

Abstract
Nanostructured semiconductors emit light from electronic states known as excitons1. For organic materials, Hund’s rules2 state that the lowest-energy exciton is a poorly emitting triplet state. For inorganic semiconductors, similar rules3 predict an analogue of this triplet state known as the ‘dark exciton’4. Because dark excitons release photons slowly, hindering emission from inorganic nanostructures, materials that disobey these rules have been sought. However, despite considerable experimental and theoretical efforts, no inorganic semiconductors have been identified in which the lowest exciton is bright. Here we show that the lowest exciton in caesium lead halide perovskites (CsPbX3, with X = Cl, Br or I) involves a highly emissive triplet state. We first use an effective-mass model and group theory to demonstrate the possibility of such a state existing, which can occur when the strong spin–orbit coupling in the conduction band of a perovskite is combined with the Rashba effect5,6,7,8,9,10. We then apply our model to CsPbX3 nanocrystals11, and measure size- and composition-dependent fluorescence at the single-nanocrystal level. The bright triplet character of the lowest exciton explains the anomalous photon-emission rates of these materials, which emit about 20 and 1,000 times faster12 than any other semiconductor nanocrystal at room13,14,15,16 and cryogenic4 temperatures, respectively. The existence of this bright triplet exciton is further confirmed by analysis of the fine structure in low-temperature fluorescence spectra. For semiconductor nanocrystals, which are already used in lighting17, lasers18 and displays19, these excitons could lead to materials with brighter emission. More generally, our results provide criteria for identifying other semiconductors that exhibit bright excitons, with potential implications for optoelectronic devices.
Similar content being viewed by others
Main
An exciton involves an electron in the conduction band that is bound Coulombically to a hole in the valence band. Its energy depends in part on the spin configuration of these two charge carriers. In organic semiconductors, the lowest-energy exciton is a triplet state in which these two carriers have parallel spins. For the electron and hole to recombine and emit light, one spin must flip simultaneously with the release of the photon to satisfy the Pauli exclusion principle. Because this coordinated process is unlikely, triplet excitons are poorly emitting.
In addition to spin, the exciton energy depends on the atomic orbitals that constitute the conduction and valence bands. In many inorganic semiconductors, the orbital motion and spin of the carriers are strongly coupled. Spin is no longer conserved, and the total angular momentum of the electron and hole (Je and Jh) must be considered. Further, the exchange interaction mixes these momenta so that only the total exciton momentum J = Je + Jh is conserved. Owing to these and other effects, each exciton state is split into several energy sublevels, known as fine structure. Studies on various materials have found that the lowest-energy sublevel is ‘dark’, meaning that optical transitions to the ground state are dipole-forbidden. Emission, if it occurs, is very slow. For example, in CdSe, recombination of the lowest exciton requires a change of two units of angular momentum4. Because the photon carries one unit, light cannot be emitted unless another unit is dissipated simultaneously, another unlikely process. The lowest excitons in all known inorganic semiconductors behave similarly, leading to the common belief that such states must be dark.
We show that this belief is incorrect by examining CsPbX3 (X = Cl, Br or I) perovskites. Crystals of these perovskites comprise corner-sharing PbX6 octahedra with Cs+ ions filling the voids in between (Fig. 1a). We first approximate the lattice as cubic and calculate band structures (Methods) for CsPbBr3 (Fig. 1b), CsPbCl3 and CsPbI3 (Extended Data Fig. 1). The bandgap occurs at the R point in the Brillouin zone, near which the valence and conduction bands are well described within the effective-mass model (see Supplementary Table 1). The top of the valence band arises from a mixture of Pb 6s and Br 4p atomic orbitals, with an overall s symmetry20,21. Thus, including spin, the hole can occupy one of two s-like Bloch states with Jh = 1/2: |↑〉h = |S〉|↑〉 or |↓〉h = |S〉|↓〉, using standard notation22. The conduction band consists of Pb 6p orbitals, leading to three possible orthogonal spatial components for the Bloch function20,21: |X〉, |Y〉 or |Z〉. Because of strong spin–orbit coupling, these components are mixed with spin to obtain a doubly degenerate Je = 1/2 state for the electron at the bottom of the conduction band:

When the momentum of the electron and hole states are then combined, the exciton splits as a result of electron–hole exchange into a J = 0 singlet state

and a threefold degenerate J = 1 triplet state

where each  is labelled with Jz, the z projection of J. The probability of light emission due to electron–hole recombination from these excitons can then be calculated (Supplementary Information section 1). We find a probability of zero for
is labelled with Jz, the z projection of J. The probability of light emission due to electron–hole recombination from these excitons can then be calculated (Supplementary Information section 1). We find a probability of zero for  and of non-zero for
and of non-zero for  , indicating a dark singlet and a bright triplet.
, indicating a dark singlet and a bright triplet.

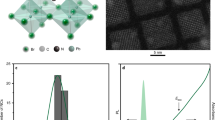
a, Orthorhombic crystal structure of CsPbBr3 (Pnma space group, unit cell shown as a frame), which differs from the idealized cubic perovskite by an octahedral tilting. b, Calculated band structure of cubic CsPbBr3 perovskite. The inset shows the first Brillouin zone of the cubic crystal lattice. The electronic bandgap is indicated in the band structure at the R point. The valence (conduction) band maximum (minimum) has  (
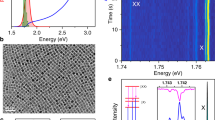
( ) symmetry. c, The expected fine structure of the band-edge exciton considering short-range electron–hole exchange (middle) and then including the Rashba effect (right) under orthorhombic symmetry. The latter splits the exciton into three bright states with transition dipoles oriented along the orthorhombic symmetry axes (labelled x, y and z) and a higher-energy dark state (labelled ‘d’). The energetic order of the three lowest sublevels is determined by the orthorhombic distortion. The orthorhombic unit cell (bottom) and the resulting sublevel order is shown for CsPbBr3. d, Transmission electron micrograph of an individual CsPbBr3 nanocrystal with an edge length of L = 14 nm.
) symmetry. c, The expected fine structure of the band-edge exciton considering short-range electron–hole exchange (middle) and then including the Rashba effect (right) under orthorhombic symmetry. The latter splits the exciton into three bright states with transition dipoles oriented along the orthorhombic symmetry axes (labelled x, y and z) and a higher-energy dark state (labelled ‘d’). The energetic order of the three lowest sublevels is determined by the orthorhombic distortion. The orthorhombic unit cell (bottom) and the resulting sublevel order is shown for CsPbBr3. d, Transmission electron micrograph of an individual CsPbBr3 nanocrystal with an edge length of L = 14 nm.
These selection rules are confirmed by group theory. At the R point, the band-edge electron and hole states transform as irreducible representations  and
and  , respectively (with the superscript denoting parity)23. Exchange then splits the exciton into a dark singlet (
, respectively (with the superscript denoting parity)23. Exchange then splits the exciton into a dark singlet ( ) and a bright triplet (
) and a bright triplet ( ); see Supplementary Information section 2 and Supplementary Table 3.
); see Supplementary Information section 2 and Supplementary Table 3.
Detailed calculations (see Supplementary Information section 1) reveal the energetic order of these levels. If only short-range exchange is included, then the singlet lies below the triplet (Fig. 1c). However, CsPbX3 perovskites should also exhibit a large Rashba effect5. This occurs in semiconductors with strong spin–orbit coupling and an inversion asymmetry. For the closely related hybrid organic–inorganic perovskites, the impact of this effect on photovoltaic and spintronic devices has been discussed extensively6,7,8,9. Although the cause of the inversion asymmetry (cation positional instabilities24 or surface effects9) remains unknown, the Rashba effect should alter the fine structure. Indeed, the bright triplet exciton can be lowered below the dark singlet exciton.
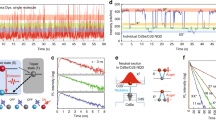
To examine this possibility, we studied colloidal nanocrystals of CsPbX3 (Methods). Compared to bulk crystals, nanocrystals enable the additional effect of system size to be investigated. Such particles are roughly cube-shaped with edge lengths of L = 8–15 nm (Fig. 1d). Before these nanocrystals were introduced11, all technologically relevant semiconductor nanocrystals exhibited slow, sub-microsecond radiative lifetimes at cryogenic temperatures, owing to the lowest exciton being dark4. By contrast, CsPbX3 nanocrystals emit about 1,000 times faster (with sub-nanosecond lifetimes)12. In Fig. 2a we show photoluminescence decays for individual CsPbI3, CsPbBr3 and CsPbBr2Cl nanocrystals at cryogenic temperatures. The decay times are 0.85 ns, 0.38 ns and 0.18 ns, respectively, decreasing with increasing emission energy. The photoluminescence quantum yield for the fastest of these samples, the CsPbBr2Cl nanocrystals (L = 14 ± 1 nm; throughout we quote the mean value and standard deviation from several measurements), was measured to be near unity (88% ± 14%) at 5 K (Extended Data Fig. 2), which indicates that these decay times can be related directly to radiative lifetimes. In Fig. 2b we present a larger set of decay times (squares) for individual CsPbI3, CsPbBr3 and CsPbBr2Cl nanocrystals. All are much shorter than those reported for CdSe, CdS, CdTe, InAs, InSb, InP, PbSe, PbS and PbTe nanocrystals13,14,15,16, consistent with the lowest exciton being the bright triplet.

a, Photoluminescence decays (open circles) measured from single CsPbI3 (L = 14 nm), CsPbBr3 (L = 11 nm) and CsPbBr2Cl (L = 14 nm) perovskite nanocrystals. By fitting the data with an exponential decay function (red lines), radiative decays of 0.85 ns, 0.38 ns and 0.18 ns were obtained for CsPbI3, CsPbBr3 and CsPbBr2Cl perovskite nanocrystals, respectively. b, Calculated radiative lifetimes of the bright triplet exciton versus transition energy for CsPbX3 nanocrystals with X = Cl, Br or I. The theoretical results (circles) are divided into three size regimes (labels on individual points give the edge lengths, L, of the cube-shaped nanocrystals): strong (orange), intermediate (blue) and weak (green) exciton confinement. These values are compared with measured photoluminescence decays from individual perovskite nanocrystals (squares; sizes of the CsPbI3, CsPbBr3 and CsPbBr2Cl crystals as in a). A data point for an ensemble of CsPbCl3 nanocrystals (L = 10 nm) is also shown. Measured values are consistent with calculations in the intermediate confinement regime, which include electron–hole correlations. c, d, Detected photon counts (left panels) versus time from individual CsPbI3 (c) and CsPbBr3 (d) nanocrystals (sizes as in a). Traces show ‘A-type’ blinking from the nanocrystals25. These data can be analysed to separate contributions to the photoluminescence decay from exciton and trion emission (right panels). The targeted temperature in all experiments was 5 K, but may be higher (10–20 K; see Fig. 3). e, Calculated distribution of the z component of the electric field normalized to the applied field (along the z direction) at infinite distance,  . This quantity is plotted versus position z across the centre line of spherical (dashed lines) or cube-shaped (solid lines) nanocrystals for various ratios of the dielectric constant inside (εin) and outside (εout) the nanocrystals (see legend). The field inside the nanocrystal is essentially always lower for the cube than for the sphere. Upper inset, calculated two-dimensional distribution of
. This quantity is plotted versus position z across the centre line of spherical (dashed lines) or cube-shaped (solid lines) nanocrystals for various ratios of the dielectric constant inside (εin) and outside (εout) the nanocrystals (see legend). The field inside the nanocrystal is essentially always lower for the cube than for the sphere. Upper inset, calculated two-dimensional distribution of  inside a cube-shaped nanocrystals plotted on the x–z mid-plane for εin/εout = 6. Lower inset, calculated ratio
inside a cube-shaped nanocrystals plotted on the x–z mid-plane for εin/εout = 6. Lower inset, calculated ratio  of radiative decay times for spherical and cubical nanocrystals with the same volume versus εin/εout for strong (blue) and weak (red) confinement.
of radiative decay times for spherical and cubical nanocrystals with the same volume versus εin/εout for strong (blue) and weak (red) confinement.
However, fast decays could also indicate emission from trions (charged excitons). Trions are optically active, but suffer from rapid non-radiative Auger recombination. They should therefore exhibit quicker but weaker decays than excitons. In our single-nanocrystal experiments discussed above, trion contributions are reduced by spectral filtering (Extended Data Fig. 3). However, to test the role of trions explicitly, we analysed the photon stream from individual nanocrystals without filtering (Fig. 2c, d, left). The correlation of emission intensity with lifetime allows the strong exciton and weak trion contributions to be separated (Fig. 2c, d, right)25. We confirm fast exciton lifetimes for CsPbI3 and CsPbBr3 nanocrystals of 1.2 ns and 0.4 ns, respectively, consistent with ensemble measurements (Extended Data Fig. 4).
To compare with theory, we calculated radiative lifetimes for perovskite nanocrystals within the effective-mass model. In addition to the wavefunctions in equations (2) and (3), exciton confinement within the nanocrystal must be included via envelope functions for the electron and hole. If CsPbX3 nanocrystals were spherical, excitonic lifetimes could be calculated using previously described methods (Supplementary Information section 3). However, for cubes, the electric field of a photon not only changes across the boundary of the nanocrystal, owing to dielectric screening (as in spherical nanocrystals), but also becomes inhomogeneous (Fig. 2e, Extended Data Figs 5, 6). We included this inhomogeneity, along with the Rashba effect and the orthorhombic lattice distortion in CsPbX3 nanocrystals in our calculations26. For simplicity, we assumed the nanocrystals were cube-shaped. Only when the Rashba effect was included could a self-consistent model for CsPbX3 nanocrystals be obtained, as we describe below.
The Rashba coefficient was estimated from low-temperature photoluminescence spectra (see below). If the effective Rashba field is parallel to one of the orthorhombic symmetry axes of the nanocrystal (see Supplementary Information section 1 for details and other cases), then the bright triplet exciton (J = 1) is split into three non-degenerate sublevels

which lie below the dark singlet (Fig. 1c). The triplet states represent three linear dipoles polarized along the orthorhombic symmetry axes (x, y, z). Transitions from these three sublevels have the same oscillator strength. Moreover, in cube-shaped nanocrystals, these states still emit as linear dipoles despite the inhomogeneous field (Supplementary Information sections 1 and 3).
The radiative lifetime of the triplet exciton τex is evaluated from

with ω the angular transition frequency, n the refractive index of the surrounding medium, m0 the free-electron mass, c the speed of light, Ep = 2P2/m0 the Kane energy (Extended Data Fig. 7) and P the Kane parameter22. I‖ is an overlap integral that includes the envelope functions of the electron and hole and the field-averaged transition-dipole moment (Supplementary Information section 3).
In Fig. 2b we present the τex calculated for CsPbX3 nanocrystals (circles). The results can be divided into three regimes, depending on the size of the nanocrystal. When the nanocrystal is smaller than the Bohr radius of the exciton aB (strong exciton confinement, orange circles), the predicted radiative lifetime decreases from 2 ns to 1 ns with increasing emission energy. For large nanocrystals in the opposite limit (weak exciton confinement, green circles), the lifetime should be even shorter because weakly confined excitons exhibit larger oscillator strengths27. In this size regime (L ≈ 15–25 nm), the calculated lifetimes decrease below 100 ps for CsPbBr3 and CsPbCl3 nanocubes. The lifetime would be decreased further in spheres of the same volume (Fig. 2e, lower inset).
The measured photoluminescence decays in Fig. 2b (squares) lie between those predicted for strong and weak confinement. Because the size of the nanocrystals and aB are comparable, the electron and hole motion is correlated. If this effect is taken into account (intermediate exciton confinement, blue circles), then calculations for L ≈ 4–16 nm (Supplementary Information section 3, Extended Data Fig. 8) agree well with the experiment.
The order of exciton levels used above depends on the values and relative signs of the Rashba coefficients for the electron and hole. If they have the same sign, then the angular-momentum texture (that is, how the orientation of the angular momentum varies with wavevector) exhibits the same helicity at the valence-band maximum and conduction-band minimum7. Optical transitions between these bands are allowed when the helicity is preserved (owing to their s and p symmetry, respectively.) Thus, for this case, the lowest exciton sublevel should be bright; see Supplementary Information section 1.E for details. A similar situation exists in transition-metal dichalcogenide monolayers28.
We estimated the values of the Rashba coefficients from photoluminescence spectra of individual nanocrystals, which reveal the fine structure directly. Our nanocrystals exhibit one, two or three peaks, all with near-linear polarization (Fig. 3a–c, Extended Data Figs 9, 10). This is consistent with the three non-degenerate exciton sublevels in equation (4) under orthorhombic symmetry, which should emit as orthogonal linear dipoles. For simplicity, we assume that the electron and hole Rashba coefficients are equal. The value (0.38 eV Å) required to fit the observed splittings (about 1 meV) is reasonable, lying between those for conventional III-V quantum wells and organic–inorganic perovskites (see Supplementary Information section 1.F). We note that for nanocrystals with tetragonal symmetry, |Ψx〉 and |Ψy〉 in equation (4) remain degenerate (Supplementary Information section 1.E), which explains recently observed two-peak spectra from individual CsPbBr3 nanocrystals29.

a–c, Photoluminescence spectra (points) of individual nanocrystals (L = 14 ± 1 nm) that exhibit a single peak (a), two peaks (b) and three (c) peaks. Single- and multi-Lorentzian-function fits are displayed as solid lines (grey lines are the cumulative fits).The targeted temperature was 5 K; however, the quantitative fits of the relative peak intensities, based on a Boltzmann distribution (Supplementary Information section 4), required higher temperatures (10–20 K). This may indicate a warmer sample temperature due to imperfect thermal contact and/or laser heating. Alternatively, deviations from a Boltzmann distribution may be present. The insets show the polarization of each of the spectral features. For the spectra, a linear polarizer was placed in the detection path. The angle of this polarizer was adjusted so that the relative intensity of the features in the spectra matched the polarization dependence in the insets. d–f, Simulated spectra and polarizations for nanocrystal orientations that match the experimental results in a–c; see Supplementary Information section 4 for details. Each panel lists the required observation direction relative to the orthorhombic unit-cell axes. g, Experimental statistics for the observation of single-, two- and three-peak spectra from individual nanocrystals with L = 7.5–14 nm (51 spectra with 35 splittings in total). h, i, Experimental fine-structure splitting measured for the two- (h) and three-peak (i) spectra. The average splitting  in each case is provided.
in each case is provided.
Emitting dipoles that are perpendicular (parallel) to the observation direction should show strong (no) emission. Thus, the intensity from each bright triplet sublevel is explained by both its thermal population and the orientation of the nanocrystal. Single-line spectra (Fig. 3a) arise when the two upper sublevels are unpopulated. Strong linear polarization from this single line (Fig. 3a, inset) supports this interpretation. If the sublevel splitting in this nanocrystal were instead spectrally unresolved, then the line would be unpolarized. From the expected three orthogonal dipoles, we calculated the relative intensity of the photoluminescence peaks and their polarization for arbitrary observation directions (Supplementary Information section 4, Extended Data Fig. 11). We then determined (Fig. 3d–f) the nanocrystal orientations that are consistent with the spectra and polarizations in Fig. 3a–c. Again, good agreement is obtained.
In Fig. 3g we present the experimental statistics for one-, two- and three-line spectra. One is most common, suggesting that only the lowest sublevel is populated. For the two- and three-line spectra, the measured energy splittings are plotted in Fig. 3h, i. Given three sublevels separated by energies Δ1 and Δ2 (Fig. 3i, inset), the average splitting  is
is  , with bars denoting averages. However, two-line spectra can involve any two of the three features, leading to the average
, with bars denoting averages. However, two-line spectra can involve any two of the three features, leading to the average  . We therefore predict a ratio of 1.33 for average measured splittings in two- versus three-line spectra. The experimentally determined ratio of 1.42 ± 0.12 again supports our model.
. We therefore predict a ratio of 1.33 for average measured splittings in two- versus three-line spectra. The experimentally determined ratio of 1.42 ± 0.12 again supports our model.
While we have used cryogenic temperatures to confirm the existence of the bright triplet exciton, its influence on emission remains important at room temperature. Although the splittings are small compared to the thermal energy, the three triplet states (from four sublevels in total) are dipole-allowed and thermally populated, unlike in other nanocrystals13,14,15,16. For example, in CdSe nanocrystals only three of eight band-edge sublevels are bright, and these can be poorly populated even at room temperature. This and other effects (Supplementary Information section 5) explain why room-temperature emission from CsPbX3 perovskite nanocrystals is 20 times faster than in other systems. The emission should be even faster for nanowires and nanoplatelets. Such shapes can further decrease the radiative lifetime, owing to diminished dielectric screening and smaller one- or two-dimensional excitons30.
Although CsPbX3 nanocrystals are oxidatively stable, their long-term stability may be limited in warm, bright and moist environments without encapsulation to provide thermal and environmental stability. Moreover, the discovery that their lowest exciton is bright reveals criteria for obtaining this phenomenon in other materials. Potential semiconductors should lack inversion symmetry, and one band edge should have s symmetry and the other p, with the latter affected by strong spin–orbit coupling such that Je,h = 1/2. Finally, the Rashba coefficient for the electron and the hole must be non-zero with the same sign.
References
Scholes, G. D. & Rumbles, G. Excitons in nanoscale systems. Nat. Mater. 5, 683–696 (2006)
Hund, F. Concerning the interpretation of complex spectra, especially the elements scandium to nickel. Z. Phys. 33, 345–371 (1925)
Onodera, Y. & Toyozawa, Y. Excitons in alkali halides. J. Phys. Soc. Jpn 22, 833–844 (1967)
Nirmal, M. et al. Observation of the “dark exciton” in CdSe quantum dots. Phys. Rev. Lett. 75, 3728–3731 (1995)
Bychkov, Yu. A. & Rashba, E. I. Oscillatory effects and the magnetic susceptibility of carriers in inversion layers. J. Phys. C 17, 6039–6045 (1984)
Kim, M., Im, J., Freeman, A. J., Ihm, J. & Jin, H. Switchable S=1/2 and J=1/2 Rashba bands in ferroelectric halide perovskites. Proc. Natl Acad. Sci. USA 111, 6900–6904 (2014)
Zheng, F., Tan, L. Z., Liu, S. & Rappe, A. M. Rashba spin–orbit coupling enhanced carrier lifetime in CH3NH3PbI3. Nano Lett. 15, 7794–7800 (2015)
Kepenekian, M. et al. Rashba and Dresselhaus effects in hybrid organic–inorganic perovskites: from basics to devices. ACS Nano 9, 11557–11567 (2015)
Mosconi, E., Etienne, T. & De Angelis, F. Rashba band splitting in organohalide lead perovskites: bulk and surface effects. J. Phys. Chem. Lett. 8, 2247–2252 (2017)
Isarov, M. et al. Rashba effect in a single colloidal CsPbBr3 perovskite nanocrystal detected by magneto-optical measurements. Nano Lett. 17, 5020–5026 (2017)
Protesescu, L. et al. Nanocrystals of cesium lead halide perovskites (CsPbX3, X=Cl, Br, and I): novel optoelectronic materials showing bright emission with wide color gamut. Nano Lett. 15, 3692–3696 (2015)
Rainò, G. et al. Single cesium lead halide perovskite nanocrystals at low temperature: fast single-photon emission, reduced blinking, and exciton fine structure. ACS Nano 10, 2485–2490 (2016)
Crooker, S. A., Barrick, T., Hollingsworth, J. A. & Klimov, V. I. Multiple temperature regimes of radiative decay in CdSe nanocrystal quantum dots: Intrinsic limits to the dark-exciton lifetime. Appl. Phys. Lett. 82, 2793–2795 (2003)
Wuister, S. F., van Houselt, A., de Mello Donegá, C., Vanmaekelbergh, D. & Meijerink, A. Temperature antiquenching of the luminescence from capped CdSe quantum dots. Angew. Chem. Int. Ed. 43, 3029–3033 (2004)
Du, H. et al. Optical properties of colloidal PbSe nanocrystals. Nano Lett. 2, 1321–1324 (2002)
Bischof, T. S., Correa, R. E., Rosenberg, D., Dauler, E. A. & Bawendi, M. G. Measurement of emission lifetime dynamics and biexciton emission quantum yield of individual InAs colloidal nanocrystals. Nano Lett. 14, 6787–6791 (2014)
Shirasaki, Y., Supran, G. J., Bawendi, M. G. & Bulović, V. Emergence of colloidal quantum-dot light-emitting technologies. Nat. Photon. 7, 13–23 (2013)
Dang, C. et al. Red, green and blue lasing enabled by single-exciton gain in colloidal quantum dot films. Nat. Nanotechnol. 7, 335–339 (2012)
Kim, T. H. et al. Full-colour quantum dot displays fabricated by transfer printing. Nat. Photon. 5, 176–182 (2011)
Bir, G. L. & Pikus, G. E. Symmetry and Strain-Induced Effects in Semiconductors Ch. 23 (Wiley, 1974)
Even, J., Pedesseau, L., Jancu, J.-M. & Katan, C. Importance of spin–orbit coupling in hybrid organic/inorganic perovskites for photovoltaic applications. J. Phys. Chem. Lett. 4, 2999–3005 (2013)
Kane, E. O. in Semiconductors and Semimetals Vol. 1 (eds Willardson, R. K. & Beer, A. C. ) 75–100 (Academic Press, 1966)
Koster, G. F., Dimmock, J. O., Wheeler, R. G. & Statz, H. Properties of the Thirty-Two Point Groups (MIT Press, 1963)
Yaffe, O. et al. Local polar fluctuations in lead halide perovskite crystals. Phys. Rev. Lett. 118, 136001 (2017)
Galland, C. et al. Two types of luminescence blinking revealed by spectroelectrochemistry of single quantum dots. Nature 479, 203–207 (2011)
Cottingham, P. & Brutchey, R. L. On the crystal structure of colloidally prepared CsPbBr3 quantum dots. Chem. Commun. 52, 5246–5249 (2016)
Rashba, E. I. & Gurgenishvili, G. E. Edge absorption theory in semiconductors. Sov. Phys. Solid State 4, 759–760 (1962)
Dery, H. & Song, Y. Polarization analysis of excitons in monolayer and bilayer transition-metal dichalcogenides. Phys. Rev. B 92, 125431 (2015)
Fu, M. et al. Neutral and charged exciton fine structure in single lead halide perovskite nanocrystals revealed by magneto-optical spectroscopy. Nano Lett. 17, 2895–2901 (2017)
Rodina, A. V. & Efros, Al. L. Effect of dielectric confinement on optical properties of colloidal nanostructures. J. Exp. Theor. Phys. 122, 554–566 (2016)
Yakunin, S. et al. Low-threshold amplified spontaneous emission and lasing from colloidal nanocrystals of caesium lead halide perovskites. Nat. Commun. 6, 8056 (2015)
Smyder, J. A. et al. The influence of continuous vs. pulsed laser excitation on single quantum dot photophysics. Phys. Chem. Chem. Phys. 16, 25723–25728 (2014)
Mehl, M. J. et al. The AFLOW library of crystallographic prototypes: part 1. Comput. Mater. Sci. 136 (Suppl.), S1–S828 (2017)
Kresse, G. Ab Initio Molekular Dynamik für Flüssige Metalle. PhD thesis, Technische Universität Wien (1993)
Kresse, G. & Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 48, 13115–13118 (1993)
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994)
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994)
Perdew, J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008); erratum 102, 039902 (2009)
Krukau, A. V., Vydrov, O. A., Izmaylov, A. F. & Scuseria, G. E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 125, 224106 (2006)
Acknowledgements
We thank F. Krieg for providing large CsPbBr3 nanocrystals, S. Yakunin and J. Jagielski for assistance with absolute quantum-yield measurements, and E. Ivchenko, M. Glazov and E. Rashba for discussions. M.A.B., G.R., T.S., M.V.K. and R.F.M. acknowledge the European Union’s Horizon-2020 programme through the Marie-Skłodowska Curie ITN network PHONSI (H2020-MSCA-ITN-642656) and the Swiss State Secretariat for Education Research and Innovation (SERI). J.G.M., S.G.L., N.B., J.L.L. and Al.L.E. acknowledge support from the US Office of Naval Research (ONR) through the core funding of the Naval Research Laboratory. R.V. was funded by ONR grant N0001416WX01849. A.S. acknowledges support from the Center for Advanced Solar Photophysics (CASP), an Energy Frontier Research Center (EFRC) funded by BES, OS, US DOE. D.J.N. and M.V.K. acknowledge partial financial support from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC grant agreement number 339905 (QuaDoPS Advanced Grant) and number 306733 (NANOSOLID Starting Grant), respectively.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks A. Meijerink, E. Rabani and M. Saba for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Electronic structure for CsPbCl3 and CsPbI3 perovskites.
a, Calculated band structure of cubic perovskite CsPbCl3. b, Calculated band structure of cubic perovskite CsPbI3. See Methods for details about the calculations.
Extended Data Figure 2 Measurements to estimate the low-temperature quantum yield for our CsPbBr2Cl nanocrystals.
a, b, Photoluminescence spectra (a) and decays (b) for CsPbBr2Cl nanocrystals (L = 14 ± 1 nm) embedded in a PMMA film at 295 K (red) and 5 K (black). Data for the two temperatures are plotted on the same intensity scale. For the same sample, a calibrated integrating sphere was used to measure the photoluminescence quantum yield at 295 K (43% ± 1%). To obtain the quantum yield at 5 K, the photoluminescence and optical absorption for several spots at 295 K and 5 K under constant weak excitation (at 3.06 eV) were measured. The photoluminescence increased substantially, as seen in both the spectra and decay signal, whereas the absorption stayed nearly constant (data not shown). From these results, the quantum yield at 5 K was estimated to be 88% ± 14%. The photoluminescence decays in b are plotted on both a linear and a logarithmic (inset) intensity scale, with decay times of 1.60 ns (295 K) and 0.23 ns (5 K). The decrease in decay time at low temperature is clearly accompanied by an increase in the total emitted intensity (area under the decay traces).
Extended Data Figure 3 Exciton and trion emission from an individual CsPbBr2Cl nanocrystal.
a, Photoluminescence spectrum of a single CsPbBr2Cl nanocrystal, showing exciton peaks at 2.5158 eV (red) and 2.5175 eV (blue) and a trion peak (black) that is redshifted by 15–17 meV. The targeted temperature was 5 K (see Fig. 3 caption). b, Polarization properties of the exciton (left) and trion (right) emission peaks. The normalized area of a Lorentzian-peak fit for the two exciton peaks (red and blue) and the trion peak (black) are shown as a function of the linear polarizer angle (placed in front of the spectrograph). Both exciton peaks show a dominantly linear polarization, with the main axis indicated by the blue and red lines. The trion emission is unpolarized. See Supplementary Section 4 for further discussion.
Extended Data Figure 4 Composition-dependent ensemble photoluminescence decay measurements of lead halide perovskite nanocrystals.
a, Typical streak-camera measurement of the photoluminescence from an ensemble of CsPbBr2Cl nanocrystals at 5 K. In this example, the nanocrystals have L = 14 nm. The emission peak is centred at 2.51 eV, and the exponential decay time is 210 ps, as extracted by summing over all energies, which is in good agreement with the results for single CsPbBr2Cl nanocrystals of the same size. The ensemble decay spectrum is slightly asymmetric (being faster at higher energies), which might originate from the activation of an energy-transfer process from smaller to larger nanocrystals. To account for this effect, we considered only the long component of the decay curve. b, Photoluminescence lifetimes at 5 K extracted for ensemble samples of nanocrystals (NCs) of various compositions and sizes (as labelled). The ensemble data (solid circles) are compared with single-nanocrystal measurements (open circles). The good agreement between the two datasets is further evidence that the measured single-nanocrystal photoluminescence decays are due to fast exciton radiative lifetimes and not to trions, because the ensemble data are acquired at very low excitation power, at which photo-generated charging is not observed.
Extended Data Figure 5 Calculation of the interior electric field in cube-shaped nanocrystals.
a, Line plot of the electric potential φ along the centre line between the capacitor plates (see inset and Supplementary Information section 3.B). b, Line plot of the normalized electric-field magnitude  along the centre line between the capacitor plates (see inset).
along the centre line between the capacitor plates (see inset).
Extended Data Figure 6 Contour plots of normalized electric-field magnitude across a cube-shaped nanocrystal.
a–d, Contour plots of  for four different ratios (4, 6, 8 and 10, respectively) of the dielectric constant inside the nanocrystal (εin) and of the surrounding medium (εout) (see Supplementary Information section 3.B). The plots depict the x–z mid-plane of the cube and are valid for the symmetry-equivalent y–z mid-plane. e, Contour plot of
for four different ratios (4, 6, 8 and 10, respectively) of the dielectric constant inside the nanocrystal (εin) and of the surrounding medium (εout) (see Supplementary Information section 3.B). The plots depict the x–z mid-plane of the cube and are valid for the symmetry-equivalent y–z mid-plane. e, Contour plot of  on the x–z mid-plane of the cube for εin/εout = 9. The
on the x–z mid-plane of the cube for εin/εout = 9. The  distribution on the y–z mid-plane is identical. In all panels, the z direction is vertical and the perturbations near the corners of the plots are artefacts of the interpolation resolution used by the software that we used to construct them.
distribution on the y–z mid-plane is identical. In all panels, the z direction is vertical and the perturbations near the corners of the plots are artefacts of the interpolation resolution used by the software that we used to construct them.
Extended Data Figure 7 Extraction of the Kane energy Ep for the lead halide perovskites.
The Kane energy, defined according to equation (5), can be extracted for CsPbCl3, CsPbBr3 and CsPbI3 from the band structures presented in Fig. 1b and Extended Data Fig. 1 near the band edges. The slope of the red line is used, according to a procedure described in Supplementary Information section 1.B.
Extended Data Figure 8 Variational calculations related to the determination of the exciton radiative lifetime in cube-shaped nanocrystals within the intermediate-confinement regime.
a, b, Dimensionless electron–hole correlation constant (b = βL, where β is the value of the variational parameter that minimizes the energy; a) and the square modulus of the ratio of I‖ for intermediate and strong confinement (b) as a function of the size of the nanocrystal relative to the Bohr radius of an electron (L/ae), for the three materials studied; me and mh are the electron and hole effective masses, respectively. The inset in b shows the square modulus of I‖ in the strong-confinement regime for several different dielectric constants, εin/εout. See Supplementary Information section 3.D for details.
Extended Data Figure 9 Representative two-peak spectra for individual CsPbBr2Cl nanocrystals.
a–i, Photoluminescence spectra of different single nanocrystals that exhibit two emission peaks at a targeted temperature of 5 K (see Fig. 3 caption). Each spectrum was recorded with a linear polarizer in the detection path. Therefore, the relative intensities displayed cannot be used to determine the relative (potentially thermal) population within the fine-structure multiplet. The linear polarizer was used here because it can be rotated to resolve all spectral features. Without the polarizer, the low-energy peak typically dominates in intensity.
Extended Data Figure 10 Representative three-peak spectra for individual CsPbBr2Cl nanocrystals.
Details as for Extended Data Fig. 9, but for nanocrystals that exhibit three emission peaks.
Extended Data Figure 11 Predicted exciton spectra and polarization properties for individual perovskite nanocrystals.
The plots show the expected exciton fine structure in photoluminescence spectra from three orthogonal dipoles of the lowest-energy exciton. The dipoles are oriented along the orthorhombic symmetry axes. The insets show the emission probability for the dipoles as a function of the polarization angle. a–d, Expected fine structure for observation in the [010], [001], [011] and [312] directions with respect to the orthorhombic symmetry axes, respectively. The temperature effect on the population of the sublevels is not considered (that is, the populations of the sublevels are assumed to be equal). e–h, As in a–d, but taking the temperature effect on the population of the sublevels into consideration. The temperature is assumed to be comparable to the fine-structure splitting: kBT ≈ Δ1 = Δ2, where kB is the Boltzmann constant and T is temperature. See Supplementary Information section 4 for further details.
Supplementary information
Supplementary Information
This file contains supplementary text S1-S5. (PDF 1240 kb)
Rights and permissions
About this article

Cite this article
Becker, M., Vaxenburg, R., Nedelcu, G. et al. Bright triplet excitons in caesium lead halide perovskites. Nature 553, 189–193 (2018). https://doi.org/10.1038/nature25147
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature25147
- Springer Nature Limited
This article is cited by
-
Coherent phenomena and dynamics of lead halide perovskite nanocrystals for quantum information technologies
Nature Materials (2024)
-
Dark states of electrons in a quantum system with two pairs of sublattices
Nature Physics (2024)
-
Hidden triplet states at hybrid organic–inorganic interfaces
Nature Reviews Materials (2024)
-
Theory of coherent phonons coupled to excitons
npj 2D Materials and Applications (2024)
-
Engineering colloidal semiconductor nanocrystals for quantum information processing
Nature Nanotechnology (2024)