
Abstract
Background/Objectives:
Epidemics of obesity and diabetes are escalating. High-calorie/high-fat food is a major cause for these global health issues, but molecular mechanisms underlying high-fat, diet-induced obesity are still not well understood. The aryl hydrocarbon receptor (AhR), a transcription factor that acts as a xenobiotic sensor, mediates environmental toxicant-induced obesity, insulin resistance and development of diabetes. AhR also influences lipid metabolism and diet-induced obesity. The effects of AhR deficiency on diet-induced obesity, hepatic steatosis and insulin resistance were examined.
Methods:
Male wild-type (WT), AhR null (AhR−/−) and AhR heterozygote (AhR+/−) mice were fed a normal chow diet (NCD, 10% kcal from fat) or a high-fat diet (HFD, 60% kcal from fat) for up to 14 weeks. Adiposity, adipose and liver morphology, insulin signaling, metabolic parameters and gene profiles were assessed.
Results:
AhR deficiency protected against HFD-induced obesity, hepatic steatosis, insulin resistance and inflammation. Moreover, AhR deficiency preserved insulin signaling in major metabolic tissues. These protective effects result from a higher energy expenditure in AhR-deficient mice compared with WT. Levels of transcript for both the thermogenic gene, uncoupling protein 1 (Ucp1), in brown adipose tissue and mitochondrial β-oxidation genes in muscle were significantly higher in AhR−/− and AhR+/− mice compared with WT.
Conclusions:
This work documents a physiologically relevant function for AhR in regulation of body weight, hepatic fat deposition, insulin sensitivity and energy expenditure under HFD exposure, suggesting that AhR signaling may be developed as a potential therapeutic target for treatment of obesity and metabolic disorders.
Similar content being viewed by others


Introduction
More than one-third of adults and 17% of youth in the United States are obese.1 Obesity is a complex metabolic disease that is a major risk factor for development of type 2 diabetes and cardiovascular diseases. Although high calorie intake and decreased energy expenditure are causative, other, ill-defined factors contribute to obesity and metabolic disorders; the identity of specific genes that impact the balance of energy intake and expenditure remains elusive.2 Several nuclear receptors, such as peroxisome proliferator activated receptors (PPARs) and the xenobiotic receptor, pregnane X receptor (PXR), have been identified as modulators of lipid metabolism that are important in the development of obesity and type 2 diabetes.3, 4, 5, 6
Although aryl hydrocarbon receptor (AhR) was initially characterized as a xenobiotic sensing, ligand-dependent transcription factor that mediates toxicological responses, many studies suggest that AhR is an integral, physiological regulator of energy metabolism, and contributes to the development of obesity and metabolic disorders. Epidemiological studies indicate that activation of the AhR by environmental pollutants such as dioxins is associated with an increased risk of obesity and type 2 diabetes.7, 8, 9, 10 Experimental studies further document that AhR overactivation promotes obesity, hepatic steatosis, nonalcoholic steatohepatitis and insulin resistance.11, 12, 13, 14 In addition, a recent study reports that AhR signaling may be activated by the modern western diet.15 However, the function of AhR in diet-induced obesity, insulin resistance and metabolic disorders remains under investigated.
In this study, AhR deficiency created in AhR null (AhR−/−) and AhR heterozygous (AhR+/−) mice protects against diet-induced obesity and its metabolic complications. AhR deficiency promotes increased energy expenditure when mice are fed a high-fat diet (HFD, 60% fat diet) but not the normal chow diet (NCD, 10% fat diet). This correlates with higher uncoupling protein 1 (Ucp1) transcript levels in brown adipose tissue (BAT), elevated mitochondrial β-oxidation and respiratory chain gene transcript levels in muscle, and enhanced insulin signaling. These data highlight AhR signaling as a potential therapeutic target for treatment of obesity and type 2 diabetes.
Materials and methods
Schematic time line of experimental procedures
Six-week-old male wild-type (WT), AhR+/− or AhR−/− (Bradfield strain)16 mice were exposed to NCD or HFD for up to 14 weeks (see Supplementary Figure 1). Body weight was measured each week. During weeks 5–7 mice were housed individually; food intake and physical activity were measured by an infrared beam interruption (Coulbourn Instruments, Whitehall, PA, USA) during weeks 6–7. Glucose tolerance test (GTT) was performed at week 10 and insulin tolerance test (ITT) at week 12. At week 13, blood samples were collected from live mice to assay triglycerides, insulin, leptin and adiponectin. At week 14, mice were killed for harvest of epididymal white adipose tissue (eWAT), liver and skeletal muscle (right gastrocnemius). eWAT was used as a surrogate measure for visceral fat; subcutaneous fat was not assessed. To quantify eWAT, epididymal fat was carefully dissected after mice were killed. eWAT weight was normalized to total body weight. An acute insulin stimulation assay was performed on a subset of mice killed at week 14.
Metabolic measurements
For the GTT, mice were fasted for 16 h, a drop of blood from the snipped tail was used to test the glucose concentration using a glucometer (TRUEtrack, Walgreens, Springfield, IL, USA) and then mice were injected intraperitoneally with 1 g of glucose per kilogram of body weight. Blood glucose was subsequently measured at 15, 30, 60, 90 and 120 min post injection. For the ITT, mice were fasted for 6 h, a drop of blood from the snipped tail was used to measure glucose and then mice were injected intraperitoneally with 0.75 units insulin per kilogram of body weight. Blood glucose was subsequently measured at 15, 30, 60 and 90 min post injection.
Total and resting metabolic rates were measured at week 6 by indirect calorimetry using the Oxymax Indirect Calorimetry System/Metabolic Cage from Columbus Instruments (CLAMS, Columbus, OH, USA) in which zirconia and infared sensors are used to record O2 and CO2, respectively. Mice were individually housed in respiratory chambers. All comparisons were based on mice studied simultaneously in eight different respiratory chambers connected to the same O2 and CO2 sensor to minimize the effect of environmental variation and instrument calibration on data. Mice had free access to food and water during the measurements and were adapted to metabolic cages for 24 h before data collection. WT and AhR+/− mice on NCD (n=8 mice for each) or HFD (n=8 for each) were tested; gas samples were collected and analyzed every 5 min per animal; hourly averages were calculated. Output data from the software include O2 consumption (VO2) (ml kg−1 per minute), CO2 production (VCO2) (ml kg−1 per minute), respiratory quotient (RQ=VCO2/VO2) and heat production (heat=CV × VO2; CV=3.815+(1.232 × RQ)). Please refer to Columbus Instruments\Equations for Energy Expenditure for details (http://www.colinst.com/brief.php?id=61). At the end of the calorimetry study, these animals were killed to collect interscapular BAT and skeletal muscle (right gastrocnemius).
Histological analysis
Tissues were fixed in 10% formalin, embedded in paraffin, sectioned and stained with hematoxylin-eosin (H&E) at the Histology Department of Memorial Medical Center, Springfield IL. Quantification of adipocyte size was done on H&E-stained sections using ImageTool software (http://compdent.uthscsa.edu/dig/itdesc.html).
Biochemical assays
Blood samples were collected from mice after a 16-h fast. Serum insulin (Crystal Chem, Downers Grove, IL, USA), leptin (Crystal Chem) and adiponectin (Invitrogen, Grand Island, NY, USA) were measured by ELISA. Triglycerides (Pointe Scientific, Inc., Canton, MI, USA) were measured using a commercial kit, according to the manufacturer’s instructions.
Real-time PCR and western-blot analysis
For real-time PCR analysis, reverse transcription was performed with a kit from Promega (Madison, WI, USA). SYBR green-based real-time reverse transcriptase PCR was carried out on an ABI StepOne Plus real-time PCR system (Grand Island, NY, USA). PCR data were normalized to β-2 microglobulin (B2M). Primer sequences for real-time PCR are listed in Supplementary Table 1. Those not listed in Supplementary Table 1 were purchased from Qiagen (Valencia, CA, USA), and those sequences are proprietary. Western blot analysis was performed on samples from WT or AhR−/− mice after 14 weeks of HFD feeding to assess nuclear factor kappa of light polypeptide gene enhancer in B cells inhibitor alpha (IκBα) and phospho-c-Jun N-terminal kinase (pJNK) (Cell Signaling, Beverly, MA, USA). β-Actin was used as a loading control for western blot experiments.
For the in vivo acute insulin stimulation assay performed at week 14, mice were fasted for 6 h, anesthetized with a cocktail of Ketamine/Xylazine, and then injected with 0.75 units of insulin per kilogram of body weight via the vena cava. Liver, skeletal muscle (gastrocnemius) and eWAT were collected 3–7 min following insulin injection. Protein samples were subjected to western blotting to assess insulin-stimulated phospho-Akt (Ser473, Thr308; Cell Signaling). Total Akt (Cell Signaling) and β-actin (Sigma, St Louis, MO, USA) were used as internal controls as described previously.17
Statistical analysis
Data are expressed as mean±s.e.m. IBM SPSS11.0 (http://www-01.ibm.com/software/analytics/spss/products/statistics/downloads.html) was used to perform statistical analysis. Independent t-test and one-way or two-way ANOVA with Tukey’s post-hoc analysis were utilized to compare differences and P<0.05 was considered as statistically significant.
Results
AhR deficiency protects against HFD-induced adiposity
To explore the function of AhR in energy metabolism including fat metabolism and adiposity, male WT, AhR−/− and AhR+/− mice were fed either NCD or HFD for 14 weeks starting at 6 weeks of age. When fed the NCD, there were no significant differences among the three genotypes of mice in body weight, total eWAT weight and the ratio of eWAT to body weight (Figures 1a and b). eWAT was selected to represent abdominal fat, a major contributor to obesity and predictor of metabolic dysfunction.

AhR deficiency alleviates HFD-induced adiposity. Male WT, AhR−/− and AhR+/− mice were fed NCD or HFD for 14 weeks. (a) Body weight gain during 14-week feeding and appearance (NCD, WT, n=20, AhR−/−, n=8, AhR+/−, n=6; HFD, WT, n=13, AhR−/−, n=12, AhR+/− HFD, n=14). (b) Epididymal fat-pad weight and its percentage of total body weight in NCD-fed or HFD-fed mice at week 14 (NCD, n=4–6; HFD, WT, n=11, AhR−/−, n=7, AhR+/−, n=12). (c, d) Representative images of H&E-stained eWAT sections from mice with 14 week HFD (c) and adipocyte size eWAT from mice after 14 weeks NCD or HFD (d). (e) Liver was extracted from mice after 14 weeks NCD or HFD, real-time PCR was performed to assess AhR transcript levels (NCD, n=4–6; HFD, WT, n=13, AhR−/−, n=8, AhR+/−, n=6). *P<0.05, **P<0.001 by two-way ANOVA followed by one-way ANOVA (a), or one-way ANOVA with Turkey’s post hoc comparison (b, d).
Beginning at 8 weeks after initiation of HFD feeding, AhR−/− and AhR+/− mice exhibited significantly lower body weight than WT (Figure 1a). After 14 weeks on the HFD, total eWAT weight as well as the ratio of eWAT to body weight was lower in AhR−/− and AhR+/− mice compared with their WT counterparts (Figure 1b). Adipocytes in H&E-stained sections from the eWAT of AhR−/− and AhR+/− mice were smaller in size compared with WT after 14 weeks of HFD feeding (Figures 1c and d). Since total eWAT weight in the AhR−/− and AhR+/− mice was less than eWAT weight in WT, this quantitation indicates that hyperplasia of adipocytes cannot account for the smaller adipocyte size in the AhR-deficient mice.
AhR transcripts in liver were undetectable in AhR−/− mice and decreased to 30% of WT in AhR+/− mice (Figure 1e). HFD feeding did not alter AhR transcript levels in any of the mice. Levels of Cyp1A1, a classical target for AhR, were also not changed with HFD feeding compared with NCD (data not shown). These results suggest that AhR deficiency protects against diet-induced adiposity even though HFD feeding itself does not alter AhR transcript levels or classical Cyp1A1 activity within the individual AhR genotypes.
AhR deficiency protects against HFD-induced hepatic steatosis
HFD feeding induces hepatic steatosis in animal models. H&E staining revealed that HFD induced an accumulation of lipids in liver sections of WT mice, but not in HFD-fed AhR−/− and AhR+/− mice (Figure 2a). In the liver, transcript levels of CD36/fatty acid translocase, as well as hepatic lipogenic genes including the transcription factor, sterol regulatory element-binding protein 1c, and its substrates, fatty acid synthase (FAS) and acetyl Coenzyme A carboxylase (ACC), were elevated in HFD-fed WT mice compared with NCD-fed WT mice (Figure 2b, data presented as HFD/NCD ratios, where a value of >1 represents increases in HFD compared with NCD). CD36, FAS and ACC were significant lower in the AhR−/− and AhR+/− mice compared with WT after HFD feeding.

AhR deficiency protects against HFD-induced hepatic steatosis. WT, AhR−/− and AhR+/− mice were killed after NCD or HFD feeding for 14 weeks. (a) Livers were sectioned and stained with H&E, and representative sections are presented for each group, Scale bar: 200 μm (NCD, n=6–8; HFD, WT, n=13, AhR−/−, n=8, AhR+/−, n=12). (b, c) Total RNA was isolated from liver and mRNA levels of CD36, sterol regulatory element-binding protein 1c (SREBP1c), FAS, ACC, PPARα, CPT1b, PEPCK, G6Pase and PDK4 genes were examined by real-time PCR. Each gene was normalized against the amount of B2M in each sample (n=8–12 for each group). Data are expressed as the ratio of levels in HFD animals to NCD animals to accentuate changes associated with HFD feeding. *P<0.05, by one-way ANOVA with Tukey’s post hoc comparison.
Similarly, in WT mice fed HFD, PPARα and fatty acid β-oxidation genes (carnitine palmitoyltransferase 1b, CPT1b; acetyl CoA oxidase, ACO), gluconeogenesis genes (phosphoenolpyruvate carboxykinase, PEPCK; glucose-6-phosphatase, G6Pase) and the glucose oxidation gene (pyruvate dehydrogenase lipoamide kinase 4, PDK4) were elevated compared with NCD, but this increase was significantly suppressed in AhR−/− and AhR+/− mice fed HFD (Figure 2c, data shown as HFD/NCD). In addition, all mice had increased serum triglyceride levels on the HFD compared with the NCD; there were no differences in triglycerides among the genotypes on the same diet (Table 1). These data suggest that the protective effects of AhR deficiency on HFD-induced hepatic steatosis may be primarily due to impaired entry of fat into the liver (lower CD36 levels), but also to decreased synthesis of fat and glucose (lower FAS, ACC, PEPCK and G6Pase), accompanied by lower fat and glucose oxidation (lower CPT-1, ACO and PDK4).
AhR deficiency improves HFD-induced insulin resistance, hyperglycemia and hyperinsulinemia
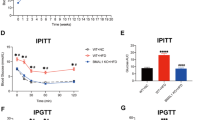
Obesity can induce insulin resistance, metabolic syndrome and type 2 diabetes. To explore whether the decreased adiposity and hepatic steatosis in AhR−/− or AhR+/− mice are concurrent with improved metabolic function, such as improved insulin sensitivity and reduced hyperglycemia, GTT (week 10) and ITT (week 12) were performed (see Supplementary Figure 1). GTT and ITT revealed that both AhR−/− and AhR+/− mice exhibited significantly improved glucose tolerance and insulin sensitivity compared with WT mice on HFD (Figures 3a and b). Moreover, although fasting glucose was not changed, fed blood glucose and fasting serum insulin were significantly lower in AhR−/− and AhR+/− mice compared with WT mice fed the HFD (Figure 3c and Table 1).

AhR deficiency improves HFD-induced insulin resistance, hyperglycemia and hyperinsulinemia. (a, b) GTT after 10 weeks of NCD or HFD feeding (a) and ITT after 12 weeks of NCD or HFD feeding (b) in WT, AhR−/− and AhR+/− mice. (c) Fasted and fed blood glucose were measured from the same mice in (a and b) (NCD, n=7–14; HFD, WT, n=17, AhR−/−, n=13, AhR+/−, n=13 for a–c). (d) After 14 weeks of HFD feeding, some mice received an injection of insulin (0.75 U kg−1) through inferior vena cava, tissues were harvested at pre-injection and post-injection (3 min for liver, 5 min for gastrocnemius muscle, 7 min for eWAT). Western-blot analysis was performed to assess insulin-stimulated phospho-Akt (Ser308 and Ser473), total Akt and β-actin were used to normalize. n=3–5 for each group. Representative blots were shown for each group. *P<0.05, **P<0.001, by two-way ANOVA followed by one-way ANOVA with Tukey’s post hoc comparison.
Insulin signaling is a determinant of insulin sensitivity, and obese subjects commonly have compromised insulin signaling. An acute insulin response assay was performed at week 14 to assess insulin signaling; insulin signaling in liver, skeletal muscle and eWAT was substantially better in AhR−/− and AhR+/− mice, as revealed by increased insulin-stimulated phospho-Akt (both Ser473 and Thr308; Figure 3d). These data support the findings of systemically improved insulin sensitivity demonstrated by GTT and ITT (Figures 3a and b).
AhR deficiency alters chronic inflammation and adipokine levels
Obesity is commonly associated with chronic low-grade inflammation. Chronic excesses in nutrient availability activate inhibitor κB kinase and JNK through toll-like receptor, leading to increased expression of inflammatory mediators, and ultimately to disruption of the insulin signaling pathway.18, 19 Consequently, inflammatory markers were examined after 14 weeks of HFD feeding. Real-time PCR revealed no changes in eWAT transcript levels of the cytokines IL1β and IL6 in any of the mice after HFD feeding and no differences due to genotype (Figure 4a, data shown as HFD/NCD). However, the macrophage markers CD68 and F4/80, as well as the proinflammatory cytokine, TNFα, were significantly elevated by HFD feeding in the eWAT of WT mice; these same genes increased to a much lower extent in the eWAT of HFD-fed AhR−/− and AhR+/− mice (Figure 4a), suggesting that AhR deficiency reduced HFD-induced inflammation in eWAT to levels approaching those in NCD fed mice (Figure 4a, HFD/NCD ratios approach 1).

AhR deficiency alters chronic inflammation and adipokine levels. Mice were killed after 14 weeks of NCD or HFD feeding. eWAT and liver were harvested and total RNA and protein were extracted. Real-time PCR analysis for the macrophage marker genes CD68 and F4/80; cytokines TNFα, IL-1β, IL-6, and IL10 in eWAT (a) and liver (b). Levels of mRNA in (a) and (b) are expressed as a ratio of HFD to NCD within the same genotype to accentuate changes associated with HFD feeding. Western-blot analysis was performed for p-JNK and IκBα expression in HFD-fed mice in eWAT (c) and liver (d). Transcript levels in eWAT were determined for the adipokines, leptin (e) and adiponectin (f). PCR data in (a), (b), (e) and (f) were normalized against the amount of B2M; western-blot data in (c) and (d) were normalized to β-actin. n=8–12 for each group, *P<0.05 and **P<0.001 by one-way ANOVA with Tukey’s post hoc comparison.
In the liver, transcripts for CD68, TNFα, IL1β and IL6 were increased by HFD in all genotypes (Figure 4b, data shown as HFD/NCD). However, the increase in CD68, TNFα and IL1β was attenuated in the liver of AhR−/− and AhR+/− mice compared with WT when all animals were on the HFD (Figure 4b). F4/80 levels were only increased by HFD in the liver of WT mice. The anti-inflammatory cytokine, IL10, was actually suppressed by HFD feeding in the liver of WT mice, but not in AhR-deficient mice fed HFD (Figure 4b).
JNK phosphorylation was largely reduced in eWAT and slightly decreased in the liver of AhR−/− mice (Figures 4c and d). Inhibitor κB kinase can phosphorylate and degrade its inhibitor, IκBα, leading to activation of nuclear factor κB. AhR−/− mice had much higher IκBα expression in eWAT and liver (Figures 4c and d). Increased IκBα may stabilize nuclear factor κB and lead to the subsequent decreased synthesis of cytokines and inflammation in AhR−/− mice.
HFD increased leptin transcripts, as well as serum leptin, in all three genotypes when compared with mice fed the NCD (Figure 4e and Table 1). Although overall serum leptin concentrations were significantly lower in NCD than in HFD for all groups, the increase in serum leptin following HFD feeding was significantly less for AhR−/− and AhR+/− mice compared with WT after HFD feeding (Table 1). This difference likely results from the substantially reduced amount of eWAT in the AhR-deficient mice.
HFD decreased adiponectin transcript levels only in WT mice (Figure 4f). However, serum adiponectin was not lower in HFD-fed WT mice (Table 1), perhaps because of the increase in eWAT. HFD did not change adiponectin transcript levels in the AhR−/− and AhR+/− mice compared with genotype-matched NCD controls. However, AhR−/− and AhR+/− mice exhibited higher serum adiponectin (Table 1) and higher adiponectin transcript levels (Figure 4f) compared with WT mice after 14 weeks on the HFD. Decreased inflammation and increased adiponectin in AhR−/− and AhR+/− mice may be a cause of the observed decrease in HFD-induced insulin resistance. Adiponectin can be anti-inflammatory and contribute to insulin sensitization, as well as modulate energy balance through effects on components of the mitochondrial respiratory chain.20
AhR deficiency increases energy expenditure
To evaluate physiological mechanisms of decreased HFD-induced obesity in the AhR-deficient mice, food intake and physical activity were measured at 6–7 weeks after initiation of the experiment, and indirect calorimetry was performed to measure energy expenditure. These experiments were performed at weeks 6–7 to minimize the effects that might be caused by differences in adiposity and body weight, which were not observed until week 8. Differences in body weight among WT, AhR−/− and AhR+/− mice were not significantly different at weeks 6–7 (Figure 1a). Because AhR+/− and AhR−/− are not different, and we were limited by the number of metabolic chambers available for these studies, we used only AhR+/− mice for these experiments.
Energy expenditure for WT and AhR+/− mice was similar on NCD (Figure 5a). However, AhR+/− mice fed HFD had higher VO2, VCO2 (Table 2) and energy expenditure than WT mice on the same diet (Figure 5b), although the RQ was not different between genotypes on the same diet (Table 2). AhR+/− mice fed HFD had higher energy expenditure per hour, normalized for weight, than either WT animals fed the same diet or AhR+/− fed the NCD (Figure 5c). Food intake and activity in WT and AhR+/− mice were very similar to each other in both diet conditions (Figures 5d and e). These data suggest that AhR deficiency may increase metabolic rate under HFD conditions, and thereby protect against diet-induced adiposity.

AhR ablation and attenuation increases energy expenditure. WT (n=8) and AhR+/− (n=8) mice were fed NCD or HFD for 6 weeks. (a, b) Hourly energy expenditure over a 24-h period was calculated from VO2 and VCO2 using the equation described in the Materials and methods section based on the Columbus Instruments Comprehensive Lab Animal Monitoring System (CLAMS) (b). (c) Summary of 24 h energy expenditure. Caloric intake, as calculated by food intake per mouse per week × 5.2 kcal g−1 (d), and total locomotor activity (e) were monitored for 1 weeks (weeks 6–7 of the HFD feeding). *P<0.05, one-way ANOVA with Tukey’s post hoc comparison.
AhR deficiency increases levels of thermogenic genes in BAT and β-oxidation genes in skeletal muscle
BAT dissipates energy expenditure though production of heat, and skeletal muscle specializes in energy expenditure through regulation of fatty acid β-oxidation. The thermogenic gene, Ucp1, was significantly higher in BAT of AhR+/− mice compared with WT mice after 7 weeks of HFD feeding; moreover, key upstream regulators of Ucp1, PPARγ coactivator 1α (PGC1α) and PR domain containing 16 (PRDM16), were also increased in the BAT of AhR+/− mice (Figure 6a, data shown as HFD/NCD). Importantly, levels of these genes were not different between WT and AhR+/− when mice were fed the NCD. HFD feeding increased their levels only in the BAT of AhR+/− mice and produced no effect on their levels in WT mice (Figure 6a).

Gene profiles for BAT and muscle fatty acid β-oxidation. WT and AhR+/− mice fed HFD or NCD for 6 weeks were fasted overnight and killed. BAT and muscle were harvested and total RNA was extracted. Real-time PCR measured transcripts of Ucp1, Pgc1α and Prdm16 in BAT (a) and transcripts of Pparδ, Pgc1α, Aco, Cpt1b Ucp2 and Ucp3 in muscle (b). n=8 for each group. Data are presented as a ratio of HFD/NCD to accentuate changes associated with HFD feeding. *P<0.05, **P<0.001, one-way ANOVA with Tukey’s post hoc comparison.
In skeletal muscle, the fatty acid β-oxidation genes Aco and Cpt1b, and the mitochondrial uncoupling proteins Ucp2 and Ucp3 were significantly higher in AhR+/− mice, consistent with the increase in levels of their transcriptional regulators, PPARδ and PGC1α (Figure 6b, data shown as HFD/NCD). For the most part, this difference resulted from a downregulation of these genes in the WT mice fed the HFD. There were no differences in levels of these genes between the genotypes in mice on the NCD (data not shown). The one exception was Ucp2, which was upregulated in skeletal muscle of both WT and AhR+/− on the HFD (Figure 6b). Increased expression of thermogenic genes in BAT and mitochondrial β-oxidation genes in muscle is consistent with the notion of increased energy expenditure in the AhR+/− mice.
Discussion
AhR is important in the development of toxicant-induced obesity, insulin resistance, hepatic steatosis, metabolic syndrome and type 2 diabetes.14, 21, 22 However, whether AhR contributes to HFD-induced metabolic dysfunction remains unclear. The present study, consistent with previous observations15, 23 reveals a novel role for AhR in fat metabolism and glucose homeostasis. The lean phenotype in the AhR-deficient mice may primarily be due to an increase in energy expenditure, resulting from increased metabolism in BAT and skeletal muscle, independent of food intake and activity that specifically occurs when these animals are challenged with the HFD. The combination of increased BAT and skeletal muscle metabolism and reduced transfer of lipids from the blood to the liver in the AhR-deficient mice may offset, and thus serum triglycerides were not different among the groups of mice. Furthermore, AhR deficiency resulted in decreased fed glucose and fasted insulin, and better insulin responsiveness compared with WT controls. These results suggest that AhR deficiency protects against HFD-induced obesity, insulin resistance and hepatic steatosis.
Obesity can be characterized as a chronic low-grade inflammatory disease. The chronic presence of excess nutrients such as fatty acids can stimulate an inflammatory response by activating toll-like receptors, leading to activation of certain kinase pathways, such as JNK and inhibitor κB kinase, to promote transcription of inflammatory mediators and consequently induce chronic inflammation.18, 19 Immune cells, including macrophages, T cells, B cells, neutrophils and mast cells, are involved in the HFD-triggered pathogenesis of obesity, insulin resistance and dyslipidemia.24, 25, 26, 27, 28 AhR, as an essential mediator of the innate immune system, regulates mast cell differentiation, growth and infiltration;29 genetic deficiency of mast cells or their pharmacological stabilization reduces HFD-induced obesity and diabetes in mice.26
To explore the effects of AhR deficiency on HFD-induced inflammation and metabolic dysfunction, we examined several important inflammatory genes. After 14 weeks of HFD feeding, the two AhR-deficient mouse strains displayed prominent differences in inflammatory genes compared with WT. AhR−/− and AhR+/− mice developed reduced transcript levels of key pro-inflammatory genes, yet elevated transcript levels of the anti-inflammatory gene, IL-10 under these conditions. However, the fact that the AhR−/− and AhR+/− mice fed HFD were significantly leaner than WT suggests that increased inflammation and the resulting insulin resistance and hyperinsulinemia in HFD-fed WT may be secondary effects resulting from a phenotype of increased abdominal adiposity.
PPARα, as a fat metabolism sensor, is central to the control of mitochondrial fatty acid β-oxidation in the liver.30 PPARα expression was increased in the liver of HFD-fed mice as an adaptive response to attenuate fat accumulation in the liver.31 In a previous study, we demonstrated that PPARα is regulated by AhR.24 Thus, it is not surprising that AhR deficiency prevents HFD-induced PPARα expression. It therefore seems logical that PPARα target genes, including CPT1b and ACO, key enzymes in the regulation of fatty acid β-oxidation, are also decreased after HFD feeding in AhR-deficient animals compared with WT.
Fatty acid translocase/CD36, which is a target gene of both PPARα and AhR,22 is also lower in AhR-deficient mice compared with WT mice after 14 weeks of HFD feeding, which may provide the primary explanation for protection against HFD-induced hepatic steatosis; decreased levels of CD36 would reduce transfer of fat from the blood into the liver. In addition, the regulators of liver lipogenesis, FAS and ACC, were not increased by HFD in AhR-deficient mice as they were in WT controls. This may be a consequence of improved insulin sensitivity in the two strains of AhR-deficient mice. These data suggest that the protection from hepatic steatosis by AhR deficiency may result from decreased uptake of fatty lipids, consistent with previous reports that demonstrated increased mobilization of lipids from the blood is a key for the development of hepatic steatosis, while the increased de novo synthesis of fat is compensated for by increased fatty acid β-oxidation.32
AhR-deficient mice fed HFD expend more energy, despite unchanged activity and food intake, than WT mice under the same dietary conditions. This increased energy consumption could result from physiological differences in BAT and skeletal muscle in AhR+/− mice fed HFD. BAT dissipates chemical energy in the form of heat as a defense against obesity through activity of the gene UCP1. Transcriptional regulators PGC1α and PRDM16 determine brown adipocyte development and function.33, 34 In this study, we found that Ucp1 transcripts are higher in AhR+/− mice compared with WT mice. In addition, the transcripts of Pgc1α and Prdm16 are higher in AhR+/− mice, suggesting that increased heat production in AhR+/− mice may be at least partially explained by increased activity of BAT. Additionally, AhR+/− mice also show increased β-oxidation in skeletal muscle, the major site for using free fatty acids as fuel source, which may also contribute to their higher energy expenditure. β-Oxidation was assessed by examining levels of key enzymes, including CPT1b and ACO, as well as mitochondrial uncoupling proteins UCP2 and UCP3. The results indicate higher expression of all of these genes in AhR+/− mice, as well as increased levels of related transcription factors, PPARδ and PGC1α. Thus, although the mechanism by which AhR regulates PRDM16, PPARδ and PGC1α remains a mystery, interaction of AhR with other transcription factors may alter energy expenditure and contribute to HFD-induced pathology including obesity, insulin resistance and hepatic steatosis.
When considering weight gain and energy expenditure, absorption of nutrients by the gut may also be an important contributory factor. AhR is widely expressed including in the large and small intestine. However, a previous study found that AhR does not affect energy absorption by the intestine; absorption of nutrients in animals that express a low affinity AhR is not different from animals with a high affinity AhR.15 Therefore, this study did not investigate absorption of nutrients by the gut. Gross observation of feces produced by the various strains of mice under NCD or HFD conditions did not reveal any obvious differences among the genotypes. Further experimentation is required to discern whether AhR deficiency might alter absorption of nutrients by the gut.
Other physiological mechanisms contributing to the leanness phenotype in AhR-deficient mice require further investigation. Because AhR may be activated by dietary components including fats and fat derivatives,35 components of HFD could activate AhR leading to altered transcription of target genes related to energy metabolism, resulting in decreased energy metabolism in the HFD-fed WT mice. However, if this is the case, the downstream effects of AhR activation are likely mediated by mechanisms that are non-classical, as the prototypical AhR target genes, including Cyp1a1, were not increased in HFD-fed WT mice in this study (data not shown).
It is also intriguing that AhR+/− mice that express approximately 30% of the AhR levels compared with the WT seem better protected than the AhR−/− mice. The reasons for this are not entirely clear. However, AhR−/− mice have several developmental defects including some liver abnormalities that might alter systemic metabolism, especially as animals age. Whether these effects might compromise the lean phenotype remians currently unknown. Furthermore, there is ample evidence in the literature to demonstrate that AhR can either protect from or contribute to pathologies such as cancer and inflammatory disease.36, 37, 38 This remains controversial. Thus, one might argue that the AhR+/− mice are a better model for these studies; there may be a ‘dose effect’ in the AhR heterozygote mice, where a lower level of expression prevents the detrimental developmental effects and preserves certain physiological functions, including the ability to process xenobiotics. AhR underexpression then prevents the effects of overactivation that may occur due to either excessive nutrients or the presence of an AhR agonist in the HFD. Notably, AhR−/− mice have improved insulin sensitivity when on NCD.23
Alternatively, AhR−/− and AhR+/− mice may develop higher energy expenditure due to alteration of the sympathetic nervous system, which was not explored in this investigation. To distinguish these possible mechanisms, tetracycline-inducible, Cre/loxp recombinase systems could be utilized to develop tissue-specific AhR-deficiency models to assess whether the lean phenotype results from AhR redundancy itself or developmental defects as a result of early AhR reduction. Also conditional knockout mice can discern the relative contributions of various tissues to the leanness phenotype.
Overall this study identifies AhR deficiency as a factor that can protect from the detrimental metabolic effects of HFD feeding in mice. AhR deficiency prevented weight gain and abdominal obesity, most likely through increased activity of BAT and increased β-oxidation in skeletal muscle, resulting in a lean phenotype. Leanness was likely the cause for protection from increased inflammation and insulin resistance. Reduced uptake of fat into the liver in the AhR-deficient mice protected from HFD-induced hepatic steatosis. Perhaps the most intriguing finding is that AhR knockdown (AhR+/−) was just as effective as AhR ablation (AhR−/−) in providing metabolic protection. If suppressing AhR activity, or at least preventing its overactivation, can enhance metabolic function to prevent obesity and its sequelae, then the AhR and its downstream signaling pathways may be novel targets for the development of pharmaceuticals, independent of food intake, to combat obesity and diabetes.
References
Ogden CL, Carroll MD, Kit BK, Flegal KM . Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA 2014; 311: 806–814.
Wright SM, Aronne LJ . Causes of obesity. Abdom Imaging 2012; 37: 730–732.
Berger J, Moller DE . The mechanisms of action of PPARs. Annu Rev Med 2002; 53: 409–435.
Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H et al. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003; 113: 159–170.
He J, Gao J, Xu M, Ren S, Stefanovic-Racic M, O'Doherty RM et al. PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes 2013; 62: 1876–1887.
Spruiell K, Richardson RM, Cullen JM, Awumey EM, Gonzalez FJ, Gyamfi MA . Role of pregnane X receptor in obesity and glucose homeostasis in male mice. J Biol Chem 2014; 289: 3244–3261.
Henriksen GL, Ketchum NS, Michalek JE, Swaby JA . Serum dioxin and diabetes mellitus in veterans of Operation Ranch Hand. Epidemiology 1997; 8: 252–258.
Fujiyoshi PT, Michalek JE, Matsumura F . Molecular epidemiologic evidence for diabetogenic effects of dioxin exposure in U.S. Air force veterans of the Vietnam war. Environ Health Perspect 2006; 114: 1677–1683.
Warner M, Mocarelli P, Brambilla P, Wesselink A, Samuels S, Signorini S et al. Diabetes, metabolic syndrome, and obesity in relation to serum dioxin concentrations: The Seveso Women's Health Study. Environ Health Perspect 2013; 121: 906–911.
Taylor KW, Novak RF, Anderson HA, Birnbaum LS, Blystone C, Devito M et al. Evaluation of the association between persistent organic pollutants (POPs) and diabetes in epidemiological studies: a national toxicology program workshop review. Environ Health Perspect 2013; 121: 774–783.
Lee HK . Mitochondrial dysfunction and insulin resistance: the contribution of dioxin-like substances. Diabetes Metab J 2011; 35: 207–215.
Nishiumi S, Yoshida M, Azuma T, Yoshida K, Ashida H . 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs an insulin signaling pathway through the induction of tumor necrosis factor-alpha in adipocytes. Toxicol Sci 2010; 115: 482–491.
He J, Hu B, Shi X, Weidert ER, Lu P, Xu M et al. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol Cell Biol 2013; 33: 2047–2055.
Arsenescu V, Arsenescu RI, King V, Swanson H, Cassis LA . Polychlorinated biphenyl-77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ Health Perspect 2008; 116: 761–768.
Kerley-Hamilton JS, Trask HW, Ridley CJ, Dufour E, Ringelberg CS, Nurinova N et al. Obesity is mediated by differential aryl hydrocarbon receptor signaling in mice fed a Western diet. Environ Health Perspect 2012; 120: 1252–1259.
Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA . Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci USA 1996; 93: 6731–6736.
Ueki K, Yballe CM, Brachmann SM, Vicent D, Watt JM, Kahn CR et al. Increased insulin sensitivity in mice lacking p85beta subunit of phosphoinositide 3-kinase. Proc Natl Acad Sci USA 2002; 99: 419–424.
Olefsky JM, Glass CK . Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010; 72: 219–246.
Gregor MF, Hotamisligil GS . Inflammatory mechanisms in obesity. Annu Rev Immunol 2011; 29: 415–445.
Tarantino G, Finelli C . What about non-alcoholic fatty liver disease as a new criterion to define metabolic syndrome? World J Gastroenterol 2013; 19: 3375–3384.
Ruzzin J, Petersen R, Meugnier E, Madsen L, Lock EJ, Lillefosse H et al. Persistent organic pollutant exposure leads to insulin resistance syndrome. Environ Health Perspect 2010; 118: 465–471.
Lee JH, Wada T, Febbraio M, He J, Matsubara T, Lee MJ et al. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 2010; 139: 653–663.
Wang C, Xu CX, Krager SL, Bottum KM, Liao DF, Tischkau SA . Aryl hydrocarbon receptor deficiency enhances insulin sensitivity and reduces PPAR-alpha pathway activity in mice. Environ Health Perspect 2011; 119: 1739–1744.
Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009; 15: 914–920.
Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 2009; 15: 921–929.
Liu J, Divoux A, Sun J, Zhang J, Clement K, Glickman JN et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med 2009; 15: 940–945.
Talukdar S, Oh da Y, Bandyopadhyay G, Li D, Xu J, McNelis J et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 2012; 18: 1407–1412.
Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 2011; 17: 610–617.
Zhou Y, Tung HY, Tsai YM, Hsu SC, Chang HW, Kawasaki H et al. Aryl hydrocarbon receptor controls murine mast cell homeostasis. Blood 2013; 121: 3195–3204.
Ferre P . The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes 2004; 53: S43–S50.
Lee Y, Wang MY, Kakuma T, Wang ZW, Babcock E, McCorkle K et al. Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J Biol Chem 2001; 276: 5629–5635.
Chakravarthy MV, Pan Z, Zhu Y, Tordjman K, Schneider JG, Coleman T et al. ‘New’ hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab 2005; 1: 309–322.
Kajimura S, Seale P, Spiegelman BM . Transcriptional control of brown fat development. Cell Metab 2010; 11: 257–262.
Kajimura S, Saito M . A new era in brown adipose tissue biology: molecular control of brown fat development and energy homeostasis. Annu Rev Physiol 2014; 76: 225–249.
McMillan BJ, Bradfield CA . The aryl hydrocarbon receptor is activated by modified low-density lipoprotein. Proc Natl Acad Sci USA 2007; 104: 1412–1417.
Julliard W, Fechner JH, Mezrich JD . The aryl hydrocarbon receptor meets immunology: friend or foe? A little of both. Front Immunol 2014; 5: 458.
Murray IA, Patterson AD, Perdew GH . Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer 2014; 14: 801–814.
Wang C, Zhang ZM, Xu CX, Tischkau SA . Interplay between Dioxin-mediated signaling and circadian clock: a possible determinant in metabolic homeostasis. Int J Mol Sci 2014; 15: 11700–11712.
Acknowledgements
We gratefully acknowledge Dr Andrzej Bartke and Cristal Hill (Southern Illinois University School of Medicine Geriatrics Research Laboratory) for helping the indirect calorimetry testing and analysis. This work was supported by grant from NIEHS (ES017774) to SAT.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on International Journal of Obesity website
Supplementary information
Rights and permissions
About this article

Cite this article
Xu, CX., Wang, C., Zhang, ZM. et al. Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. Int J Obes 39, 1300–1309 (2015). https://doi.org/10.1038/ijo.2015.63
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2015.63
- Springer Nature Limited
This article is cited by
-
Type 1 diabetes and diet-induced obesity predispose C57BL/6J mice to PM2.5-induced lung injury: a comparative study
Particle and Fibre Toxicology (2023)
-
Plasma aryl hydrocarbon receptor associated with epicardial adipose tissue in men: a cross-sectional study
Diabetology & Metabolic Syndrome (2023)
-
Total parenteral nutrition impairs glucose metabolism by modifying the gut microbiome
Nature Metabolism (2023)
-
Knockdown of IL4I1 Improved High Glucose-evoked Insulin Resistance in HepG2 Cells by Alleviating Inflammation and Lipotoxicity Through AHR Activation
Applied Biochemistry and Biotechnology (2023)
-
Insight into the physiological and pathological roles of the aryl hydrocarbon receptor pathway in glucose homeostasis, insulin resistance, and diabetes development
Cellular & Molecular Biology Letters (2022)