expressed in enteric neurons and nerve fibers in the ileum and colon [8, 198]. Accordingly, CB1 inverse agonist increased electrically induced contractions in mouse ileum, concomitantly with whole gut transit under physiological conditions [165, 198]. Consequently, total CB1–KO mice showed an accelerated gastrointestinal transit compared to control animals during in vivo transit experiments using charcoal feeding [222].
FAAH is also expressed in the enteric nervous system [8]. Thus, pharmacological FAAH blockade (with AM3506) caused a reduction in electrically evoked contractions in the ileum of LPS-treated mice [8]. In vivo, LPS was able to increase upper gastrointestinal transit in wild-type but not in FAAH-deficient mice [8]. Furthermore, Izzo and colleagues [85] studied the impact of the DIO model in intestinal motility and eCB levels in the small intestine. They proposed that DIO-induced AEA reduction in the small intestine might correlate to enhanced gastrointestinal transit in obese rats [85].
In line with the above findings, AEA acutely decreased blood glucose levels during an oral glucose tolerance test (GTT) [210]. However, AEA promoted glucose intolerance during an intraperitoneal GTT, when this route of glucose application excluded an involvement of the GI tract [210]. Consequently, glucose levels were lower after AEA treatment during a duodenal GTT, when glucose was directly loaded into the duodenum, in wild-type mice but not in total CB1-KO mice [210]. In addition, AEA treatment also delayed gastrointestinal motility during a charcoal meal test. Therefore, authors concluded that AEA was able to reduce small intestinal motility and gastric emptying, and to delay glucose absorption via CB1 signalling in the gastrointestinal tract.
Piomelli and colleagues proposed a role of the eCB system in the gut in the regulation of fat intake [46, 48]. They extensively studied the function of the eCB system in cephalic reflexes in the upper gut. Cephalic phase responses are anticipatory responses that enable the animal to respond efficiently to feeding behaviour. Importantly, these responses have a significant effect on meal size. Cephalic responses can be studied using sham feeding experiments, where animals can ingest foods, but the foods are not digested or absorbed. In sham fed rats, fat intake increased the levels of AEA and 2-AG in the small intestine, whereas sham sugar and protein intake did not [46, 48]. Importantly, sham fat intake did not modify the eCB levels in other peripheral and central tissues, including other parts of the GI tract [46]. In addition, the intraduodenal administration of a fat emulsion did not induce any significant effect on jejunal 2-AG levels [48]. The elevated eCB tone in jejunum, after sham fat feeding, was blocked by rimonabant as well as vagotomy, suggesting the engagement of CB1 in the vagal nerves [46]. Notably, local rimonabant administration into the duodenum caused a decrease in fat sham intake, suggesting a positive feedback mechanism to control food intake [46, 48]. Therefore, these data indicate that the presence of fat in the oral cavity induces the cephalic responses in the small intestine via the vagal nerve, resulting in the increased eCB tone in the small intestine that controls in turn fat intake.
Furthermore, 24-h food deprivation was also able to increase 2-AG levels in the rat small intestine and serum, but not in other peripheral tissues [49]. Refeeding quickly normalized 2-AG levels. Similar to the cephalic reflex of fat intake, the response was blunted after surgical denervation of vagal nerve, suggesting the role of cholinergic synaptic transmission. To further confirm the involvement of the cholinergic pathway, intraduodenal administration of M3 muscarinic antagonist decreased jejunal 2-AG levels concomitantly with food deprivation induced refeeding.
Another study also reported that starvation induced a significant AEA increase selectively in the small intestine but not in the brain or stomach [69]. Refeeding was able to reverse intestinal AEA levels. Interestingly, capsaicin-induced sensory denervation abolished the hyperphagic effect of CB1 agonists [69], which supports the positive feedback mechanism of gut CB1 signalling controlling feeding behaviour proposed by Piomelli’s findings.
To sum up, these findings suggest that eCB signalling via CB1 in the small intestine facilitates energy absorption via several mechanisms, such as a potent hunger signal promoting food intake as well as by reducing gut motility via inhibition of cholinergic transmission. Acetylcholine release from efferent vagal nerves is directly involved in 2-AG synthesis in the gut through the activation of M3 muscarinic receptors expressed in the jejunum mucosa. Intestinal AEA levels are also able to regulate the gastrointestinal transit as well as feeding behaviour.
Microbiota
During the last years, evidence has supported that the gut microbiota is a contributing factor to obesity development [5, 31, 56]. Microbial composition regulates host metabolism, affecting adipocyte and liver functions [5, 31]. Microbiota plays a critical role to process dietary polysaccharides. Thus, the composition of gut microbiome, which is in turn affected by diet, regulates gut permeability promoting plasma intestinal-generated LPS levels (termed metabolic endotoxemia) [5, 56], which have been associated with inflammation and metabolic dysfunction in obesity.
Several studies support that gut microbiota regulates intestinal eCB tone [31, 56, 148, 178]. Cani and colleagues studied the potential link between the eCB system and gut microbiota in the control of whole-body metabolism. First, they found that changes in gut microbiota composition control CB1 mRNA expression in the colon, but not in the small intestine [148]. Thus, prebiotic treatment decreased selectively AEA levels and CB1 gene expression in the colon of ob/ob mice, which correlated with a decreased plasma LPS levels [148], suggesting that intestinal eCB system might regulate gut-barrier function. Accordingly, intraperitoneal pharmacological treatments targeting the eCB system changed gut permeability and the distribution of tight junction proteins in cell cultures of colonic epithelial monolayer cells [148]. Interestingly, prebiotics reduced significantly fat mass by blocking CB1 signalling in adipose tissue [148]. In cultured adipose tissue explants, LPS treatment completely abolished cannabinoid-mediated adipogenesis. A potential underlying mechanism might be that LPS modulates eCB metabolism in immune cells [113, 224]. Therefore, these findings propose that microbiota modulates the intestinal eCB system by controlling gut-barrier function via CB1, which affects adipocyte physiology through LPS-eCB regulatory loop [148].
However, other findings suggest a protective role of 2-AG in the regulation of gut-barrier function [3, 56] The abundance of a particular gut bacteria (Akkermansia muciniphila) inversely correlates with body weight in rodents and humans [56]. A. muciniphila treatment counteracted obesity-related metabolic dysfunctions, including increased fat mass, metabolic endotoxemia, and adipose tissue inflammation [56]. Strikingly, A. muciniphila colonization enhanced intestinal eCB levels (2-AG, 2-oleoylglycerol, and 2-palmitoylglycerol) and restored gut-barrier dysfunction during obesity. In addition, in a mouse model of colitis, elevated 2-AG levels by a selective MAGL inhibitor (JZL184, via i.p.) improved morphological changes in the colon associated with the disease, reduced endotoxemia, as well as peripheral and central inflammation [3]. Both CB1 and CB2 antagonists abolished the beneficial effects of MAGL inhibition [3].
Supporting the relevance of microbiota-induced intestinal eCB tone in obesity, engineered NAPE-producing E. coli administration in drinking water had beneficial effects in a DIO model [31]. A strain of E. coli was generated to express N-acyltransferase that catalyzes NAPE formation, which is the immediate precursor of NAEs (Fig. 1). Treatment with NAPE-producing E. coli for a week induced a selective twofold increase in NAPE levels in the colon, which restored obesity-induced increased body weight, food intake, glucose intolerance, and hepatic steatosis for at least 4 weeks after the treatment [31]. Feeding increased the abundance of NAPE in the small intestine, concomitantly with an elevated NAPE–PLD activity and expression [60, 61, 68]. Therefore, it is tentative to speculate about the conversion of NAPE by NAPE–PLD in the small intestine, for example, into the anorectic OEA or others members of NAE family, which could regulate feeding behaviour, among other physiological responses.
Strikingly, changes in eCB tone in adipose tissue are also able to regulate gut microbiota composition promoting obesity, suggesting an intriguing positive feedback mechanism. Thus, the adipocyte-specific NAPE–PLD deletion induced changes in the gut microbiota [67] through an unknown mechanism. Interestingly, the microbiota transplantation from mutant mice to germ-free mice induced an obese phenotype, recapitulating the phenotype observed in the adipocyte-specific NAPE–PLD–KO mice, indicating a main contribution of changes in the gut microbiota composition as responsible for the obese phenotype of these adipocyte-specific NAPE–PLD–KO mice.
Overall, based on the above evidence, AEA disrupts the gut-barrier function, while 2-AG has a protective role in gut-barrier dysfunction associated with certain pathological conditions. In addition, gut microbiota can influence the intestinal eCB tone that controls gut-barrier function via CB1 and CB2 [3, 56, 148], although further studies will have to elucidate the underlying mechanism of microbiota-regulated eCB activity in the gut permeability and their contribution to the development of obesity, as well as to decipher the microbiota-adipocyte crosstalk described in these studies, where eCB system plays a key regulatory function.
Oral cavity
Remarkably, elevated eCB levels were found in the saliva in obese subjects [134], which may be an attractive biomarker. Thus, it is tentative to speculate about the role of eCBs in the oral cavity in obesity, because some authors reported that CB1 in the mouse tongue are able to enhance sweet taste responses [221]. In the tongue, CB1 is coexpressed with sweet-umami T1r3 receptors in about 70% of taste cells [221]. Intraperitoneal administration of 2-AG (1 mg/kg) enhanced gustatory nerve responses to sweeteners, but not to the other four basic tastes (salty, bitter, sour, and umami) [221]. In addition, intraperitoneal injection of AEA and 2-AG selectively increased behavioural responses to a sweet–bitter mixture in wild-type mice, but not in total CB1–KO mice [221]. Therefore, eCBs can modulate the orosensory information and taste perception of sweet foods in the oral cavity, and these responses may be altered by the obesity-related overactive eCB system. This mechanism might fuel the proposal that the tongue is an attractive target to reduce overconsumption of high caloric food through a peripheral mechanism [221].
Peripheral restricted CB1 antagonist
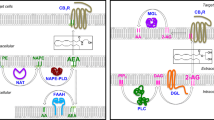
In this review, compelling evidence supporting peripheral CB1 blockade as potential new therapeutic target to tackle obesity epidemic were described (Fig. 2). After the withdrawal of rimonabant from the market, the design and characterization of different peripheral CB1 blockers has been further intensified [34, 186, 202, 203]. These peripheral CB1 antagonists are designed to contain limited penetrance to cross the blood-brain barrier (BBB), thereby strongly reducing CNS-mediated side effects as seen with rimonabant.

Endocannabinoid-mediated responses in different tissues regulating the body’s metabolism and energy balance. Schematic representation of the main responses in central and peripheral tissues, and microbiota-regulated effects in the gut. Overall, the endocannabinoid system promotes energy storage and increases vulnerability to the development of metabolic disorders. Direction of arrows indicates simulation and inhibition, respectively, after the activation of endocannabinoid signalling
The effects of two peripheral CB1 blockers (AM6545 and JD5037) in the context of obesity have been studied in particular [34, 202,203,204]. AM6545 is a CB1 neutral antagonist, while JD5037 is a CB1 inverse agonist as demonstrated in GTPγS-binding assays [202, 203]. Both compounds showed low brain penetrance, high affinity, and selectivity for CB1, oral bioavailability, and no central effects in behavioural tests. AM6545 was able to significantly reduce the body weight in diet-induced obese mice, but it was not as effective as rimonabant [34, 202]. In contrast, JD5037 had similar efficiency as a central CB1 inverse agonist (SLV319) in alleviating diet-induced metabolic impairments [203]. JD5037 decreased body weight, food intake, fat mass, dyslipidaemia, glucose intolerance, insulin resistance, hepatic steatosis, and increased total energy expenditure in DIO mice. Importantly, JD5037 did not have any effect in body weight and food intake in total CB1–KO mice, indicating the involvement of CB1 in these responses [203]. The treatment of JD5037 was also ineffective in ob/ob and db/db mice, which strongly pointed out to the role of leptin resensitization as the beneficial effect of this treatment. Indeed, DIO mice were leptin resistant, and JD5037 treatment restored leptin sensitivity in the hypothalamus by decreasing leptin secretion in adipose tissue and increasing leptin clearance in the kidney [203]. Interestingly, JD5037 restored leptin hypothalamic signalling in POMC neurons by reactivating melanocortin signalling, which led to decreased food intake in JD5037-treated DIO mice [204]. As a result, peripheral CB1 blockade induced hypophagia was abolished in melanocortin MCR4-KO mice as well as by treatment with a potent MCR4 antagonist [204]. In summary, peripheral CB1 antagonists alleviate obesity and obesity-related complications by reversing leptin resistance.
Current studies have focused on a new generation of peripheral CB1 antagonists with low BBB penetration by increasing the polar surface area and lowering its relative hydrophobicity and lipophilicity [186]. In addition to peripheral CB1 blocker, other potential alternatives might be CB1 partial agonists, drugs targeting eCB synthesis or degradation, or negative CB1 allosteric modulators. Endogenous molecules can also inhibit CB1 activation, acting as negative allosteric modulators [51, 73, 212]. In fact, the neurosteroid pregnenolone blocked THC-promoted feeding behaviour both in sated Wistar rats as well as in 24-h fasting mice, although pregnenolone did not have any effect when it was administrated alone [212]. In addition, hemopressin is a small eCB-like peptide, called pepcans, and it has been demonstrated to reduce food intake in rats [51, 57, 106, 175]. Further experiments are required to determine the effects of such negative CB1 allosteric modulators in the context of obesity and their use as potential anti-obesity treatment.
As mentioned above, the eCB system is involved in gastrointestinal motility and secretion. Cannabinoid agonists slowed gastrointestinal transit, and peripheral CB1 blockers (AM6545 and JD5037) significantly reversed CB1-induced decrease in gastrointestinal motility [34, 202, 203]. Several evidences have demonstrated that the eCB system is tonically active in the gut (reviewed in [131, 185]. Therefore, it has been suggested that overactive eCB tone may be protective against intestinal inflammation in animal models of colitis, human inflammatory bowel syndrome, and human colorectal carcinoma (reviewed in [131, 185]. In addition, inhibitory effect of cannabinoids in gastric acid secretion could prevent gastric ulcer formation (reviewed in [71, 131]. In line with these findings, we have seriously to consider that long-lasting peripheral CB1 blockade contains the possible risk to increase intestinal motility, to reduce intestinal permeability, and thereby may cause side effects, such as gastric ulcer and inflammatory bowel disease (IBD). IBD symptoms include diarrhoea, abdominal pain, fatigue, and weight loss, and, finally, it can lead to life-threatening complications.
Conclusion
In this review, we have exposed the complex and diverse roles of the eCB system in the regulation of energy metabolism, by discussing studies using genetically modified mutant mice and pharmacological approaches (Fig. 2). Considering the clinical translation of these findings to humans, eCB levels can easily be assessed in the saliva and blood/plasma, suggesting attractive biomarkers for diagnosis and prevention of metabolic diseases or eating disorders. In addition, genetic studies could be useful to identify those obese patients who can be benefit from treatments targeting the eCB system. Genetic studies reported several mutations and polymorphisms in the eCB system associated with obesity [9, 19, 136, 190]. In this scenario, human subjects with FABP1 T94A variant showed a higher risk of NAFLD [163] as well as hepatic lipid accumulation and altered hepatic eCB levels [139, 130].
To summarize, a plethora of data showed that the eCB system is a key player in the regulation of energy homeostasis by promoting energy intake and storage, whereby both central and peripheral tissues are involved in. In addition, the eCB system participates in rewarding properties of highly palatable food. Therefore, an overactivation of the eCB system promotes obesity and metabolic syndrome. In consequence, eCB blockade will also increase resilience to eating disorders associated with unlimited access to calorie-enriched food in modern societies, although CNS effects have to be taken into consideration. Finally, the peripheral eCB system seems to be an attractive pharmacological target to treat obesity and obesity-related disorders, indicating the therapeutic usefulness of peripherally restricted CB1 blockers.
References
Addy C, Wright H, Van Laere K, Gantz I, Erondu N, Musser BJ, Lu K, Yuan J, Sanabria-Bohórquez SM, Stoch A, Stevens C, Fong TM, De Lepeleire I, Cilissen C, Cote J, Rosko K, Gendrano IN 3rd, Nguyen AM, Gumbiner B, Rothenberg P, de Hoon J, Bormans G, Depré M, Eng WS, Ravussin E, Klein S, Blundell J, Herman GA, Burns HD, Hargreaves RJ, Wagner J, Gottesdiener K, Amatruda JM, Heymsfield SB (2008) The acyclic CB1R inverse agonist taranabant mediates weight loss by increasing energy expenditure and decreasing caloric intake. Cell Metab 7:68–78
Agudo J, Marin M, Roca C, Molas M, Bura AS, Zimmer A, Bosc F, Maldonado R (2010) Deficiency of CB2 cannabinoid receptor in mice improves insulin sensitivity but increase food intake and obesity with age. Diabetologia 53:2629–2640
Alhouayek M, Lambert DM, Delzene NM, Cani PD (2011) Increasing endogenous 2-arachidonoylglycerol levels counteracts colitis and related systemic inflammation. FASEB J 25:2711–2721
Alvheim AR, Malde MK, Osei-Hyiaman D, Lin YH, Pawlosky RJ, Madsen L, Kristiansen K, Froyland L, Hibbeln JR (2012) Dietary linoleic acid elevates endogenous 2-AG and anandamide and induces obesity. Obesity 20:194–1994
Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101:15718–15723
Bacci A, Huguernard JR, Prince DA (2004) Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature 431:312–316
Barutta F, Corbelli A, Mastrocola R, Gambino R, Di Marzo V, Pinach S, Rastaldi MP, Perin PC, Gruden G (2010) Cannabinoid receptor 1 blockade ameliorates albuminuria in experimental diabetic nephrophaty. Diabetes 59:1046–1054
Bashashati M, Storr MA, Wood JT, Godlewski G, Liu J, Ho W, Keenan CM, Zhang H, Alapafuja SO, Cravatt BF, Lutz B, Mackie K, Kunos G, Patel KD, Makriyannis A, Davison JS, Sharkley KA (2012) Inhibiting fatty acid amide hydrolase normalizes endotoxin-induced enhanced gastrointestinal motility in mice. Br J Pharmacol 165:1556–1571
Baye TM, Zhang Y, Smith E, Hillard CJ, Gunnell J, Mykleust J, James R, Kissebah AH, Olivier M, Wilke RA (2008) Genetic variation in cannabinoid receptor 1 (CNR1) is associated with derangements in lipid homeostasis, independent of body mass index. Pharmacogenomics 9:1647–1656
Belendiuk KA, Baldini LL, Bonn-Miller OMO (2015) Narrative review of the safety and efficacy of marijuana for the treatment of commonly state-approved medical and psychiatric disorders. Addict Sci Clin Pract 10:1–10
Bellocchio L, Cervino C, Vicennati V, Pasquali R, Pagotto U (2008) Cannabinoid type 1 receptor: another arrow in the adipocytes’ bow. J Neuroendocrinol 1:130–138
Bellocchio L, Lafenetre P, Cannich A, Cota D, Puente N, Grandes P, Chaouloff F, Piazza PV, Marsicano G (2010) Bimodal control of stimulated food intake by endocannabinoid system. Nat Neurosci 13:281–283
Bénard G, Massa F, Puente N, Lourenço J, Bellocchio L, Soria-Gómez E, Matias I, Delamarre A, Metna-Laurent M, Cannich A, Hebert-Chatelain E, Mulle C, Ortega-Gutiérrez S, Martín-Fontecha M, Klugmann M, Guggenhuber S, Lutz B, Gertsch J, Chaouloff F, López-Rodríguez ML, Grandes P, Rossignol R, Marsicano G (2012) Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat Neurosci 15:558–564
Bensaid M, Gary-BoboM Esclangon A, Maffrand JP, Le Fur G, Oury-Donat F, Soubrie P (2003) The cannabinoid CB1 receptor antagonist SR141716 increases Acrp30 mRNA expression in adipose tissue of obese fa/fa rats and in cultured adipocyte cells. Mol Pharmacol 63:908–914
Berger WT, Ralph BP, Kaczocha M, Sun J, Balius TE, Rizzo RC, Haj-Dahmane S, Ojima I, Deutsch DG (2012) Targeting FABP anandamide transporters—a novel strategy for development of anti-inflammatory and anti-nociceptive drugs. PLoS One 7:e50968
Bermúdez-Silva FJ, Suárez J, Baixeras E, Cobo N, Bautista D, Cuesta-Muñoz AL, Fuentes E, Juan-Pico P, Castro MJ, Milman G, Mechoulam R, Nadal A, Rodríguez de Fonseca F (2008) Presence of functional cannabinoid receptors in human endocrine pancreas. Diabetologia 51:476–487
Blankman JL, Simon GM, Cravatt BF (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 14:1347–1356
Blüher M, Engeli S, Klöting N, Berndt J, Fasshauer M, Batkai S, Pacher P, Schön MR, Jordan J, Stmvoll M (2006) Dysregulation of the peripheral and adipose tissue endocannabinoid system in human abdominal obesity. Diabetes 55:3053–3060
Bordicchia M, Battistoni I, Mancinelli L, Giannini E, Refi G, Minardi D, Muzzonigro G, Mazzucchelli R, Montironi R, Piscitelli F, Petrosino S, Dessì-Fulgheri P, Rappelli A, Di Marzo V, Sarzani R (2010) Cannabinoid CB1 receptor expression in relation to visceral adipose depots, endocannabinoid levels, microvascular damage, and the presence of the Cnr1 A3813G variant in humans. Metabolism 59:734–741
Bouaboula M, Hilariet S, Marchand J, Fajas L, Le Fur G, Casellas P (2005) Anandamide induced PPAR-gamma transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur J Pharmacol 517:174–181
Boyd AM, Sturgill JF, Poo C, Isaacson JS (2012) Cortical feedback control of olfactory bulb circuits. Neuron 76:1161–1174
Bowles NP, Karatsoreos IN, Li X, Vemuri VK, Wood JA, Li Z, Tamashiro KI, Schwartz GJ, Makriyannis AM, Kunos G, Hillard CJ, McEwen BS, Hill MN (2015) A peripheral endocannabinoid mechanism contributes to glucocorticoid-mediated metabolism syndrome. Proc Natl Acad Sci USA 112:285–290
Brestoff JR, Artis D (2015) Immune regulation of metabolic homeostasis in health and disease. Cell 161:146–160
Busquets-Garcia A, Gomis-González M, Srivastava RK, Cutando L, Ortega-Alvaro A, Ruehle S, Remmers F, Bindila L, Bellocchio L, Marsicano G, Lutz B, Maldonado R, Ozaita A (2016) Peripheral and central CB1 cannabinoid receptors control stress-induced impairment of memory consolidation. Proc Natl Acad Sci USA 113:9904–9909
Cardinal P, Bellocchio L, Clark S, Cannich A, Klugman M, Lutz B, Marsicano G, Cota D (2012) Hypothalamic CB1 cannabinoid receptors regulate energy balance in mice. Endocrinology 153:1–8
Cardinal P, Andre C, Quarta C, Bellocchio L, Clark S, Elie M, Leste-Laserre T, Maire M, Gonzales D, Cannich A, Pagotto U, Marsicano G, Cota D (2014) CB1 cannabinoid receptor on SF-1-expressing neurons of the ventromedial hypothalamus determines metabolic responses to diet and leptin. Mol Metab 3:705–716
Cardinal P, Bellocchio L, Guzman-Quevedo O, Andre C, Clark S, Elie M, Leste-Laserre T, Maire M, Gonzales D, Cannich A, Marsicano G, Cota D (2015) Cannabinoid type 1 (CB1) receptor in Sim1-expressing neurons regulate energy expenditure in male mice. Endocrinology 156:411–418
Cavuoto P, McAinch AJ, Hatzinikolas G, Janovska A, Game P, Wittert GA (2007) The expression of receptors for endocannabinoids in human and rodent skeletal muscle. Biochem Biophys Res Comm 364:105–110
Cavuoto P, McAinch AJ, Hatzinikolas G, Cameron-Smith D, Wittert GA (2007) Effects of cannabinoid receptors on skeletal muscle oxidative pathways. Mol Cell Endocrinol 267:63–69
Chan O, Sherwin RS (2012) Hypothalamic regulation of glucose-stimulated insulin secretion. Diabetes 61:564–565
Chen Z, Guo L, Zhang Y, Walzem RL, Pendergast JS, Printz R, Morris LC, Matafonova E, Sien X, Kang L, Coulon D, McGuinness OP, Niswender KD (2014) Incorporation of therapeutically modified bacteria into gut microbiota inhibits obesity. J Clin Invest 8:3391–3406
Choi Y, Fujikawa T, Lee J, Reuter A, Kim KW (2013) Revisiting the ventral medial nucleus of the hypothalamus of the roles of SF-1 neurons in energy homeostasis. Fronti Neurosci 7:1–9
Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A (2007) Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet 370:1706–1713
Cluny NL, Vemuri VK, Chambers AP, Limebeer CL, Bedard H, Wood JT, Lutz B, Zimmer A, Parker LA, Makriyannis A, Sharkey KA (2010) A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br J Pharmacol 161:629–642
Cone RD (2005) Anatomy and regulation of the central melanocortin system. Nat Neurosci 8:571–578
Cota D, Marsicano G, Tschöp M, Grübler Y, Flachskamm C, Schubert M, Auer D, Thöne-ReineckeC Ortmann S, Cervino C, Linthorst A, Pasquali R, Lutz B, Stalla GK, Pagotto U (2003) Decreased fat mass in mice deficient for cannabinoid receptor 1 is due to decreased orexigenic drive and impaired adipocyte differentiation. J Clin Invest 112:423–431
Cote M, Matias I, Lemieux I, Petrosino S, Almeras N, Despres JP, Di Marzo V (2007) Circulating endocannabinoid levels, abdominal adiposity and related cardiometabolic risk factors in obese men. Int J Obes 31:692–699
Coutts AA, Pertwee RG (1997) Inhibition by cannabinoid receptor agonists of acetylcholine release from guinea-pig myenteric plexus. Br J Pharmacol 121:1557–1566
Craft R, Marusich JA, Wiley JL (2013) Sex differences in cannabinoid pharmacology: a reflection of differences in endocannabinoid system? Life Sci 19:476–481
Crespillo A, Suárez J, Bermúdez-Silva FJ, Rivera P, Vida M, Alonso M, Palomino A, Lucena MA, Serrano A, Pérez-Martín M, Macias M, Fernández-Llébrez P, Rodríguez de Fonseca F (2011) Expression of the cannabinoid system in muscle. Effects of a high-fat diet and CB1 receptor blockade. Biochem J 433:175–185
D’Eon TM, Pierce KA, Roix JJ, Tyler A, Chen H, Teixeiras SR (2008) Therole of adipocyte insulin resistance in the pathogenesis of obesity-related elevations in endocannabinoids. Diabetes 57:1262–1268
Despres JP, Golay A, Sjostrom L (2005) Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med 353:2121–2134
Devane WA, Hanus L, Breuer A, Pertwee RG, Sevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949
Di Marzo V, Goparajau SK, Wang L, Liu J, Batkai S, Jarai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunos G (2001) Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 410:822–825
Di Marzo V, De Petrocellis L (2010) Endocannabinoids as regulators of transient receptor potential (TRP) channels. A further opportunity to develop new endocannabinoid-based therapeutic drugs. Curr Med Chem 17:1430–1449
Di Patrizio NV, Astarita G, Schwartz G, Li X, Piomelli D (2011) Endocanabinoid signal in the gut controls dietary fat intake. Proc Natl Acad Sci USA 108:12904–12908
Di Patrizio NV, Piomelli D (2012) The thrifty lipids: endocannabinoids and the neural control of energy conservation. Trends in Neurosci 35:403–411
Di Patrizio NV, Joslin A, Jung KM, Piomelli D (2013) Endocanabinoid signal in the gut mediates preference for dietary unsaturated fats. FASEB J 27:2513–2520
Di Patrizio NV, Igarashi M, Narayanaswami V, Murray C, Gancayco J, Russell A, Jung KM, Piomelli D (2015) Fasting stimulates 2-AG biosynthesis in the small intestine: role of cholinergic pathways. Am J Physiol Regul Integr Comp Physiol 309:R805–R813
Dhopeshwarkar A, Mackie K (2014) CB2 Cannabinoid receptors as a therapeutic target-what does the future hold? Mol Pharmacol 86:430–437
Dodd GT, Mancini G, Lutz B, Luckman SM (2010) The peptide hemopressin acts through CB1 cannabinoid receptors to reduce food intake in rats and mice. J Neurosci 30:7369–7376
Eckardt K, Sell H, Taube A, Koenen M, Platzbecker B, Cramer A, Horrighs A, Lehtonen M, Tennagels N, Eckel J (2009) Cannabinoid type 1 receptors in human skeletal muscle cells participate in the negative crosstalk between fat and muscle. Diabetologia 52:664–674
Engeli S, Lehmann AC, Kaminski J, Hass V, Janke J, Zoerner AA, Luft FC, Tsikas D, Jordan J (2014) Influence of dietary fat intake on the endocannabinoid system in lean and obese subjects. Obesity 22:E70–E76
Esposito I, Proto MC, Gazzerro P, Laezza C, Miele C, Alberobello AT, D’Esposito V, Beguinot F, Formisano P, Rifulco M (2008) The cannabinoid CB1 receptor antagonist rimonabant stimulates 2-Deoxyglucose uptake in skeletal muscle cells by regulating the expression of phosphatidylinositol-3-kinase. Mol Pharmacol 74:1678–1686
Esposito G, Capoccia E, Turco F, Palumbo I, Lu J, Steardo A, Cuomo R, Sarnelli G, Steardo L (2014) Palmitoyethanolamide improves colon inflammation through an enteric glial/Toll like receptor 4-dependent PPAR-alpha activation. Gut 63:1300–1312
Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD (2013) Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA 110:9066–9071
Ferrante C, Recinella L, Leone S, Chiavaroli A, Di Nisio C, Martinotti S, Mollica A, Macedonio G, Stefanucci A, Dvorácskó S, Tömböly C, De Petrocellis L, Vacca M, Brunetti L, Orlando G (2017) Anorexigenic effects induced by RVD-hemopressin(α) administration. Pharmacol Rep 69:1402–1407
Fischer K, Ruiz HH, Jhun K, Finan B, Oberlin DJ, van der Heide V, Kalinovich AV, Petrovic N, Wolf Y, Clemmensen C, Shin AC, Divanovic S, Brombacher F, Glasmacher E, Keipert S, Jastroch M, Nagler J, Schramm KW, Medrikova D, Collden G, Woods SC, Herzig S, Homann D, Jung S, Nedergaard J, Cannon B, Tschöp MH, Müller TD, Buettner C (2017) Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat Med 23:623–630
Fisette A, Tobin S, Decarie-Spain L, Bouyakdan K, Peyot ML, Madiraju M, Prentki M, Fulton S, Alquier T (2014) α/β-hydrolase domain 6 in the ventromedial hypothalamus controls energy metabolism flexibility. Cell Rep 17:1217–1226
Fu J, Astarita G, Gaetani S, Kim J, Cravatt BJ, Mackie K, Piomelli D (2007) Food intake regulates oleoylethanolamide formation and degradation in the proximal small intestine. J Biol Chem 282:1518–1528
Fu J, Di Patrizio NV, Guijarro A, Schwartz GJ, Li Gaetani S, Astarita G, Piomelli D (2011) Sympathetic activity controls fat-induced oleoylethanolamide signalling in small intestine. J Neurosci 31:5730–5736
Fuss J, Steinle J, Bindila L, Auer MK, Kirchherr H, Lutz B, Gass P (2015) A runner’s high depends on cannabinoid receptors in mice. Proc Natl Acad Sci USA 112:13105–13108
Gary-Bobo M, Elachouri G, Scatton B, Le Fur G, Oury-Donat F, Bensaid M (2006) The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits cell proliferation and increases markers of adipocyte maturation in cultured mouse 3T3 F442A preadipocytes. Mol Pharmacol 69:471–478
Gary-Bobo M, Elachouri G, Gallas JF, Janiak P, Marini P, Ravinet-Trillou C, Chabbert M, Cruccioli N, Pfersdorff C, Roque C, Arnone M, Croci T, Soubrie P, Oury-Donat F, Maffrand JP, Scatton B, Lacheretz F, LeFur G, Herbert JM, Bensaid M (2007) Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa rats. Hepatology 46:122–129
Gatta-Cherifi B, Matias I, Vallee M, Tabarin A, Marsicano G, Piazza PV, Cota D (2012) Simultaneous postprandial deregulation of the orexigenic endocannabinoid anandamide and anorexigenic peptide YY in obesity. Int J Obes 36:880–885
Gatta-Cherifi B, Cota D (2016) New insights on the role of the endocannabinoid system in the regulation of energy balance. Int J Obes 40:210–219
Geurts L, Everard A, Van Hul M, Essaghir A, Duparc T, Matamoros S, Plovier H, Castel J, Denis RG, Bergiers M, Druart C, Alhouayek M, Delzenne NM, Muccioli GG, Demoulin JB, Luquet S, Cani PD (2015) Adipose tissue NAPE-PLD control fat mass development by altering the browning process and gut microbiota. Nat Commun 6:1–15
Gillum MP, Zhang D, Zhang XM, Erion DM, Jamison RA, Choi C, Dong J, Shanabrough M, Duenas HR, Frederick DW, Hsiao JJ, Horvath TL, Lo CM, Cline GW, Shulman GI (2008) N-acylphosphatidylethanolamine, a gu-derived circulating factor induced by fat ingestion, inhibits food intake. Cell 135:813–824
Gomez R, Navarro M, Ferrer B, Trigo JM, Bilbao A, Del Arco IO, Cippielli A, Nava F, Piomelli D, Rodriguez de Fonseca F (2002) A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J Neurosci 22:9612–9617
Gutiérrez-Rodríguez A, Bonilla-Del Río I, Puente N, Gómez-Urquijo SM, Fontaine CJ, Egaña-Huguet J, Elezgarai I, Ruehle S, Lutz B, Robin LM, Soria-Gómez E, Bellocchio L, Padwal JD, van der Stelt M, Mendizabal-Zubiaga J, Reguero L, Ramos A, Gerrikagoitia I, Marsicano G, Grandes P (2018) Localization of the cannabinoid type-1 receptor in subcellular astrocyte compartments of mutant mouse hippocampus. Glia 66:1417–1431
Gyires K, Zadori ZS (2016) Role of cannabinoids in gastrointestinal mucosa defense and inflammation. Curr Neuropharmacol 14:935–951
Häring M, Enk V, Aparisi Rey A, Loch S, Ruiz de Azua I, Weber T, Bartsch D, Monory K, Lutz B (2015) Cannabinoid type-1 receptor signalling in central serotonergic neurons regulates anxiety-like behavior and sociability. Front Behav Neurosci 3:235
Heimann AS, Gomes I, Dale CS, Pagano RL, Gupta A, de Souza LL, Luchessi AD, Castro LM, Giorgi R, Rioli V, Ferro ES, Devi LA (2007) Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc Natl Acad Sci USA 104:20588–20593
Herling AW, Kilp S, Elvert R, Haschke G, Kramer W (2008) Increased energy expenditure contributes more to the body weight-reducing effect of rimonabant than reduced food intake in candy-fed Wistar rats. Endocrinology 149:2557–2566
Herling AW, Kilp S, Juretschke HP, Neumann-Haefelin C, Gerl M, Kramer W (2008) Reversal of visceral adiposity in candy-diet fed female Wistar rats by the CB1 receptor antagonist rimonabant. Int J Obes 32:1363–1372
Heyman E, Gamelin FX, Goekint M, Di Piscitelli F, Roelands B, Leclair E, Di Marzo V, Meeusen R (2012) Intense exercise increases circulating endocannabinoid and BDNF levels in humans-possible implications for reward and depression. Psychoneuroendocrinology 37:844–851
Higgs S, Barber DJ, Cooper AJ, Terry P (2005) Differential effects of two cannabinoid receptor agonists on progressive ratio responding for food and free-feeding in rats. Behav Pharmacol 16:389–393
Hinden L, Udi S, Drori A, Gammal A, Nemirovski A, Hadar R, Baraghitghy S, Permyakova A, Geron M, Cohen M, Tsytkin-Kirschenzweig A, Riahi Y, Leibowitz G, Nahmias Y, Priel A, Tam J (2018) Modulation of renal GLUT2 by the cannabinoid-1 receptor: implications for the treatment of diabetic nephrophaty. J Am Soc Nephrol 29:434–448
Hons IM, Storr MA, Mackie K, Lutz B, Pittman QJ, Mawe GM, Sharkey KA (2012) Plasticity of mouse enteric synapses mediated through endocannabinoid and purinergic signalling. Neurogastroenterol Motil 24:e113–e124
Hotamisligil G (2017) Inflammation, metaflammation and immunometabolic disorders. Nature 177:177–185
Hu SS, Mackie K (2015) Distribution of the endocannabinoid system in the central nervous system. Handb Exp Pharmacol 231:59–93
Huang H, McIntosh AL, Martin GG, Landrock D, Chung S, Landrock KK, Dangott LJ, Li S, Kier AB, Schroeder F (2016) FABP1: a novel hepatic endocannabinoid and cannabinoid binding protein. Biochemistry 55:5243–5255
Huang L, Ramirez J, Frampton GA, Golden LE, Quinn MA, Pae HY, Horvat D, Liang L, DeMorrow S (2011) Anandamide exerts its antiproliferative actions on cholangiocarcinoma by activation of the GPR55 receptor. Lab Invest 91:1007–1017
Hui X, Gu P, Zhang J, Nie T, Pan Y, Wu D, Feng T, Zhong C, Wang Y, Lam KS, Xu A (2015) Adiponectin enhances cold-induced browning of subcutaneous adipose tissue via promoting M2 macrophage proliferation. Cell Metab 22:279–290
Izzo AA, Piscitelli F, Capasso R, Aviello G, Romano B, Borrelli F, Petrosino S, Di Marzo V (2009) Peripheal endocannabinoid dysregulation in obesity: relation to intestinal motility and energy processing induced by food deprivation and re-feeding. B J Pharmacol 158:451–461
Izzo AA, Deutsch DG (2011) Unique pathway for anandamide synthesis and liver regeneration. Proc Natl Acad Sci USA 108:6339–6340
Jamshidi N, Taylor DA (2001) Anandamiede administration into the ventromedial hypothalamus stimulates appetite in rats. Br J Pharmacol 134:1151–1154
Jeffrey E, Berry R, Church CD, Yu S, Shook BA, Horsley V, Rosen ED, Rodeheffer MS (2014) Characterization of Cre recombinase models for the study of adipose tissue. Adipocyte 3:206–211
Jenkin KA, McAinch AJ, Zhang Y, Kelly DK, Hryciw DH (2015) Elevated cannabinoid receptor 1 and G protein-coupled receptor 55 expression in proximal tubule cells and whole kidney exposed to diabetic conditions. Clin Exp Pharmacol Physiol 42:256–262
Jeong W, Osei-Hyiaman D, Park O, Liu J, Batkai S, Mukhopadhyay P, Horiguchi N, Harvery-White J, Marsicano G, Lutz B, Gao B, Kunos G (2008) Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab 7:227–235
Jourdan T, Godlewski G, Cinar E, Bertola A, Szanda G, Liu J, Tam J, Han T, Mukhopadhayay B, Skarulis MC, Ju C, Aouadi M, Czech MP, Kunos G (2013) Activation of the Nlpr3 inflammasome in infiltrating macrophages by endocannabinoids mediate beta cell loss in type 2 diabetes. Nat Med 19:1132–1140
Jourdan T, Szanda G, Rosenberg AZ, Tam J, Earley BJ, Godlewski G, Cinar E, Liu Z, Liu J, Ju C, Pacher P, Kunos G (2014) Overactive cannabinoid 1 receptor in podocytes drives type 2 diabetic nephrophaty. Proc Natl Acad Sci USA 24:5420–5428
Jourdan T, Szanda G, Cinar E, Godlewski G, Holovac DJ, Park JK, Nicoloro S, Shen Y, Liu J, Rosenberg AZ, Liu Z, Czech MP, Kunos G (2017) Developmental role of macrophage cannabinoid-1 receptor signalling in type 2 diabetes. Diabetes 66:994–1007
Jourdan T, Nicoloro S, Zhou Z, Shen Y, Liu J, Goffey NJ, Cinar E, Godlewski G, Gao V, Aouadi M, Czech MP, Kunos G (2017) Decreasing CB1 receptor signalling in Kupffer cells improve insulin sensitivity in obese mice. Mol Metab 6:1517–1528
Jung KM, Clapper JR, Fu J, D’Agostino G, Guijarro A, Thongkham D, Avanesian A, Astarita G, DiPatrizio NV, Frontini A, Cinti S, Diano S, Piomelli D (2012) 2-Arachidonoylglyceraol signaling in forebrain regulates systemic energy metabolism. Cell Metab 15:299–310
Kaczocha LM, Glaser ST, Deutsch DG (2009) Identification of intracellular carriers for the endocannabinoid anandamide. Proc Natl Acad Sci USA 106:6375–6380
Kaczocha M, Vivieca S, Sun J, Glaser ST, Deutsch DG (2012) Fatty acid binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J Biol Chem 287:3415–3424
Kaczocha M, Rebecchi MJ, Ralph BP, Teng YHG, Berger WT, Galbavy W, Elmes MW, Glaser ST, Wang L, Rizzo RC, Deutsch DG, Ojima I (2014) Inhibition of fatty acid binding protein elevates brain anandamide levels and produces analgesia. PLoS One 9:e94200
Karaliota S, Siafaka-Kapadai A, Gontinou C, Psarra K, Mavri-Vavayanni M (2009) Anadamide increases the differentiation of rat adipocytes and causes PPARgamma and CB1 receptor upregulation. Obesity 17:1830–1838
Kirkham TC, WilliamsCM Fezza F, Di Marzo V (2002) Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: stimulation of eating by 2-arachidonoyl glycerol. Br J Pharmacol 136:550–557
Koch M, Varela L, Kim JG, Kim JD, Hernandez-Nuno F, Simonds SE, Castorena CM, Vienna CR, Elmquist JK, Morozov YM, Rakic P, Bechmann I, Cowley MA, Szigeti-Buck K, Dietrich MO, Gao XB, Diano S, Horvath TL (2015) Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature 519:45–50
Kozak KR, Crews BC, Morrow JD, Wang LH, Ma YH, Weinander R, Jakobsson PJ, Marnett LJ (2002) Metabolism of the endocannabinoids, 2-arachidonoylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol ester and ethanolamides. J Biol Chem 277:44877–44885
Krowicki ZK, Moerschbaecher JM, Winsauer PJ, Digavalli SV, Hornby PJ (1999) Δ9-tetrahydrocannabinol inhibits gastric motility in the rat through cannabinoid CB1 receptors. Eur J Pharmacol 371:187–196
Larkins RG, Dunlop ME (1992) The link between hyperglycaemia and diabetic nephrophaty. Diabetologia 35:499–504
Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, Kahn BB, Kahn CR (2013) Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 62:864–874
Leone S, Ferrante C, Recinella L, Chiavaroli A, Mollica A, Tömböly C, Stefanucci A, Dimmito MP, Dvorácskó S, Verratti V, De Petrocellis L, Orlando G, Brunetti L (2018) Effects of RVD-hemopressin (α) on feeding and body weight after standard or cafeteria diet in rats. Neuropeptides 72:38–46
Leung D, Saghatelian A, Simon GM, Cravatt BF (2006) Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry 45:4720–4726
Li Y, Kim J (2015) Neuronal expression of CB2 cannabinoid receptor mRNAs in the mouse hippocampus. Neuroscience 311:253–267
Liedhegner ES, Vogt CD, Sem DS, Cunninham CW, Hillard CJ (2014) Sterol carrier protein-2: binding protein for endocannabinoids. Mol Neurobiol 50:149–158
Lim JC, Lim SK, Han HJ, Park SH (2010) Cannabinoid receptor 1 mediates palmitic acid-induced apoptosis via endoplasmic reticulum stress in human renal proximal tubular cells. J Cell Physiol 225:654–663
Lim JC, Lim SK, Park MJ, Kim GY, Han HJ, Park SH (2011) Cannabinoid receptor 1 mediates high glucose-induced apoptosis via endoplasmic reticulum stress in primary cultured rat mesangial cells. Am J Physiol Renal Physiol 301:F179–F188
Lindborg KA, Teachey MK, Jacob S, Henriksen EJ (2010) Effects of in vitro antagonism of endocannabinoid-1 receptors on the glucose transport system in normal and insulin-resistant rat skeletal muscle. Diabetes Obes Metab 12:722–730
Liu J, Batkai S, Pacher P, Harvey-White J, Wagner JA, Cravatt BF, Gao B, Kunos G (2003) Lipopolysaccharide induces anandamide synthesis in macrophages via CD14/MAPK/phosphoinositide 3-kinase/NF-kB independently of platelet-activating factor. J Biol Chem 278:45034–45039
Liu YL, Connoley IP, Wilson CA, Stock MJ (2005) Effects of the cannabinoid CB1 receptor antagonist SR141716 on oxygen consumption and soleus muscle glucose uptake in Lep (ob)/Lep(ob) mice. Int J Obes 29:183–187
Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim H, Kunos G (2006) A biosynthetic pathway for anandamide. Proc Natl Acad Sci USA 103:13345–13350
Lumeng CN, Bodzin JL, Saltiel AR (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117:175–184
Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR (2007) Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 56:16–23
Luo Z, Ma L, Zhao Z, He H, Yang D, Feng X, Ma S, Chen X, Zhu T, Cao T, Liu D, Nilius B, Huang Y, Yan Z, Zhu Z (2012) TRPV1 activation improves exercise endurance and energy metabolism through PGC-1α upregulation in mice. Cell Res 22:551–564
Maccioni P, Pes D, Carai MAM, Gessa GL, Colombo G (2008) Supression by the cannabinoid receptor antagonist, rimonabant, of the reinforcing and motivational properties of a chocolate-flavoured beverage in rats. Behav Pharmacol 19:197–209
Malenczyk K, Jazurek M, Keimpema E, Silvestri C, Janikiewicz J, Mackie K, Di Marzo V, Redowicz MJ, Harkany T, Dobrzyn A (2013) CB1 cannabinoid receptors couple to focal adhesion kinase to control insulin release. J Biol Chem 288:32685–32699
Malenczyk K, Keimpema E, Piscitelli F, Calvigioni D, Björklund P, Mackie K, Di Marzo V, Hökfelt TG, Dobrzyn A, Harkany T (2015) Fetal endocannabinoids orchestrate the organization of pancreatic islet microarchitecture. Proc Natl Acad Sci USA 112:E6185–E6194
Marinelli S, Pacioni S, Bisogno T, Di Marzo V, Prince DA, Huguenard JR, Bacci A (2008) The endocannabinoid 2-arachidonoylglycerol is responsible for the slow self-inhibition in neocortical neurons. J Neurosci 28:13532–13541
Marinelli S, Pacioni S, Cannich A, Marsicano A, Bacci A (2009) Self-modulation of neocortical pyramidal neurons by endocannabinoids. Nat Neurosci 12:1488–1490
Maroso M, Szabo GG, Kim HK, Alexander A, Bui AD, Lee SH, Lutz B, Soltesz I (2016) Cannabinoid control of learning and memory through HCN channels. Neuron 89:1059–1073
Markopoulos F, Rokini D, Gire DH, Murthy VN (2012) Functional properties of cortical feedback projections to the olfactory bulb. Neuron 76:1175–1188
Martin GG, Chung S, Landrock D, Landrock KK, Huang H, Dangott LJ, Peng X, Kaczocha M, Seeger DR, Murphy EJ, Golovko MY, Kier AB, Schroeder F (2016) FABP1 gene ablation impacts brain endocannabinoid system in male mice. J Neurochem 138:407–422
Martin GG, Chung S, Landrock D, Landrock K, Dangott LJ, Peng X, Kaczocha M, Murphy EJ, Kier AB, Schroeder F (2016) Female mice are resistant to Fabp1 gene ablation induced alterations in brain endocannabinoid levels. Lipids 51:1007–1020
Martin GG, Landrock D, Chung S, Dangott LJ, McIntosh AL, Mackie JT, Kier AB, Schroeder F (2017) Loss of fatty acid binding protein-1 alters the hepatic endocannabinoid system response to a high fat diet. J Lip Res 58:2114–2126
Martin GG, Landrock D, Chung S, Dangott LJ, Seeger DR, Murphy EJ, Golovko MY, Kier AB, Schroeder F (2017) Fabp1 gene ablation inhibits high fat diet-induced increase in brain endocannabinoids. J Neurochem 140:294–306
Martin GG, Landrock D, Dangott LJ, McIntosh AL, Kier AB, Schroeder F (2018) Human liver fatty acid binding FABP1 T94A variant, nonalcohol fatty liver disease, and hepatic endocannabinoid system. Lipids 53:27–40
Massa F, Monory K (2006) Endocannabinoids and the gastrointestinal tract. J Endocrinol Invest 29:47–57
Matias I, Gonthier MP, Orlando P, Martiadis V, De Petrocellis L, Cervino C, Petrosino S, Hoareau L, Festy F, Pasquali R, Roche R, Maj M, Pagotto U, Monteleone P, Di Marzo V (2006) Regulation, function, and dysregulation of endocannabinoids in models of adipose and beta-pancreatic cells and in obesity and hyperglycemia. J Clin Endocrinol Metab 91:3171–3180
Matias I, Di Marzo V (2007) Endocannabinoids and the control of energy balance. Trends Endocrinol Metab 18:27–37
Matias I, Gatta-Cherifi B, Tabarin A, Clark S, Leste-Lasserre T, Marsicano G, Piazza PV, Cota D (2012) Endocannabinoids measurement in human saliva as potential biomarker of obesity. PLoS One 7:e42399
Marks J, Carvou NJ, Debnam ES, Srai SK, Unwin RJ (2003) Diabetes increases facilitative glucose uptake and GLUT2 expression at the rat proximal tubule brush border membrane. J Physiol 553:137–145
Martins C, Genelhu V, Pimentel M, Celoria B, Mangia R, Aveta T, Silvestri C, Di Marzo V, Francischetti E (2015) Circulating endocannabinoids and the polymorphism 385C > A in fatty acid amide hydrolase (FAAH) Gene may identify the obesity phenotype related to cardiometabolic risk: a study conducted in a Brazilian population of complex interethnic admixture. PLoS ONE 10:e0142728
Matsuda LA, Lolait SJ, Browntein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564
Mattace Raso G, Satoro A, Russo R, Simeoli R, Paciello O, Di Carlo C, Diano S, Calignano A, Meli R (2014) Palmitoyethanolamide prevents metabolic alterations and restores leptin sensitivity in ovariectomized rats. Endocrinology 155:1291–1301
McIntosh AL, Huang H, Storey SM, Landrock K, Landrock D, Petrescu AD, Gupta S, Atshaves BP, Kier AB, Schroeder F (2014) Human FABP1 T94A variant impacts fatty acid metabolism and PPAR-activation in cultured human female hepatocytes. Am J Physiol Gastrointest Liver Physiol 307:G164–G176
McIntosh AL, Huang H, Landrock D, Martin G, Li S, Kier AB, Schroeder F (2018) Impact of Fabp1 gene ablation on uptake and degradation of endocannabinoids in mouse hepatocytes. Lipids 53:561–580
McIntosh AL, Martin GG, Huang H, Landrock D, Kier AB, Schroeder F (2018) Delta-9-tetrahydrocannabinol induces endocannabinoid accumulation in mouse hepatocytes: antagonism by Fabp1 gene ablation. J Lipid Res 59:646–657
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almong S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z (1992) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Phamacol 50:83–90
Mendizabal-Zubiaga J, Melser S, Bernard G, Ramos A, Reguero L, Arrabal S, Elezgarai I, Gerrikagoitia I, Suarez J, Rodiguez De Fonseca F, Puene N, Marsicano G, Grandes P (2016) Cannabinoid CB1 receptor are localized in striated muscle mitochondria and regulate mitochondrial respiration. Front Physiol 7:1–10
Metna-Laurent M, Marsicano G (2015) Rising stars: modulation of brain functions by astroglial type-1 cannabinoid receptors. Glia 63:353–364
Monory K, Massa F, Egertová M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wölfel B, Dodt HU, Zieglgänsberger W, Wotjak CT, Mackie K, Elphick MR, Marsicano G, Lutz B (2006) The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron 51:455–466
Moreira FA, Grieb M, Lutz B (2009) Central side-effects of therapies based on CB1 cannabinoid receptor agonists and antagonists: focus on anxiety and depression. Best Pract Res Clin Endocrinol Metab 23:133–144
Muccioli GG (2010) Endocannabinoid biosynthesis and inactivation, from simple to complex. Drug Discov Today 15:474–483
Muccioli GG, Naslain D, Bäckhed F, Reigstad CS, Lambert DM, Delzenne NM, Cani PD (2010) The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol 6:1–15
Mukhopadhyay B, Cinar R, Yin S, Liu J, Tam J, Godlewski G, Harvey-White J, Mordi I, Cravatt BF, Lotersztajn S, Gao B, Yuan Q, Schuebel K, Goldman D, Kunos G (2011) Hyperactivation of anandamide synthesis and regulation of cell-cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proc Natl Acad Sci USA 108:6323–6328
Mullican SE, Tomaru T, Gaddis CA, Peed LC, Sundaram A, Lazar MA (2013) A novel adipose-specific gene deletion model demonstrates potential pitfalls of existing methods. Mol Endocrinol 27:127–134
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65
Nam DH, Lee MH, Kin JE, Song HK, Kang YS, Lee JE, Kim HW, Cha JJ, Hyun YY, Kim SH, Han KH, Han JY, Cha DR (2012) Blockade of cannabinoid receptor 1 improves insulin resistance, lipid metabolism, and diabetic nephropathy in db/db mice. Endocrinology 153:1387–1396
Naughton SS, Mathai ML, Hryciw DH, McAinch AJ (2013) Fatty acid modulation of the endocannabinoid system and the effect on food intake and metabolism. Int J Endocrinol 2013:1–11
Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A (2011) Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480:104–108
Njoo C, Agarwal N, Lutz B, Kuner R (2015) The cannabinoid receptor CB1 interacts with the WAVE1 complex and plays a role in actin dynamics and structural plasticity in neurons. PLoS Biol 13:e1002286
Nyiri G, Szabadits E, Cserep C, Mackie K, Shigemoto R, Freund TF (2005) GABAB and CB1 cannabinoid receptor expression identified two types of septal cholinergic neurons. Eur J Neurosci 21:3034–3042
Oddi S, Fezza F, Pasquariello N, D’Agostino A, Catanzaro G, De Simone C, Rapino C, Finazzi-Agro A, Macarrone (2009) Molecular identification of albumin and Hsp70 as cytosolic anandamide-binding proteins. Chem Biol 16:624–632
Osei-Hyiaman D, DePetrillo M, Patcher P, Liu J, Radaeva S, Bakai S, Harvey-White K, Mackie K, Offertaler L, Wang L, Kunos G (2005) Endocannabinoids activation at hepatic CB1 receptor stimulates fatty acid synthesis and contributes to diet induced obesity. J Clin Invest 115:1298–1305
Osei-Hyiamann D, Liu J, Zhou L, Godlewski G, Harvey-White J, Joeng W, Batkai S, Marsicano G, Lutz B, Buettner C, Kunos G (2008) Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest 118:3160–3169
Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R (2006) The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev 27:73–100
Parsons LH, Hurd YL (2015) Endocannabinoid signalling in reward and addiction. Nat Rev Neurosci 16:579–594
Pekala P, Kawakami M, Vine M, Lane MD, Cerami A (1983) Studies of insulin resistance in adipocytes induced by macrophage mediator. J Exp Med 157:1360–1365
Peng XE, Wu YL, Lu QQ, Hu ZJ, Lin X (2012) Two genetic variants in FABP1 and susceptibility to non-alcoholic fatty liver disease in a Chinese population. Gene 500:54–58
Pertwee RG (2001) Cannabinoids in the gastrointestinal tract. Gut 48:859–867
Pertwee RG (2005) Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci 76:1307–1324
Piazza V, Cota D, Marsicano G (2017) The CB1 receptor as the cornerstone of exostasis. Neuron 93:1252–1274
Pirzgalska RM, Seixas E, Seidman JS, Link VM, Martinez Sanchez N, Mahu I, Mendes R, Gres V, Kubasova N, Morris I, Arus BA, Larabee CM, Vasques M, Tortosa F, Sousa AL, Anandan S, Tranfield E, Hahn MK, Iannacone M, Spann NJ, Glass CK, Domingos A (2017) Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med 23:1309–1318
Pi-Sunyer FX, Aronne LJ, Heshmati HM, Devin J, Rosenstock J (2006) Effec of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: IO, North America: a randomized controlled trial. JAMA 295:761–775
Quarta C, Bellocchio L, Mancini G, Mazza R, Cervino C, Braulke LJ, Fekete C, Latorre R, Nanni C, Bucci M, Clemens LE, Heldmaier G, Watanabe M, Leste-Lassere T, Maitre M, Tedesco L, Fanelli F, Reuss S, Klaus S, Srivastava RK, Monory K, Valerio A, Grandis A, De Giorgio R, Pasquali R, Nisoli E, Cota D, Lutz B, Marsicano G, Pagotto U (2010) CB1 signalling in forebrain and sympathetic neurons is a key determinant of endocannabinoid actions on energy balance. Cell Metab 11:273–285
Qiu Y, Nguyen KD, Odergaard JI, Cui X, Tian X, Locksley RM, Palmiter RD, Chawla A (2014) Eosinophils and type 2 cytokine signalling in macrophages orchestrate development of functional beige fat. Cell 157:1292–1308
Ramírez-López MT, Vázquez M, Bindila L, Lomazzo E, Hofmann C, Blanco RN, Alén F, Antón M, Decara J, Ouro D, Orio L, Suarez J, Lutz B, Rodríguez de Fonseca F, Gómez de Heras R (2016) Exposure to a highly caloric palatable diet during progestational and gestational periods affects hypothalamic and hippocampal endocannabinoid levels at birth and induces adiposity and anxiety-like behaviors in male rat offspring. Front Behav Neurosci 9:339
Ramírez-López MT, Vázquez M, Bindila L, Lomazzo E, Hofmann C, Blanco RN, Alén F, Antón M, Decara J, Arco R, Ouro D, Orio L, Suárez J, Lutz B, Gómez de Heras R, Rodríguez de Fonseca F (2016) Maternal caloric restriction implemented during the preconceptional and pregnancy period alters hypothalamic and hippocampal endocannabinoid levels at birth and induces overweight and increased adiposity at adulthood in male rat offspring. Front Behav Neurosci 10:208
Rao RR, Long JZ, White JP, Svensson KJ, Lou J, Lokurkar J, Jedrchowski MP, Ruas JL, Wrann CD, Lo JC, Camera DM, Lachey J, Gygi S, Seehra J, Hawley JA, Spiegelman BM (2014) Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 157:1279–1291
Ravinet Trillou C, Delgorge C, Menet C, Arnone M, Soubrie P (2004) CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int J Obes Relat Metab Disord 28:640–648
Recinella L, Chiavaroli A, Ferrante C, Mollica A, Macedonio G, Stefanucci Dimmito MP, Dvorácskó S, Tömböly C, Brunetti L, Orlando G, Leone S (2018) Effects of central RVD-hemopressin(α) administration on anxiety, feeding behavior and hypothalamic neuromodulators in the rat. Pharmacol Rep 70:650–657
Roche R, Hoareau L, Bes-Houtmann S, Gonthier MP, Laborde C, Braon JF, Haffaf Y, Cesari M, Festy F (2007) Presence of the cannabinoid receptors, CB1 and CB2, in human and subcutaneous adipocytes. Histochem Cell Biol 4:1–11
Rodriguez de Fonseca F, Navarro M, Gomez R, Escudero L, Nava F, Fu J, Murillo-Rodriguez E, Giuffrida A, LoVerme J, Gaetani S, Katuria S, Gall C, Piomelli D (2001) An anorexic lipid mediator regulated by feeding. Nature 414:189–196
Rousseaux C, Thuru X, Gelot A, Barnich N, Neut C, Dubuquoy L, Dubuquoy C, Merour E, Geboes K, Chamaillard M, Ouwehand A, Leyer G, Carcano D, Colombel JF, Ardid D, Desreumaux P (2007) Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat Med 13:35–37
Rubino T, Parolaro D (2011) Sexually dimorphic effects of cannabinoid compounds on emotion and cognition. Front Behav Neurosci 5:64
Ruiz de Azua I, Mancini G, Srivastava RK, Rey AA, Cardinal P, Tedesco L, Zingaretti CM, Sassmann A, Quarta C, Schwitter C, Conrad A, Wettschureck N, Vemuri VK, Makriyannis A, Hartwig J, Mendez-Lago M, Bindila L, Monory K, Giordano A, Cinti S, Marsicano G, Offermanns S, Nisoli E, Pagotto U, Cota D, Lutz B (2017) Adipocyte cannabinoid receptor CB1 regulates energy homeostasis and alternatively activated macrophages. J Clin Invest 127:4148–4162
Salem N, Pawlosky R, Wegher B, Hibbeln J (1999) In vivo conversion of linoleic acid to arachidonic acid in human adults. Prostaglandins Leukot Esent Fatty acids 60:407–410
Sarzani R, Bordicchia M, Marcucci P, Bedetta S, Santini S, Giovagnoli A, Scappini L, Minardi D, Muzzonigro G, Dessi-Fulgheri P, Rappelli A (2009) Altered pattern of cannabinoid type 1 receptor expression in adipose tissue of dysmetabolic and overweight patients. Metabolism 58:361–367
Sassman A, Offermanns S, Wettschureck N (2010) Tamoxifen-inducible Cre-mediated recombination in adipocytes. Genes 48:618–625
Scheen AJ, Finer N, Hollander P, Jensen MD, Van Gaal LF, For the RIO-Diabetes Study Group (2006) Efficacy and tolerability of rimonabant in overweight or obese patients with type 2 diabetes: a randomised controlled study. Lancet 368:1660–1672
Sharkey KA, Wiley JW (2016) The role of the endocannabinoid system in the brain-gut axis. Gastroenterology 151:252–266
Sharma MK, Machhi J, Murumkar P, Yadav MR (2018) New role of phenothiazine derivatives as peripherally acting CB1 receptor antagonizing anti-obesity agents. Sci Rep 8:1–17
Siegmund SV, Qian T, de Minicis S, Harvey-White J, Kunos G, Vinod KY, Hungund B, Scwabe RF (2007) The endocannabinoid 2-arachidonoyl glycerol induces death of hepatic stellate cells via mitochondrial reactive oxygen species. FASEB J 21:2798–2806
Simon GM, Cravatt BF (2010) Characterization of mice lacking candidate N-acyl ethanolamide biosynthetic enzymes provides evidence for multiple pathways that contribute to endocannabinoid production in vivo. Mol BioSyst 6:1411–1418
Simon V, Cota D (2017) Endocannabinoids and metabolism: past, present and future. Eur J Endocrinol 176:R309–R324
Sipe JC, Scott TM, Murray S, Harismendy O, Simon GM, Cravatt BF, Waalen J (2010) Biomarkers of endocannabinoid system activation in severe obesity. PLoS One 5:e8792
Silvestri C, Di Marzo V (2013) The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disordes. Cell Metab 17:475–490
Solinas M, Goldberg SR (2005) Motivational effects of cannabinoids and opioids on food reinforcement depend on simultaneous activation of cannabinoid and opioid systems. Neuropsychopharmacology 30:2035–2040
Solinas M, Justinova Z, Goldberg SR, Tanda G (2006) Anandamide administration alone and after inhibition of fatty acid amide hydrolase (FAAH) increases dopamine levels in the nucleus accumbens shell in rats. J Neurochem 98:408–419
Soria-Gomez E, Matias I, Rueda-Orozco PE, Cisneros M, Petrosino S, Navarro L, Di Marzo V, Prospero-Garcia O (2007) Pharmacological enhancement of the endocannabinoid system in the nucleus accumbens shell stimulates food intake and increased c-Fos expression in the hypothalamus. Br J Pharmacol 151:1109–1116
Soria-Gómez E, Bellocchio L, Reguero L, Lepousez G, Martin C, Bendahmane M, Ruehle S, Remmers F, Desprez T, Matias I, Wiesner T, Cannich A, Nissant A, Wadleigh A, Pape HC, Chiarlone AP, Quarta C, Verrier D, Vincent P, Massa F, Lutz B, Guzmán M, Gurden H, Ferreira G, Lledo PM, Grandes P, Marsicano G (2014) The endocannabinoid system control food intake via olfactory processes. Nat Neurosci 17:407–415
Sparling PB, Giuffrida A, Piomelli D, Rosskopf L, Dietrich A (2003) Exercise activates the endocannabinoid system. NeuroReport 14:2209–2211
Starowicz KM, Cristino L, Matias I, Capasso R, Izzo AA, Di Marzo V (2008) Endocannabinoid dysregulation in the pancreas and adipose tissue of mice fed with hig-fat diet. Obesity 16:553–565
Storr MA, Bashashati M, Hirota C, Vemuri VK, Keenan CM, Duncan M, Lutz B, Mackie K, Makriyannis A, Macnaughton WK, Sharkey KA (2010) Differential effects of CB1 neutral antagonists and inverse agonists on gastrointestinal motility in mice. Neurogastroenterol Motil 22:787–796
Suarez J, Rivera P, Arrabal S, Crespillo A, Serrano A, Baixeras E, Pavon FJ, Cifuentes M, Nogueiras R, Ballesteros J, Dieguez C, Rodriguez de Fonseca F (2014) Oleoylethanolamide enhances β-adrenergic-mediated thermogenesis and white-to-brown adipocyte phenotype in epididymal white adipose tissue in rats. Dis Model Mech 7:129–141
Suarez-Zamorano N, Fabbiano S, Chevalier C, Stojanovic O, Colin DJ, Stevanovic A, Veyrat-Durebex C, Tarallo V, Rigo D, Germain S, Ilievska M, Montet X, Seimbille Y, Hapfelmeier S, Trajkovski M (2015) Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat Med 12:1497–1501
Sumithran P, Prendergast LA, Dlebridge E, Purcell K, Shulkes A, Kriketos A, Proietoo J (2011) Long-term persistence of hormonal adaptations to weight loss. N Engl J Med 365:1597–1604
Tam J, Vemuri K, Liu J, Batkai S, Mukhopadhyay B, Godlewski G, Osei-Hyiaman D, Ohnuma S, Ambudkar SV, Pickel J, Makriyannis A, Kunos G (2010) Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest 120:2953–2966
Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T, Szanda G, Mukhopadhyay B, Chedester L, Liow JS, Innis RB, Cheng K, Rice KC, Deschamps JR, Chrovat RJ, McElroy JF, Kunos G (2012) Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab 16:167–179
Tam J, Szanda G, Drori A, Liu Z, Cinar R, Kashiwaya Y, Reitman ML, Kunos G (2017) Peripheral cannabinoid-1 receptor blockade restores hypothalamic leptin signalling. Mol Metab 6:1113–1125
Tedesco L, Valerio A, Cervino C, Cardile A, Pagano C, Vettor R, Pasquali R, Carruba MO, Marsicano G, Lutz B, Pagotto U, Nisoli E (2008) Cannabinoid type 1 receptor blockade promotes mitochondrial biogenesis through endothelial nitric oxide synthase expression in white adipocytes. Diabetes 57:2028–2036
Tedesco L, Valerio A, Dossena M, Cardile A, Ragni M, Pagano C, Pagotto U, Carruba MO, Vettor R, Nisoli E (2010) Cannabinoid receptor stimulation impairs mitochondrial biogenesis in mouse white adipose tissue, muscle, and liver: the role of eNOS, p38 MAPK, and AMPK pathways. Diabetes 59:2826–2836
Thorens B (2010) Central control of glucose homeostasis: the brain-endocrine pancreas axis. Diabetes Metab 36:S45–S49
Tolson KP, Genmelli T, Meyer D, Yazdani U, Kozlitina J, Zinn AR (2014) Inducible neuronal ablation of Smi1 in adult mice causes hyperphagic obesity. Endocrinology 155:2436–2444
Tontonoz P, Hu E, Spiegelman BM (1994) Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 79:1147–1156
Troy-Fioramonti S, Demizieux K, Gresti J, Muller T, Verges B, Degrace P (2015) Acute activation of cannabinoid receptors by anandamide reduces gastrointestinal motility and improves postprandial glycemia in mice. Diabetes 64:808–818
Udi S, Hinden L, Earley B, Drori A, Reuveni N, Hadar R, Gammal A, Cinar R, Nemirovski A, Tam J (2017) Proximal tubular cannabinoid-1 receptor regulates obesity-induced CKD. J Am Soc Nephrol 28:3518–3532
Vallée M, Vitiello S, Bellocchio L, Hébert-Chatelain E, Monlezun S, Martin-Garcia E, Kasanetz F, Baillie GL, Panin F, Cathala A, Roullot-Lacarrière V, Fabre S, Hurst DP, Lynch DL, Shore DM, Deroche-Gamonet V, Spampinato U, Revest JM, Maldonado R, Reggio PH, Ross RA, Marsicano G, Piazza PV (2014) Pregnenolone can protect the brain from cannabis intoxication. Science 343:94–98
Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rossner S, For the RIO-Europe Sudy Group (2005) Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO_Europe study. Lancet 365:1389–1397
Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makryannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pitman QJ, Patel KD, Sharkey KA (2005) Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 310:329–332
Verty AN, McGregor IS, Mallet PE (2005) Paraventricular hypothalamic CB1 cannabinoid receptors are involved in the feeding stimulatory effects of d9-tetrahydrocannabinol. Neuropharmacology 49:1101–1109
Verty AN, Stefanidis A, McAinch AJ, Hryciw DH, Oldfield B (2015) Anti-obesity effect of the CB2 receptor agonist JWH-015 in diet-induced obese mice. PLoS One 10:e0140592
Ward SJ, Dykstra LA (2005) The role of CB1 receptors in sweet versus fat reinforcement: effect of CB1 receptor deletion, CB1 receptor antagonism (SR141716A) and CB1 receptor agonism (CP-55940). Behav Pharmacol 16:381–388
Wolf Y, Boura-Halfon S, Cortese N, Haimon Z, Sar Shalom H, Kuperman Y, Kalchenko V, Brandis A, David E, Segal-Hayoun Y, Chappell-Maor L, Yaron A, Jung S (2017) Brown-adipsoe-tissue macrophages control tissue innervation and homeostatic energy expenditure. Nat Immunol 18:665–674
Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW (2013) Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classical activation. Cell Metab 18:816–830
Yan ZC, Liu DY, Zhang LL, Shen CY, Ma QL, Cao TB, Wang LJ, Nie H, Zidek W, Tepel M, Zhu ZM (2007) Exercise reduces adipose issue via cannabinoid receptor type 1 which is regulated by peroxisome proliferator-activated receptor-δ. Biochem Biophys Res Commun 354:427–433
Yoshida R, Ohkuri T, Jyotaki M, Yasuo T, Horio N, Yasumatsu K, Sanematsu K, Shigemura N, Yamamoto T, Margolskee RF, Ninomiya Y (2010) Endocannabinoids selectively enhance sweet taste. Proc Natl Acad Sci USA 107:935–939
Yuece B, Sibaev A, Broedl UC, Marsicano G, Göke B, Lutz B, Allescher HD, Storr M (2007) Cannabinoid type 1 receptor modulates intestinal propulsion by an attenuation of intestinal motor responses within the myenteric part of the peristaltic reflex. Neurogastroenterol Motil 19:744–753
Zelber-Sagi S, Azar S, Nemirovski A, Webb M, Halpern Z, Shibolet O, Tam J (2017) Serum levels of endocannabinoids are independently associated with nonalcoholic fatty liver disease. Obesity 25:94–101
Zhu C, Solorzano C, Sahar S, Realini N, Fung E, Sassone-Corsi P, Piomelli D (2011) Proinflammatory stimuli control N-acylphosphatidylethanolamine-specific phospholipase D expression in macrophages. Mol Pharmacol 79:786–792
Acknowledgements
I. R. de A. was partly funded the Boehringer Ingelheim Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ruiz de Azua, I., Lutz, B. Multiple endocannabinoid-mediated mechanisms in the regulation of energy homeostasis in brain and peripheral tissues. Cell. Mol. Life Sci. 76, 1341–1363 (2019). https://doi.org/10.1007/s00018-018-2994-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2994-6