
Abstract
Background/objectives
Obesity is a global epidemic and the underlying basis for numerous comorbidities. We report that the aryl hydrocarbon receptor (AHR) plays a key role in the metabolism of obesity. The AHR is a promiscuous, ligand-activated nuclear receptor primarily known for regulating genes involved in xenobiotic metabolism and T cell polarization. The aims of the work reported here were to understand the underlying mechanism of AHR-based obesity and to determine whether inhibition of AHR activity would reverse obesity.
Methods
Mice were fed control (low fat) and Western (high fat) diets with and without the AHR antagonist alpha-naphthoflavone (aNF). Gene expression of identified AHR-regulated genes from liver and adipose tissue was characterized. To determine the role of the AHR in obesity reversal, selected mice in control and Western diet regimens were switched at midpoint to the respective control and Western diets containing aNF, and the identified AHR-regulated genes characterized.
Results
AHR inhibition prevented obesity in mice on a 40-week diet regimen. The likely AHR-regulated and cross-regulated downstream effectors of AHR-based obesity were shown to be CYP1B1, PPARα-target genes, SCD1, and SPP1 (osteopontin). Western diet caused an increase of mRNA and protein expression of the Cyp1b1, Scd1, and Spp1, and PPARα-target genes in the liver, and inhibition of the AHR maintained expression of these genes near control levels. The body weight of obese mice on Western diet switched to Western diet containing aNF decreased to that of mice on control diet concurrently with a reduction in the expression of liver CYP1B1, PPARα-target genes, SCD1, and SPP1. AHR inhibition prevented hypertrophy and hyperplasia in visceral adipose tissue and limited expression levels of CYP1B1 and SPP1 to that of mice on control diet.
Conclusions
AHR inhibition prevents and reverses obesity by likely reducing liver expression of the Cyp1b1, Scd1, Spp1, and PPARα-target genes; and the AHR is a potentially potent therapeutic target for the treatment and prevention of obesity and linked diseases.
Similar content being viewed by others


Introduction
Childhood and adult obesity are an ever-growing epidemic, in which one in six children ages 2–19 are obese, and over two-thirds of adults are overweight or obese [1]. Obesity contributes to numerous medical maladies including inflammation [2, 3], diabetes and metabolic syndrome [4], cardiovascular disease [5], and cancer [6] as well as incurring huge medical care costs [7]. The causes of obesity are based on complex gene–environment interactions dependent on lifestyle factors [8], and many identified genetic loci are associated with the risk of obesity [9, 10]. A prominent lifestyle choice associated with obesity is the consumption of the high-caloric, high-fat Western diet [11].
We have accumulated evidence showing that the aryl hydrocarbon receptor (AHR) plays a key role in obesity [12,13,14]. The AHR is a promiscuous, ligand-activated nuclear receptor [15, 16] primarily known for regulating genes involved in xenobiotic metabolism [17, 18] and in innate and adaptive immunity [19,20,21]. We showed in C57BL/6J (B6) mice that obesity and adiposity were significantly reduced [12, 13] and that liver steatosis improved to near control levels [13] when fed Western diet containing the mechanistically different AHR antagonists α-naphthoflavone (aNF) or CH-223191 [22,23,24]. Although there were some minor differences, we also showed that inhibition of the AHR was effective in preventing obesity and liver steatosis in male and female mice [12].
Based on studies with B6 mice and a mouse hepatocyte cell line [13], we found that Western diet-derived oxidized low density lipoproteins induced toll-like receptor 2/4 signaling to initiate downstream signaling events through NFκB that ultimately activated indoleamine 2,3-dioxygenase 1 (IDO1). IDO1 metabolizes tryptophan (Trp) to kynurenine (Kyn), a known AHR agonist [19, 25,26,27]. We proposed that the sustained increased levels of Kyn-induced, AHR-directed transcription [13], including the canonical AHR-regulated Cyp1b1 gene [12], caused diet-induced obesity. In turn, obesity was prevented by AHR inhibition using the antagonist aNF or CH-223191 [12, 13].
Pharmacological and behavioral modification approaches for the treatment of obesity for the most part have been ineffectual [28]. As a result, we sought to determine the mechanism by which the Western diet-activated AHR causes diet-induced obesity in mice, and second, to determine whether inhibition of AHR signaling would be an effective means to reverse obesity. We surmised from the work reported here that the AHR may be the hub of a network of genes that includes cytochrome P450 1b1 (Cyp1b1) and other key AHR-regulated and cross-talking downstream genes, i.e., peroxisome proliferator-activated receptor alpha (Ppara), stearoyl-coA desaturase 1 (Scd1), and secreted phosphoprotein (Spp1), also known as osteopontin, that encode proteins that influence lipid metabolism, hepatic steatosis, and obesity. Lastly, we show that obese mice on Western diet, when switched to a Western diet containing the AHR inhibitor aNF, gradually lose and maintain body mass at control levels.
Materials and methods
Mice
Male B6 mice (strain C57BL/6J, stock #000664) of ~5 weeks of age were purchased from The Jackson Laboratory (Bar Harbor, ME). The mice were reared in 12-h light/dark cycles, and to minimize exposure to exogenous toxicants, cages were bedded with chemical-free shredded paper (Pure-o-cell, The Andersons Lab Bedding, Maumee, OH). Food consumption was determined midway during the diet regimens with four to eight mice from each experimental group, in which the mice were housed in cages containing a chow-measuring apparatus. Control diet contained 3.8 kcal/g, and Western diet contained 4.6 kcal/g. Food consumption and thus aNF consumption was ad libitum. We found in a dose–response experiment that chow containing 2% aNF by mass was the minimal effective dose for preventing obesity in mice fed a Western diet [13]. The calculated amount of consumed aNF and CH-223191 was ~90 mg/kg body mass/day and ~10 mg/kg body mass/day, respectively. The calculations were based on the estimated consumption of ~4.5 g of chow per mouse per day (http://www.researchdiets. com/) and on an adult mean body mass of 30 g. Diet regimens of 5-, 20-, and 40-week durations were carried out. The body mass of each mouse was measured and recorded weekly for the duration of each regimen. If obviously sick or dead, the mouse was eliminated from the study. For the IDO1/2 inhibition study, mice were given 1-methyl-L-tryptophan (1MLT; Sigma‐Aldrich, St. Louis, MO; CAS Number 21339–55–9) in their drinking water (5 mg/ml, pH 11.0) or water adjusted to pH 11.0 for the control group [29]. The study was not blinded as the different chows were color coded. We did not observe any unusual differences in lifespan or disease incidence within the confines of the studies. All animals were treated humanely following the regulations and specifications of the Dartmouth IACUC.
Diet
Mouse chow was purchased from Research Diets, Inc. (New Brunswick, NJ). The custom low-fat control diet, (D12450B) contained 20% kcal protein, 70% kcal carbohydrates (35% kcal from sucrose), 10% kcal fat (4.5% kcal from lard). The custom Western diet (D12071702) contained 20% kcal protein, 35% kcal carbohydrates (17.5% kcal from sucrose), 45% kcal fat (40% kcal from lard), and 2% cholesterol. The ingredients for the control and Western diets are listed in [12]. The diets contained no detectable phytoestrogens or xenobiotics according to the manufacturer. The aNF (CAS Number 604-59-1, ≥98% purity) and CH-223191 (CAS Number 301326-22-7) were purchased from Sigma-Aldrich. The aNF or CH-223191 was added directly to the mouse food during production and comprised 2% and 0.22%, respectively, of the total mass.
Histology
Histology procedures were carried out by the Dartmouth Pathology Shared Resource. Liver and adipose tissue samples taken at sacrifice were fixed in 10% neutral-buffered formalin. The tissue was processed, paraffin-embedded, and sectioned at a thickness of 5 µm and adhered onto glass slides. Liver tissue was stained using hematoxylin and eosin. Immunohistochemistry for CYP1B1 (Abcam, Cambridge, MA) was performed using a Leica Bond Rx (Leica Biosystems, Buffalo Grove, IL). Four sections from each liver were examined, and each section was at least 30 µm apart with representative sections selected. The sectioned tissue was examined at 40× magnification using an Olympus BX51 microscope (Waltham, MA). Images were generated using identical settings with a QImaging Micro Publisher 5.0 RTV camera (Surrey, British Columbia, Canada).
Determination of arachidonic acid and aNF concentrations
At sacrifice, plasma was obtained by centrifugation from collected blood samples and stored at −80 °C. High-performance liquid chromatography (HPLC) was carried out by the Dartmouth Clinical Pharmacology Shared Resource. HPLC-grade acetonitrile was purchased from MilliporeSigma (Burlington, MA). Ultrapure water was provided using a Barnstead purification system. C57BL/6 plasma was purchased from Innovative Research (Novi, MI). For arachidonic acid, arachidonic acid-d8 (AA-d8) was used as an internal standard. Arachidonic acid and arachidonic acid-d8 stocks were dissolved in DMSO and stored at −4 °C, and working dilutions were made daily in 70% acetonitrile. Calibrators and quality control solutions were made in 45 mg/ml human albumin in phosphate-buffered saline (PBS) and processed as plasma samples. Plasma samples (50 μl) were processed by adding 2.5 μl of a 100 μg/ml internal standard (AA-d8) and 100 μl of 3% ammonium hydroxide and briefly vortexed and centrifuged for 30 s at 6000 × g. Oasis MAX SPE cartridges were used to purify samples according to manufacturer instructions, and eluent was collected in 600 μl 3% formic acid in acetonitrile. Samples were dried under nitrogen at 45 °C and suspended in 50 μl 70% acetonitrile for injection onto the LC-MS/MS system. HPLC separation was achieved on a Dionex Ultimate 3000 HPLC system with a Phenomenex Luna Omega C18 2.1 × 50 mm, 1.6 micron column with 2.1 × 10 mm C18 guard at 40 °C. Isocratic separation utilized 30% 5 mM ammonium acetate and 70% acetonitrile with a flow rate of 0.3 μl/min. A TSQ Vantage mass spectrometer was operated in negative ion mode with a collision pressure of 1.4 mTorr to measure arachidonic acid (303.206→259.26 m/z) and arachidonic acid-d8 (311.255→267.29 m/z) with collision energies of 16 and 15, respectively. The ESI source was operated with a spray voltage of 500 V, vaporizer temperature of 409 °C, capillary temperature of 257 °C, and sheath and auxiliary gases at 30 and 5 arbitrary units, respectively. The quantitative range was 0.2–50 μg/ml with inter- and intraday accuracies of 90–100% and 84–105%, respectively, across three quality control levels.
HPLC methods for aNF detection [30] utilized aNF (7,8-benzoflavone) and 8-methylflavone purchased from Indofine Chemical Company (Hillsborough, NJ). Protein precipitation was performed by adding 80 µl of acetonitrile to a 40 µl-plasma sample. Liver samples were sonicated in PBS, and the cellular debris was pelleted by centrifugation. Four microliters of a 100 µg/ml 8-methylflavone solution was added as the internal standard. The samples were vortexed for 1 min and centrifuged at 6000 × g for 10 min. Thirty microliters of supernatant was injected into the HPLC system for analysis.
The HPLC analysis was performed on the Dionex Ultimate 3000 system described above. The workstation used ChromeleonTM and XCaliburTM softwares to conduct the experiments. The separation was performed using a Luna C18 column (100 × 4.6 mm, 3-µm particle size). The mobile phase was 80/20 (v/v) acetonitrile/water and was delivered isocratically with a flow rate of 1 ml/min. The UV absorbance was measured at 280 nm. The internal standard 8-methylflavone and aNF appeared on the chromatograph at ~2.37 and 2.88 min, respectively, with no interfering peaks. The lower limit of quantification of aNF was 1 μg/ml. The standard curve was linear over the concentration range of 0.01–20 µg/ml with a regression coefficient >0.99. Recovery from the sample extraction was 102%. Quality control samples were included at the concentrations of 0.03, 0.3, and 3 µg/ml. The inter- and intra-day accuracy was 97–112%. The precision expressed as CV% was 4.5–9.3%.
Western blotting
Liver and adipose tissue were homogenized on ice using RIPA buffer. Proteins were pelleted by centrifugation and concentrations measured. Proteins were resolved by SDS-PAGE under reduced conditions and transferred to polyvinylidene difluoride membrane (EMD Millipore, Burlington, MA). Primary rabbit antibodies to CYP1B1, CYP4A, ELOVL5, FADS1, and FGF21 (Abcam, Cambridge, MA); and to SCD1 and Vinculin (Cell Signaling Technology, Danvers, MA) were incubated with the membrane-bound proteins overnight. Secondary antibodies (fluorescence or HRP-conjugated; Cell Signaling Technologies, Danvers, MA) were detected either on the Licor Odyssey CLx (Li-Cor Biosciences, Lincoln, NE) or by electrochemiluminescence (ECL, Thermofisher, Waltham, MA). Densitometry readings of bands from Western blots were normalized to viniculin.
Enzyme-linked immunosorbent assays
Blood was collected from mice via cardiac puncture at termination. Plasma was separated via Ficoll (GE Life Sciences, Pittsburgh, PA) gradients and assayed using an ELISA for SPP1 (R&D Systems, Bio-Techne, Minneapolis, MN).
Nanostring
Total RNA isolated from liver and adipose tissue (100 ng) was used as input for the nCounter PlexSet system (Nanostring Technologies, Seattle, WA) which can digitally detect and count specific mRNAs in a multiplexed format. A custom panel, including three housekeeping genes (GusB, Rpl19, and Vcl), was designed and implemented.
Microarrays
Three to four biological replicates per experimental condition were carried out for the microarray studies. Total RNA was isolated and purified from mouse liver (sliced into small pieces) and adipose tissue homogenized in Tri-Reagent (Sigma-Aldrich, St. Louis, MO). RNA purity, quantity, and quality were determined using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). The mRNA gene expression microarray experiments were carried out by the Dartmouth Genomics & Molecular Biology Shared Resource using the Mouse 430A or Mouse 430A 2.0 Arrays (Affymetrix, Santa Clara, CA). Approximately 100 ng of total RNA per sample was labeled for each array using the GeneChip 3′ IVT Reagent Kit (Applied Biosystems, Foster City, CA). Labeled RNA was incubated with the arrays and stained using the corresponding kit on the GeneChip Fluidics Station. The arrays were scanned using the Affymetrix GeneChip Scanner 3000.
Adipocyte tissue proliferation and growth
Adipocyte growth and proliferation was examined by an adipocyte-specific pulse-chase experiment in a B6 mouse model using a tamoxifen-inducible, fused adiponectin-cre estrogen receptor (Adiponectin-creER) with a dual fluorescent reporter vector [31, 32]. Induction of cre recombinase causes the fluorescent reporter to undergo an expression switch from the Tomato (mT, red fluorescence) tag, which locates to the plasma membrane, to the plasma membrane-targeted green fluorescent protein (GFP) (mG, green fluorescence) at ~95% recombination efficiency [32], which identifies cre-expressing cells [33, 34]. To observe adipocyte proliferation, daily IP injections of tamoxifen in vegetable oil was given to 8-week old male mice at 50 mg/kg for 5 days followed by a 1 week recovery (pulse), and then placed on a control or Western ± aNF diet for 8 weeks (chase). At termination, visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT) were sectioned and prepared for fluorescence microscopy.
To determine adipocyte size, an adipocyte area was determined using the ImageJ plugin Adiposoft. Representative images at 40× magnification were taken of sectioned VAT stained with hematoxylin and eosin (three images per animal, nine images per experimental group). Adiposoft was used on “Auto” mode to detect adipocytes with a diameter >20 μm and <100 μm. Graphpad Prism was used to identify and exclude outliers and in carrying out a one-way ANOVA with multiple comparisons.
Statistical analyses
Standard student t test was employed to determine p values in the mouse studies. The raw data from the Nanostring studies were processed through nSolver software, background corrected, and normalized first by using the geometric mean of the top three positive controls followed by a second normalization based on expression of the housekeeping genes. Class comparisons were generated through 1-way ANOVA analysis using Partek Genomics Suite software (Partek Incorporated, St. Louis, MO). Microarray analyses were performed using BRB-Array Tools Version 4.5 (Biometric Research Branch of the Division of Cancer Treatment & Diagnosis of the National Cancer Institute under the direction of Dr. Richard Simon). Probe set summaries from the imported CEL files were computed using the RMA method, which applies a background correction on the PM (Perfect Match) data and a quantile normalization, and summarizes the probe set information using Tukey’s median polish algorithm [35]. Differentially expressed genes were identified using a random-variance t test [36] and multiple testing correction methods [37].
Results
CYP1B1 expression in AHR-based obesity
We have shown that AHR inhibition effectively prevents obesity and fatty liver in B6 females and males in diet regimens of 20- and 26-week durations [12, 13]. We wanted to investigate whether AHR inhibition by aNF was an effective obesity preventative in a diet regimen of longer duration and to determine the mechanism by which the blocking of AHR signaling prevented Western diet-induced obesity. Male B6 mice were fed control and Western diets with and without aNF (Fig. 1a, b). As observed in the shorter-term diet regimens [12, 13], total body mass gain of mice fed Western + aNF diet to that of mice fed Western diet alone was significantly less and near the mean body mass of mice fed control diet. There were no significant differences in the amount of food consumed for a given diet ± aNF (Fig. 1c). Based on differential gene expression profiles from microarray data of liver from B6 mice fed a Western diet to those fed a control diet, the predicted active cellular pathways were associated with increased inflammation and decreased lipid metabolism (Table S1). In turn, in liver of mice fed Western + aNF diet to that of mice fed Western diet alone, the predicted cellular pathways were redirected to reduced inflammation and oxidative phosphorylation and increased levels of protein synthesis and transport (Table S2).

a Male B6 mice (n = 4 mice per experimental group) were fed control and Western diets with and without aNF (~90 mg/kg/day) ad libitum for 40 weeks beginning at weaning. b Mean body mass gain of each experimental group at week 40. c Consumption of food per mouse for each experimental group (n = 4). d Liver mass/body mass ratios were determined by weighing at the conclusion of the 40-week diet regimen. e Representative liver sections (n = 3) stained with hematoxylin and eosin (40× magnification). f Total RNA isolated from liver was subjected to microarray analysis. g Proteins isolated from liver at termination were resolved by Western blotting. Vinculin served as a loading control (n = 3). h Representative sections of liver tissue (n = 3) stained with anti-CYP1B1 antibody at termination of the 40-week diet regimen (50× magnification) with inset (200× magnification) showing a central vein. p value: *≤0.05; **≤0.01; ***≤0.001. Error bars represent standard error of means (SEM).
In the shorter-term studies [12, 13], liver steatosis was not evident in male and female mice fed Western + aNF diet, but the aNF caused liver enlargement independent of diet. In contrast here, the longer-term diet regimen resulted in more moderate outcomes for the liver in that no significant levels of hepatomegaly was observed in mice treated with aNF fed either the control or Western diet (Fig. 1d). Second, the liver of mice fed Western diet for the 40-week diet regimen had a visibly reduced level of fat-containing vesicles per unit area (Fig. 1e, lower left) to that of livers from mice fed Western diet from the reported shorter-term diet regimens [12, 13]. Nonetheless, aNF prevented fat storage in mice fed Western diet (Fig. 1e, lower right) as had been previously observed [12, 13]. The results suggest that AHR inhibition ameliorates hepatic steatosis regardless of the duration of high-fat diet intake.
Our working hypothesis is that the AHR-CYP1B1 axis comprises the first steps in the pathway of AHR-based obesity. Knockout of the Ahr gene prevents obesity and liver steatosis [13, 38, 39] as does knockout of the Cyp1b1 gene [40,41,42]. We carried out microarray and Western blot assays with mRNA and protein isolated from liver of mice on the 40-week diet regimen. Mice on Western diet showed a significant increase in Cyp1b1 mRNA (Fig. 1f) and protein (Fig. 1g) levels relative to that of mice on control diet, while mice on the Western + aNF diet had Cyp1b1 mRNA and protein levels near or below that of the mice on control diet.
We next asked in what liver cell type(s) the AHR-CYP1B1 axis is active. Hepatocytes are the parenchymal cell type of the liver, and the hepatocyte-specific knockout of the Ahr renders female mice obesity resistant [43]. Hepatocytes in the liver are organized into hexagonal physico-functional units called lobules, where many metabolic and xenobiotic enzymes are differentially distributed along a periportal–perivenous axis [44]. Hepatocytes located in either of the two hepatic regions often carry out complementary functions, e.g., glycolysis in perivenous hepatocytes and gluconeogenesis in periportal hepatocytes [45]. The AHR is known to be expressed and active in perivenous hepatocytes of the liver lobule [46], and CYP1B1 is expressed in the same hepatocyte subset after chronic 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure [47]. Liver sections from mice on the 40-week regimen of control and Western diets with and without aNF were stained using anti-CYP1B1 antibody (Fig. 1h). CYP1B1 staining in liver sections from mice fed Western diet was markedly increased in hepatocytes surrounding the central veins but diminished to near control levels in mice treated with aNF. These results show that Western diet-induced, AHR-regulated CYP1B1 expression in the liver occurs primarily in perivenous hepatocytes.
Expression of PPAR alpha-target genes is reduced by AHR inhibition
The nuclear receptor PPARα is a critical regulator of lipid metabolism in the liver and regulates genes involved in fatty acid transport and mitochondrial fatty acid β-oxidation [48]. Microarray results showed that relative to mice fed Western diet alone in a 5-week diet regimen, liver mRNA levels of numerous PPARα target genes were affected similarly by aNF and CH-223191 (Table S3), two mechanistically different acting AHR antagonists [22,23,24]. Further, 19 of 22 PPARα target genes examined behaved similarly to that of the Cyp1b1 knockout mouse [40] (exceptions: Ms4a7, Pctp, and Pdk4). Western blots of liver protein of selected PPARα target genes from mice on the 40-week diet regimen (Fig. 2) reflected the expression levels of the corresponding mRNAs. The protein levels of ELOVL5, CYP4A, FADS1, and FGF21 from mice fed chow containing aNF were decreased relative to mice fed Western diet alone. The results suggest that the AHR-CYP1B1 axis can be extended to include PPARα.

Male B6 mice were fed control and Western diets with and without aNF (~90 mg/kg/day) ad libitum for 40 weeks beginning at weaning. Proteins isolated from liver at termination were resolved by Western blotting. Vinculin served as a loading control.
Scd1 mRNA levels are decreased in liver by AHR antagonists
Mice deficient in the Scd1 gene, which encodes Stearoyl-CoA desaturase 1, the rate-limiting enzyme that reduces saturated fatty acids to monounsaturated fatty acids, are resistant to diet-induced obesity and become insulin sensitive [49, 50]. Microarrays were carried out with mRNA from liver of B6 male mice on the 5-week and 40-week diet regimens to determine whether AHR inhibition suppressed Scd1 mRNA levels. For the 5-week diet regimen (Fig. 3a), Western diet caused Scd1 mRNAs to rise nearly 11-fold relative to that of mice on control diet, whereas, mice on Western diet containing aNF or CH-223191 showed a significant decrease in Scd1 mRNA gene expression. The mouse groups on the 40-week regimen (Fig. 3b) showed much less disparity among diets, but nonetheless, AHR inhibition by aNF caused Scd1 mRNA levels to be near that of mice on control diet. A Western blot of liver protein from mice on the 40-week diet regimen showed that inhibition of AHR signaling by aNF caused a decrease in SCD1 protein levels relative to that of mice fed the corresponding diets (Fig. 3c). The results support the hypothesis that blocking AHR activity in turn decreases SCD1 expression leading at least in part to the prevention of obesity.

a Total RNA isolated from liver was analyzed using microarrays from male B6 mice (n = 4 mice per experimental group) fed control and Western diets with and without aNF (~90 mg/kg/day) or CH-223191 (~10 mg/kg/day) ad libitum for 5 weeks beginning at weaning and b from male B6 mice (n = 3 mice per experimental group) fed control and Western diets with and without aNF (~90 mg/kg/day) ad libitum for 40 weeks beginning at weaning. c Proteins isolated from liver were analyzed by Western blotting from male B6 mice fed control and Western diets with and without aNF (~90 mg/kg/day) ad libitum for 40 weeks beginning at weaning. d Plasma arachidonic acid concentrations were determined by HPLC from male B6 mice (n = 4 mice per experimental group) fed control and Western ± aNF diets (aNF at ~90 mg/kg/day) ad libitum for 10 weeks beginning at weaning. e Total RNA isolated from liver was analyzed using microarrays from male B6 mice (n = 3 mice per experimental group) fed control and Western diets with and without aNF (~90 mg/kg/day) ad libitum for 40 weeks beginning at weaning. C Control diet, W Western diet; WNF Western + aNF diet, WCH Western + CH-223191 diet. p value: *≤0.05; **≤0.01; ***≤0.001. Error bars represent SEM.
Arachidonic acid causes a decrease in both Scd1 mRNA and activity levels [51] and is a substrate of CYP1B1 [52]. To investigate whether arachidonic acid may play a role in reducing Scd1 mRNA levels, arachidonic acid plasma concentrations (Fig. 3d) and relative mRNA levels of Pla2g4a (Fig. 3e), which encodes the phospholipase that generates endogenous arachidonic acid from membranes [53], were determined. AHR inhibition by aNF caused a significant drop in arachidonic acid plasma concentrations while Pla2g4a mRNA was maintained at control levels, suggesting that arachidonic acid is not complicit in the modulation of Scd1 gene expression and SCD1 activity.
AHR inhibition causes drop in expression of the obesity biomarker osteopontin
Secreted phosphoprotein 1 (Spp1 or osteopontin) gene expression is regulated by the AHR [54], and SPP1 protein synthesis and secretion in vascular pericytes are regulated by CYP1B1 [55]. Knockout of the Spp1 gene causes obesity resistance [56] demonstrating that SPP1 participates in metabolism and is a possible participant in AHR-based obesity. We found that relative to mice fed Western diet, AHR inhibition by aNF resulted in a significant drop in mouse liver Spp1 mRNA levels (Fig. 4a) and in plasma levels of secreted SPP1 (Fig. 4b). The results are in alignment with other studies demonstrating links among the AHR, CYP1B1, and SPP1 and are consistent with a model depicting SPP1 as part of a downstream gene network regulated by the AHR [54, 55, 57].

Male B6 mice (n = 4 mice per experimental group) were fed control and Western diets with and without aNF (~90 mg/kg/day) ad libitum for 40 weeks beginning at weaning. a Total RNA isolated from liver was analyzed by fluorescence-based digital counting using Nanostring technology to determine differential Spp1 mRNA levels. b SPP1 plasma levels were measured by carrying out ELISA with anti-SPP1 antibodies. p value: *≤0.05; **≤0.01. Error bars represent SEM.
AHR inhibition in adipose tissue reduces Cyp1b1 and Spp1 but increases PPARα-target gene expression
A primary function of adipose tissue is the storage of excess fat. The Cyp1b1 gene is expressed in adipose tissue at higher levels relative to liver and relative to Cyp1a1 [42, 58]. Thus, we examined in adipose tissue the expression of the same downstream AHR-regulated genes identified in liver. As in liver, expression of CYP1B1 mRNA (Fig. S1A) and protein (Fig. S1B) levels increased in VAT of mice fed Western diet relative to control levels. Treatment with aNF caused Cyp1b1 mRNA transcript numbers and CYP1B1 protein levels to be near or below that of mice fed control diet.
In contrast to liver, most of the identified PPARα target genes in VAT, with the exceptions of Scd1 and Scd2, were unaffected by Western diet or by AHR inhibition (Table S4 and Fig. S1C). Further, the Scd1 and Scd2 genes generated results opposite to that in liver, in which Western diet produced a decrease in mRNA expression relative to control VAT, and AHR inhibition increased mRNA levels relative to that of VAT from Western diet-fed mice. The results suggest that the influence of PPARα in VAT in the context of obesity is relatively inconsequential, and that the AHR may act as a transcriptional repressor in the regulation of the Scd1 and Scd2 genes.
However, similar to what was observed in liver, microarray results showed mRNA levels of the obesity biomarker Spp1 [59] increased over 18 fold in VAT of mice fed Western diet versus control diet and plummeted to a ratio of 0.08 in mice fed Western + aNF diet to those fed Western diet alone (a differential of ~225 fold) (Fig. S1D). These results were confirmed using Nanostring fluorescence-based digital counting, in which Western diet caused a significant increase in Spp1 transcript number and AHR inhibition caused a significant reduction (Fig. S1E). The data suggest that the AHR-CYP1B1 axis in adipose tissue shares some similarity with liver, with PPARα, Scd1, and Scd2 acting as exceptions.
AHR inhibition suppresses cell proliferation and growth in visceral adipose tissue
We investigated adipocyte growth and proliferation in response to Western diet with and without aNF. We carried out an adipocyte-specific pulse-chase experiment in B6 mice using an adipocyte-specific, tamoxifen-inducible, and fused adiponectin-cre estrogen receptor vector with dual fluorescence reporters [31, 32]. Induction of cre recombinase by tamoxifen causes a switch from Tomato (red fluorescence) to GFP (green fluorescence) expression at ~95% recombination efficiency [32]. Cells with red plasma membrane indicate a proliferative state. Using fluorescence microscopy, we observed that Western diet induced cell proliferation in VAT but not in SAT, and that cell proliferation in VAT was nearly abolished by treatment with aNF (Fig. S1F). Sectioned VAT from mice of each experimental group was stained with hematoxylin and eosin (Fig. S1G) and used to quantify cell size (Fig. S1H). Western diet caused the mean diameter of adipocytes to increase 2.5-fold relative to adipocytes of mice fed control diet, and addition of aNF caused adipocytes to remain near control levels indicating that AHR inhibition prevented adipocytes from storing increased amounts of fat. As in VAT, SAT cell size also appeared to increase in size in Western diet-fed mice relative to the cell size in control mice. Whereas VAT cell size in mice fed Western + aNF diet approached that of mice on control diet, in contrast, SAT cells were larger in mice fed Western + aNF diet relative to cells from control diet-fed mice. In summary, Western diet caused hypertrophy in SAT and hypertrophy and hyperplasia in VAT. Only VAT responded to AHR inhibition by remaining the size and proliferative status of control cells.
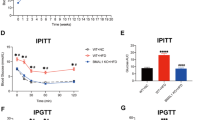
AHR inhibition reverses obesity
We have shown here and in previous studies that inhibition of the AHR prevents obesity. However, the foremost therapeutic value in blocking AHR signaling would be to use the AHR as a vehicle to treat and reverse obesity. To that end, male B6 mice were placed on a 20-week diet regimen, in which at week 10, six mice on control and Western diets were switched to control and Western diets containing aNF (Fig. 5a, b). We found that obese mice on Western diet switched to a Western + aNF diet, steadily lost and then partially gained body mass over the following 21 days to attain a body mass near that of mice on control diet throughout the regimen and remained as such for the remainder of the study. Food consumption was measured for the mice switched from Western to Western + aNF diet, and although food consumption dropped for days 3–5 post switch, consumption resumed to typical levels by post-switch day 6 (Fig. 5c).

a At week 10 (denoted by arrow) of a 20-week diet regimen, six mice on respective control (C) and Western (W) diets were switched to control + aNF (CNF) and Western + aNF (WNF) diets (~90 mg/kg/day) (n = 6 per final experimental group). Mice were fed ad libitum beginning at weaning. b Mean body mass gain of each experimental group at week 10 (n = 12) and 20 (n = 6). c Food consumption for mice (n = 4) switched from Western diet to Western + aNF diet was determined over an 18-day period during the diet switch at week 10 of the 20-week diet regimen. d Subcutaneous adipose tissue (SAT), gonadal or visceral adipose tissue (VAT), and brown adipose tissue (BAT) mass/total body mass (BM) ratios were determined by weighing at the end of the 20-week diet regimen (n = 6/experimental group). e Liver mass/total body mass (BM) ratios were determined by weighing at the end of the 20-week diet regimen (n = 6/experimental group). f Representative liver sections stained with hematoxylin and eosin at termination of the 20-week diet regimen (40× magnification). Total RNA isolated from liver at termination of the 20-week diet regimen was analyzed by microarrays to determine differential mRNA levels for Scd1 (g) and Spp1 (h). i Proteins isolated from liver at termination of the 20-week diet regimen were resolved by Western blotting. Vinculin served as a loading control (n = 3). j Plasma levels of aNF were determined by HPLC at 20-week termination from mice (n = 3) fed control, control to control + aNF, Western, and Western to Western + aNF diets, in which is shown a representative HPLC profile at week 20 of a mouse switched from Western diet to Western + aNF diet. k At week 10 of a 20-week diet regimen, six mice on Western diet were switched to control diet (W-C) (n = 6 per final experimental group). Mice were fed ad libitum beginning at weaning. l Food consumption for the experimental group switched from Western to control diet (n = 4) was determined over an 8-day period at near week 10 during the 20-week diet regimen. m Plasma arachidonic acid concentrations were determined by HPLC from male B6 mice (n = 4 mice per experimental group) fed control and Western diets with and without aNF (~90 mg/kg/day) ad libitum for 20 weeks beginning at weaning. p values: *≤0.05; **≤0.01; ***≤0.001. Error bars represent SEM.
We found that the body mass loss in the mice switched to Western + aNF diet occurred in all the fat depots (Fig. 5d). We observed that heptamegaly developed in the mice on control and Western diets containing aNF (Fig. 5e), confirming that aNF in diet regimens of shorter durations causes more severe liver enlargement in contrast to those of a longer durations (see Fig. 1d). Although hepatomegaly was evident in mice on control + aNF diet, as similarly observed in mice on the 40-week diet regimens (Fig. 1d), there was no apparent effect observed histologically (Fig. 5f). Hepatic steatosis was eliminated at the terminal 20-week timepoint in mice switched to Western + aNF diet compared with mice on Western diet alone (Fig. 5f). Liver Cyp1b1 mRNA levels were not significantly changed by treatment with aNF (data not shown) but liver Scd1 and Spp1 mRNA levels were significantly reduced by aNF exposure (Fig. 5g, h). The mRNA levels of PPARα target genes were for the most part increased by consumption of Western diet and lowered by AHR inhibition (Table S5). CYP1B1 and SCD1 protein levels rose in mice on the Western diet relative to mice on control diet, but the PPARα-regulated Cyp4a gene showed a relative decrease (Fig. 5i). Mice switched from Western diet to Western + aNF diet also displayed a modest decrease in CYP1B1 protein levels and a substantial drop in SCD1 and CYP4A protein levels. The lower CYP1B1, SCD1, CYP4A, and SPP1 protein and mRNA levels from aNF treatment (Fig. 5g–i) is consistent with our model depicting metabolism as dependent on an AHR-CYP1B1-PPARα-SCD1-SPP1 axis.
In vitro assays using rat hepatic cytosol have shown that lower concentrations of aNF antagonizes AHR signaling while concentrations of ≥10 μM exhibit AHR agonist activity [60]. We measured aNF concentrations in plasma and liver lysates to determine whether aNF was likely working in vivo in mice as an antagonist or agonist. Concentration levels of aNF were measured by HPLC at termination of the 20-week diet regimens (Fig. 5j). The aNF concentrations were not quantifiable, i.e., <1 μg/ml, in the liver lysates and in plasma from mice on the control, Western, and control to control + aNF diet regimens. In three mice from the Western to Western + aNF diet regimen, the aNF plasma concentrations were 0.999, 0.967, and 1.012 μg/ml, which is ~3.67 μM. These results indicate that aNF as administered acts as an antagonist in vivo, and suggest that the higher CYP1B1 levels in mice fed control + aNF to that of the other experimental groups (Fig. 5i) was not due to activated AHR signaling.
To determine whether the switch from Western diet to Western + aNF diet may be similar in effect as would a switch from Western diet to low-fat control diet, at week 10 of a 20-week diet regimen, six male B6 mice were switched from Western to control diet. We observed that the mice switched to control diet gained significantly less weight than the mice remaining on Western diet (Fig. 5k) and that fat storing vesicles in the liver were nearly eliminated (Fig. 5f), However, the switched mice never dropped to the body mass levels of mice continuously on control diet, as did the mice fed Western diet and treated with aNF. Food consumption indicated that the eating habits of the switched mice were not overtly affected (Fig. 5l). Similar to earlier results shown in Fig. 3d, AHR inhibition by aNF in mice on the switched diet regimen caused arachidonic acid plasma concentrations to drop significantly relative to mice on Western diet (Fig. 5m). In contrast, mice switched from Western diet to control diet showed a significant increase in plasma concentrations of arachidonic acid (Fig. 5m). Furthermore, the same mice switched from Western to control diet displayed CYP1B1 protein levels that were generally unaffected while SCD1 protein levels were relatively higher (Fig. 5i). The results suggest that aNF causes a more immediate and complete blockage of AHR signaling resulting in a steeper and greater drop in body mass and AHR-directed transcription, whereas in mice switched from Western to control diet, Western-diet derived AHR agonists remain available to prolong AHR signaling, induce AHR-directed gene expression, and elevate the body mass baseline.
Discussion
Our hypothesis is that the AHR is the regulatory hub of a downstream network of genes that underlies obesity (Fig. 6). Mice on a 40-week diet regimen fed a Western diet became obese and developed fatty liver, much of which we attribute to the upregulation of liver Cyp1b1, PPARα target genes, Scd1, and Spp1; while mice on Western diet containing the AHR antagonist aNF were obesity resistant, possessed livers devoid of fat vesicles, and displayed decreased levels of mRNA and protein of CYP1B1, PPARα target genes, SCD1, and SPP1 near that of mice on low-fat control diet (Figs. 1–4 and Tables S1–S3).

a The AHR, activated by Western diet-derived components, initiates the collective and cooperative upregulation of CYP1B1, PPARα target genes, SCD1, and SPP1 to cause obesity and fatty liver. b Inhibition of the AHR by the antagonist aNF inverts the expression levels of CYP1B1, PPARα target genes, SCD1, and SPP1 to prevent and reverse obesity and fatty liver.
The proposal that the AHR may serve as a metabolic hub is well supported by work of others. Knockout of any one of the genes in the proposed AHR network, including Ahr [13, 39], Cyp1b1 [40,41,42], Ppara [61], Scd1 [62, 63], or Spp1 [56] results in reduced obesity and ameliorated liver steatosis for B6 mice fed a high-fat diet. Extensive crosstalk in the gene network is well documented, in which the AHR regulates the gene expression of Cyp1b1 [64], Ppara [38], Scd1 [65], and Spp1 [54]; CYP1B1 the gene expression of Ppara [40, 66], Scd1 [41], and Spp1 [55]; and PPARα the regulation of Scd1 [67] and Spp1 [68]. The inhibition of AHR signaling reduced PPARα mRNA and protein in mouse hepatocyte cell lines [38]. In Ahr [38] and Cyp1b1 [40, 41] gene knockout mice, Ppara and Scd1 gene expression and/or protein activity are reduced; and the adenoviral expression of human SCD1 in Cyp1b1 knockout mice restored body mass to wild type levels [41]. Our work supports, complements, and extends these studies.
In vitro assays have shown that at concentrations of 10 μM and greater, aNF acts as an AHR agonist [60]. We reported that aNF was not at measureable levels in liver and at 3.67 μM in plasma (Fig. 5j) indicating that aNF as used in our studies acted as an antagonist [60]. Other findings from our work here support the contention that aNF worked as an antagonist. Profiles of liver mRNA from mice in the 40-week diet regimen showed that Western diet caused Cyp1b1 mRNA levels to increase (2.38-fold), while Cyp1a1 and Cyp1a2 mRNA levels were unchanged; and in liver of mice fed Western + aNF diet, only Cyp1b1 mRNA showed a significant drop (0.73-fold) while Cyp1a1 and Cyp1a2 mRNA levels were again unchanged. The results are compatible with the notion that a Western diet-derived component (Kyn) acted as an AHR agonist in liver such that Cyp1b1 (of the Cyp1 gene family) was specifically induced and that aNF acted as an antagonist to block that activity.
We asked how inhibition of the AHR and the resulting decreased expression and activity of CYP1B1, PPARα, SCD1, and SPP1 prevented and reversed obesity and fatty liver. The observed decrease in liver mRNA and protein levels of these genes by AHR inhibition may phenotypically copy, at least in part, the loss of any one of these genes. It was proposed [41] that the loss of the canonical AHR-regulated Cyp1b1 gene prevents obesity [40, 66] via arachidonic acid, a polyunsaturated fatty acid (20:4 omega-6) that is the precursor for numerous AHR signaling and inflammatory molecules [69]. Arachidonic acid is an endogenous substrate of CYP1B1 [52] and causes Scd1 mRNA levels and SCD1 activity to drop [51]. Thus, it could be surmised that a drop in CYP1B1 levels by AHR inhibition would lead to an accumulation of arachidonic acid to cause a decrease in SCD1 levels and activity. Instead, plasma levels of arachidonic acid dropped significantly in mice fed Western diet containing aNF (Fig. 3d and Fig. 5m) when CYP1B1 levels are relatively lower. The decreased arachidonic acid levels may be due to a decrease in the expression of phospholipase A2s, a cytosolic phospholipase that releases arachidonic acid from cellular membranes, a major arachidonic acid source [53]. The Pla2g4a gene, which encodes phospholipase A2s, is transcriptionally regulated by the AHR [70]. We observed that Western diet caused a significant increase in Pla2g4a mRNA levels and that AHR inhibition caused a significant decrease (Fig. 3e).
Hence for SCD1, which supplies monounsaturated fats required for obesity [41, 42, 71], the observed reduction in mRNA and protein levels is likely not due to an influence by arachidonic acid but rather by a decrease in Scd1 gene induction. AHR [65], CYP1B1 [41], and PPARα [67] all positively regulate Scd1 gene expression. Thus, a working hypothesis is that a repressed AHR-CYP1B1-PPARα axis downregulates Scd1 expression to impact AHR-based obesity. Furthermore, Scd1 gene loss causes higher energy expenditure from increased fatty acid β-oxidation in the liver [72], and inhibition of the AHR may also lead to a SCD1-dependent increase in energy expenditure.
As has been observed, Scd1 gene regulation by the AHR may be ligand and tissue dependent. The potent AHR ligand 2,3,7,8-tetrachlorodibenzodioxin induced hepatic Scd1 mRNA expression and steatosis [65] while the AHR agonist β-naphthoflavone repressed Scd1 expression [73], the latter outcome compatible to the results observed here in adipose tissue, in which Scd1 mRNA levels rose in mice treated with aNF (Fig. S1C). The repression of Scd1 by β-naphthoflavone in the liver was independent of DNA binding by the AHR and consistent with the espoused notion that the AHR may bind and interfere with the transcription factor sterol element binding protein 1c (SREBP1c) [73], a key regulator of genes involved in fatty acid biosynthesis. Although liver Scd1 gene expression did not follow a pattern of SREBP1c interference (Fig. 3a, b), Fasn, another key gene in fatty acid biosynthesis that was repressed after β-naphthoflavone treatment and regulated independently of DNA binding by the AHR [73], here showed a significant increase in liver mRNA levels in mice fed Western + aNF diet to that of mice fed Western diet alone (1.85-fold), suggesting that the AHR is not interacting with SREBP1c.
In regard to PPARα, a ligand-activated master regulator of fat metabolism [74], significant changes were seen in numerous PPARα-regulated genes as a result of AHR inhibition (Table S3), similar to what was observed in mice with knockout of either the Ahr [38] or Cyp1b1 gene [40, 66]. However, we observed little or no change in PPARα mRNA and protein levels among mice fed the different diets suggesting that PPARα activity rather than expression was affected by AHR inhibition. A possible explanation is that PPARα activity was reduced by a drop in the availability of endogenous PPARα ligands, which include eicosanoids [75]. As described above, plasma levels of arachidonic acid, from which eicosanoids are derived [76], were significantly lower in mice fed Western + aNF diet to that of mice fed control and Western diets.
SPP1 or osteopontin is a secreted protein that has key roles in extracellular matrix remodeling, biomineralization, cell migration, and regulation of cytokine production but little is understood how it contributes to obesity [77, 78], yet, the impact of SPP1 on obesity is evident in that deletion of the AHR-regulated Spp1 gene [54] protects mice on a high-fat diet from extreme weight gain [56]. SPP1 is often used as a biomarker for obesity [59] as SPP1 levels rise dramatically in obese mice and humans [77]. It is known that SPP1 enables obesity-related inflammation by facilitating the recruitment of monocytes/macrophages to adipose tissue by acting as a chemotactic molecule to the extracellular matrix [79, 80].
The Trp metabolite Kyn is a known AHR agonist [19, 25,26,27]. We had proposed that in the context of obesity, amplified AHR activity was not due to increased levels of Trp, which is of equal amounts in the Western and control diets, but rather to increased IDO1 activity to produce surplus Kyn from the available dietary Trp [13]. In our model, Western diet, in contrast to control diet, provides a greater quantity of oxidized-low density lipoproteins, which induce the TLR2/4 pathway, and in turn, increases IDO1 levels and activity [13]. Because Kyn is an AHR agonist, and the AHR is a positive transcription factor for the Ido1 gene [19], a positive feedback loop is in place to contribute to an obese state. Nonetheless, the effect of AHR inhibition by aNF to prevent and reverse obesity seems to be primarily downstream of the AHR rather than upstream for the following reasons. Although it has been reported that Ido1 (and Ido2) gene expression is regulated in an AHR dependent manner [19], liver Ido1 and Ido2 mRNA levels from the studies carried out here were unaffected by diet and aNF treatment. Further, of the identified genes upstream of the AHR in obesity, only Tlr2 mRNA levels were affected (2.2 fold increase in Western diet to control diet, aNF had no effect). In contrast as reported here, Western diet and Western + aNF diet caused a respective significant rise and drop in mRNA and protein levels in liver of the identified downstream genes in AHR-based obesity.
Although other organs/tissues play major roles in obesity, e.g., adipose [81] (Fig. S1), we hypothesize that the liver, specifically hepatocytes, are the primary site for AHR-based obesity. First, conditional knockout of the Ahr gene in hepatocytes causes obesity resistance in female mice [43]; second, AHR levels and activity are greatest by far in hepatocytes [82, 83]; third, the majority of fat metabolism occurs in the liver [84] some of which is dependent on the AHR [73, 85]; and fourth, as shown in Fig. 1i, diet-based, AHR-regulated expression of CYP1B1 is in a predicted subset of hepatocytes. That is, the AHR is located and active primarily in perivenous hepatocytes where xenobiotic metabolism, lipogenesis, and glycolysis mainly occur [46, 86] and where CYP1B1 has been shown to be exclusively expressed [47].
It is often misstated in the literature that the Cyp1b1 gene is not expressed in hepatocytes but rather chiefly in non-parenchymal liver cells, e.g., stellate cells. However, our data (Fig. 1i), in agreement with the study cited above [47], clearly demonstrate that CYP1B1 is expressed specifically in perivenous hepatocytes and that expression is inducible by Western diet and downregulated by AHR inhibition. Based on gene expression profiles, some major metabolic pathways known to be active in the perivenous region, e.g., lipogenesis and glycolysis, are predicted to coexist with the AHR in mice fed Western diet (Table S1) and which are silenced by AHR inhibition (Table S2).
We have observed that aNF in shorter term diet regimens of 20 and 26 weeks caused significant hepatomegaly regardless of diet [12, 13]. In the 40-week diet regimen reported here, aNF caused a milder, statistically non-significant level of hepatomegaly in mice on the control + aNF diet and no hepatomegaly in mice on the Western + aNF diet (Fig. 1d). The results suggest that over sufficient time, the liver may adapt to the disruptive impact of aNF on liver-to-body ratio via an adjustment of the hematostat, a mechanism proposed to maintain the liver-to-body-weight ratio near at near normal proportions at all times to preserve body and metabolic homeostasis [87]. The process seems unique to the liver and encompasses several regenerative pathways and cell proliferation pathways.
In closing, we propose a new and unprecedented role for the AHR based on our results showing that AHR inhibition in mice can prevent and reverse diet-induced obesity and fatty liver. Our overarching goal is to determine whether AHR signaling may play a role in human obesity and fatty liver disease similar to that seen in mice, hence, identifying the AHR as a therapeutic target may ultimately fulfill an unmet clinical need for obesity prevention and treatment. Behavioral modification approaches and drug interventions to treat human obesity have been disappointing, especially over longer time periods [88]. Literature on the long-term effects of interventional drug treatments for human obesity is scarce [28], and our studies support the idea that anti-AHR drugs over an extended time span may be effective. Clinical trials testing the controlled modulation of the AHR with more specific and potent drugs than aNF could provide exciting new therapeutic avenues, which spotlights a real need for the systematic screening of novel drugs that can specifically inhibit AHR activity. Finally, there are still many questions regarding the clinical treatment of obesity via the AHR, including how AHR inhibiton may affect gut microbiota [89], the re-setting of metabolic baselines, liver metabolism and liver size, adipose and muscle metabolism, and energy homeostasis.
Data availability
Any data and materials not in the public domain that were generated from these reported studies will be made available upon request.
References
Hales CM, Fryar CD, Carroll MD, Freedman DS, Ogden CL. Trends in obesity and severe obesity prevalence in us youth and adults by sex and age, 2007–2008 to 2015–2016. JAMA. 2018;319:1723–5.
De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–9.
Naukkarinen J, Rissanen A, Kaprio J, Pietilainen KH. Causes and consequences of obesity: the contribution of recent twin studies. Int J Obes. 2012;36:1017–24.
Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet. 2011;378:815–25.
Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association scientific statement on obesity and heart disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2006;113:898–918.
van den Brandt PA, Spiegelman D, Yaun S-S, Adami H-O, Beeson L, Folsom AR, et al. Pooled analysis of prospective cohort studies on height, weight, and breast cancer risk. Am J Epidemiol. 2000;152:514–27.
Cawley J, Meyerhoefer C. The medical care costs of obesity: an instrumental variables approach. J Health Econ. 2012;31:219–30.
Rask-Andersen M, Karlsson T, Ek WE, Johansson Å. Gene-environment interaction study for BMI reveals interactions between genetic factors and physical activity, alcohol consumption and socioeconomic status. PLoS Genet. 2017;13:e1006977.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–48.
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206.
Hill JO, Peters JC. Environmental contributions to the obesity epidemic. Science. 1998;280:1371–4.
Moyer BJ, Rojas IY, Kerley-Hamilton JS, Nemani KV, Trask HW, Ringelberg CS, et al. Obesity and fatty liver are prevented by inhibition of the aryl hydrocarbon receptor in both female and male mice. Nutr Res. 2017;44:38–50.
Moyer BJ, Rojas IY, Kerley-Hamilton JS, Hazlett HF, Nemani KV, Trask HW, et al. Inhibition of the aryl hydrocarbon receptor prevents Western diet-induced obesity. Model for AHR activation by kynurenine via oxidized-LDL, TLR2/4, TGFbeta, and IDO1. Toxicol Appl Pharmacol. 2016;300:13–24.
Kerley-Hamilton JS, Trask HW, Ridley CJ, Dufour E, Ringelberg CS, Nurinova N, et al. Obesity is mediated by differential aryl hydrocarbon receptor signaling in mice fed a Western diet. Environ Health Perspect. 2012;120:1252–9.
Lahvis GP, Lindell SL, Thomas RS, McCuskey RS, Murphy C, Glover E, et al. Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc Natl Acad Sci USA. 2000;97:10442–7.
Nebert DW, Puga A, Vasiliou V. Role of the Ah receptor and the dioxin-inducible [Ah] gene battery in toxicity, cancer, and signal transduction. Ann NY Acad Sci. 1993;685:624–40.
Guo J, Sartor M, Karyala S, Medvedovic M, Kann S, Puga A, et al. Expression of genes in the TGF-beta signaling pathway is significantly deregulated in smooth muscle cells from aorta of aryl hydrocarbon receptor knockout mice. Toxicol Appl Pharmacol. 2004;194:79–89.
Puga A, Sartor MA, Huang M, Kerzee JK, Wei Y, Tomlinson CR, et al. Gene expression profiles of mouse aorta and cultured vascular muscle cells are widely different yet show common responses to dioxin exposure. Cardiovasc Toxicol. 2004;4:385–404.
Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–8.
Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65.
Quintana FJ, Sherr DH. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev. 2013;65:1148–61.
Kim SH, Henry EC, Kim DK, Kim YH, Shin KJ, Han MS, et al. Novel compound 2-methyl-2H-pyrazole-3-carboxylic acid (2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) prevents 2,3,7,8-TCDD-induced toxicity by antagonizing the aryl hydrocarbon receptor. Mol Pharmacol. 2006;69:1871–8.
Zhao B, DeGroot DE, Hayashi A, He G, Denison MS. CH223191 is a ligand-selective antagonist of the Ah (dioxin) receptor. Toxicol Sci. 2010;117:393–403.
Smith KJ, Murray IA, Tanos R, Tellew J, Boitano AE, Bisson WH, et al. Identification of a high-affinity ligand that exhibits complete aryl hydrocarbon receptor antagonism. J Pharmacol Exp Ther. 2011;338:318–27.
Nguyen NT, Hanieh H, Nakahama T, Kishimoto T. The roles of aryl hydrocarbon receptor in immune responses. Int Immunol. 2013;25:335–43.
Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci USA. 2010;107:19961–6.
Veldhoen M, Hirota K, Christensen J, O'Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–9.
Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. J Am Med Assoc. 2014;311:74–86.
Takamatsu M, Hirata A, Ohtaki H, Hoshi M, Ando T, Ito H, et al. Inhibition of indoleamine 2,3-dioxygenase 1 expression alters immune response in colon tumor microenvironment in mice. Cancer Sci. 2015;106:1008–15.
Wang X, Wang Q, Morris ME. Pharmacokinetic interaction between the flavonoid luteolin and gamma-hydroxybutyrate in rats: potential involvement of monocarboxylate transporters. AAPS J. 2008;10:47–55.
Jeffery E, Berry R, Church CD, Yu S, Shook BA, Horsley V, et al. Characterization of Cre recombinase models for the study of adipose tissue. Adipocyte. 2014;3:206–11.
Jeffery E, Church CD, Holtrup B, Colman L, Rodeheffer MS. Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol. 2015;17:376.
Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605.
Berry R, Rodeheffer MS. Characterization of the adipocyte cellular lineage in vivo. Nat Cell Biol. 2013;15:302.
Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15.
Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–55.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300.
Wang C, Xu C-X, Krager SL, Bottum KM, Liao D-F, Tischkau SA. Aryl hydrocarbon receptor deficiency enhances insulin sensitivity and reduces PPAR-α pathway activity in mice. Environ Health Perspect. 2011;119:1739–44.
Xu CX, Wang C, Zhang ZM, Jaeger CD, Krager SL, Bottum KM, et al. Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. Int J Obes. 2015;39:1300–9.
Larsen MC, Bushkofsky JR, Gorman T, Adhami V, Mukhtar H, Wang S, et al. Cytochrome P450 1B1: an unexpected modulator of liver fatty acid homeostasis. Arch Biochem Biophys. 2015;571:21–39.
Li F, Jiang C, Larsen MC, Bushkofsky J, Krausz KW, Wang T, et al. Lipidomics reveals a link between CYP1B1 and SCD1 in promoting obesity. J Proteome Res. 2014;13:2679–87.
Liu X, Huang T, Li L, Tang Y, Tian Y, Wang S, et al. CYP1B1 deficiency ameliorates obesity and glucose intolerance induced by high fat diet in adult C57BL/6J mice. Am J Transl Res. 2015;7:761–71.
Girer NG, Carter D, Bhattarai N, Mustafa M, Denner L, Porter C, et al. Inducible loss of the aryl hydrocarbon receptor activates perigonadal white fat respiration and brown fat thermogenesis via fibroblast growth factor 21. Int J Mol Sci. 2019; 20:e950.
Jungermann K, Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev Nutr. 1996;16:179–203.
Jungermann K, Katz N. Functional specialization of different hepatocyte populations. Physiol Rev. 1989;69:708–64.
Braeuning A, Ittrich C, Köhle C, Hailfinger S, Bonin M, Buchmann A, et al. Differential gene expression in periportal and perivenous mouse hepatocytes. FEBS J. 2006;273:5051–61.
Walker NJ, Crofts FG, Li Y, Lax SF, Hayes CL, Strickland PT, et al. Induction and localization of cytochrome P450 1B1 (CYP1B1) protein in the livers of TCDD-treated rats: detection using polyclonal antibodies raised to histidine-tagged fusion proteins produced and purified from bacteria. Carcinogenesis. 1998;19:395–402.
Ferre P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53(Suppl 1):S43–50.
Flowers MT, Ntambi JM. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr Opin Lipidol. 2008;19:248–56.
Liu X, Miyazaki M, Flowers MT, Sampath H, Zhao M, Chu K, et al. Loss of Stearoyl-CoA desaturase-1 attenuates adipocyte inflammation: effects of adipocyte-derived oleate. Arterioscler Thromb Vasc Biol. 2010;30:31–38.
Sessler AM, Kaur N, Palta JP, Ntambi JM. Regulation of stearoyl-CoA desaturase 1 mRNA stability by polyunsaturated fatty acids in 3T3-L1 adipocytes. J Biol Chem. 1996;271:29854–8.
Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB. Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P4501B1. Drug Metab Dispos. 2004;32:840–7.
Murakami M, Kudo I. Phospholipase A2. J Biochem. 2002;131:285–92.
Chuang CY, Chang H, Lin P, Sun SJ, Chen PH, Lin YY, et al. Up-regulation of osteopontin expression by aryl hydrocarbon receptor via both ligand-dependent and ligand-independent pathways in lung cancer. Gene. 2012;492:262–9.
Palenski TL, Sorenson CM, Jefcoate CR, Sheibani N. Lack of Cyp1b1 promotes the proliferative and migratory phenotype of perivascular supporting cells. Lab Investig. 2013;93:646–62.
Lancha A, Rodriguez A, Catalan V, Becerril S, Sainz N, Ramirez B, et al. Osteopontin deletion prevents the development of obesity and hepatic steatosis via impaired adipose tissue matrix remodeling and reduced inflammation and fibrosis in adipose tissue and liver in mice. PLoS One. 2014;9:e98398.
Shehin SE, Stephenson RO, Greenlee WF. Transcriptional regulation of the human CYP1B1 gene: evidence for involvement of an aryl hydrocarbon receptor response element in constitutive expression. J Biol Chem. 2000;275:6770–6.
Ellero S, Chakhtoura G, Barreau C, Langouet S, Benelli C, Penicaud L, et al. Xenobiotic-metabolizing cytochromes p450 in human white adipose tissue: expression and induction. Drug Metab Dispos. 2010;38:679–86.
Catalan V, Gomez-Ambrosi J, Rodriguez A, Ramirez B, Izaguirre M, Hernandez-Lizoain JL, et al. Increased obesity-associated circulating levels of the extracellular matrix proteins osteopontin, chitinase-3 like-1 and tenascin c are associated with colon cancer. PLoS ONE. 2016;11:e0162189.
Santostefano M, Merchant M, Arellano L, Morrison V, Denison MS, Safe S. alpha-Naphthoflavone-induced CYP1A1 gene expression and cytosolic aryl hydrocarbon receptor transformation. Mol Pharmacol. 1993;43:200–6.
Guerre-Millo M, Rouault C, Poulain P, André J, Poitout V, Peters JM, et al. PPAR-α–null mice are protected from high-fat diet–induced insulin resistance. Diabetes. 2001;50:2809–14.
Sampath H, Flowers MT, Liu X, Paton CM, Sullivan R, Chu K, et al. Skin-specific deletion of stearoyl-CoA desaturase-1 alters skin lipid composition and protects mice from high fat diet-induced obesity. J Biol Chem. 2009;284:19961–73.
Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, et al. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc Natl Acad Sci USA. 2002;99:11482–6.
Zhang L, Savas U, Alexander DL, Jefcoate CR. Characterization of the mouse Cyp1B1 gene. Identification of an enhancer region that directs aryl hydrocarbon receptor-mediated constitutive and induced expression. J Biol Chem. 1998;273:5174–83.
Angrish MM, Jones AD, Harkema JR, Zacharewski TR. Aryl hydrocarbon receptor-mediated induction of stearoyl-CoA desaturase 1 alters hepatic fatty acid composition in TCDD-elicited steatosis. Toxicol Sci. 2011;124:299–310.
Bushkofsky JR, Maguire M, Larsen MC, Fong YH, Jefcoate CR. Cyp1b1 affects external control of mouse hepatocytes, fatty acid homeostasis and signaling involving HNF4alpha and PPARalpha. Arch Biochem Biophys. 2016;597:30–47.
Miller CW, Ntambi JM. Peroxisome proliferators induce mouse liver stearoyl-CoA desaturase 1 gene expression. Proc Natl Acad Sci USA. 1996;93:9443–8.
Nakamachi T, Nomiyama T, Gizard F, Heywood EB, Jones KL, Zhao Y, et al. PPARalpha agonists suppress osteopontin expression in macrophages and decrease plasma levels in patients with type 2 diabetes. Diabetes. 2007;56:1662–70.
Bennett M, Gilroy DW. Lipid mediators in inflammation. Microbiol Spectr. 2016;4, https://doi.org/10.1128/microbiolspec.MCHD-0035-2016.
Kinehara M, Fukuda I, Yoshida K, Ashida H. Aryl hydrocarbon receptor-mediated induction of the cytosolic phospholipase A(2)alpha gene by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mouse hepatoma Hepa-1c1c7 cells. J Biosci Bioeng. 2009;108:277–81.
Miyazaki M, Flowers MT, Sampath H, Chu K, Otzelberger C, Liu X, et al. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 2007;6:484–96.
Dobrzyn P, Dobrzyn A, Miyazaki M, Cohen P, Asilmaz E, Hardie DG, et al. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc Natl Acad Sci USA. 2004;101:6409–14.
Tanos R, Murray IA, Smith PB, Patterson A, Perdew GH. Role of the Ah receptor in homeostatic control of fatty acid synthesis in the liver. Toxicol Sci. 2012;129:372–9.
Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015;62:720–33.
Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci USA. 1997;94:4312–7.
Subhash PK, David SG, David RJ, Letts LG. Eicosanoids in inflammation: biosynthesis, pharmacology, and therapeutic frontiers. Curr Top Med Chem. 2007;7:311–40.
Kahles F, Findeisen HM, Bruemmer D. Osteopontin: a novel regulator at the cross roads of inflammation, obesity and diabetes. Mol Metab. 2014;3:384–93.
Icer MA, Gezmen-Karadag M. The multiple functions and mechanisms of osteopontin. Clin Biochem. 2018;59:17–24.
Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Investig. 2007;117:2877–88.
Lund SA, Giachelli CM, Scatena M. The role of osteopontin in inflammatory processes. J Cell Commun Signal. 2009;3:311–22.
Korenblat KM, Fabbrini E, Mohammed BS, Klein S. Liver, muscle and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology. 2008;134:1369–75.
Henderson Colin J, McLaughlin Lesley A, Osuna-Cabello M, Taylor M, Gilbert I, McLaren Aileen W, et al. Application of a novel regulatable Cre recombinase system to define the role of liver and gut metabolism in drug oral bioavailability. Biochem J. 2015;465:479–88.
Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, et al. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci USA. 2000;97:779–82.
Nguyen P, Leray V, Diez M, Serisier S, Le Bloc'h J, Siliart B, et al. Liver lipid metabolism. J Anim Physiol Anim Nutr. 2008;92:272–83.
Tanos R, Patel RD, Murray IA, Smith PB, Patterson AD, Perdew GH. Aryl hydrocarbon receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner. Hepatology. 2012;55:1994–2004.
Bazotte RB, Silva LG, Schiavon FP. Insulin resistance in the liver: deficiency or excess of insulin? Cell Cycle. 2014;13:2494–500.
Michalopoulos GK. Hepatostat: liver regeneration and normal liver tissue maintenance. Hepatology. 2017;65:1384–92.
Grandone A, Di Sessa A, Umano GR, Toraldo R, Miraglia del Giudice E. New treatment modalities for obesity. Best Pract Res Clin Endocrinol Metab. 2018;32:535–49.
Hubbard TD, Liu Q, Murray IA, Dong F, Miller C, Smith PB, et al. Microbiota metabolism promotes synthesis of the human Ah receptor agonist 2,8-dihydroxyquinoline. J Proteome Res. 2019;18:1715–24.
Acknowledgements
We thank the editors and reviewers for their thoughtful comments. The authors acknowledge the following core facilities: Genomics & Molecular Biology Shared Resource, Clinical Pharmacology Shared Resource, Irradiation, Pre-clinical Imaging & Microscopy Shared Resource, and Pathology Shared Resource at the Norris Cotton Cancer Center at Dartmouth with NCI Cancer Center Support Grant 5P30CA023108-40.
Funding
This work was supported by funding from NCI 5P30CA023108-40, NIH-NCRR award 5P20RR024475-02, NIH-NIGMS award 8P20GM103534-02, and a NCCC Prouty Pilot Award.
Author information
Authors and Affiliations
Contributions
IYR, BJM, and CRT conceived and designed the studies. IYR, CSR, and BJM performed the experiments and acquired the data. IYR, CSR, and CRT ran the computations and analyzed the data. IYR and CRT wrote and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
The studies with mice (Mus musculus) were conducted using an animal protocol approved by the Dartmouth Hitchcock Medical Center Institutional Animal Care and Use Committee, IACUC PROTOCOL NUMBER toml.cr.1#2(m5ar5), ASSURANCE NUMBER A3259-01.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rojas, I.Y., Moyer, B.J., Ringelberg, C.S. et al. Reversal of obesity and liver steatosis in mice via inhibition of aryl hydrocarbon receptor and altered gene expression of CYP1B1, PPARα, SCD1, and osteopontin. Int J Obes 44, 948–963 (2020). https://doi.org/10.1038/s41366-019-0512-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-019-0512-z
- Springer Nature Limited
This article is cited by
-
Identification of potential key lipid metabolism-related genes involved in tubular injury in diabetic kidney disease by bioinformatics analysis
Acta Diabetologica (2024)
-
Plasma aryl hydrocarbon receptor associated with epicardial adipose tissue in men: a cross-sectional study
Diabetology & Metabolic Syndrome (2023)
-
Insight into the physiological and pathological roles of the aryl hydrocarbon receptor pathway in glucose homeostasis, insulin resistance, and diabetes development
Cellular & Molecular Biology Letters (2022)
-
Intestinal microbial metabolites in human metabolism and type 2 diabetes
Diabetologia (2020)