
Abstract
The DEK oncogene is highly expressed in cells from most human tissues and overexpressed in a large and growing number of cancers. It also fuses with the NUP214 gene to form the DEK-NUP214 fusion gene in a subset of acute myeloid leukemia. Originally characterized as a member of this translocation, DEK has since been implicated in epigenetic and transcriptional regulation, but its role in these processes is still elusive and intriguingly complex. Similarly multifaceted is its contribution to cellular transformation, affecting multiple cellular processes such as self-renewal, proliferation, differentiation, senescence and apoptosis. Recently, the roles of the DEK and DEK-NUP214 proteins have been elucidated by global analysis of DNA binding and gene expression, as well as multiple functional studies. This review outlines recent advances in the understanding of the basic functions of the DEK protein and its role in leukemogenesis.
Similar content being viewed by others

Introduction
The DEK gene was originally discovered as a fusion partner in the (6;9)(p23;q34) chromosomal translocation in acute myeloid leukemia (AML), described in detail below.1 Since then, DEK has been shown to be expressed in most human cells and tissues and overexpressed in tumors of different origin, including but not limited to those of the skin, liver, breast, ovaries, brain, bladder and colon.2, 3, 4, 5, 6, 7, 8, 9 DEK has also generally been considered to be upregulated in AML, based on increased expression in a majority of patients in two independent studies.10, 11 We also recently showed that DEK protein levels are increased by multiple leukemia-associated fusion proteins.12 Contrarily, another study has shown the downregulation of DEK in pediatric AML and a recent analysis of two large data sets showed lower expression of DEK in adult AML than in normal bone marrow.13, 14 However, its well-established function in the proliferation, differentiation and self-renewal of hematopoietic cells as well as its multiple roles in carcinogenesis suggest that DEK may be a driver and possible therapeutic target also in leukemia.15
DEK and DNA binding
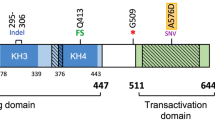
The DEK gene encodes a conserved and structurally unique protein with orthologs in most higher eukaryotes but without known human paralogs.16 The protein is 43 kDa in size and contains 375 amino acids, of which all but 26 are included in the DEK-NUP214 fusion protein.17 The domain structure is still incompletely defined but certain structures have been related to specific functions. The only part of DEK with homology to other proteins is the SAP domain, which is located in the middle of the protein sequence. This domain contains a helix–turn–helix motif that resembles the Hox protein homeodomain and mediates binding to DNA.18 SAP domains are found in DNA-binding proteins with diverse functions in processes such as cell signaling, DNA repair and chromosomal organization.19 The binding of DEK to DNA is mediated both by the SAP domain and by a second DNA-binding structure in the C-terminal end of the protein (Figure 1).18 The specificity of the binding between DEK and DNA has been investigated in several studies, demonstrating that it depends on either the sequence or the structure of the chromatin and that it correlates with the transcriptional activity of the gene. It has been widely noted that the binding of DEK to DNA depends on the structure rather than the sequence of the DNA, based on the findings that DEK accumulates at specific chromatin structures such as four-way DNA junctions and binds to several different DNA sequences with similar affinity.20 DEK has also been shown to bind DNA of various sequences in the absence of other proteins.21, 22 Sequence-specific binding has however been demonstrated to the peri-ets site of the HIV-2 enhancer by showing that DEK binds preferentially to this sequence over unrelated sequences and that the binding is abolished upon mutation of an essential nucleotide.23 In addition, DEK has been shown to bind to different sequence variants of the class II major histocompatibility complex promoter with varying affinity. Also, this binding is abrogated by the introduction of a specific mutation in the DNA.24 The distribution of the DEK protein throughout the genome was recently determined by chromatin immunoprecipitation sequencing in the myeloid U937 cell line.25 In this study, we demonstrated that DEK accumulates at the transcription start sites of genes that are highly and ubiquitously expressed across different cell types and tissues. The accumulation of DEK protein at specific sites does not appear to be determined by a specific DEK-binding motif but DEK-binding sites are enriched for motifs for certain transcription factors, including PU.1 and SP1, supporting the notion that such transcription factors may provide the specificity in the interaction between DEK and DNA.

Schematic structures of the DEK and NUP214 proteins. ‘SAP’ denotes the SAP (SAF-A/B, acinus and PIAS) domain, one of the two identified DNA-binding domains in the DEK protein (gray). ‘CC’ denotes the coiled-coil domains that localize NUP214 to the nuclear pore complex. ‘FG repeats’ denote the recurring sequences of phenylalanine and glycine that mediate nucleocytoplasmic transport in wild-type NUP214. The vertical dashed line indicates the breakpoint in the (6;9)(p23;q34) chromosomal translocation, which fuses almost the entire DEK protein with the carboxy terminal two-thirds of the NUP214 protein. Density of posttranslational modification sites was calculated based on previously assembled data.15
DEK and gene regulation
DEK is strongly implicated in gene regulation but its precise role has remained elusive. Over the past two decades, several studies have provided valuable insights but the results are still paradoxical at best and contradictory at worst. Regardless, we are still far from a comprehensive view of the role of DEK in transcriptional regulation. Immunofluorescent imaging has consistently localized DEK to euchromatin.26, 27, 28 Immunoprecipitation studies have confirmed that DEK associates with activating histone modifications such as H3K4me2/3 rather than repressive modifications such as H3K9me3.27 DEK also displayed higher enrichment at the promoter of the complement receptor 2 (CR2) gene in a cell line expressing the CR2 gene than in a comparable cell line in which it was silent. Induction of gene expression in the silent cell line by treatment with the demethylation agent 5-aza-2’-deoxycytidine conferred accumulation of DEK at the promoter.29 Conversely, the binding of DEK to the topoisomerase 1 promoter was lost upon transcriptional repression.26 DEK also coactivates the ecdysone nuclear receptor in Drosophila melanogaster by serving as a histone chaperone, incorporating histones with activating modifications into the chromatin at gene regulatory sites.27 Furthermore, DEK enhances the activity of the transcriptional activators AP-2α and C/EBPα.30, 31 DEK also interacts with the transcriptional activator MLLT3 and promotes its transcriptional expression.32 There are thus many indications that DEK is associated with transcriptional activation. However, DEK also associates with heterochromatin binding protein 1α (HP1α) and strengthens its binding to H3K9me3, thus preserving heterochromatin. Consequently, knockdown of DEK markedly reduces the distribution of constitutive heterochromatin.33 In addition, DEK associates with the chromatin remodeling complex B-WICH, which is involved in the replication of heterochromatin.34 Consistent with these findings, the deposition of DEK onto chromatin inhibits the access of endonucleases and the DNA replication machinery.21 DEK has also been shown to inhibit several activating histone acetylations, including those of H3K14 and H3K16. This action prevents transcriptional activation by the histone acetyltransferases p300 and PCAF.35 Specific inhibition of activating acetylations in the promoter region appears to be the mechanism by which DEK represses the transcription of the peroxiredoxin 6 gene.36 Additionally, DEK has been identified as a member of a transcriptional repression complex with Daxx and has been shown to antagonize transcription promoted by nuclear factor-κB and tumor necrosis factor α.37, 38 Thus, ample evidence suggests that DEK has a role not only in the activation but also in the repression of gene expression. We recently addressed the complex role of DEK in transcriptional regulation by combining genome-wide DNA-binding and gene expression analysis.25 Based on these data, we could conclude that the binding of DEK to a target gene may confer either transcriptional activation or repression, thus consolidating the contradictory reports on the role of DEK in gene regulation. However, the factors that determine whether DEK serves to increase or decrease gene expression in any given context remain unknown and their identification should be a focus of future research.
DEK and cellular function
Much like its complex role in transcriptional regulation, DEK is involved in multiple cellular functions with implications for cancer biology, including proliferation, differentiation, senescence and apoptosis. Consistent with its well-documented role as an oncogene, expression of DEK favors proliferation over differentiation. DEK expression is generally high in rapidly proliferating cells and decreases with differentiation.3, 39, 40 Our previous work has confirmed this notion in primary hematopoietic cells.41 Depletion of DEK by short hairpin RNA reduces cellular proliferation, whereas overexpression promotes proliferation and prevents differentiation of both keratinocytes and multiple breast cancer cell lines.39, 40 In the hematopoietic system, DEK contributes to the maintenance of long-term hematopoietic stem cells.42 Presumably, the maturation and accompanying proliferation of these cells is what leads to the increase in colony-forming capacity that results from DEK depletion.31 DEK has also been identified as a senescence inhibitor as DEK expression is reduced during replicative senescence, while overexpression of DEK prolongs the lifespan of both primary and transformed keratinocytes.43 Several studies have examined the role of DEK in apoptosis, assigning it antiapoptotic properties, although by different mechanisms. Knockdown of DEK leads to apoptosis in HeLa cells through p53 stabilization and a subsequent increase in p53-mediated transcription.44 Studies in melanoma cells have shown that DEK depletion can cause apoptosis independently of p53. In these cells, DEK instead exerts its antiapoptotic activity by promoting the transcription of the antiapoptotic protein MCL-1.45 Consistent with these findings, reduced DEK expression sensitizes cells from various tissues to apoptosis induced by genotoxic agents.45, 46 This may also be related to the most recently discovered function of DEK as a cofactor in DNA damage repair. DEK depletion leads to a decrease in non-homologous end-joining, activation of the DNA damage response and enhanced consequences of genotoxic stress.47 This finding may explain the early observation that DEK enhances genome stability and reduces the rates of spontaneous mutation and recombination in ataxia telangiectasia cells.48 Given these functions, it is not surprising that DEK contributes to cellular transformation. Overexpression of DEK in human keratinocytes in combination with the HRAS and human papilloma virus E6 and E7 oncogenes increases the formation of colonies in soft agar and tumors upon transplantation into mice. Interestingly, these tumor cells are more sensitive than the surrounding normal tissue to depletion of DEK by injection of short hairpin RNA.14 In combination with the finding that DEK knockout mice appear to be healthy but less prone to develop tumors, this suggests that DEK may be a promising target for cancer therapy.14
DEK as an extracellular protein
Surprisingly for a chromatin-associated factor, DEK has been found to be actively secreted by macrophages. Released in exosomes or as free protein, extracellular DEK is proinflammatory and functions as a chemotactic factor that attracts neutrophils, natural killer cells and cytotoxic T cells.49 Strikingly, DEK is also internalized by neighboring cells and translocated to the nucleus, where it has been demonstrated to perform at least some of its regular functions. Such uptake reverses the chromatin alterations and DNA repair deficiencies that result from DEK depletion.50 The addition of recombinant DEK protein also recaptures the effect of endogenous DEK on the colony-forming capacity of hematopoietic progenitor cells.42
The regulation of DEK
The regulation of DEK has been far less studied than its effects on other genes, proteins and functions. The high expression of DEK in rapidly proliferating cells may in part be explained by the activity of the E2F-1, NF-Y, YY-1 and estrogen receptor α transcription factors. These transcriptional activators are highly active in cancer and normal cells with high proliferation rates and are the only factors known to directly modulate the transcription of the DEK gene.3, 51, 52 On the posttranslational level, DEK is regulated by phosphorylation, acetylation and poly(ADP-ribosyl)ation. Phosphorylation of DEK is performed by casein kinase 2 and peaks at the G1 phase of the cell cycle, but has not been demonstrated to affect cell cycle regulation or progression.53 Phosphorylation reduces the affinity of the binding between DEK and DNA, but DEK remains bound to chromatin through dimerization with unphosphorylated DEK.53 However, casein kinase 2-mediated phosphorylation is a prerequisite for the binding of DEK to histones and the histone chaperone activity.27 Thus, phosphorylation could be a mechanism by which the different actions of the DEK protein are balanced. Acetylation of DEK also reduces its binding to DNA and relocalizes the protein to interchromatin granule clusters containing the RNA processing machinery.54 Accordingly, some studies have reported that DEK associates with splicing factors and is essential for intron removal.55, 56, 57 However, the specificity of the DEK antibodies used in these studies has been challenged and the concept remains questionable.16, 58 Finally, DEK is modified by poly(ADP-ribose) polymerase 1. Poly(ADP-ribosyl)ation of DEK accumulates during apoptosis, leading to the removal of DEK from chromatin and its subsequent exocytosis.46, 59 This posttranslational modification may be of special importance in inflammation as extracellular DEK can serve as an antigen to generate autoantibodies against the protein, which have been identified in both juvenile rheumatoid arthritis, systemic lupus erythematosus and other inflammatory diseases.60, 61, 62
The DEK-NUP214 fusion gene
The (6;9)(p23;q34) chromosomal translocation was originally identified in small subsets of patients with AML.63, 64 Recent assessments have estimated that about 1% of all AMLs carry this specific rearrangement.65, 66, 67 It is found in both adult and pediatric AML, but the latter form dominates with a mean age of diagnosis of 23 years.66 The t(6;9)(p23;q34) has traditionally been associated with poor prognosis, although a recent retrospective study suggests that the outcome for pediatric patients with this translocation may be more similar to that of other childhood AML.66, 67 Patients are generally treated with either chemotherapy or allogeneic hematopoietic stem cell transplantation, with a slightly more favorable prognosis for the latter group.67 In 1992, the (6;9)(p23;q34) translocation was characterized as a fusion between specific introns in the gene encoding the chromatin architectural protein DEK and the gene encoding the nucleoporin NUP214 (originally termed CAN).17 The translocation is reciprocal but the NUP214-DEK fusion does not produce a transcript, leaving DEK-NUP214 as the sole gene product of the translocation.1 The fusion protein includes almost the entire DEK protein and the carboxy terminal two-thirds of the NUP214 protein (Figure 1), resulting in a large protein of ~165 kDa.1
Despite its identification more than two decades ago, the role of the DEK-NUP214 fusion protein still remains largely unknown. It resides in the nucleus, likely due to the nuclear localization signal in the DEK protein. However, the staining pattern of DEK-NUP214 differs from that of DEK, suggesting that the localization and thus the function of DEK-NUP214 is qualitatively different from that of DEK.68 Another indication of this is our previous finding that expression of DEK-NUP214 increases the protein synthesis of myeloid cells, as this effect was not achieved by expression of the DEK protein or any of the six DEK-NUP214 deletion mutants, but rather required all the major domains of the fusion protein.69 We also show increased phosphorylation of the translational regulator eukaryotic initiation factor 4E, suggesting that DEK-NUP214 affects the regulation of protein synthesis.69 However, DEK-NUP214 also appears to directly interact with the DEK protein and interfere with its function. When the DEK-NUP214 protein was expressed in 293T cells, it co-immunoprecipitated with DEK and abolished the binding between DEK and other factors in the identified histone chaperone complex.27 Among these was casein kinase 2, which has been previously shown to mediate a phosphorylation of DEK that alters its association with chromatin.53 This dominant-negative effect of DEK-NUP214 on DEK function lead to altered expression of genes bound by the histone chaperone complex and was suggested as a mechanism by which DEK-NUP214 contributes to leukemogenesis. It is however unlikely that this is a major role, as DEK is a bona fide oncogene that is generally upregulated in cancer and interference with DEK would thus be expected to counter rather than promote leukemogenesis.
The leukemogenic potential of DEK-NUP214 has been established in a murine model, where DEK-NUP214 was found to induce leukemia when transduced to long-term but not short-term repopulating stem cells.70 The resulting leukemia could however be maintained by more mature cells, suggesting that there is a difference between leukemia-initiating and -maintaining cells in DEK-NUP214-induced leukemia. The finding that DEK-NUP214, as opposed to, for example, PML-RARα, only has the potential to initiate leukemia from very immature cells also suggests that it may be a first hit rather than a secondary event during leukemogenesis. The contribution of DEK-NUP214 to the leukemogenic process has however not been fully characterized. Expression of DEK-NUP214 has no effect on the terminal differentiation of human U937 cells as induced by vitamin D3 or that of primary murine Sca+/Lin− cells induced by GM/G-CSF.70, 71 Neither does it prolong the colony-forming capacity of murine progenitor cells, an in vitro assay of self-renewal capacity. However, the expression of DEK-NUP214 does increase the number of colonies formed both in vitro and in vivo, an effect that is similar in magnitude to that of PML-RARα.70 This suggests that DEK-NUP214 may affect the proliferation rather than the differentiation or self-renewal of hematopoietic cells. We confirmed this notion by introducing DEK-NUP214 in the myeloid U937 cell line, where expression of the fusion gene lead to increased proliferation by upregulation of the mammalian target of rapamycin (mTOR) protein and a subsequent increase in mTORC1 but not mTORC2 signaling. The proliferative effect was reversed by treatment with the mTORC1 inhibitor everolimus, suggesting that leukemias with the (6;9)(p23;q34) translocation may be susceptible to treatment with the emerging classes of mTOR inhibitors.72
The t(6;9)(p23;q34) is usually the only cytogenetic aberration in these leukemias, but one of the most consistent findings of leukemic cells with the DEK-NUP214 fusion gene is the concomitant mutation of the FLT3 gene. Internal tandem duplications that cause constitutive activation of the FLT3 tyrosine kinase are one of the most common genetic aberrations in AML. However, whereas 20–30% of all AML patients carry an FLT3-ITD mutation, the incidence among patients with the (6;9)(p23;q34) translocation is around 60%.65, 66, 67, 73, 74 Preliminary results from Martin Ruthardt’s research group suggest that FLT3-ITD promotes leukemia induction by DEK-NUP214 in a murine model of disease (Heinssmann et al. ASH Annual Meeting abstract, 2012). However, a synergistic effect to explain the high coincidence of the two mutations has yet to be demonstrated.
Conclusion
Our understanding of DEK biology has greatly increased in recent years but so has the complexity of its function. DEK mainly binds to highly expressed genes but can act to either promote or repress their transcription. The mechanisms underlying this dual role are not yet understood and should be a primary focus of future studies. DEK also affects crucial oncogenic processes such as cell proliferation, differentiation, senescence and apoptosis. And as a bona fide oncogene, it contributes to cellular transformation both in vitro and in vivo. A major challenge for future research will be to not only continue characterizing the role of DEK in such cellular processes but to also determine common mechanisms that could explain multiple effects of altered DEK expression and possibly also consolidate the seemingly contradictory functions of the DEK protein in epigenetic and transcriptional regulation. Furthermore, it will be important to investigate the effect of DEK inhibition on these functions in both healthy and malignant cells to assess the potential of DEK as a drug target in cancer.
References
von Lindern M, Fornerod M, Soekarman N, van Baal S, Jaegle M, Hagemeijer A et al. Translocation t(6;9) in acute non-lymphocytic leukaemia results in the formation of a DEK-CAN fusion gene. Bailliere's Clin Haematol 1992; 5: 857–879.
Abba MC, Sun H, Hawkins KA, Drake JA, Hu Y, Nunez MI et al. Breast cancer molecular signatures as determined by SAGE: correlation with lymph node status. Mol Cancer Res 2007; 5: 881–890.
Carro MS, Spiga FM, Quarto M, Di Ninni V, Volorio S, Alcalay M et al. DEK expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle (Georgetown, TX) 2006; 5: 1202–1207.
Han S, Xuan Y, Liu S, Zhang M, Jin D, Jin R et al. Clinicopathological significance of DEK overexpression in serous ovarian tumors. Pathol Int 2009; 59: 443–447.
Kappes F, Khodadoust MS, Yu L, Kim DS, Fullen DR, Markovitz DM et al. DEK expression in melanocytic lesions. Hum Pathol 2011; 42: 932–938.
Kondoh N, Wakatsuki T, Ryo A, Hada A, Aihara T, Horiuchi S et al. Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res 1999; 59: 4990–4996.
Kroes RA, Jastrow A, McLone MG, Yamamoto H, Colley P, Kersey DS et al. The identification of novel therapeutic targets for the treatment of malignant brain tumors. Cancer Lett 2000; 156: 191–198.
Orlic M, Spencer CE, Wang L, Gallie BL . Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosomes Cancer 2006; 45: 72–82.
Sanchez-Carbayo M, Socci ND, Lozano JJ, Li W, Charytonowicz E, Belbin TJ et al. Gene discovery in bladder cancer progression using cDNA microarrays. Am J Pathol 2003; 163: 505–516.
Casas S, Nagy B, Elonen E, Aventin A, Larramendy ML, Sierra J et al. Aberrant expression of HOXA9, DEK, CBL and CSF1R in acute myeloid leukemia. Leuk Lymphoma 2003; 44: 1935–1941.
Larramendy ML, Niini T, Elonen E, Nagy B, Ollila J, Vihinen M et al. Overexpression of translocation-associated fusion genes of FGFRI, MYC, NPMI, and DEK, but absence of the translocations in acute myeloid leukemia. A microarray analysis. Haematologica 2002; 87: 569–577.
Sanden C, Nilsson HJ, Gullberg U . The DEK oncoprotein is upregulated by multiple leukemia-associated fusion genes. Blood Cells Mol Dis 2014; 54: 284–285.
Logan GE, Mor-Vaknin N, Braunschweig T, Jost E, Schmidt PV, Markovitz DM et al. DEK oncogene expression during normal hematopoiesis and in Acute Myeloid Leukemia (AML). Blood Cells Mol Dis 2015; 54: 123–131.
Wise-Draper TM, Mintz-Cole RA, Morris TA, Simpson DS, Wikenheiser-Brokamp KA, Currier MA et al. Overexpression of the cellular DEK protein promotes epithelial transformation in vitro and in vivo. Cancer Res 2009; 69: 1792–1799.
Broxmeyer HE, Mor-Vaknin N, Kappes F, Legendre M, Saha AK, Ou X et al. Concise review: role of DEK in stem/progenitor cell biology. Stem Cells 2013; 31: 1447–1453.
Waldmann T, Scholten I, Kappes F, Hu HG, Knippers R . The DEK protein—an abundant and ubiquitous constituent of mammalian chromatin. Gene 2004; 343: 1–9.
von Lindern M, Fornerod M, van Baal S, Jaegle M, de Wit T, Buijs A et al. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell Biol 1992; 12: 1687–1697.
Kappes F, Scholten I, Richter N, Gruss C, Waldmann T . Functional domains of the ubiquitous chromatin protein DEK. Mol Cell Biol 2004; 24: 6000–6010.
Aravind L, Koonin EV . SAP—a putative DNA-binding motif involved in chromosomal organization. Trends Biochem Sci 2000; 25: 112–114.
Waldmann T, Baack M, Richter N, Gruss C . Structure-specific binding of the proto-oncogene protein DEK to DNA. Nucleic acids Res 2003; 31: 7003–7010.
Alexiadis V, Waldmann T, Andersen J, Mann M, Knippers R, Gruss C . The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes Dev 2000; 14: 1308–1312.
Waldmann T, Eckerich C, Baack M, Gruss C . The ubiquitous chromatin protein DEK alters the structure of DNA by introducing positive supercoils. J Biol Chem 2002; 277: 24988–24994.
Fu GK, Grosveld G, DEK Markovitz DM . DEK, an autoantigen involved in a chromosomal translocation in acute myelogenous leukemia, binds to the HIV-2 enhancer. Proc Natl Acad Sci USA 1997; 94: 1811–1815.
Adams BS, Cha HC, Cleary J, Haiying T, Wang H, Sitwala K et al. DEK binding to class II MHC Y-box sequences is gene- and allele-specific. Arthritis Res Ther 2003; 5: R226–R233.
Sanden C, Jarvstrat L, Lennartsson A, Brattas PL, Nilsson B, Gullberg U . The DEK oncoprotein binds to highly and ubiquitously expressed genes with a dual role in their transcriptional regulation. Mol Cancer 2014; 13: 215.
Hu HG, Scholten I, Gruss C, Knippers R . The distribution of the DEK protein in mammalian chromatin. Biochem Biophys Res Commun 2007; 358: 1008–1014.
Sawatsubashi S, Murata T, Lim J, Fujiki R, Ito S, Suzuki E et al. A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes Dev 2010; 24: 159–170.
Takata H, Nishijima H, Ogura S, Sakaguchi T, Bubulya PA, Mochizuki T et al. Proteome analysis of human nuclear insoluble fractions. Genes Cells 2009; 14: 975–990.
Hu HG, Illges H, Gruss C, Knippers R . Distribution of the chromatin protein DEK distinguishes active and inactive CD21/CR2 gene in pre- and mature B lymphocytes. Int Immunol 2005; 17: 789–796.
Campillos M, Garcia MA, Valdivieso F, Vazquez J . Transcriptional activation by AP-2alpha is modulated by the oncogene DEK. Nucleic Acids Res 2003; 31: 1571–1575.
Koleva RI, Ficarro SB, Radomska HS, Carrasco-Alfonso MJ, Alberta JA, Webber JT et al. C/EBPalpha and DEK coordinately regulate myeloid differentiation. Blood 2012; 119: 4878–4888.
Shibata T, Kokubu A, Miyamoto M, Hosoda F, Gotoh M, Tsuta K et al. DEK oncoprotein regulates transcriptional modifiers and sustains tumor initiation activity in high-grade neuroendocrine carcinoma of the lung. Oncogene 2010; 29: 4671–4681.
Kappes F, Waldmann T, Mathew V, Yu J, Zhang L, Khodadoust MS et al. The DEK oncoprotein is a Su(var) that is essential to heterochromatin integrity. Genes Dev 2011; 25: 673–678.
Cavellan E, Asp P, Percipalle P, Farrants AK . The WSTF-SNF2h chromatin remodeling complex interacts with several nuclear proteins in transcription. J Biol Chem 2006; 281: 16264–16271.
Ko SI, Lee IS, Kim JY, Kim SM, Kim DW, Lee KS et al. Regulation of histone acetyltransferase activity of p300 and PCAF by proto-oncogene protein DEK. FEBS Lett 2006; 580: 3217–3222.
Kim DW, Chae JI, Kim JY, Pak JH, Koo DB, Bahk YY et al. Proteomic analysis of apoptosis related proteins regulated by proto-oncogene protein DEK. J Cell Biochem 2009; 106: 1048–1059.
Hollenbach AD, McPherson CJ, Mientjes EJ, Iyengar R, Grosveld G . Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J Cell Sci 2002; 115: 3319–3330.
Sammons M, Wan SS, Vogel NL, Mientjes EJ, Grosveld G, Ashburner BP . Negative regulation of the RelA/p65 transactivation function by the product of the DEK proto-oncogene. J Biol Chem 2006; 281: 26802–26812.
Privette Vinnedge LM, McClaine R, Wagh PK, Wikenheiser-Brokamp KA, Waltz SE, Wells SI . The human DEK oncogene stimulates beta-catenin signaling, invasion and mammosphere formation in breast cancer. Oncogene 2011; 30: 2741–2752.
Wise-Draper TM, Morreale RJ, Morris TA, Mintz-Cole RA, Hoskins EE, Balsitis SJ et al. DEK proto-oncogene expression interferes with the normal epithelial differentiation program. Am J Pathol 2009; 174: 71–81.
Ageberg M, Gullberg U, Lindmark A . The involvement of cellular proliferation status in the expression of the human proto-oncogene DEK. Haematologica 2006; 91: 268–269.
Broxmeyer HE, Kappes F, Mor-Vaknin N, Legendre M, Kinzfogl J, Cooper S et al. DEK regulates hematopoietic stem engraftment and progenitor cell proliferation. Stem Cells Dev 2012; 21: 1449–1454.
Wise-Draper TM, Allen HV, Thobe MN, Jones EE, Habash KB, Munger K et al. The human DEK proto-oncogene is a senescence inhibitor and an upregulated target of high-risk human papillomavirus E7. J Virol 2005; 79: 14309–14317.
Wise-Draper TM, Allen HV, Jones EE, Habash KB, Matsuo H, Wells SI . Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol 2006; 26: 7506–7519.
Khodadoust MS, Verhaegen M, Kappes F, Riveiro-Falkenbach E, Cigudosa JC, Kim DS et al. Melanoma proliferation and chemoresistance controlled by the DEK oncogene. Cancer Res 2009; 69: 6405–6413.
Kappes F, Fahrer J, Khodadoust MS, Tabbert A, Strasser C, Mor-Vaknin N et al. DEK is a poly(ADP-ribose) acceptor in apoptosis and mediates resistance to genotoxic stress. Molecular and cellular biology 2008; 28: 3245–3257.
Kavanaugh GM, Wise-Draper TM, Morreale RJ, Morrison MA, Gole B, Schwemberger S et al. The human DEK oncogene regulates DNA damage response signaling and repair. Nucleic Acids Res 2011; 39: 7465–7476.
Meyn MS, Lu-Kuo JM, Herzing LB . Expression cloning of multiple human cDNAs that complement the phenotypic defects of ataxia-telangiectasia group D fibroblasts. Am J Hum Genet 1993; 53: 1206–1216.
Mor-Vaknin N, Punturieri A, Sitwala K, Faulkner N, Legendre M, Khodadoust MS et al. The DEK nuclear autoantigen is a secreted chemotactic factor. Mol Cell Biol 2006; 26: 9484–9496.
Saha AK, Kappes F, Mundade A, Deutzmann A, Rosmarin DM, Legendre M et al. Intercellular trafficking of the nuclear oncoprotein DEK. Proc Natl Acad Sci USA 2013; 110: 6847–6852.
Privette Vinnedge LM, Ho SM, Wikenheiser-Brokamp KA, Wells SI . The DEK oncogene is a target of steroid hormone receptor signaling in breast cancer. PLoS One 2012; 7: e46985.
Sitwala KV, Adams K, Markovitz DM . YY1 and NF-Y binding sites regulate the transcriptional activity of the dek and dek-can promoter. Oncogene 2002; 21: 8862–8870.
Kappes F, Damoc C, Knippers R, Przybylski M, Pinna LA, Gruss C . Phosphorylation by protein kinase CK2 changes the DNA binding properties of the human chromatin protein DEK. Molecular and cellular biology 2004; 24: 6011–6020.
Cleary J, Sitwala KV, Khodadoust MS, Kwok RP, Mor-Vaknin N, Cebrat M et al. p300/CBP-associated factor drives DEK into interchromatin granule clusters. J Biol Chem 2005; 280: 31760–31767.
Le Hir H, Izaurralde E, Maquat LE, Moore MJ . The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon–exon junctions. EMBO J 2000; 19: 6860–6869.
McGarvey T, Rosonina E, McCracken S, Li Q, Arnaout R, Mientjes E et al. The acute myeloid leukemia-associated protein, DEK, forms a splicing-dependent interaction with exon–product complexes. J Cell Biol 2000; 150: 309–320.
Soares LM, Zanier K, Mackereth C, Sattler M, Valcarcel J . Intron removal requires proofreading of U2AF/3' splice site recognition by DEK. Science (New York, NY) 2006; 312: 1961–1965.
Reichert VL, Le Hir H, Jurica MS, Moore MJ . 5' Exon interactions within the human spliceosome establish a framework for exon junction complex structure and assembly. Genes Dev 2002; 16: 2778–2791.
Gamble MJ, Fisher RP . SET and PARP1 remove DEK from chromatin to permit access by the transcription machinery. Nat Struct Mol Biol 2007; 14: 548–555.
Dong X, Wang J, Kabir FN, Shaw M, Reed AM, Stein L et al. Autoantibodies to DEK oncoprotein in human inflammatory disease. Arthritis Rheum 2000; 43: 85–93.
Sierakowska H, Williams KR, Szer IS, Szer W . The putative oncoprotein DEK, part of a chimera protein associated with acute myeloid leukaemia, is an autoantigen in juvenile rheumatoid arthritis. Clin Exp Immunol 1993; 94: 435–439.
Wichmann I, Respaldiza N, Garcia-Lozano JR, Montes M, Sanchez-Roman J, Nunez-Roldan A . Autoantibodies to DEK oncoprotein in systemic lupus erythematosus (SLE). Clin Exp Immunol 2000; 119: 530–532.
Schwartz S, Jiji R, Kerman S, Meekins J, Cohen MM . Translocation (6;9)(p23;q34) in acute nonlymphocytic leukemia. Cancer Genet Cytogenet 1983; 10: 133–138.
Vermaelen K, Michaux JL, Louwagie A, Van den Berghe H . Reciprocal translocation t(6;9)(p21;q33): a new characteristic chromosome anomaly in myeloid leukemias. Cancer Genet Cytogenet 1983; 10: 125–131.
Oyarzo MP, Lin P, Glassman A, Bueso-Ramos CE, Luthra R, Medeiros LJ . Acute myeloid leukemia with t(6;9)(p23;q34) is associated with dysplasia and a high frequency of flt3 gene mutations. Am J Clin Pathol 2004; 122: 348–358.
Slovak ML, Gundacker H, Bloomfield CD, Dewald G, Appelbaum FR, Larson RA et al. A retrospective study of 69 patients with t(6;9)(p23;q34) AML emphasizes the need for a prospective, multicenter initiative for rare 'poor prognosis' myeloid malignancies. Leukemia 2006; 20: 1295–1297.
Sandahl JD, Coenen EA, Forestier E, Harbott J, Johansson B, Kerndrup G et al. t(6;9)(p22;q34)/DEK-NUP214 rearranged pediatric myeloid leukemia: an international study on 62 patients. Haematologica 2014.
Fornerod M, Boer J, van Baal S, Jaegle M, von Lindern M, Murti KG et al. Relocation of the carboxyterminal part of CAN from the nuclear envelope to the nucleus as a result of leukemia-specific chromosome rearrangements. Oncogene 1995; 10: 1739–1748.
Ageberg M, Drott K, Olofsson T, Gullberg U, Lindmark A . Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes Chromosomes Cancer 2008; 47: 276–287.
Oancea C, Ruster B, Henschler R, Puccetti E, Ruthardt M . The t(6;9) associated DEK/CAN fusion protein targets a population of long-term repopulating hematopoietic stem cells for leukemogenic transformation. Leukemia 2010; 24: 1910–1919.
Boer J, Bonten-Surtel J, Grosveld G . Overexpression of the nucleoporin CAN/NUP214 induces growth arrest, nucleocytoplasmic transport defects, and apoptosis. Mol Cell Biol 1998; 18: 1236–1247.
Sanden C, Ageberg M, Petersson J, Lennartsson A, Gullberg U . Forced expression of the DEK-NUP214 fusion protein promotes proliferation dependent on upregulation of mTOR. BMC Cancer 2013; 13: 440.
Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368: 2059–2074.
Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002; 99: 4326–4335.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Sandén, C., Gullberg, U. The DEK oncoprotein and its emerging roles in gene regulation. Leukemia 29, 1632–1636 (2015). https://doi.org/10.1038/leu.2015.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2015.72
- Springer Nature Limited
This article is cited by
-
Exosomal DEK removes chemoradiotherapy resistance by triggering quiescence exit of breast cancer stem cells
Oncogene (2022)
-
Unclassified Neuroendocrine Tumor with a Novel CHD4::AFF2 Fusion: Expanding the Family of AFF2-Rearranged Head and Neck Malignancies
Head and Neck Pathology (2022)
-
Transformation of human CD34+ hematopoietic progenitor cells with DEK-NUP214 induces AML in an immunocompromised mouse model
Oncogene (2016)