
Abstract
Early high-dose therapy (HDT), consisting of high-dose melphalan and autologous stem cell transplantation following doublet or triplet novel agent induction, is a preferred management strategy for transplant-eligible myeloma patients. We set out to examine the utility of the current fluorescence in situ hybridization (FISH)-based risk stratification in a homogenously treated population of transplant-eligible myeloma patients receiving novel induction regimens and early HDT with or without posttransplant maintenance therapy. FISH was available in 409 patients at the time of diagnosis for patients receiving HDT within 12 months of diagnosis. We present comprehensive outcomes for chromosome 14 translocations and 17p abnormalities that both support and refute current risk stratification models. In contrast to its current classification as a marker of 'standard risk' (SR), t(11;14) was associated with inferior overall survival (OS) when compared with the classical SR cohort. The use of novel agent maintenance therapy (bortezomib or lenalidomide) following early HDT ameliorates the negative prognostic value of high-risk (HR) cytogenetic markers. HR patients who received maintenance following early HDT had similar OS compared with the SR cohort at 5 years.
Similar content being viewed by others

Introduction
Among transplant-eligible multiple myeloma (MM) patients, treatment with high-dose therapy (HDT) and autologous stem cell transplantation (ASCT) early, after a defined period of initial induction therapy, is a preferred management strategy, even in the novel agent era.1, 2, 3, 4 Risk stratification in MM, currently based primarily on cytogenetic abnormalities, is critical for long-term counseling of transplant-eligible patients and application of risk adapted treatment algorithms to maximize clinical outcomes while minimizing therapy related toxicities.5, 6 The tools available for risk assessment of newly diagnosed MM are expanding. Conventional cytogenetics, given low plasma cell proliferation rates and hence low sensitivity of conventional metaphase karyotyping, have been surpassed in clinical application by interphase and cytoplasmic immunoglobulin fluorescence in situ hybridization (FISH). Gene expression profiling may further expand to provide additional discernment in prognosis in newly diagnosed myeloma; however, gene expression profiling has limitations that currently preclude its widespread use. For the majority of patients currently diagnosed with MM, FISH in combination with other patient-related clinical and laboratory markers (plasma cell proliferative index, International Staging System (ISS), lactate dehydrogenase and so on) remains the de facto standard for determining initial risk.7
Translocations involving the immunoglobulin heavy chain (IgH) gene locus on chromosome 14 and 5 common partner chromosomes are present in approximately one-third of newly diagnosed patients and are associated with a variable and often inferior clinical outcome.8 The Mayo Stratification for Myeloma and Risk Adapted Therapy (mSMART) guidelines are similar in construct to IMWG (International Myeloma Working Group) and National Comprehensive Cancer Network criteria and have been adapted widely for use in clinical practice. Current mSMART guidelines deem translocations of t(14;16) and t(14;20) as high risk (HR), while t(4;14) is considered intermediate risk and t(11;14) and t(6;14) are classified as standard risk (SR).9, 10 In addition to IgH translocations, the loss of the short arm of chromosome 17 (del(17p)), classified by mSMART as HR, is thought to be related to the loss of one p53 allele and is observed in approximately 6% of newly diagnosed myeloma patients. Del(17p) has been associated with an inferior outcome even in the context of HDT.11, 12, 13 Together, specific IgH translocations and del(17p) found on FISH at diagnosis are the most common mechanism for classifying newly diagnosed MM as adverse risk.14 The clinical reality for the transplant-eligible patient deemed HR based on FISH abnormalities is a paucity of comparative effectiveness data leading to uncertainty as to the optimal treatment strategy following doublet or triplet induction and early HDT. Consolidation regimens, maintenance strategies, tandem transplantation, observation with second application of HDT at disease recurrence and nonmyeloablative allogeneic stem cell transplantation are all being investigated in the context of ongoing clinical trials.
Although there is good clinical data and little controversy as to the negative impact of t(4;14) translocations and del(17p) deletions at diagnosis, heterogeneity in clinical outcomes does exist within these groups. Outcome heterogeneity is certainly related to host-specific factors as well as the variable approach in management these patients receive following upfront therapy.15 Also, recent data from the MD Anderson Cancer Center group has suggested inferior outcomes for t(11;14) patients treated with ASCT compared with a standard cytogenetic risk cohort.16 This raises the question of the accuracy of risk stratification schemes based on historical data prior to the frequent use of novel agents. Our aim in performing this current study was to comprehensively describe the clinical outcomes of newly diagnosed MM patients with IgH translocations or del(17p) findings on FISH in the context of the homogenous application of early HDT as this fills a void in the current literature.
Patients and methods
Patients
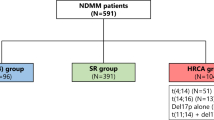
Following approval of the Mayo Clinic Institutional Review Board and in accordance with the Declaration of Helsinki, 941 patients evaluated and treated with HDT at Mayo Clinic Rochester for newly diagnosed MM between 2003 and 2012 were retrospectively identified. In order to ensure a homogenous patient population for analysis and interpretation, patients were excluded if they did not undergo early HDT (defined as ASCT within 12 months of initial diagnosis) or if they did not have a bone marrow FISH evaluation with sufficient plasma cells for analysis from within 6 months of diagnosis. Following these exclusion criteria, the final cohort consisted of 409 patients, of which 95% received at least one novel agent in the course of initial induction therapy.
FISH studies
FISH assays were performed on isolated plasma cells from bone marrow aspirates utilizing standard methods previously described, including immunofluorescent detection of cytoplasmic immunoglobulin light chains. Probes against 14q32(5′IGH,3′IGH), 17p13.1 (p53) and 17cen (D17Z1) were used to screen for abnormalities, and when indicated, confirmatory probes were utilized to confirm specific IgH translocations. Sensitivity thresholds determined by the standards set forth from our reference laboratory were used, and the probe was considered normal if the number of abnormal signals was under the following percentage for each probe; 14q32(5′IGH,3′IGH)—5.0%; t(11;14), t(4;14), t(14;16), t(6;14) and t(14;20)—3.0%; 17p13.1 (p53)—7.0%; and 17cen (D17Z1)—9.0%. For the purposes of this analysis, patients were classified as HR if they had del(17p), t(4;14), t(14;20) or t(14;16); patients were classified as SR if they had any other FISH abnormality (chromosome 1q abnormalities were not evaluated), except for t(11;14), which as a group were evaluated separately. If a patient had more than one cytogenetic abnormality, they were classified into the HR or SR group on the basis of the most adverse abnormality.
Treatment approaches
Patients received a variety of induction regimens for the initial treatment of their myeloma. Our standard practice has been to use bortezomib-containing regimens for patients with HR cytogenetics and lenalidomide and dexamethasone for those with SR myeloma. Patients who presented to us after initiation of their induction therapy by their referring physician were continued on the therapy as long as they were tolerating the regimen and the disease was responding and no changes were systematically made. Peripheral blood stem cell collection was performed typically with granulocyte colony-stimulating factor alone with plerixafor added for lack of adequate mobilization with chemotherapy-based mobilization limited to those who fail this approach or have inadequate disease control with the initial therapy. Conditioning regimen typically consisted of melphalan 200 mg/m2 (87% patients), with dose reduction of melphalan to 140 mg/m2 (12% patients) considered for those >70 years, significant comorbidities or renal insufficiency. Eight patients in the study cohort received zevalin and melphalan for conditioning as they were enrolled in an ongoing clinical trial evaluating that regimen. In general, maintenance treatments were recommended in patients with HR disease or had residual disease (less complete remission) but were left to the physician to discuss with the patients. The typical regimens consisted of lenalidomide 10–15 mg daily or bortezomib 1.3–1.6 mg/m2 every other week.
Statistical methods
Descriptive statistics were used to characterize the patient cohort. One-way analysis of variance test was used to compare baseline continuous variables in more than two groups when a parametric distribution could reasonably be assumed. Pearson’s chi-square test was used to compare baseline nominal variables between two or more groups. Response and disease progression were defined per IMWG criteria.17 Overall survival (OS) and progression-free survival (PFS) were calculated from time of ASCT day 0, or where indicated from diagnosis, to death or defined disease progression. Survival curves were calculated per Kaplan–Meier estimates and compared using the log-rank test. JMP Version 10.0 software (2012 SAS Institute, Cary, NC, USA) was used for analysis.
Results
Of the 409 patients in the final study cohort who had evaluable FISH from within 6 months of diagnosis and underwent HDT and ASCT within 12 months of diagnosis, the median estimated follow-up was 43.0 (95% confidence interval (CI) 40.8-46.5) months from the time of diagnosis, with 283 patients (69%) alive at the last follow-up. The median age at presentation was 59 (range 23–75) years, and 249 (61%) were men. Induction regimens included a novel agent (immunomodulators, including thalidomide and lenalidomide, or proteasome inhibitors, including bortezomib) in 95% of patients prior to HDT and ASCT. Partial response or better was obtained prior to HDT in 328 patients (80%), while 46 patients (11%) had primary refractory disease in that they did not have measurable response prior to HDT, and 34 patients (8%) had relapsed disease even though they underwent HDT within 12 months of diagnosis.
Overall, 202 patients (49.4%) had an abnormal probe to 14q32(5′IGH,3′IGH). The specific chromosome 14 translocation breakdown was as follows: 41 (10.0%) patients with t(4;14), 2 (0.5%) patients with t(6;14), 78 (19.1%) patients with t(11;14), 13 (3.2%) patients with t(14;16), 5 (1.2%) patients with t(14;20), and 63 (15.4%) patients with an otherwise undefined abnormality of the 14q32(5′IGH,3′IGH) loci not corresponding to one of the five described common translocations. Fifty patients (12.2%) had either del(17p) or monosomy 17. The baseline characteristics of all patients as well as pertinent baseline comparisons at diagnosis for each FISH category with n>30 studied are detailed in Tables 1 and 2. Myeloma bone disease was less common among those with t(4;14) disease as compared with the entire cohort (73% versus 86%, P=0.044). There were no differences in age, plasma cell labeling index, B2-microglobulin absolute values or serum creatinine at diagnosis between the entire cohort and individual FISH groups described above with n>30 patients.
We then comprehensively evaluated and compared outcomes in the entire cohort and FISH-based subgroups. We specifically compared the outcomes for each of the common cytogenetic type with the entire cohort to estimate the relative significance of the abnormality. For the entire cohort of 409 patients, median PFS following early HDT was 24 (95% CI 21.6–25.4) months, while median OS was 92 (95% CI 80.1–99.8) months. For the 41 patients with t(4;14) disease, median PFS following early HDT was 23.5 (95% CI 12.9–25.1) months, while median OS was not reached (NR) (95% CI 53.8 months–NR). There was no statistical difference in PFS (P=0.294) or OS between the full cohort and the t(4;14) group (Figure 1a). We then stratified the patients by ISS stage and compared t(4;14) patients to the general cohort (Figure 1b). Among ISS stage I–II patients, there was no difference in PFS or OS between the groups. For ISS stage III patients, those with t(4;14) had shorter PFS compared with the general cohort (12 versus 20.5 months, P=0.0455); there was no difference in OS; however; sample size was small (five patients with only one death).

(a) PFS and OS of t(4;14) patients compared with the general cohort. (b) PFS of t(4;14) patients stratified by ISS stage at diagnosis.
For the 78 patients with t(11;14) disease, PFS and OS were 21.9 (95% CI 16.8–25.2) months and 56.9 (95% CI 45.7–80.4) months, respectively, from ASCT day 0. Compared with the general cohort, there was no difference in PFS, while there was a late difference in OS, with t(11;14) showing inferior survival (P=0.0201; Figure 2a). When examined by the ISS stage, the impact of t(11;14) on PFS remained the same. The OS difference was preserved among the ISS stage I and II patients, while there was no OS difference observed within ISS stage III patients (Figure 2b).

(a) PFS and OS of t(11;14) patients compared with the general cohort. (b) OS of t(11;14) patients stratified by ISS stage at diagnosis.
Outcomes in the 63 patients with an undefined chromosome 14 translocation revealed PFS following early HDT of 26.6 (95% CI 21.0–32.5) months and OS of 92.8 (95% CI 92.0–not calculable) months. Neither were significantly different from the comparison general cohort (P=0.564 and P=0.444, respectively). Comparison after stratification by ISS stage did not reveal a significant difference in either PFS or OS outcomes.
For the 50 patients with del(17p) or monosomy 17 disease, PFS and OS were 16.9 (95% CI 13.8–27.7) months and 48.3 (95% CI 33.1–64.0) months, respectively. Both were statistically inferior when compared with the general cohort (P=0.050 and P=0.0006, respectively; Figure 3a). Analyzing PFS when stratified into ISS categories, those patients with ISS I/II disease had no statistical difference in PFS (22.7 versus 24.4 months P=0.348), whereas those patients with ISS III disease continued to see inferior PFS following early ASCT (14.9 versus 20.5 months, P=0.026) (Figure 3b). ISS stratification revealed no change when analyzing OS, with 17p abnormal patients having inferior OS whether they were ISS I/II or ISS III at diagnosis (Figure 3c). A summary of outcomes in each of the above groups as well as those patients with t(14;16) and t(14;20) is presented in Table 3. When stratified by risk group, the median PFS for HR, t(11;14) and SR patients from diagnosis was 24.9 (23,30), 28.1 (21,31) and 30.4 (28,34) months, respectively (P=0.034). The median OS for HR, t(11;14) and SR patients from diagnosis was 60.5 (46,71), 73.4 (54,89) and 103 (98,113) months, respectively (P<0.0001; Figure 4a).

(a) PFS and OS of del(17p) patients compared with the general cohort. (b) PFS of del(17p) patients stratified by ISS stage at diagnosis. (c) OS of del(17p) patients stratified by ISS stage at diagnosis.

(a) OS of HR, SR and t(11;14). (b) OS of HR and SR patients receiving maintenance therapy following early HDT.
The use of maintenance or consolidation immunochemotherapy was also assessed following early HDT. Based on the documentation of the treating hematologist at day +100 after ASCT, plans for the use of maintenance or consolidation therapy, typically with lenalidomide or bortezomib with or without a corticosteroid, were observed in 30.3% of patients in the entire cohort. Most commonly used in these settings were lenalidomide (n=74 patients) and bortezomib (n=39 patients) as single agents. As expected, the use of maintenance or consolidation therapies was seen more frequently in patients with t(4;14) (46.3%) or del(17p)/monosomy 17 (68%) disease as compared with the entire cohort (Pearson P-value <0.0001 for each). We did not evaluate for the length of maintenance or consolidation therapy, toxicities or dose reductions, only the clinical intent to treat and with which agents at day +100. We also did not perform a comprehensive evaluation of subsequent therapies at the time of first relapse following early HDT. In contrast to the general cohort, at 60 months of follow-up from diagnosis, there was no statistical difference between HR patients and SR patients among those who received post-ASCT maintenance therapy (P=0.19; Figure 4b). On multivariate analysis, HR FISH, t(11;14) and relapsed disease at the time of early HDT and ASCT were all independently associated with shorter OS. For multivariate analysis of PFS, HR FISH, relapsed disease at the time of early HDT and ASCT and not receiving maintenance therapy following HDT were all independently associated with shorter PFS (Table 4).
Discussion
This study fills an important void in the current literature in that it provides important and current prognostic data for newly diagnosed MM patients with chromosome 14 translocations or 17p abnormalities detected on FISH who undergo early HDT following novel agent induction. Data on cytogenetic risk stratification in MM should always be interpreted in the context of patient-specific and treatment-related factors, which have previously been demonstrated and confirmed by our data showing additional discernment in outcome prognostication, that is, ISS stratification within FISH categories. A particular strength of our current data is the homogenous application of novel induction regimens and early HDT. We did not analyze secondary therapies beyond the use of maintenance or consolidation regimens after initial HDT. Potential confounding factors to keep in mind are our exclusion of patients with insufficient plasma cells for FISH analysis, as it is unknown whether these patients may be predisposed to an alternate outcome compared with the general cohort.
The French Myeloma Group (IFM) experience, published in 2007, allows for perspective by comparison.9 The 936 patients they were able to analyze for FISH abnormalities were diagnosed between 2000 and 2003; the majority were not treated with novel-agent induction regimens. There was also significant heterogeneity in the upfront transplant conditioning regimens used compared with our cohort, and the timing of HDT in relation to diagnosis is not as clearly defined. The incidence of translocations t(11;14), t(4;14) and loss of 17p were relatively similar to our data. Several important outcome similarities and differences should be noted. Regarding PFS, the IFM reported results from time of diagnosis, whereas we landmarked from ASCT. However, for t(4;14) and del(17p) patients their results (20.6 months and 15 months respectively) are similar to ours for median PFS. Median OS for del(17p) patients in the IFM experience was 22 months, whereas we found median OS of 48 months for this group. The IFM did not note a difference in OS when analyzing t(11;14), whereas we found patients with t(11;14) had inferior OS, with median survival of 56.9 compared with 92 months.
Current schema classify t(11;14) as a SR genetic marker for newly diagnosed myeloma patients.18 Historically, there has been variation in how t(11;14)(q13;q32) has been interpreted in terms of its associated prognostic value. Early studies had associated a favorable prognosis with t(11;14).19 A previously published analysis from our group showed no difference in the t(11;14) group compared with those patients without the abnormality, with PFS of 20.1 versus 15.3 months and OS of 36.6 versus 34.8 months.10 The MD Anderson Cancer Center group has, as previously mentioned, published on their experience with t(11;14) newly diagnosed MM and ASCT.16 Several differences in populations are worth noting. The MD Anderson Cancer Center cohort consisted of 18 patients with isolated t(11;14) findings compared with 69 patients in our cohort. Additionally, a greater proportion of our patients received novel-agent induction regimens (95% versus 78%), and the MD Anderson Cancer Center group did not restrict to early HDT and includes t(11;14) patients whose initial ASCT took place as much as 60 months following initial diagnosis. Notably, our data show similar PFS results for the t(11;14) group and overall improvement in median OS to 56.9 months. However, compared with the general cohort in our current study with median OS of 92 months, these t(11;14) patients had inferior OS compared with our updated general cohort. This suggests the potential need for further analysis of t(11;14) patients for other factors correlating with this observed outcome heterogeneity, such as a tendency for clinicians to pursue less aggressive therapy at early biochemical relapse, and the potential need to reconsider its position as a SR genetic marker.
Maintenance therapy following HDT and ASCT in MM has been shown to improve PFS in large multicenter trials using lenalidomide or bortezomib.20, 21, 22, 23 Our retrospective data show that the use of either lenalidomide or bortezomib maintenance following early HDT is independently associated with improved PFS. Our group has adopted a risk-adapted therapy strategy for post-ASCT maintenance. Patients who received maintenance therapy after ASCT were analyzed by FISH stratified risk cohort, and those patients with HR cytogenetics at initial diagnosis did not have inferior OS as compared with those patients with SR cytogenetics. These data support validation of our risk-adapted maintenance strategy (http://www.msmart.org), and future trials evaluating maintenance immunochemotherapy in MM should report on outcomes in distinct cytogenetic risk groups.
Our data demonstrate an expected improvement in OS in the novel agent era compared with previous reports on myeloma outcomes following HDT. We confirmed the 'HR' prognostic significance of 17p abnormalities as well as the 'intermediate-risk' classification of t(4;14) disease. The ability to draw conclusions from our t(6;14), t(14;16) and t(14;20) was limited by small sample size, but the 'HR' classification of t(14;16) and t(14;20) is supported by inferior median OS outcomes, albeit not to a level of significance. The results from our current study call into question the 'SR' assignment of t(11;14) disease in the setting of novel induction and early HDT, suggesting that the t(11;14) abnormality should be in the intermediate-risk group. Finally, the application of maintenance therapy in HR patients seems to be validated as these patients had non-inferior OS at 5 years when compared with SR patients who also received post-HDT novel-agent maintenance therapy.
References
Boccadoro M, Cavallo F, Gay FM, Di Raimondo F, Nagler A, Montefusco V et al. Melphalan/prednisone/lenalidomide (MPR) versus high-dose melphalan and autologous transplantation (MEL200) plus lenalidomide maintenance or no maintenance in newly diagnosed multiple myeloma (MM) patients. J Clin Oncol 2013; Suppl (ASCO abstracts): abstract 8509.
Palumbo A, Gay F, Spencer A, Di Raimondo F, Zdenek A, Larocca A et al. A phase III study of ASCT vs cyclophosphamide-lenalidomide-dexamethasone and lenalidomide-prednisone maintenance vs lenalidomide alone in newly diagnosed myeloma patients. Blood 2013; 122 (ASH abstracts): 763.
Moreau P, Attal M . All transplantation-eligible patients with myeloma should receive ASCT in first response. Hematology 2014; 2014: 250–254.
Rchardson PG, Laubach JP, Munshi NC, Anderson KC . Early or delayed transplantation for multiple myeloma in the era of novel therapy: does one size fit all? Hematology 2014; 2014: 255–261.
Chng WJ, Dispenzieri A, Chim C-S, Fonseca R, Goldschmidt H, Lentzsch S et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014; 28: 269–277.
Kapoor P, Fonseca R, Rajkumar SV, Sinha S, Gertz MA, Stewart AK et al. Evidence for cytogenetic and fluorescence in situ hybridization risk stratification of newly diagnosed multiple myeloma in the era of novel therapies. Mayo Clin Proc 2010; 85: 532–537.
Avet-Loiseau H. The future of FISH for the Diagnosis of Presenting Myeloma. EHA-SWG Scientific Meeting on Multiple Myeloma. September 2014. European Hematological Association: Barcelona, Spain, Abstract/Education book, pp 16–17.
Kumar S, Fonseca R, Ketterling RP, Dispenzieri A, Lacy MQ, Gertz MA et al. Trisomies in multiple myeloma: impact on survival in patients with high-risk cytogenetics. Blood 2012; 119: 2100–2105.
Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood 2007; 109: 3489–3495.
Gertz MA, Lacy MQ, Dispenzieri A, Greipp PR, Litzow MR, Henderson KJ et al. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and -17p13 in myeloma patients treated with high-dose therapy. Blood 2005; 106: 2837–2840.
Chang H, Qi C, Yi QL, Reece D, Stewart AK . p53 gene deletion detected by fluorescence in situ hybridization is an adverse prognostic factor for patients with multiple myeloma following autologous stem cell transplantation. Blood 2005; 105: 358–360.
Drach J, Ackermann J, Fritz E, Krömer E, Schuster R, Gisslinger H et al. Presence of a p53 gene deletion in patients with multiple myeloma predicts for short survival after conventional-dose chemotherapy. Blood 1998; 92: 802–809.
Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003; 101: 4569–4575.
Mikhael JR, Dingli D, Roy V, Reeder CB, Buadi FK, Hayman SR et al. Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) Consensus Guidelines 2013. Mayo Clin Proc 2013; 88: 360–376.
Moreau P, Attal M, Garban F, Hulin C, Facon T, Marit G et al. Heterogeneity of t(4;14) in multiple myeloma. Long-term follow-up of 100 cases treated with tandem transplantation in IFM99 trials. Leukemia 2007; 21: 2020–2024.
Sasaki K, Lu G, Saliba RM, Bashir Q, Hosing C, Popat U et al. Impact of t(11;14)(q13;q32) on the outcome of autologous hematopoietic cell transplantation in multiple myeloma. Biol Blood Marrow Transplant 2013; 19: 1227–1232.
Durie BG, Harousseau JL, Miguel JS, Bladé J, Barlogie B, Anderson K et al. International uniform response criteria for multiple myeloma. Leukemia 2006; 20: 1467–1473.
Rajkumar SV . Multiple myeloma: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol 2014; 89: 999–1009.
Soverini S, Cavo M, Cellini C, Terragna C, Zamagni E, Ruggeri D et al. Cyclin D1 overexpression is a favorable prognostic variable for newly diagnosed multiple myeloma patients treated with high-dose chemotherapy and single or double autologous transplantation. Blood 2003; 102: 1588–1594.
Attal M, Lauwers-Cances V, Marit G, Caillot D, Moreau P, Facon T et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med 2012; 366: 1782–1791.
McCarthy PL, Owzar K, Hofmeister CC, Hurd DD, Hassoun H, Richardson PG et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med 2012; 366: 1770–1781.
Palumbo A, Cavallo F, Gay F, Di Raimondo F, Ben Yehuda D, Petrucci MT et al. Autologous transplantation and maintenance therapy in multiple myeloma. N Engl J Med 2014; 271: 895–905.
Sonneveld P, Schmidt-Wolf IGH, van der Holt B, El Jarari L, Bertsch U, Salwender H et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: results of the randomized phase III HOVON-65/ GMMG-HD4 trial. J Clin Oncol 2012; 30: 2946–2955.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
GP Kaufman designed the research, collected and analyzed data and wrote the paper. SK Kumar designed the research, analyzed data and contributed to the writing of the paper. The other authors were involved in writing the manuscript.
Rights and permissions
About this article

Cite this article
Kaufman, G., Gertz, M., Dispenzieri, A. et al. Impact of cytogenetic classification on outcomes following early high-dose therapy in multiple myeloma. Leukemia 30, 633–639 (2016). https://doi.org/10.1038/leu.2015.287
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2015.287
- Springer Nature Limited
This article is cited by
-
Multiple myeloma with t(11;14): impact of novel agents on outcome
Blood Cancer Journal (2023)
-
High-risk disease in newly diagnosed multiple myeloma: beyond the R-ISS and IMWG definitions
Blood Cancer Journal (2022)
-
Increasing genomic discovery in newly diagnosed multiple myeloma: defining disease biology and its correlation to risk
Annals of Hematology (2022)
-
Venetoclax in combination with carfilzomib and dexamethasone in relapsed/refractory multiple myeloma harboring t(11,14)(q13;q32): two case reports and a review of the literature
Journal of Medical Case Reports (2020)
-
Long-term survival and polyclonal immunoglobulin reconstitution after allogeneic stem cell transplantation in multiple myeloma
Annals of Hematology (2020)