
Abstract
Ethanol is an important risk factor for the occurrence of several brain disorders that depend on the amount, period and frequency of its consumption. Chronic use of ethanol often leads to the development of neurodegenerative syndromes, which cause morphological and functional impairments such as foetal alcohol syndrome in newborns exposed to ethanol during pregnancy, Wernicke–Korsakoff Syndrome and, more rarely, Marchiafava–Bignami disease (MBD). MBD is characterized by primary degeneration of the corpus callosum, without inflammation and is associated with oxidative stress and hypovitaminosis, as well as altered mental status, to mention dementia, seizures, depression and so on. This review discusses MBD and poor nutrition as a risk factor for the development of such alcoholic syndrome, with focus on diagnosis, pathogenic aspects, signs and symptoms, as well as therapeutic perspectives. On the basis of the inclusion/exclusion criteria adopted, the performed search in scientific databases (Pubmed, Scielo and Google Scholar) resulted in 100 studies that are being presented and discussed in the present work. Review, case–control and cohort studies on alcoholism-associated hypovitaminosis, oxidative stress, MBD and ethanol metabolism pathways were admitted as relevant. We highlight that MBD is a poorly described, diagnosed, insidious and progressive condition, for which evidence suggests a synergism between ethanol-induced neurotoxic effects and hypovitaminosis B. Present treatment consists of vitamin B1(thiamine) supplementation. Nonetheless, other strategies such as the inclusion of antidepressants or steroidal anti-inflammatories as add-on therapies have been employed as an attempt to improve the damage. Indeed, both the diagnosis and treatment are difficult, and death occurs within few years.
Similar content being viewed by others

Introduction
Chronic ingestion of ethanol promotes important morphological and physiological alterations in the organism, mostly affecting the central nervous system (CNS),1, 2 and particularly in the brain. In fact, even in the CNS, there are areas or cell populations that are more vulnerable than others, such as the prefrontal cortex, hippocampus, cerebellum, corpus callosum and glial cells.3, 4, 5 The gastrointestinal tract may also be affected, causing a dietary deficiency that may contribute to further damage.1, 6, 7
Wernicke–Korsakoff syndrome, for example, is a worldwide health problem, characterized by memory loss or confusion and cerebellar dysfunction caused by severe deficiency of thiamine (vitamin B1) and concomitant alcoholism.8, 9 Furthermore, severe and rare neurological complications associated with vitamin deficiency, such as Marchiafava–Bignami disease (MBD), are also reported as a result of chronic ethanol-driven neurotoxicity, which is marked by focal necrosis and corpus callosum demyelination.10
Vitamin deficiencies, mainly vitamin B1, and malnourishment are common characteristics in chronic alcoholics that may lead to MBD, due to liver damage-induced metabolic disturbance ultimately causing a B1-hypovitaminosis.11 Therefore, in this review, we will outline current concepts on the neurologic complications of MBD and the poor nutrition as a risk factor for alcoholic syndromes, focusing on diagnosis, pathogenic mechanisms, signs and symptoms, as well as available therapeutic perspectives.
General and clinical features
First reported in 1903 in Italian alcoholics, MBD is a rare neurological syndrome, associated with patients who have a history of alcoholism and malnutrition. MBD can occur among both women and men; however, it is most frequently diagnosed in men between 40 and 60 years of age,12, 13, 14, 15 and is characterized by degeneration, necrosis and atrophy of the corpus callosum, the major white matter commissure connecting the cerebral hemispheres, which facilitates cognitive, sensory and motor interhemispheric exchange of information.16 Although the differences in gender-dependent alcohol-induced toxic effects have been discussed, conclusive data that reveal the variability in gender-dependent susceptibility for the development of MBD does not exist.17 As a matter of fact, the pathogenesis of MBD as well as its progression remain unclear.18, 19
MBD can also appear in non-alcoholic patients, where it is associated with carbon monoxide (CO) poisoning, sepsis, sickle cell disease, Plasmodium falciparum infection during cerebral malaria,20 cardiac carcinoma surgery21 and diabetes.22, 23, 24, 25
Clinically, MBD leads to psychotic and emotional disorders, such as apathy, depression and aggressive behaviour, with ‘interhemispheric disconnection syndrome’ related to apraxia, ataxia, visual dyslexia, loss of consciousness, seizures, mental confusion and psychosis during the acute and subacute form and progressive dementia as the major clinical manifestation of the chronic form.18, 26, 27, 28 A few reports of visual hallucinations and auditory delusions are associated with chronic alcohol consumption in female patients.29, 30
Diagnosis and classification
MBD diagnosis is difficult, but can be obtained by magnetic resonance imaging (MRI), where knowledge of pathognomonic signs is essential: hyper-intense lesions without mass effect within the corpus callosum (hypodensity) accompanied by swelling of the genu and splenium.16 However, other diseases involving alterations in the corpus callosum, namely, recurrent artery infarction of Heubner, neoplastic diseases (astrocytoma or lymphoma), demyelinating diseases (multiple sclerosis, encephalitis, lymphoma, infarction and astrocytoma) must also be considered for a proper differential diagnosis.31
In general, MBD is characterized by middle portion (middle lamina) degeneration, usually symmetric, of the myelinated fibre tracts from the corpus callosum accompanied by relative sparing of both thin upper and lower edges.32 In some cases, the anterior and posterior commissures, centrum semiovale and other white matter tracts may also be affected.33, 34 However, evidence suggests the presence of disconnections beyond the commissure fibres. Changes in extracallosal projections, cortico-cortical and cortico-subcortical connecting fibres also promote transneural depression.35 In this sense, Bhat et al.36 reported disruption of fibre tracts in the corpus callosum and Yang et al.37 reported the first case of a MBD patient with asymmetric extracallosal lesions confined to one side of the brain and highlighted diagnostic MRI methods for better prognosis prediction.
Because of how relevant thiamine deficiency is for the development of neurological diseases such as MBD, its plasma levels could be suggested as a biological marker for proper diagnosis.38 Nevertheless, technical limitations must be considered. Thiamine serum concentration, for example, is not an accurate marker for tissue samples.39 Erythrocyte transketolase activation assay and thiamine pyrophosphate concentration evaluation by high performance liquid chromatography are more useful and reliable for the study and diagnosis of MBD.39, 40 However, due to quick vitamin replacement these values are rapidly normalized; therefore, their application should be limited to initial diagnosis, as an add-on to MRI-based evaluation.39
Brion et al.41 postulated three identifiable forms of MBD: acute, subacute and chronic. Acute MBD is characterized by a sudden onset of altered consciousness and seizures; mutism may include muscle stiffness and spread with dysphagia, poor diagnosis and death within a few days. The subacute form is characterized by a sudden onset of dementia that eventually progresses to a vegetative state, possibly progressing steadily to death in a few months. In chronic cases, more frequently recognized by technological advances, little consensus exists about the clinical findings that characterize this phase, limited to progressive dementia, behavioural abnormalities and signs of interhemispheric disconnection.28, 42, 43
Another pattern classification, which considers clinical status and injury detectable by MRI, distinguishes two types. The first, type ‘A’, displays a large deficit of consciousness, seizures, dysarthria and hemiparesis. Often, a hyper-intense swelling of the corpus callosum is observed, associated with extracallosal injuries. In the second type, ‘B’, there is less disturbance of consciousness with partial callosal lesions on MRI. Nonetheless, dysarthria, gait disorders and interhemispheric signal disconnection are also present. Patients who present the type B pattern often evolve with a better prognosis.44
Role of hypovitaminosis B and oxidative stress in ethanol-induced neurotoxicity
The exact mechanism that underlies MBD is unknown, but evidence from studies with humans (brain tissue) or animals (rodents) suggests a synergism between ethanol-induced neurotoxic effects and hypovitaminosis B, especially thiamine, which is an essential B complex vitamin needed in both neurons and glial cells. Vitamin B deficiency is associated with other injuries in the CNS, such as alcohol-induced dementia and Wernicke–Korsakoff syndrome.45
He et al.33 in a rat model of alcoholism reported that the pathology of the corpus callosum resulted from synergistic effects of alcohol intoxication and thiamine depletion. Alcohol intake per se impairs neuronal plasticity, interferes with lipid metabolism and affects the expression of proteins responsible for binding cytoskeletal elements in the human brain white matter.46 On the other hand, thiamine deficiency produces neurological impairment, as observed in rodent animal models.47
Thiamine (Vitamin B1) deficiency and neuropathy
Thiamine is converted to its active form, thiamine pyrophosphate, using magnesium as a cofactor. In turn, thiamine pyrophosphate acts as a coenzyme for the pyruvate dehydrogenase complex (PDH) and alpha-ketoglutarate dehydrogenase in both the Kreb’s cycle and pentose-phosphate pathway. These enzyme systems have key roles in the carbohydrate metabolism process, generating adenosine triphosphate (ATP) to energetically sustain cellular processes and reactions in the CNS, which have been demonstrated in both human and animal tissues (rodents).48 Reduced ATP induces inhibition of catechol-O-methyl transferase activity, increasing the levels of the neurotransmitter dopamine, which in turn can produce delirium, hallucinations and delusions.
Another neurotransmitter involved is acetylcholine, reduced levels of which have been linked to cognitive impairment (dementia) and delirium, whereas demyelinating diseases are related to thiamine deficiency syndromes, such as MBD. Other neurotransmitters, such as glutamate, aspartate and gamma-aminobutyric acid, also have their levels reduced following thiamine deficiency. Furthermore, an increase in the precursor glutamine, due to hypoactivity of alpha-ketoglutarate dehydrogenase, produces excitation and delirium in humans.49
Considering the brain areas that are characterized by intense metabolic activity and their need for large amounts of thiamine, a drop on thiamine-dependent enzyme activity affects local metabolism and energy production. This framework promotes neuronal/glial loss and CNS atrophy, being reported for both human as well as for rodent animal model.50 The thiamine-dependent enzyme transketolase catalyzes reactions that produce ribose-5-phosphate and reduced nicotinamide adenine dinucleotide phosphate (NADPH) from glucose-6-phosphate in the pentose-phosphate pathway, and presents reduced activity during thiamine-deficient states. This leads to reduced levels of essential molecules that participate in glutathione synthesis, which is responsible for converting hydrogen peroxide (H2O2) into H2O. Impaired production of glutathione results in H2O2 accumulation, lipid peroxidation, protein oxidation and neuronal damage. Most of these observations derive from animal (rodent) studies, that were later confirmed relevant after analysis of human brain tissue, and used to justify previously reported behavioural abnormalities (apathy and social indifference, superficial and labile emotions, and lack of goal-oriented spontaneous activity).51
Thiamine deficiencies reflect failures in the processes involved in the chronic alcoholic state, such as lack of dietary intake, intestinal malabsorption, altered hepatic uptake and metabolism, reduced re-absorption by renal tubular cells, increased skeletal and visceral protein catabolism and abnormal lipid metabolism.52 Thus, such deficiency in the tissues leads to difficulty in assembly and proper functioning of various enzymes that are important for the breakdown, or the metabolism of sugar molecules. Therefore, in the brain, thiamine is required in neurons and glia cells; thus the inadequate functioning of the thiamine-using enzymes (that is, pyruvate dehydrogenase—PDH and alpha-ketoglutarate dehydrogenase -α-KGDH) can lead to failure in critical biochemical reactions, such as the synthesis of neurotransmitters, as acetylcholine, glutamate, gamma-aminobutyric acid and aspartate. In addition, the inadequate PDH functioning may lead to failure in myelin and glutathione synthesis, thereby inhibiting the ability of these neurons to conduct signals and body’s defence against oxidative stress, respectively, as previously demonstrated both in humans and rodents.45, 53
The diversity of synergistic mechanisms involving alcohol and thiamine deficiency extend beyond their action on the CNS. As previously reported in humans, they also interact on a pharmacokinetic level, as ethanol inhibits carrier proteins present in duodenal cells that are essential for the absorption of B complex vitamins.54, 55 Because of thiamine’s cofactor activity on PDH, thiamine deficiency interferes with myelin synthesis and therefore, compromises the corpus callosum, a structure known to have high myelin content, as reported in studies performed in humans and animals (that is, rodents).21, 45 In fact, long-term malnutrition or nutrient deficiency due to alcohol ingestion may be related to the pathological consequences of MBD.
Ethanol metabolism and oxidative brain damage
Ethanol’s contribution to brain damage permeates its pharmacological properties, as it alters neurotransmitter activity in the CNS. However, said contribution is also closely related to ethanol metabolism. Despite being mainly metabolized by the liver (up to 90%), ethanol can also undergo biotransformation in other anatomic structures, including the CNS, which was observed in a rat model of alcoholism.56 Data from studies using human samples and rodent models provide evidence that ethanol metabolism takes place through an oxidative process, which mediates the production of acetaldehyde (AA) by three enzymes: alcohol dehydrogenase (ADH), catalase and cytochrome P450 2E1 (CYP2E1), which are also present in the CNS and, therefore, exert their activity on ethanol.57, 58 AA is ultimately converted into acetate by the aldehyde dehydrogenase enzymes, as demonstrated in a mice model.59
Aldehyde dehydrogenase (ADH) pathway
ADH3 (in humans and rodents) and ADH4 (only in humans) are the main isoforms of ADH responsible for ethanol metabolism in the brain, especially in the cerebellum and hippocampus.57 ADH enzymes are known to use nicotinamide adenine dinucleotide (NAD+) as a cofactor in ethanol oxidation, reducing it to NADH. This reaction consequently increases the NADH/NAD+ ratio, leading to an altered cellular redox state, which has been associated with the development of oxidative stress disorders of the CNS, both in human and animal (rodents) models.60, 61 As a matter of fact, the AA produced by ADH turns xanthine dehydrogenase into xanthine oxidase, ultimately increasing the production of reactive oxygen species (ROS) that can lead to oxidative stress-mediated blood–brain barrier injury, as previously demonstrated in primary human neurons.62
Despite the important role of ADH, evidence accumulated over the years points to catalase as the main ethanol metabolizing enzyme in the brain, according to studies performed in both mice and rats, as well as human prenatal cephalic tissues.59, 63, 64, 65 In the periphery, catalase is located within peroxisomes, metabolizing approximately 2% of the circulating ethanol. It has been shown in rat, cattle and pig liver tissues that such process is not NAD+ dependent and converts ethanol into AA after forming a complex with H2O2, leading to water and oxygen production.66 In the brain, studies developed with acatalasemic or catalase inhibitor-treated mice have demonstrated that catalase mediates up to 50% of ethanol metabolism.58
Catalase pathway
Concerning oxidative stress, catalase is also known as an antioxidant enzyme, and was found to have a reduced activity in both the cerebellum and hypothalamus of rats chronically fed with ethanol.67, 68 It has been proposed that such a reduction in catalase activity is due to the disruptive action of ethanol in the nrf2-mediated pathway, which is responsible for gene transcription of antioxidant enzymes such as catalase, thus increasing ROS levels and contributing to the establishment of an oxidative stress state, which has been reported in a study conducted in postnatal day 7 rat pups treated with resveratrol (100 mg/kg).69
Controversially, acute as well as chronic ethanol intake can promote an increase in catalase activity in the CNS, which will specifically affect the spinal cord, striatum, synaptosomes and microsomes, as previously shown in rat brain subcellular fractions.70 It has been reported for both human and animal models (rodents) that enhanced catalase activity within the CNS is dangerous due to the resulting increase in local AA concentrations. Because of its high reactivity, AA interacts with a wide variety of cell structures and biomolecules, such as mitochondria and microsomal proteins, respectively. Such interactions lead to biological dysfunction and have been reported to contribute to lipid peroxidation, ROS generation and oxidative stress.71, 72
Cytochrome P450 2E1 (CYP2E1) pathway
CYP2E1 converts ethanol to AA and ROS, a reaction that demands both oxygen and reduced NADPH, as it was shown in human and bovine brain microvascular endothelial cells, as well as mice and rat models.73, 74 As previously mentioned, although mainly expressed in the liver, CYP2E1 is also expressed in neurons from the cerebral cortex, granule cells and Purkinje cells of the cerebellum, and pyramidal neurons of the hippocampus (CA1, CA2 and CA3 regions) in the brains of rats and humans.62 Furthermore, studies in rats have demonstrated that the expression of CYP2E1 induced by ethanol consumption is associated with an increase in its activity up to 20-fold. In humans, similar effects were found in ethanol abusers, as well as moderate consumers.75 As a result, increased expression and activity of CYP2E1 promotes large increases in local concentrations of AA and ROS, leading to oxidative stress and neuronal damage in the CNS of both humans and rodents.76 These effects result in the installation of mitochondrial dysfunction, linked to overproduction of ROS (H2O2, O2−), and reduction in the respiratory rate and ATP synthesis, also mediating DNA damage, as observed in human and rodent models.77
Pro-oxidative mechanisms
Excessive generation of ROS and AA in the CNS of animals (rodents) and humans adds to the increased expression of inducible nitric oxide synthase (iNOS), and thus the synthesis of nitric oxide (NO) is induced by chronic alcohol consumption. The reaction of NO with ROS (H2O2, O2) leads to the formation of peroxynitrite, a highly harmful radical.73, 74
More recently, in a study using human neuroblastoma cells and 7-day pulp mice brain tissue, NADPH oxidase (NOX) has been implicated in oxidative damage to the CNS generated by ethanol. In fact, ethanol promotes the increase of NOX activity via a CDC42-dependent pathway. NOX, in turn, promotes intense production of ROS.78 Additionally, most of the enzymes involved in CNS-mediated ethanol metabolism, mentioned earlier, have been linked to the onset of oxidative stress, lipid peroxidation and apoptosis.79 Such events generate characteristic damage to the corpus callosum (mainly demyelination, axonal degeneration and necrosis) of human chronic consumers of ethanol, which is a prominent feature found in MBD.70, 80
Besides stimulating pro-oxidant mechanisms, ethanol also triggers oxidative stress in the CNS of humans by reducing the activity of the enzyme-based antioxidant system, which is composed of superoxide dismutase, catalase, glutathione peroxidase and glutathione reductase.80
Superoxide dismutase is the rate-limiting enzyme responsible for the conversion of H2O2 into O2-, but in chronic alcoholic intoxication, their cytosolic and mitochondrial isoforms (in the cerebral cortex and cerebellum, respectively) exhibit reduced activity, according to data reported in a study using a rat model.68 A similar effect has been observed in the spinal cord for both acute and chronic ethanol intoxication, which was attributed to superoxide dismutase inactivation, causing an accumulation of superoxide radicals in rats brains.81
Glutathione peroxidase, an enzyme found mainly in the cytosol and mitochondrial matrix, mediates the reduction of peroxides with the participation of glutathione, which is accompanied by the formation of glutathione disulphide in the human brain.82 In the context of alcoholic intoxication, a reduction of its activity due to the interaction with lipid peroxidation end-products, such as malondialdehyde (MDA) and 4-hydroksynonenalm, has been previously reported in rats chronically fed with ethanol.83
Glutathione reductase, present in the cytosol and mitochondria of most cells, is another enzyme affected by ethanol. Glutathione reductase regenerates reduced glutathione at the expense of NADPH oxidation in the murine brain.84 Acute exposure to ethanol decreases the activity of this enzyme, especially in the cerebral cortex, whereas chronic intoxication is marked by low levels of reduced glutathione, which is consumed to reduce ethanol intoxication poisoning in rodents and humans (Figure 1).67, 85

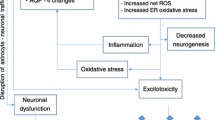
Graphical representation of major CNS damage mechanisms triggered by excessive consumption of ethanol or by thiamine deficiency related to the occurrence of MBD. Ethanol-induced damage to the (CNS) involves its direct action on cellular metabolism, increasing the activity of enzymes producing ROS, such as NOX. The ethanol metabolite, acetaldehyde, formed in the CNS by three major pathways (alcohol dehydrogenase (ADH3,4), catalase and CYP2E1), in turn also produces damage, as it raises the formation of ROS, induces the xanthine oxidase pathway and nitric oxide synthase (iNOS)-inducible enzymes. The formation of acetaldehyde is further increased due to the inductive effect of ethanol on catalase and CYP2E1. Both mechanisms contribute to oxidative stress and neurodegeneration. In addition, the consumption of ethanol compromises the absorption of thiamine, contributing to reduced thiamine levels in the CNS, one of the factors linked to MBD. Thiamine deficiency, also generated by low intake or absorption disorders, impairs thiamine pyrophosphate-dependent enzyme activity. Reduced activities of PDH and alpha-ketoglutarate dehydrogenase diminish ATP production, which can affect cell metabolism. Moreover, the reduction of transketolase activity compromises the production of glutathione, favouring the development of oxidative damage. Such mechanisms described above produce damage to the corpus callosum, which results in interhemispheric disconnection syndrome. Furthermore, thiamine deficiency reduces catechol-O-methyl transferase activity, which promotes, among other effects on neurotransmitter pathways, the elevation of dopamine levels that causes symptoms such as delirium and a reduction in acetylcholine levels, which leads to dementia.
MBD manifests itself with a series of nonspecific symptoms related to hemispheric disconnection, which is important for differential diagnosis.44 Evaluation of patient history is also an important step, checking especially whether there has been chronic exposure to alcohol or some other condition that led to malnutrition, as well as factors directly related to the development of MBD, combined with imaging tests of the findings for a better treatment approach.
Therapeutic perspectives
The current lack of knowledge about the mechanisms underlying demyelinating lesions of the corpus callosum associated with the low occurrence of this syndrome is a barrier to the establishment of an effective therapy. However, clinical practice suggests that treatment with vitamin B complex vitamins, such as thiamine (B1), folic acid (B9), cyanocobalamin (B12) and nicotinamide (B3) is essential, even if their levels are within normal limits, in order to avoid disease progression.86 Although there are large and controversial variations in dose (75–1000 mg/kg) and duration (1–12 weeks) of thiamine replacement therapy, the analysis of 153 cases demonstrated that his early initiation, within at least two weeks after the onset of symptoms, improves MBD prognosis and represents the only limiting factor in patient recovery.15, 86, 87 Among patients treated, about 40% presented normality or minimal disability compatible outcomes, whereas in untreated patients this rate drops to 20%.87
Thiamine replacement therapy has been well established for cases of Wernicke encephalopathy, which is generated by thiamine deficiency associated with excessive alcohol consumption. Considering the losses of synaptic activity in the CNS generated by thiamine deficiency as reviewed above, clinical intervention consists of daily parenteral administration of this vitamin until behavioural symptoms are reversed. This protocol has been used, sometimes with some adjustments (association with vitamin B12 associated to folate at dose of 5 mg/dl), for the treatment of MBD. In many cases, the protocol contributed to the total or partial recovery of the patient.71, 88 Although there is no placebo-controlled randomized trial for this application, clinical experience highlights it as most advantageous therapy as compared with others.87
In addition to its use as a thiamine replacement, thiamine treatment can also be used in conjunction with other therapeutic agents. Recent approaches have been directed towards the use of high doses of anti-inflammatory steroids and adjuvant therapies with antidepressants. Fukumoto and Suzuki28 proposed the use of the tetracyclic antidepressant mianserin at a low dose (10 mg/day) combined with standard thiamine treatment. This association proved effective in the control of neuropsychological symptoms, such as delusions and depressive behaviour.
The inclusion of steroidal anti-inflammatories in the treatment of MBD appears to have been based on the hypothesis that injury of the corpus callosum is connected to a bleeding disorder, with evidence of inflammation and oedema. Its application thus relates to control of the inflammatory process, with reduced cerebral oedema and blood–brain barrier stabilization. Furthermore, anti-inflammatory steroids prevent the demyelination process. For such a purpose, methylprednisolone (500 mg/day for 5 days or 1000 mg/day for 3 days, intravenously), prednisolone (30 mg/day) or dexamethasone (8 mg/day)22, 89 have been previously applied. This therapy, however, still has its basis in conceptual hypotheses and clinical trials. Unconditional logistic regression studies showed that the replacement of thiamine in fact improves MBD patients’ prognoses. Although some authors affirm that steroid therapy has not shown any advantage,87 corticosteroids associated with thiamine therapy have been used with complete resolution of MRI-identifiable lesions and improvement of clinical responses.90
Another supportive treatment strategy applied to a 61-year-old alcoholic patient with MBD consisted of chlormethiazole administered at low doses in association with thiamine (900 mg/day). In this case, the patient showed remarkable improvement.91 The action of this sedative agent, a thiazolic derivative, is based on the enhancement of gamma-aminobutyric acid activity and can present a possible neuroprotective effect.92
Preclinical studies have also pointed to the control of oxidative stress, plasma concentrations of ammonia and alcohol as important factors assisting in thiamine replacement therapy, to reduce the structural changes of the corpus callosum and cognitive disorders in MBD.80, 93
The prognosis tends to be worse in alcoholic patients, but can be improved in cases of early detection of the disease.19 In a clinical study, Uchiyama et al.94 reported that besides lesions in the corpus callosum, there were asymmetrical hypo-intense areas in several cortico-subcortical structures that were related to the presence of cerebral micro-haemorrhage. According to the authors, these findings were not dependent on the presence of callosal lesions, but were important in augmenting the severity of MBD-induced dementia.
Finally, medical data (1903–2001) have reported approximately 250 cases of MBD with 90% of them having poor prognosis, highlighting high levels of both morbidity and mortality: 200 patients died, 30 developed dementia and only in 20 cases did they recover.15 However, there is a high likelihood for sub-diagnosis, which accounts for an underestimated number of cases reported in the literature, as well as a lack that clarifies the susceptibility of gender-dependence in the occurrence of MBD. In fact, probably men have been more affected or diagnosed because they still consume more alcohol than women do, as reported by epidemiological surveys.95 Nevertheless, because of the lifestyle among women that has been modified, in which the consumption of alcohol has been increased,95 such epidemiological differences in the occurrence of MBD will be balanced. In this regard, new approaches for the prevention, early diagnosis and treatment of MBD are essential. Of concern, increasing alcohol consumption, which besides being the cause of nearly 5.9% of deaths worldwide,95 is responsible for the appearance of diseases that affect the CNS, such as MBD, due to its interference with the absorption of B complex vitamins, especially thiamine, which is directly linked to the development of corpus callosum damage.
References
Harper C . The neuropathology of alcohol-related brain damage. Alcohol Alcohol 2009; 44: 136–140.
Crews FT, Nixon K . Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol 2009; 44: 115–127.
Harper CG, Kril JJ, Daly J . Are we drinking our neurons away? BMJ 1987; 294: 534–536.
Harding AJ, Halliday GM, Harper CG, Kril JJ . Loss of vasopressin-immunoreactive neurons in alcoholics is dose-related and time-dependent. Neuroscience 1996; 72: 699–708.
Baker K, Harding A, Halliday G, Kril JJ, Harper C . Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke’s encephalopathy. Neuroscience 1999; 91: 429–438.
Fisher SJ, Lee IJ, Swaan PW, Eddington ND . Evaluation of the effect of ethanol’s toxic metabolite acetaldehyde on the gastrointestinal oligopeptide transporter, PEPT1: In vitro and in vivo studies. Alcohol Clin Exp Res 2008; 32: 162–170.
Mercer DF . Animal models for studying hepatitis C and alcohol effects on liver. World J Gastroenterol 2011; 17: 2515–2519.
Oscar-Berman M . Function and dysfunction of prefrontal brain circuitry in alcoholic Korsakoff’s syndrome. Neuropsychol Rev 2012; 22: 154–169.
Jung YC, Chanraud S, Sullivan EV . Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev 2012; 22: 170–180.
Mulholland PJ, Self RL, Stepanyan TD, Little HJ, Littleton JM, Prendergast MA . Thiamine deficiency in the pathogenesis of chronic ethanol-associated cerebellar damage in vitro. Neurosciense 2005; 135: 1129–1139.
Keil VC, Greschus S, Schneider C, Hadizadeh DR, Schild HH . The whole spectrum of alcohol-related changes in the CNS: practical MR and CT imaging guidelines for daily clinical use. Rofo 2015; 187: 1073–1083.
Marchiafava E, Bignami A . Sopra un alterazione del corpo calloso osservata in soggetti alcoolisti. Riv Patol Nerv Ment 1903; 8: 544–549.
Kosaka K, Aoki M, Kawasaki N, Adachi Y, Konuma I, Iizuka R . A non-alcoholic Japanese patient with Wernicke's encephalopathy and Marchiafav-Bignami disease. Clin Neuropathol 1984; 3: 231–236.
Rusche-Skolarus LE, Lucey BP, Vo KD, Snider BJ . Transient encephalopathy in a postoperative non-alcoholic female with Marchiafava-Bignami disease. Clin Neurol Neurosurg 2007; 109: 713–715.
Folescu R, Zamfir CL, Sişu AM, Motoc AG, Ilie AC, Moise M . Histopathological and imaging modifications in chronic ethanolic encephalopathy. Romanian J Morphol Embryol 2014; 55: 797–801.
Hoshino Y, Ueno Y, Shimura H, Miyamoto N, Watanabe M, Hattori N et al. Marchiafava-Bignami disease mimics motor neuron disease: case report. BMC Neurol 2013; 13: 208–212.
Mumenthaler MS, Taylor JL, O’Hara R, Yesavage JA . Gender difference in moderate drinking effects. Alcohol Res Health 1999; 23: 55–64.
Helenius J, Tatlisumak T, Soinne L, Valanne L, Kaste M . Marchiafava–Bignami disease: two cases with favourable outcome. Eur J Neurol 2001; 8: 269–272.
Carrilho PEM, Santos MBM, Piasecki L, Jorge AC . Marchiafava-Bignami disease: a rare entity with a poor outcome. Rev Bras Ter Intensiva 2013; 25: 68–72.
Boutboul D, Lidove O, Aguilar C, Klein I, Papo T . Marchiafava-Bignami disease complicating SC hemoglobin disease and Plasmodium falciparum infection. Presse Med 2010; 39: 990–993.
Cui Y, Zheng L, Wang X, Zhang W, Yuan D, Wei Y . Marchiafava-Bignami disease with rare etiology: a case report. Exp Ther Med 2015; 9: 1515–1517.
Suzuki Y, Oishi M, Ogawa K, Kamei S . A patient with Marchiafava–Bignami disease as a complication of diabetes mellitus treated effectively with corticosteroid. J Clin Neurosci 2012; 19: 761–762.
Yadala S, Luo JJ . Marchiafava-bignami disease in a nonalcoholic diabetic patient. Case Rep Neurol Med 2013; 2013: 1–4.
Pérez Álvarez AI, Carbajo CR, de la Tassa GM, Gómez JP . Diabetes mellitus mal controlada como desencadenante de un caso de enfermedad de Marchiafava-Bignami. Neurología 2015; 15: S0213–S4853.
Kilinc O, Ozbek D, Ozkan E, Midi I . Neurological and psychiatric findings of marchiafava-bignami disease in a nonalcoholic diabetic patient with high blood glucose levels. J Neuropsychiatry Clin Neurosci 2015; 27: e149–e150.
Lechevalier B, Andersson JC, Morin P . Hemispheric disconnection syndrome with a ‘crossed avoiding’ reaction in a case of Marchiafava-Bignami disease. J Neurol Neurosurg Psychiatry 1977; 40: 483–497.
Celik Y Marchiafava-Bignami disease in alcoholism. In: Preedy VR, Watso RR (eds). Comprehensive Handbook of Alcohol Related Pathology. Elsevier Academic Press: San Diego, 2004; pp 713–718.
Fukumoto J, Suzuki T . Marchiafava-Bignami disease treated by mianserin hydrochloride in short-term evaluated by neuropsychological analysis. Psychogeriatrics 2007; 7: 25–32.
Lucato LT, Freua F, Kok F . Chronic stage of Marchiafava-Bignami disease. Arq Neuropsiquiatr 2015; 73: 890.
Augusto L, Figueiredo R, Costa H, Reis C, Silva ML . Marchiafava-Bignami disease as a cause of visual hallucinations. Rev Bras Psiquiatr 2015; 37: 82–83.
Kumar KS, Challam RJN, Singh WJ . Marchiafava - bignami disease: a case report. J Clin Diagn Res 2014; 8: RD01–RD02.
Friese SA, Bitzer M, Freudenstein D, Voigt K, Küker W . Classification of acquired lesions of the corpus callosum with MRI. Neuroradiology 2000; 42: 795–802.
He X, Sullivan EV, Stankovic RK, Harper CG, Pfefferbaum A . Interaction of thiamine deficiency and voluntary alcohol consumption disrupts rat corpus callosum ultrastructure. Neuropsychopharmacology 2007; 32: 2207–2216.
Bano S, Mehra S, Yadav SN, Chaudhary V . Marchiafava-Bignami disease: role of neuroimaging in the diagnosis and management of acute disease. Neurol India 2009; 57: 649–652.
Nalini A, Kovoor JM, Dawn R, Kallur KG . Marchiafava-Bignami disease: two cases with magnetic resonance imaging and positron emission tomography scan findings. Neurol India 2009; 57: 644–648.
Bhat A, Punia V, Lee HJ, Marks D . Corpus callosum fibre disruption in Marchiafava-Bignami disease. Prac Neurol 2014; 14: 189–190.
Yang L, Qin W, Xu J, Hu W . Marchiafava-Bignami disease with asymmetric extracallosal lesions. Arch Med Sci 2015; 11: 895–898.
Davies SB, Joshua FF, Zagami AS . Wernicke's encephalopathy in a non-alcoholic patient with a normal blood thiamine level. Med J Aust 2011; 194: 483–484.
Kumar N . Neurologic presentations of nutritional deficiencies. Neurol Clin 2010; 28: 107–170.
Guilland JC . Vitamin B1 (thiamine). Rev Prat 2013; 63: 1074–1075. 1077-8.
Brion S Marchiafava-Bignami disease. In: Vinken PJ, Bruyn GW (eds). Handbook of Clinical Neurology. Oxford, Amsterdam: The Netherlands, 1977; pp 317–329.
Hoppe M, Ebner A . Transient lesion in the splenium of the corpus callosum: three further cases in epileptic patients and a pathophysiological hypothesis. J Neurol Neurosurg Psychiatry 2001; 70: 459–463.
Ménégon P, Sibon I, Pachai C, Orgogozo JM, Dousset V . Marchiafava-Bignami disease: diffusion-weighted MRI in corpus callosum and cortical lesions. Neurology 2005; 65: 475–477.
Heinrich A, Runge U, Khaw AV . Clinicoradiologic subtypes of Marchiafava-Bignami disease. J Neurol 2004; 251: 1050–1059.
Martin PR, Singleton CK, Hiller-Sturmhöfel S . The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health 2003; 27: 134–142.
Depaz I, Ito M, Matsumoto I, Niwa S, Kroon P, Wilce PA . Expression of hNP22 is altered in the frontal cortex and hippocampus of the alcoholic human brain. Alcohol Clin Exp Res 2003; 27: 1481–1488.
Zieve L . Influence of magnesium deficiency on the utilization of thiamine. Ann N Y Acad Sci 1969; 162: 732–743.
Spector R . Thiamin homeostasis in the central nervous system. Ann N Y Acad Sci 1982; 378: 344–354.
Osiezagha K, Ali S, Freeman C, Barker NC, Jabeen S, Maitra S . Thiamine deficiency and delirium’. Innov Clin Neurosci 2013; 10: 26–32.
Hazell AS, Todd KG, Butterworth RF . Mechanisms of neuronal cell death in Wernicke’s encephalopathy. Metab Brain Dis 1998; 13: 97–122.
Martin PR, Adinoff B, Weingartner H, Mukherjee AB, Eckardt MJ . Alcoholic organic brain disease: nosology and pathophysiologic mechanisms. Prog Neuropsychopharmacol Biol Psychiatry 1986; 10: 147–164.
Halsted CH . B-Vitamin dependent methionine metabolism and alcoholic liver disease. Clin Chem Lab Med 2013; 51: 457–465.
Singleton CK, Martin PR . Molecular mechanisms of thiamine utilization. Curr Mol Med 2001; 1: 197–207.
Tallaksen CM, Bell H, Bohmer T . Thiamin and thiamin phosphate ester deficiency assessed by high performance liquid chromatography in four clinical cases of Wernicke encephalopathy. Alcohol Clin Exp Res 1993; 17: 712–716.
Cravo ML, Glória LM, Selhub J . Hyperhomocysteinemia in chronic alcoholism: correlation with folate, vitamin B-12, and vitamin B-6 status. Am J Clin Nutr 1996; 63: 220–224.
Panchenko LF, Pirozhkov SV, Popova SV, Antonenkov VD . Effect of chronic ethanol treatment on peroxisomal acyl-CoA oxidase activity and lipid peroxidation in rat liver and heart. Experientia 1987; 43: 580–581.
Galter D, Carmine A, Buervenich S, Duester G, Olson L . Distribution of class I, III and IV alcohol dehydrogenase mRNAs in the adult rat, mouse and human brain. Eur J Biochem 2003; 270: 1316–1326.
Zimatkin SM, Pronko SP, Vasiliou V, Gonzalez FJ, Deitrich RA . Enzymatic mechanisms of ethanol oxidation in the brain. Alcohol Clin Exp Res 2006; 30: 1500–1555.
Vasiliou V, Ziegle TL, Bludeau P, Petersen DR, Gonzalez FJ, Deitrich RA . CYP2E1 and catalase influence ethanol sensitivity in the central nervous system. Pharmacogenet Genomics 2006; 16: 51–58.
Norberg A, Jones AW, Hahn RG, Gabrielsson JL . Role of variability in explaining ethanol pharmacokinetics: research and forensic applications. Clin Pharmacokinet 2003; 42: 1–31.
Manzo-Avalos S, Saavedra-Molina A . Cellular and mitochondrial effects of alcohol consumption. Int J Environ Res Public Health 2010; 7: 4281–4304.
Haorah J, Ramirez SH, Floreani N, Gorantla S, Morsey B, Persidsky Y . Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic Biol Med 2008; 45: 1542–1550.
Aragon C, Stotland L, Amit Z . Studies on ethanol–brain catalase interaction: evidence for central ethanol oxidation. Alcohol Clin Exp Res 15: 165–169.
Person RE, Chen H, Fantel AG, Juchau MR . Enzymic catalysis of the accumulation of acetaldehyde from ethanol in human prenatal cephalic tissues: evaluation of the relative contributions of CYP2E1, alcohol dehydrogenase, and catalase/peroxidases. Alcohol Clin Exp Res 2000; 24: 1433–1442.
Correa M, Sanchis-Segura C, Pastor R, Aragon CM . Ethanol intake and motor sensitization: the role of brain catalase activity in mice with different genotypes. Physiol Behav 2004; 82: 231–240.
Weiner H . Subcellular localization of acetaldehyde oxidation on liver. Ann N Y Acad Sci 1987; 492: 25–34.
Somani SM, Husain K, Diaz-Phillips L, Lanzotti DJ, Kareti KR, Trammell GL . Interaction of exercise and ethanol on antioxidant enzymes in brain regions of the rat. Alcohol 1996; 13: 603–610.
Calabrese V, Renis M, Calderone A, Russo A, Reale S, Barcellona ML et al. Stress proteins and SH-groups in oxidant-induced cellular injury after chronic ethanol administration in rat. Free Radic Biol Med 1998; 24: 1159–1167.
Kumar A, Singh CK, Lavoie HA, Dipette DJ, Singh US . Resveratrol restores Nrf2 level and prevents ethanol-induced toxic effects in the cerebellum of a rodent model of fetal alcohol spectrum disorders. Mol Pharmacol 2011; 80: 446–457.
Reddy SK, Husain K, Schlorff EC, Scott RB, Somani SM . Dose response of ethanol ingestion on antioxidant defense system in rat brain subcellular fractions. Neurotoxicology 1999; 20: 977–987.
Aggarwal A, Khandelwal A, Jiloha RC . A case of Marchiafava-Bignami disease: complete recovery with thiamine. J Neuropsychiatry Clin Neurosci 2011; 23: E28.
Heit C, Dong H, Chen Y, Thompson DC, Deitrich RA, Vasiliou VK . The role of CYP2E1 in alcohol metabolism and sensitivity in the central nervous system. Subcell Biochem 2013; 67: 2352–47.
Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y . Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol 2005; 78: 1223–1232.
Albano E . Alcohol, oxidative stress and free radical damage. Proc Nutr Soc 2006; 65: 278–290.
Liangpunsakul S, Kolwankar D, Pinto A, Gorski JC, Hall SD, Chalasani N . Activity of CYP2E1 and CYP3A enzymes in adults with moderate alcohol consumption: a comparison with nonalcoholics. Hepatology 2005; 41: 1144–1150.
Kumar A, Lavoie HA, Dipette DJ, Singh US . Ethanol neurotoxicity in the developing cerebellum: underlying mechanisms and implications. Brain Sci 2013; 3: 941–963.
Adam-Vizi V . Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxidants Redox Signal 2005; 7: 1140–1149.
Wang X, Ke Z, Chen G, Xu M, Bower KA, Frank JA et al. Cdc42-dependent activation of NADPH oxidase is involved in ethanol-induced neuronal oxidative stress. PLoS One 2012; 7: e38075.
Kim TE, Lee EJ, Young JB, Shin DJ, Kim JH . Wernicke encephalopathy and ethanol-related syndromes. Semin Ultrasound CT MR 2014; 35: 85–96.
Kashem MA, Etages HD, Kopitar-Jerala N . Differential protein expression in the corpus callosum (body) of human alcoholic brain. J Neurochem 2009; 110: 486–495.
Ledig M, M’Paria JR, Mandel P . Superoxide dismutase activity in rat brain during acute and chronic alcohol intoxication. Neurochem Res 1981; 6: 385–390.
Ansari KA, Bigelow D, Kaplan E . Glutathione peroxidase activity in surgical and autopsied human brains. Neurochem Res 1985; 10: 703–711.
Bosch-Morell F, Martínez-Soriano F, Colell A, Fernández-Checa JC, Romero FJ . Chronic ethanol feeding induces cellular antioxidants decrease and oxidative stress in rat peripheral nerves. Effect of S-adenosyl-L-methionine and N-acetyl-L-cysteine. Free Radic Biol Med 1998; 25: 365–368.
Knollema S, Hom HW, Schirmer H, Korf J, Ter Horst GJ . Immunolocalization of glutathione reductase in the murine brain. J Comp Neurol 1996; 373: 157–172.
Dringen R . Metabolism and functions of glutathione in brain. Prog Neurobiol 2000; 62: 649–671.
Iwai T, Matsuo K, Morii-Kitani F, Azuma F, Matsuo H, Takada M et al. Marchiafava-Bignami disease with hyperintensity on late diffusion-weighted imaging. Acta Radiol Short Rep 2014; 3: 1–4.
Hillbom M, Saloheimo P, Fujioka S, Wszolek ZK, Juvela S, Leone MA . Diagnosis and management of Marchiafava-Bignami disease: a review of CT/MRI confirmed cases. J Neurol Neurosurg Psychiatry 2014; 85: 168–173.
Staszewski J, Macek K, Stepien A . A reversible demyelinisation of corpus callosum in the course of Marchiafava-Bignami disease. Neurol Neurochir Pol 2006; 40: 156–161.
Consoli A, Pirritano D, Bosco D, Postorino P, Consoli D . Corticosteroid treatment in a patient with Marchiafava–Bignami Disease. Neurol Sci 2014; 35: 1143–1145.
Dujmović I, Nikolić I, Gavrić-Kezić M, Dačković J, Mesaroš Š, Drulović J . Teaching neuroImages: reversible widespread brain MRI lesions in Marchiafava-Bignami disease’. Neurology 2015; 84: e81–e82.
Ruiz-Martínez J, Martínez Pérez-Balsa A, Ruibal M, Urtasun M, Villanua J, Martí Massó JF . Marchiafava-Bignami disease with widespread extracallosal lesions and favourable course. Neuroradiology 1999; 41: 40–43.
Wilby MJ, Hutchinson PJ . The pharmacology of chlormethiazole: a potential neuroprotective agent? CNS Drug Rev 2004; 10: 281–294.
Kashem MA, Harper C, Matsumoto I . Differential protein expression in the corpus callosum (genu) of human alcoholics’. Neurochem Int 2008; 53: 1–11.
Uchiyama M, Kasai H, Kurokawa S, Sakae Y, Kinno R . Relationship between callosal lesions and cerebral microhemorrhage in Marchiafava-Bignami disease. Nihon Arukoru Yakubutsu Igakkai Zasshi 2014; 49: 238–248.
WHO 2014. Global status report on alcohol and health 2014. World Health Organization http://www.who.int/substance_abuse/publications/global_alcohol_report/en/. Accessed on October 15 2015.
Acknowledgements
FRB and FRO received scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil). MCM is supported by a research fellowship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Fernandes, L., Bezerra, F., Monteiro, M. et al. Thiamine deficiency, oxidative metabolic pathways and ethanol-induced neurotoxicity: how poor nutrition contributes to the alcoholic syndrome, as Marchiafava–Bignami disease. Eur J Clin Nutr 71, 580–586 (2017). https://doi.org/10.1038/ejcn.2016.267
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejcn.2016.267
- Springer Nature Limited
This article is cited by
-
Clinico-radiologic subtypes and therapeutic observation of acute Marchiafava-Bignami disease
Scientific Reports (2023)
-
Randomised trial of intravenous thiamine and/or magnesium sulphate administration on erythrocyte transketolase activity, lactate concentrations and alcohol withdrawal scores
Scientific Reports (2022)
-
Response to: “Before blaming a COVID vaccine for cytotoxic lesions of the corpus callosum all other differentials must be ruled out”
Neuroradiology (2022)
-
Radiologische Merkmale einer seltenen, alkoholassoziierten, neurologischen Erkrankung
Die Radiologie (2022)
-
Alcohol-Related Central Nervous System Disorders Associated with Vitamin B Deficiency
SN Comprehensive Clinical Medicine (2021)