
Abstract
Objective
To review data on the role of ethanol-induced alteration of Zn homeostasis in mediation of adverse effects of alcohol abuse.
Methods
The scholarly published articles on the association between Zn metabolism and alcohol-associated disorders (liver, brain, lung, gut dysfunction, and fetal alcohol syndrome) have been reviewed.
Results
It is demonstrated that alcohol-induced modulation of zinc transporters results in decreased Zn levels in lungs, liver, gut, and brain. Zn deficiency in the gut results in increased gut permeability, ultimately leading to endotoxemia and systemic inflammation. Similarly, Zn deficiency in lung epithelia and alveolar macrophages decreases lung barrier function resulting in respiratory distress syndrome. In turn, increased endotoxemia significantly contributes to proinflammatory state in alcoholic liver disease. Finally, impaired gut and liver functions may play a significant role in alcoholic brain damage, being associated with both increased proinflammatory signaling and accumulation of neurotoxic metabolites. It is also hypothesized that ethanol-induced Zn deficiency may interfere with neurotransmission. Similar changes may take place in the fetus as a result of impaired placental zinc transfer, maternal zinc deficiency, or maternal Zn sequestration, resulting in fetal alcoholic syndrome. Therefore, alcoholic Zn deficiency not only mediates the adverse effects of ethanol exposure, but also provides an additional link between different alcohol-induced disorders.
Conclusions
Generally, current findings suggest that assessment of Zn status could be used as a diagnostic marker of metabolic disturbances in alcohol abuse, whereas modulation of Zn metabolism may be a potential tool in the treatment of alcohol-associated disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alcohol abuse or alcohol use disorders are among the most widespread psychiatric disorders worldwide [1]. The prevalence of heavy drinking in the USA, UK, Germany, and China is 16.9, 28, 12.5, and 7.6%, respectively. In Russia, alcohol consumption has dramatically increased since 1990–2010, reaching the prevalence of heavy drinking of 19.1% of the total population above 15 years old [2]. The social and health impact of alcohol abuse is mediated by a tight association between alcohol intake and various diseases. Alcohol abuse is causally related to 25 chronic diseases [3], including cancer, cardiovascular diseases (hypertension, hemorrhagic stroke and atrial fibrillation), liver diseases (alcoholic hepatitis, cirrhosis, and fatty liver), and pancreatitis [4]. Human brain is one of the main targets of ethanol toxicity [5]. In particular, the most common symptoms of alcoholic brain damage include Wernicke encephalopathy and Korsakoff syndrome [6].
Although alcohol abuse is more characteristic for men, recent studies have demonstrated that male-to-female ratio of alcohol use disorders has significantly decreased from 5:1 (1980s) to 3:1 (2000s) [7]. The social and medical importance of alcohol abuse in women is also complicated by the fetal hazards of maternal alcohol exposure (growth restriction, craniofacial abnormalities, intellectual disabilities, etc.), being clustered into fetal alcohol spectrum disorder (FASD) [8]. High rate of fetal alcohol syndrome has been revealed in Croatia (43.01 per 1000), Italy (36.89 per 1000), South Africa (28.29 per 1000) [8].
Due to a tight association with disability and chronic diseases, alcoholism causes significant economic loss. In particular, the costs of heavy drinking in USA (2006) was $223.5 billion, with 72.2% from lost productivity [9]. At the same time, the costs were characterized by a increase to 2010 accounting for $249.0 billion lost in 2010 [10].
The high variability of clinical signs of alcohol abuse is related to the variety of mechanisms of ethanol toxicity [11, 12], including oxidative stress [13], inflammatory signaling [14], altered DNA methylation [15].
It has been demonstrated that ethanol toxicity is tightly associated with nutritional status [16]. In particular, trace element metabolism plays a significant role in alcohol toxicity [17]. Multiple observations have demonstrated that the most common alcohol-associated trace element disorder is zinc deficiency [18].
Zinc is an essential metal playing a significant role in functioning of the immune [19], endocrine [20], cardiovascular [21], and nervous system [22]. Biological effects of Zn are related to its cofactor, antioxidant, anti-inflammatory, and signaling function [23]. Severe Zn deficiency was first described by Prasad, being characterized by anemia, hepatosplenomegaly, hypogonadism, dwarfism, and geophagia [24]. At the same time, altered Zn metabolism was also shown to be associated with numerous disorders including type 2 diabetes mellitus, hypertension, immune deficiency, etc. [25].
Zinc is tightly associated with alcohol metabolism due to its catalytic role in alcohol dehydrogenase [26]. ADH is characterized by broad substrate specificity and oxidizes both endogenous and exogenous alcohols [12]. It has been demonstrated that maternal Zn supplementation potentiates the stimulatory effect of ethanol exposure on hepatic ADH and aldehyde dehydrogenase activity [27]. However, such a mechanistic approach cannot explain all effects of the interaction between zinc metabolism and alcohol toxicity.
Therefore, the objective of the present study was to review the mechanisms linking adverse health effects of alcohol toxicity and Zn metabolism.
Zn status in alcoholic patients
Current estimates propose that 30–50% of alcoholics are zinc deficient [28]. Multiple studies have demonstrated altered serum Zn status in various cohorts with alcohol abuse [29, 30]. Similar findings were made in alcoholic liver disease patients. In particular, patients with alcoholic liver disease were characterized by significantly lower hepatic Zn levels in comparison to the respective values in non-alcoholic liver disease [31]. The form of ALD also significantly affected Zn levels. Particularly, hepatic Zn levels were significantly decreased in patients with ALD, with the most prominent decrease in alcoholic fatty liver cases (− 57%), followed by mild alcoholic hepatitis (− 51%) and alcoholic cirrhosis (− 46%) [32]. An earlier study demonstrated that serum Zn levels were elevated in alcoholics with normal or fatty liver, whereas alcoholic hepatitis and cirrhosis were associated with low serum Zn levels. Moreover, serum Zn concentration significantly correlated with P450 content in hepatic tissue [33].
It has been demonstrated that hepatic Zn levels were significantly decreased in cirrhotic alcoholics, whereas no significant difference between non-cirrhotics and controls was detected. In turn, serum Zn was significantly lower in both cirrhotics and non-cirrhotics as compared to the control values. Correspondingly, urinary Zn losses in patients with alcoholic cirrhosis were more than twofold higher than those in both non-cirrhotic alcoholics and healthy examinees [34]. Plasma zinc levels in patients with alcoholic liver disease were significantly lower than those in the controls, with the lowest values observed in cirrhotic patients. Lower levels of plasma zinc were also associated with increased urinary Zn losses and decreased intake [35]. A recent study in Poland (Lublin) demonstrated that patients with alcoholic cirrhosis were characterized by a 30% decrease in serum Zn levels as compared to the control values. However, no significant group differences between different cirrhosis stages were observed [36]. It is interesting that Zn supplementation in alcoholics significantly increased serum metal levels, whereas hepatic Zn content remained unchanged in some patients, especially in those with cirrhosis [37], being indicative of profound alterations in hepatic Zn metabolism in ALD.
Furthermore, a significant decrease in serum zinc in parallel with nearly-significantly elevated copper levels was also detected in alcoholics [38]. It is proposed that the depression of Zn status in parallel with increased levels of redox-active Cu and Fe may result in oxidative stress, being the one of the mechanisms linking alcoholism and its effects and Zn deficiency [39].
Human observations are generally in agreement with experimental data. In particular, it has been demonstrated that ethanol feeding resulted in a significant decrease of Zn levels in testes (after 1 week), liver, hair, spleen, and kidneys (after 2 weeks), and all other tissues except bones (after 3–4 weeks of exposure) [40].
In parallel with decreased Zn status, alcohol exposure also results in altered Zn distribution. In particular, it has been demonstrated that drinking 20% ethanol results in a rapid decline in hepatic Zn stores, being the most significant in liver mitochondria (− 35% as compared to the control values) [41].
The observed changes in Zn status may be related to decreased intake or increased excretion of Zn. In particular, data on decreased Zn body burden are in agreement with the observation of increased urinary Zn levels in alcoholic patients without cirrhosis [42, 43]. Decreased Zn intake (lower than dietary reference values for the UK) was observed in 67% of alcoholics [44]. At the same time, a recent analysis of adults from USA (1999–2006) demonstrated that Zn intake is associated neither with drinking status (never, former, current), nor with the number of drinks per day [45]. Previous studies also demonstrate that 17% lower serum Zn levels in alcoholics is associated with decreased Zn-65 absorption as compared to the control group (37 vs 56%, respectively) [46, 47].
At the same time, certain studies did not confirm the presence of Zn deficiency in alcoholics. In particular, it has been demonstrated that intestinal absorption, elimination, and whole-body Zn content were significantly elevated in alcoholic cirrhosis patients in comparison to the control values, whereas hepatic Zn levels were decreased and significantly correlated with alcohol dehydrogenase activity [43]. In our recent studies we have also observed the absence of significant changes in hair [48] and serum of pregnant women with alcohol abuse and their offspring [49]. No significant changes in serum Zn levels were observed in an Italian population of male and female alcoholics [50].
Moreover, González-Reimers et al. [51] have demonstrated that chronic alcoholics are characterized by significantly higher hair Zn levels in comparison to the healthy examinees (305.89 ± 81.68 vs 219.0 ± 85.84 mg/kg, p < 0.05). A significant difference between distilled beverage and nondistilled beverage consumers was also noted (385.77 ± 98.65 vs 282.72 ± 62.54 mg/kg, p < 0.001) [51].
Generally, the existing studies demonstrate profound alterations of Zn handling in patients with alcohol abuse. The observed perturbations may at least partially mediate the toxic effects of ethanol in various tissues and organs, especially lungs, gut, liver, brain, as well as during the development of fetal alcohol syndrome. Moreover, various Zn-dependent pathways may also provide an additional link between multiorgan failure in alcoholism.
Lungs
Current estimates demonstrate that alcohol abuse significantly increases the risk of acute respiratory distress syndrome by a factor of 3–4, having a significant impact on alveolar epithelial cells and macrophages [52]. In turn, zinc was shown to play a significant role in the airways including regulation of ADAM33 metalloproteinase activity, NF-kB and inflammatory pathways, as well as redox environment [53], whereas modulation of Zn metabolism was suggested as a target for the treatment of respiratory diseases [54]. Certain studies have demonstrated the role of Zn deficiency in alcohol-induced impairment of lung functions [55]. In particular, it has been demonstrated that ethanol exposure significantly reduces intracellular zinc levels through the modulation of Zn-transporters in intestine (Zip1, Znt4, Zip4) and lung epithelial cells (Zip1). In addition, alcohol exposure was shown to be associated with a substantial decrease in Zip4, Znt1 and MT1 expression in alveolar macrophages, being associated with a significant depression of Krüppel-like factor 4 (KLF4) expression. The authors propose that the observed decrease in KLF4 and subsequent Zip4 expression may be associated with alcohol-induced alteration of GM-CSF and TGFβ1 signaling [56]. In turn, Zn supplementation significantly increased intracellular Zn levels, improved epithelial barrier function and increased GM-CSF and phagocytic activity of alveolar macrophages [57, 58].
It has been detected that alcohol exposure may cause a significant decrease in alveolar macrophage Zn deficiency without systemic Zn disorders as assessed by serum Zn levels. These changes are also associated with impaired bacterial phagocytosis and GM-SCF expression, that have been significantly reversed by treatment of alveolar macrophages with Zn [59, 60]. These findings are generally in agreement with the earlier observation of the tight interaction between Zn metabolism and GM-CSF [61], and TGFβ1 signaling [62]. Modulation of oxidative stress may also mediate the role of Zn in the development of alcoholic lung dysfunction [63], corresponding to the antioxidant effects of Zn [64].
Gut
Alcohol abuse may result in significant alterations of intestinal functions, being associated with increased gut permeability, impaired intestinal absorption of nutrients, altered gut microbiota [65]. It has been demonstrated that alcohol-induced liver damage may be mediated by gut dysfunction [66] and zinc may play a significant role in this association [67]. In turn, it is proposed that endotoxemia is the key factor contributing to hypozincemia in alcoholic liver disease [68].
In 2003, Lambert and colleagues demonstrated that Zn administration significantly reduces ethanol-induced increase in gut permeability followed by endotoxemia, elevated TNFα levels and degenerative and necrotic changes in liver [69]. The observation of Zn-induced improvement in gut permeability, endotoxemia, inflammation, and liver structure in MT-KO mice allowed to propose the presence of alternative (MT-independent) pathways of beneficial effects of Zn in alcoholic liver disease [70].
Further studies of the authors have demonstrated that ethanol exposure in mice resulted in a significant increase in ileal permeability (but not duodenal and jejunal), being associated with a significant reduction of tight junction proteins and zinc content. Moreover, zinc deprivation potentiated toxic effect of ethanol on intestinal barrier function [71]. HNF-4a also inversely correlated with alcohol-induced alteration of tight-junction proteins [72]. The protective effects of Zn supplementation against ethanol-associated gut leakiness were also investigated. In particular, alcohol exposure significantly reduced intestinal claudin-1, occludin, and zona occludens-1 (ZO-1) expression, whereas Zn supplementation in ethanol-exposed rats resulted in a significant activation of claudin-1 and ZO-1 expression in comparison to the control animals. These changes were also associated with the prevention of endotoxemia, hepatic macrophage activation and proinflammatory cytokine production [73].
Therefore, Zn deficiency may mediate increased gut permeability in alcoholics, thus contributing to endotoxemia and systemic inflammation.
Alcoholic liver disease
Zinc deficiency is believed to play a significant role in alcoholic liver disease [74]. Zn associated symptoms in ALD include skin lesions, immune dysfunction, hypogonadism, impaired wound healing, visual and mental dysfunctions [75]. It has been proposed that maintaining normal Zn intake is the one of essential components of the diet for normalization of nitrogen balance in patients with ALD [76].
Experimental studies demonstrated that alcohol consumption significantly decreases hepatic liver levels through oxidative stress-mediated alteration of hepatic zinc transporters, Zip5 and Zip 14 (down-regulation), and Zip 7 and Znt7 (up-regulation) [77]. Later study by the authors demonstrated a significant decrease in endoplasmic reticulum and mitochondrial Zn levels in association with elevated hepatic Zip13, Zip8, and Znt4 levels in ethanol-fed rats. These changes were associated with mitochondrial cytochrome c release, Bax and caspase-3 activation and apoptosis. Further in vitro studies demonstrated that ethanol-induced apoptosis, as well as ER and mitochondrial dysfunction were successfully reversed by Zn treatment [78]. At the same time, another study demonstrated that ethanol significantly increased MT and zinc transporters, SLC39A8 (Zip8) and SLC30A10 (Znt10), expression [79]. Therefore, the observed hepatic Zn deficiency in ALD is mediated through alteration of zinc transporters expression.
In vivo and in vitro experimental studies have demonstrated the mechanisms of the association between ethanol-induced Zn deficiency and liver damage. In particular, it has been also proposed that ethanol-induced Zn deficiency may significantly affect hepatic mitochondrial biogenesis and alter mitochondrial respiratory complexes I, III, and IV functioning, thus resulting in ROS overproduction and altered membrane potential [80]. Correspondingly, Zn deficiency was associated with altered HepG2 cell proliferation and regeneration, being associated with decreased production of hepatocyte growth factor (HGF), HNF-4α, insulin-like growth factor I (IGF-I), insulin-like growth factor binding protein 1 (IGFBP1), metallothionein (MT), and cyclin D1, whereas Zn supplementation improved liver regeneration, being indicative of the beneficial effect of Zn supplementation in alcoholic liver disease [81].
It has been demonstrated that alcohol consumption significantly reduced hepatic Zn levels in both Zn adequate and Zn deficient mice, being associated with more profound changes in liver. Moreover, Zn deficiency potentiated ethanol-induced inflammatory response and lipid accumulation through oxidative stress and expression of proinflammatory cytokines (IL-1β, TNFα, MCP-1, etc.) [82].
It has been noted that Zn deficiency increases IL-8 production in alcoholic liver disease through the inhibition of histone deacetylase and subsequent NF-kB activation [83]. It is also hypothesized that alcohol-induced Zn deficiency may mediate alterations in gene expression via histone modifications in ALD [84]. At the same time, it was noted that Zn and ethanol have a synergistic positive effect on autophagy in human hepatoma cells [79].
Correspondingly, certain studies have demonstrated a protective effect of Zn supplementation against ethanol-induced liver damage [85]. In particular, it has been demonstrated that Zn supplementation significantly prevented alcohol-induced decrease in hepatocyte proliferating cell nuclear antigen and hepatocyte nuclear factor 4alpha expression, being associated with decreased oxidative stress [86]. It has been demonstrated that Zn supplementation significantly reduced ethanol-induced activation of hepatic stellate cells, that are known to play a substantial role in liver fibrosis, through inhibition of MAPK, TGF-β and NF-kB signaling and antioxidant function [87].
Zn supplementation significantly reversed hepatic lipid accumulation through up-regulation of fatty acid ββ-oxidation (Acadl) and VLDL secretion genes (Mttp and Apob), being associated with the normalization of hepatocyte nuclear factor-4α and peroxisome proliferator activated receptor-α (PPAR-α) levels [88]. Zn was also capable of reduction of ethanol-induced expression of proapoptotic proteins like tumor necrosis factor (TNF)-α, TNF-R1, FasL, Fas, Fas-associated factor-1, and caspase-3 in murine liver [89]. It has been demonstrated that zinc supplementation significantly reduced ethanol-induced oxidative stress in both MT-KO and wild-type mice, being indicative of the MT-independent effects of Zn in alcoholic liver disease [90, 91].
The results of a clinical trial also demonstrated the beneficial effect of ZnSO4 supplementation on Zn status, endotoxemia, proinflammatory and fibrotic markers in alcoholic cirrhosis patients [92]. An earlier clinical study demonstrated a significant improvement in metabolic parameters including elevation of plasma prothrombin and alkaline phosphatase levels and depression of serum bilirubin concentrations in Zn-supplemented patients with alcoholic cirrhosis [93].
Finally, it is hypothesized that progressive alcoholic liver damage results in impaired removal of neurotoxic agents in parallel with Zn deficiency, ultimately leading to altered neuronal functions, thus providing a link between alcoholic liver disease and brain atrophy [94].
Brain
It has been noted that patients with alcohol brain atrophy were characterized by nearly twofold lower values of serum Zn as compared to the patients without cerebellar atrophy (971 vs 1817 μg/l) [95]. Moreover, it has been proposed that the dysbalance between zinc and selenium (both being antioxidant trace elements) and redox-active iron may also contribute to the development of brain atrophy in alcoholics [96]. These findings correspond to our earlier data indicating that chronic alcohol feeding for 8 months in rats results in a significant more than twofold decrease in brain Zn content (19.75 vs 8.62 mg/kg) [97].
Experimental studies demonstrated that Zn status may significantly affect the impact of ethanol consumption on neurotransmission. In particular, it has been demonstrated that Zn deficiency prevents ethanol-induced elevation of dopamine in nucleus accumbens. Moreover, prolonged Zn deficiency and alcohol consumption significantly affect the responsiveness of GABAA receptors [98]. At the same time, it has been demonstrated that zinc potentiates the stimulatory effect of ethanol on α1 glycine receptor [99]. Moreover, experimental data based on observations on mice with mutations in zinc-binding residue in the glycine receptor α1 indicate that Zn is essential mediator of the impact of ethanol on GlyR [100].
Our earlier data demonstrate that Zn supplementation may significantly affect alcoholic motivation, decreasing ethanol consumption by a factor of more than 2 (49.0 ± 4.6 vs 21.8 ± 5.5 ml/kg) [101]. Moreover, the protective effect of Zn supplementation against alcohol-induced changes in cortical, hippocampal, and cerebellar levels of trace elements was demonstrated [102, 103]. Zn supplementation also reversed ethanol-induced increase in NOS activity [104].
Taking into account the role of Zn in synaptic function and neurotransmitter metabolism [105], one can suppose that ethanol-induced perturbations in brain Zn handling may interfere with these processes. However, direct indications are still insufficient and further studies are required.
Correspondingly, a recent clinical trial demonstrated that Zn supplementation is effective in the treatment of hepatic encephalopathy, being significantly associated with improvement of physical component scale, but not mental component scale [106].
Fetal alcohol syndrome
Zinc is known to play a significant role in the regulation of reproduction and development, and disturbances in Zn homeostasis result in altered ontogenesis [107]. Multiple studies have proposed the role of altered Zn status in women with alcohol abuse in adverse pregnancy outcome [108]. Moreover, zinc deficiency was proposed to be a co-teratogen with alcohol abuse (IOM 1990). Correspondingly, maternal Zn supplementation in mice significantly reduced the incidence of fetal abnormalities associated with prenatal ethanol exposure [109, 110].
In particular, pregnant alcoholic women were characterized by significantly lower plasma Zn levels as compared to the control values (50.7 vs 72.2 μg/dl). In turn, cord plasma Zn in their offspring was also lower than that in children born from non-alcoholic women (65.5 vs 81.3 μg/dl) [111]. Moreover, we have detected a decrease in plasma Zn levels in Russian and Ukrainian pregnant women exposed to alcohol when compared to the healthy ones [18].
Experimental studies have demonstrated the effect of ethanol on zinc homeostasis in fetus. In particular, maternal short and long-term ethanol administration significantly decreased placental and fetal zinc uptake by 40 and 30%, respectively [112]. Moreover, Zn supplementation (10 and 40 mg/l) in ethanol-fed pregnant dams did not result in increased placental Zn transport [113]. Our previous studies also demonstrate that alcohol abuse results in Zn deficiency in both maternal and fetal organisms [114]. Perinatal ethanol exposure also resulted in the alteration of Zn distribution in offspring [115]. We have also demonstrated that not only maternal but also paternal ethanol exposure may affect Zn status in offspring. In particular, paternal or maternal alcohol feeding resulted in a significant 28% decrease in RBC Zn content, whereas combined paternal and maternal alcohol consumption caused a nearly twofold decrease in erythrocyte zinc levels as compared to the control animals [116].
It has been demonstrated that the alcohol-induced alteration of placental Zn transport is MT dependent [117]. Further studies indicate that alcohol exposure may induce MT synthesis in maternal liver, resulting in Zn sequestration and limited metal bioavailability for the fetus [118].
However, certain studies have demonstrated the absence of a significant protective effect of Zn supplementation against ethanol-induced reduction in brain weight [119]. Moreover, it has been demonstrated that alcohol exposure during the third trimester does not result in Zn deficiency in the fetal brain, and Zn supplementation did not prevent alcohol-induced Purkinje cell loss [120].
Earlier we have proposed that the potential mechanisms linking Zn deficiency and FAS may include antioxidant effect of Zn, as well as altered sonic hedgehog signaling, insulin growth factor signaling, and apoptotic stimulation [18].
Other tissues
As stated earlier, alcohol intoxication affects multiple organs and systems. There is growing evidence on the role of Zn imbalance in these processes. In particular, it has been proposed that Zn may have a protective effect in alcohol-induced bone pathology [121]. The earlier studies have demonstrated that ethanol exposure significantly reduced trabecular bone volume, whereas Zn supplementation increased osteoid area and serum vitamin D levels [95].
Zn possessed protective effects against alcoholic gastric lesions, preventing the increase in TBARS formation and ethanol-induced depression in antioxidant enzymes (superoxide dismutase and catalase) [122].
It is also proposed that Zn deficiency in alcoholics may significantly contribute to cardiac fibrosis in alcoholic cardiomyopathy [123]. Similarly to certain indications of MT-independent protective effects of Zn in alcoholic liver disease [90], it has been demonstrated that Zn supplementation prevents the formation of cardiac fibrosis but not myocardial hypertrophy in ethanol-exposed MT-KO mice [124].
It has been demonstrated that ethanol feeding results in a significant increase in thyroid-stimulating hormone levels in parallel with reduced T3 and T4 concentrations, whereas Zn supplementation reversed these changes. Moreover, Zn supplementation also prevented ethanol-induced decrease in serum Ca and elevation of Na levels [125].
However, no beneficial effect of Zn supplementation was observed in alcoholic myopathy model associated with IIa and IIb fiber atrophy, oxidative stress, and decreased IGF-1 levels [126]. Correspondingly, Zn supplementation did not improve the symptoms of alcoholic myopathy [127].
Although data on the impact of ethanol-induced Zn deficiency in damage of other tissues and organs in alcoholics are insufficient, it is expected that further studies will highlight additional mechanisms.
Conclusion
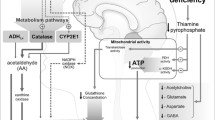
The existing data demonstrate a significant role of zinc deficiency in alcohol related organ injury. It is demonstrated that alcohol-induced modulation of zinc transporters results in decreased Zn levels in lungs, liver, gut, placenta, and brain. Zn deficiency in the gut results in increased gut permeability, ultimately leading to endotoxemia and systemic inflammation. Similarly, Zn deficiency in lung epithelia and alveolar macrophages decreases lung barrier function resulting in respiratory distress syndrome. In turn, increased endotoxemia significantly contributes to proinflammatory state in alcoholic liver disease. Finally, impaired gut and liver functions may play a significant role in alcoholic brain damage, being associated with both increased proinflammatory signaling and accumulation of neurotoxic metabolites. Moreover, ethanol-induced Zn deficiency may interfere with neurotransmission. Similar changes may take place in the fetus as a result of impaired placental zinc transfer, resulting in fetal alcoholic syndrome. Therefore, alcoholic Zn deficiency not only mediates the adverse effects of ethanol exposure, but also provides an additional link between different alcohol-induced disorders (Fig. 1).

Involvement of Zn metabolism in pathways of ethanol toxicity in gut, lungs, liver, brain and development of fetal alcoholic syndrome
Generally, current findings suggest that assessment of Zn status could be used as a diagnostic marker of metabolic disturbances in alcohol abuse, whereas modulation of Zn metabolism may be a potential tool in treatment of alcohol-associated disorders. However, potential limitations of various Zn status biomarkers should be taken into account.
References
Grant BF, Goldstein RB, Saha TD, Chou SP, Jung J, Zhang H, Pickering RP, Ruan WJ, Smith SM, Huang B (2015) Epidemiology of DSM-5 alcohol use disorder: results from the national epidemiologic survey on alcohol and related conditions III. JAMA Psychiatry 72:757–766
Organization WH (2011) Global status report on alcohol and health. World Health Organization, Geneva
Shield KD, Parry C, Rehm J (2014) Chronic diseases and conditions related to alcohol use. Alcohol Res 35:155
Parry CD, Patra J, Rehm J (2011) Alcohol consumption and non-communicable diseases: epidemiology and policy implications. Addiction 106:1718–1724
Mukherjee S (2013) Alcoholism and its effects on the central nervous system. Curr Neurovasc Res 10:256–262
Zahr NM, Pfefferbaum A (2017) Alcohol’s effects on the brain: neuroimaging results in humans and animal models. Alcohol Res Curr Rev 38:183–206
Greenfield SF, Back SE, Lawson K, Brady KT (2010) Substance abuse in women. Psychiatr Clin North Am 33:339–355
Roozen S, Peters GJY, Kok G, Townend D, Nijhuis J, Curfs L (2016) Worldwide prevalence of fetal alcohol spectrum disorders: a systematic literature review including meta-analysis. Alcohol Clin Exp Res 40:18–32
Bouchery EE, Harwood HJ, Sacks JJ, Simon CJ, Brewer RD (2011) Economic costs of excessive alcohol consumption in the US, 2006. Am J Prev Med 41:516–524
Sacks JJ, Gonzales KR, Bouchery EE, Tomedi LE, Brewer RD (2015) 2010 national and state costs of excessive alcohol consumption. Am J Prev Med 49:e73–e79
Badger TM, Ronis MJ, Seitz HK, Albano E, Ingelman-Sundberg M, Lieber CS (2003) Alcohol metabolism: role in toxicity and carcinogenesis. Alcohol Clin Exp Res 27:336–347
Cederbaum AI (2012) Alcohol metabolism. Clin Liver Dis 16:667–685
Wu D, Cederbaum AI (2003) Alcohol, oxidative stress, and free radical damage. Alcohol Res Health 27:277–284
Mandrekar P, Szabo G (2009) Signalling pathways in alcohol-induced liver inflammation. J Hepatol 50:1258–1266
Seitz HK, Stickel F (2007) Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer 7:599–612
Seitz H, Suter P (2002) Ethanol toxicity and nutritional status. In: Kotsonis, FN, Mackey, MA (eds) Nutritional toxicology, 2nd edn. Taylor & Francis, New York, pp 122–154
Aaseth J, Ringstad J, Bell H, Thomassen Y (1990) Alcohol, trace elements and liver dysfunction. In: Tomita H (ed) Trace elements in clinical medicine, pp 79–84
Keen CL, Uriu-Adams JY, Skalny A, Grabeklis A, Grabeklis S, Green K, Yevtushok L, Wertelecki WW, Chambers CD (2010) The plausibility of maternal nutritional status being a contributing factor to the risk for fetal alcohol spectrum disorders: the potential influence of zinc status as an example. Biofactors 36:125–135
Haase H, Rink L (2014) Multiple impacts of zinc on immune function. Metallomics 6:1175–1180
Li YV (2014) Zinc and insulin in pancreatic beta-cells. Endocrine 45:178–189
Foster M, Samman S (2010) Zinc and redox signaling: perturbations associated with cardiovascular disease and diabetes mellitus. Antioxid Redox Signal 13:1549–1573
Marger L, Schubert C, Bertrand D (2014) Zinc: an underappreciated modulatory factor of brain function. Biochem Pharmacol 91:426–435
Prasad AS (2014) Zinc is an antioxidant and anti-inflammatory agent: its role in human health. Front Nutr 1:14
Prasad AS, Halsted JA, Nadimi M (1961) Syndrome of iron deficiency anemia, hepatosplenomegaly, hypogonadism, dwarfism and geophagia. Am J Med 31:532–546
Chasapis CT, Loutsidou AC, Spiliopoulou CA, Stefanidou ME (2012) Zinc and human health: an update. Arch Toxicol 86:521–534
Lieber CS (2000) Alcohol: its metabolism and interaction with nutrients. Annu Rev Nutr 20:395–430
Skalny AV, Kampov-Polevoy AB, Voronin AE (2001) Influence of zinc on ethanol metabolizing enzymes activity in the offspring of rats submitted to the alcohol intoxication. Trace Elem Med (Moscow) 2:21–23
Deshpande JD, Joshi MM, Giri PA (2013) Zinc: the trace element of major importance in human nutrition and health. Int J Med Sci Public Health 2:1–6
Atukorala T, Herath C, Ramachandran S (1986) Zinc and vitamin A status of alcoholics in a medical unit in Sri Lanka. Alcohol Alcohol Suppl 21:269–275
Avaroglu D, Inal TC, Demir M, Attila G, Acartürk E, Evlice YE, Kayrin L (2005) Biochemical indicators and cardiac function tests in chronic alcohol abusers. Croat Med J 46:233–237
Kiilerich S, Dietrichson O, Loud F, Naestoft J, Christoffersen P, Juhl E, Kjems G, Christiansen C (1980) Zinc depletion in alcoholic liver diseases. Scand J Gastroenterol 15:363–367
Bode JC, Hanisch P, Henning H, Koenig W, Richter FW, Bode C (1988) Hepatic zinc content in patients with various stages of alcoholic liver disease and in patients with chronic active and chronic persistent hepatitis. Hepatology 8:1605–1609
Hartoma TR, Sotaniemi E, Pelkonen O, Ahlqvist J (1977) Serum zinc and serum copper and indices of drug metabolism in alcoholics. Eur J Clin Pharmacol 12:147–151
Rodriguez-Moreno F, González-Reimers E, Santolaria-Fernandez F, Galindo-Martin L, Hernandez-Torres O, Batista-Lopez N, Molina-Perez M (1997) Zinc, copper, manganese, and iron in chronic alcoholic liver disease. Alcohol 14:39–44
Bergheim I, Parlesak A, Dierks C, Bode J, Bode C (2003) Nutritional deficiencies in German middle-class male alcohol consumers: relation to dietary intake and severity of liver disease. Eur J Clin Nutr 57:431
Prystupa A, Błażewicz A, Kiciński P, Sak JJ, Niedziałek J, Załuska W (2016) Serum concentrations of selected heavy metals in patients with alcoholic liver cirrhosis from the Lublin region in Eastern Poland. Int J Environ Res Public Health 13:582
Zarski J, Arnaud J, Labadie H, Beaugrand M, Favier A, Rachail M (1987) Serum and tissue concentrations of zinc after oral supplementation in chronic alcoholics with or without cirrhosis. Gastroenterol Clin Biol 11:856–860
Wu C-T, Lee J-N, Shen WW, Lee S-L (1984) Serum zinc, copper, and ceruloplasmin levels in male alcoholics. Biol Psychiatry 19:1333–1338
Babenko GA, Skalny AV. Spontaneous and induced chemiluminescence of blood serum at chronic alcoholism. Trace Elem Med (Moscow) 2:46–47
Ahmed SB, Russell RM (1982) The effect of ethanol feeding on zinc balance and tissue zinc levels in rats maintained on zinc-deficient diets. J Lab Clin Med 100:211–217
Wang J, Pierson RN (1975) Distribution of zinc in skeletal muscle and liver tissue in normal and dietary controlled alcoholic rats. J Lab Clin Med 85:50–58
Sullivan JF, Lankford HG (1965) Zinc metabolism and chronic alcoholism. Am J Clin Nutr 17:57–63
Mills PR, Fell GS, Bessent RG, Nelson LM, Russell RI (1983) A study of zinc metabolism in alcoholic cirrhosis. Clin Sci (Lond) 64:527–535
Manari A, Preedy V, Peters T (2003) Nutritional intake of hazardous drinkers and dependent alcoholics in the UK. Addict Biol 8:201–210
Breslow RA, Guenther PM, Juan W, Graubard BI (2010) Alcoholic beverage consumption, nutrient intakes, and diet quality in the US adult population, 1999–2006. J Am Diet Assoc 110:551–562
Dinsmore W, Callender ME, McMaster D, Todd J, Love AH (1985) Zinc absorption in alcoholics using zinc-65. Digestion 32:238–242
Valberg L, Flanagan P, Ghent C, Chamberlain M (1985) Zinc absorption and leukocyte zinc in alcoholic and nonalcoholic cirrhosis. Dig Dis Sci 30:329–333
Skalny A, Berezkina E, Grabeklis A, Kiyaeva E, Tinkov A (2016) Hair trace elements in women with alcohol abuse and their offspring. Trace Elem Electrolytes 33:144
Skalny AV, Berezkina ES, Kiyaeva EV, Alidzhanova IE, Grabeklis AR, Tinkov AA (2016) The effect of alcohol consumption on maternal and cord blood electrolyte and trace element levels. Acta Sci Pol Technol Aliment 15:439–445
Mancinelli R, Barlocci E, Ciprotti M, Senofonte O, Fidente RM, Draisci R, Attilia ML, Vitali M, Fiore M, Ceccanti M (2013) Blood thiamine, zinc, selenium, lead and oxidative stress in a population of male and female alcoholics: clinical evidence and gender differences. Ann Ist Super Sanita 49:65–72
Gonzalez-Reimers E, Aleman-Valls M, Barroso-Guerrero F, Santolaria-Fernandez F, Lopez-Lirola A, Campelo EG-V, Jarque-Lopez A, Rodriguez-Gaspar M (2002) Hair zinc and copper in chronic alcoholics. Biol Trace Elem Res 85:269
Joshi PC, Guidot DM (2007) The alcoholic lung: epidemiology, pathophysiology, and potential therapies. Am J Physiol Lung Cell Mol Physiol 292:L813–L823
Zalewski PD (2006) Zinc metabolism in the airway: basic mechanisms and drug targets. Curr Opin Pharmacol 6(3):237–243
Zalewski PD, Truong-Tran AQ, Grosser D, Jayaram L, Murgia C, Ruffin RE (2005) Zinc metabolism in airway epithelium and airway inflammation: basic mechanisms and clinical targets. A review. Pharmacol Ther 105(2):127–149
Janssen WJ (2013) Alveolar macrophage dysfunction and chronic alcohol use. Time to think about zinc. Am J Respir Crit Care Med 188:716–723
Curry-McCoy TV, Guidot DM, Joshi PC (2013) Chronic alcohol ingestion in rats decreases Krüppel-like factor 4 expression and intracellular zinc in the lung. Alcohol Clin Exp Res 37:361–371
Joshi PC, Guidot DM (2008) Zinc deficiency suppresses alveolar macrophage function in in vivo models of both chronic alcohol ingestion and HIV-1 transgenic expression. FASEB J 22:856.823
Joshi PC, Mehta A, Jabber WS, Fan X, Guidot DM (2009) Zinc deficiency mediates alcohol-induced alveolar epithelial and macrophage dysfunction in rats. Am J Respir Cell Mol Biol 41:207–216
Mehta AJ, Yeligar SM, Brown LA, Guidot DM (2012) Zinc supplementation in vitro improves immune function and restores intracellular zinc levels in alveolar macrophages isolated from human alcoholics. Recent advances in phagocyte biology. Am Thoracic Soc:A1369–A1369
Mehta AJ, Yeligar SM, Elon L, Brown LA, Guidot DM (2013) Alcoholism causes alveolar macrophage zinc deficiency and immune dysfunction. Am J Respir Crit Care Med 188:716–723
Vignesh KS, Figueroa JAL, Porollo A, Caruso JA, Deepe GS (2013) Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity 39(4):697–710
Maywald M, Wessels I, Rink L (2017) Zinc signals and immunity. Int J Mol Sci 18(10):2222
Mehta AJ, Guidot DM (2014) Alcohol-mediated zinc deficiency within the alveolar space: a potential fundamental mechanism underlying oxidative stress and cellular dysfunction in the alcoholic lung.Alcohol use disorders and the lung. Springer, Berlin, pp 173–184
Kloubert V, Rink L (2015) Zinc as a micronutrient and its preventive role of oxidative damage in cells. Food Funct 6(10):3195–3204
Bode C, Bode JC (2003) Effect of alcohol consumption on the gut. Best Pract Res Clin Gastroenterol 17:575–592
Szabo G (2015) Gut–liver axis in alcoholic liver disease. Gastroenterology 148:30–36
Chen P, Schnabl B (2014) Host–microbiome interactions in alcoholic liver disease. Gut liver 8:237
McClain CJ, Hill DB, Song Z, Deaciuc I, Barve S (2002) Monocyte activation in alcoholic liver disease. Alcohol 27:53–61
Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ (2003) Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. J Pharmacol Exp Ther 305:880–886
Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ (2004) Preservation of intestinal structural integrity by zinc is independent of metallothionein in alcohol-intoxicated mice. Am J Pathol 164:1959–1966
Zhong W, McClain CJ, Cave M, Kang YJ, Zhou Z (2010) The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am J Physiol Gastrointest Liver Physiol 298:G625-G633
Zhou Z, Zhong W (2012) Zinc and hepatocyte nuclear factor-4α in alcohol-induced intestinal barrier dysfunction. J Epithel Biol Pharmacol 5
Zhong W, Li Q, Sun Q, Zhang W, Zhang J, Sun X, Yin X, Zhang X, Zhou Z (2015) Preventing gut leakiness and endotoxemia contributes to the protective effect of zinc on alcohol-induced steatohepatitis in rats. J Nutr 145:2690–2698
Singal AK, Kamath PS, Gores GJ, Shah VH (2014) Alcoholic hepatitis: current challenges and future directions. Clin Gastroenterol Hepatol 12:555–564
Beier JI, Arteel GE, McClain CJ (2011) Advances in alcoholic liver disease. Curr Gastroenterol Rep 13:56–64
McCullough AJ, O’Shea RS, Dasarathy S (2011) Diagnosis and management of alcoholic liver disease. J Dig Dis 12:257–262
Sun Q, Li Q, Zhong W, Zhang J, Sun X, Tan X, Yin X, Sun X, Zhang X, Zhou Z (2014) Dysregulation of hepatic zinc transporters in a mouse model of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 307:G313-G322
Sun Q, Zhong W, Zhang W, Li Q, Sun X, Tan X, Sun X, Dong D, Zhou Z (2015) Zinc deficiency mediates alcohol-induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. Am J Physiol Gastrointest Liver Physiol 308:G757-G766
Liuzzi J, Yoo C (2013) Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol Trace Elem Res 156:350–356
Sun Q, Zhong W, Zhang W, Zhou Z (2016) Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: role of zinc deficiency. Am J Physiol Gastrointest Liver Physiol 310:G205–G214
Kang X, Song Z, McClain CJ, Kang YJ, Zhou Z (2008) Zinc supplementation enhances hepatic regeneration by preserving hepatocyte nuclear factor-4α in mice subjected to long-term ethanol administration. Am J Pathol 172:916–925
Zhong W, Zhao Y, Sun X, Song Z, McClain CJ, Zhou Z (2013) Dietary zinc deficiency exaggerates ethanol-induced liver injury in mice: involvement of intrahepatic and extrahepatic factors. PloS One 8:e76522
Zhao Y, Zhong W, Sun X, Song Z, Clemens DL, Kang YJ, McClain CJ, Zhou Z (2011) Zinc deprivation mediates alcohol-induced hepatocyte IL-8 analog expression in rodents via an epigenetic mechanism. Am J Pathol 179:693–702
Moghe A, Joshi-Barve S, Ghare S, Gobejishvili L, Kirpich I, McClain CJ, Barve S (2011) Histone modifications and alcohol-induced liver disease: are altered nutrients the missing link? World J Gastroenterol 17:2465
Kotegov VP, Skalny AV, Brudastov JA, Suldin AV, Malkova JG (2012) Research of action of zinc sulphate on glucose tolerance, hypoglycemic effect of insulin and ethanol toxicity. Prob Biol Med Pharm Chem 6:63–67
Xiao M, Liu C, Sun W, Dong M, Hu G, Li J (2012) Zinc supplementation effects on alcoholic liver disease and the molecular mechanism. Zhongguo Ying Yong Sheng Li Xue Za Zhi 28:84–88
Szuster-Ciesielska A, Plewka K, Daniluk J, Kandefer-Szerszeń M (2009) Zinc supplementation attenuates ethanol-and acetaldehyde-induced liver stellate cell activation by inhibiting reactive oxygen species (ROS) production and by influencing intracellular signaling. Biochem Pharmacol 78:301–314
Kang X, Zhong W, Liu J, Song Z, McClain CJ, Kang YJ, Zhou Z (2009) Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4α and peroxisome proliferator-activated receptor-α. Hepatology 50:1241–1250
Zhou Z, Liu J, Song Z, McClain CJ, Kang YJ (2008) Zinc supplementation inhibits hepatic apoptosis in mice subjected to a long-term ethanol exposure. Exp Biol Med (Maywood) 233:540–548
Zhou Z, Sun X, Lambert JC, Saari JT, Kang YJ (2002) Metallothionein-independent zinc protection from alcoholic liver injury. Am J Pathol 160:2267–2274
Zhou Z, Wang L, Song Z, Saari JT, McClain CJ, Kang YJ (2005) Zinc supplementation prevents alcoholic liver injury in mice through attenuation of oxidative stress. Am J Pathol 166:1681–1690
Mohammad MK, Falkner KC, Song M, McClain CJ, Cave MC (2015) Low dose zinc sulfate (220 mg) supplementation for three months normalizes zinc levels, endotoxeima, pro-inflammatory/fibrotic biomarkers & improves clinical parameters in alcoholic cirrhosis-a double-blind placebo controlled-(ZAC) clinical trial. Hepatology 62:851A–852A
Weismann K, Christensen E, Dreyer V (1979) Zinc supplementation in alcoholic cirrhosis. J Intern Med 205:361–366
Liu J, Lewohl JM, Harris RA, Dodd PR, Mayfield RD (2007) Altered gene expression profiles in the frontal cortex of cirrhotic alcoholics. Alcohol Clin Exp Res 31:1460–1466
Gonzalez-Reimers E, Durán-Castellón M, Martín-Olivera R, López-Lirola A, Santolaria-Fernández F, De la Vega-Prieto M, Pérez-Ramírez A, Campelo EG-V (2005) Effect of zinc supplementation on ethanol-mediated bone alterations. Food Chem Toxicol 43:1497–1505
González-Reimers E, Santolaria-Fernández F (2011) Brain atrophy in alcoholics. Handbook of behavior, food and nutrition. Springer, Berlin, pp 2993–3010
Skal’nyi A, Kukhtina E, Ol’khovskaya I, Glushchenko N (1992) Reduction of voluntary ethanol consumption by a long-acting zinc preparation. Bull Exp Biol Med 113:506–509
Morud J, Adermark L, Ericson M, Söderpalm B (2015) Alterations in ethanol-induced accumbal transmission after acute and long-term zinc depletion. Addict Biol 20:170–181
McCracken LM, Trudell JR, Goldstein BE, Harris RA, Mihic SJ (2010) Zinc enhances ethanol modulation of the α1 glycine receptor. Neuropharmacology 58:676–681
McCracken LM, Blednov YA, Trudell JR, Benavidez JM, Betz H, Harris RA (2013) Mutation of a zinc-binding residue in the glycine receptor α1 subunit changes ethanol sensitivity in vitro and alcohol consumption in vivo. J Pharmacol Exp Ther 344:489–500
Skalny A, Kampov-Polevoy A (1988) Zinc sulfate in the prevention of alcohol motivation in rats. Biull Eksp Biol Med 105
Vyatchanina ES, Skalny AV (2007) Protectant action of zinc sulfate at intrauterine alcohol influence. Vestnik Orenburg State Univ 12:113–116
Vyatchanina ES (2011) Preconceptional alcoholic intoxication alters the distribution of metals in matured rat brain of offspring. J Trace Elem Med Biol 25:S59–S62
Maksymovych I, Kharchenko O, Chaika V, Gadilia O, Ostapchenko L (2010) Nitric oxide synthase activity in liver and brain cells under development of chronic alcoholic intoxication and zinc acetate treatment. Alcohol Clin Exp Res 34:168A
Sensi SL, Paoletti P, Koh J-Y, Aizenman E, Bush AI, Hershfinkel M (2011) The neurophysiology and pathology of brain zinc. J Neurosci 31:16076–16085
Takuma Y, Nouso K, Makino Y, Hayashi M, Takahashi H (2010) Clinical trial: oral zinc in hepatic encephalopathy. Aliment Pharmacol Ther 32:1080–1090
Uriu-Adams JY, Keen CL (2010) Zinc and reproduction: effects of zinc deficiency on prenatal and early postnatal development. Birth Defects Res B Dev Reprod Toxicol 89:313–325
May PA, Gossage JP (2011) Maternal risk factors for fetal alcohol spectrum disorders: not as simple as it might seem. Alcohol Res Health 34:15
Miller SI, Del Villano BC, Flynn A, Krumhansl M (1983) Interaction of alcohol and zinc in fetal dysmorphogenesis. Pharmacol Biochem Behav 18:311–315
Carey LC, Coyle P, Philcox JC, Rofe AM (2003) Zinc supplementation at the time of ethanol exposure ameliorates teratogenicity in mice. Alcohol Clin Exp Res 27:107–110
Flynn A, Martier S, Sokol R, Miller S, Golden N, Del Villano B (1981) Zinc status of pregnant alcoholic women: a determinant of fetal outcome. Lancet 317:572–575
Ghishan FK, Patwardhan R, Greene HL (1982) Fetal alcohol syndrome: inhibition of placental zinc transport as a potential mechanism for fetal growth retardation in the rat. J Lab Clin Med 100:45–52
Ghishan FK, Greene HL (1983) Fetal alcohol syndrome: failure of zinc supplementation to reverse the effect of ethanol on placental transport of zinc. Pediatr Res 17:529–531
Skalny AV (1987) Zinc deficiency in mothers and their offspring in alcohol abuse. Akush Gynecol 4:6–8
Murillo-Fuentes M, Reyes A, MaLuisa O, Murillo M, Carreras O (2010) Different effects on zinc redistribution if ethanol is consumed before or immediately after birth. J Trace Elem Med Biol 24:200–206
Skalny AV, Slavin FI (1988) Mineral exchange and tissue-specific cytotoxic activity of blood serum in offspring at chronic alcohol intoxication in parents. S. S. Korsakov J Neurol Psychiatry 88:107–109
Carey LC, Coyle P, Philcox JC, Rofe AM (2000) Ethanol decreases zinc transfer to the fetus in normal but not metallothionein-null mice. Alcohol Clin Exp Res 24:1236–1240
Coyle P, Martin SA, Carey LC, Summers BL, Rofe AM (2009) Ethanol-mediated fetal dysmorphology and its relationship to the ontogeny of maternal liver metallothionein. Alcohol Clin Exp Res 33:1051–1058
Samson HH, Diaz J (1981) Altered development of brain by neonatal ethanol exposure: zinc levels during and after exposure. Alcohol Clin Exp Res 5:563–569
Chen WJA, Berryhill EC, West JR (2001) Zinc supplementation does not attenuate alcohol-induced cerebellar Purkinje cell loss during the brain growth spurt period. Alcohol Clin Exp Res 25:600–605
González-Reimers E, Santolaria-Fernández F, Alvisa-Negrín J (2013) Bone changes in alcoholics: a review. Alcohol 1:16
Ineu RP, Oliveira CS, Oliveira VA, Moraes-Silva L, da Luz SCA, Pereira ME (2013) Antioxidant effect of zinc chloride against ethanol-induced gastrointestinal lesions in rats. Food Chem Toxicol 58:522–529
Awtry EH, Philippides GJ (2010) Alcoholic and cocaine-associated cardiomyopathies. Prog Cardiovasc Dis 52:289–299
Wang L, Zhou Z, Saari JT, Kang YJ (2005) Alcohol-induced myocardial fibrosis in metallothionein-null mice: prevention by zinc supplementation. Am J Pathol 167:337–344
Pathak R, Dhawan D, Pathak A (2011) Effect of zinc supplementation on the status of thyroid hormones and Na, K, and Ca levels in blood following ethanol feeding. Biol Trace Elem Res 140:208–214
Castellón MD, González-Reimers E, López-Lirola A, Olivera RM, Santolaria-Fernández F, Galindo-Martín L, Abreu-González P, González-Hernández T (2005) Alcoholic myopathy: lack of effect of zinc supplementation. Food Chem Toxicol 43:1333–1343
Jung MK, Callaci JJ, Lauing KL, Otis JS, Radek KA, Jones MK, Kovacs EJ (2011) Alcohol exposure and mechanisms of tissue injury and repair. Alcohol Clin Exp Res 35:392–399
Acknowledgements
The current investigation is supported by the Russian Foundation for Basic Research within Project No. 15-04-08621.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Skalny, A.V., Skalnaya, M.G., Grabeklis, A.R. et al. Zinc deficiency as a mediator of toxic effects of alcohol abuse. Eur J Nutr 57, 2313–2322 (2018). https://doi.org/10.1007/s00394-017-1584-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00394-017-1584-y