
Abstract
The preparation and characterization of thiol-functionalized graphene oxide–iron oxide (Fe3O4–GO–SH) nanocomposites (novel magnetic adsorbents) for the simultaneous preconcentration and determination of heavy metal ions, such as Ag(I), Pb(II), and Cd(II), from water samples were carried out in the present research. The characterization of the resultant Fe3O4–GO–SH nanocomposites was performed by SEM, TEM, EDX, XRD, FT-IR, and VSM. The preconcentration optimization of pH solution, adsorbent amount, ultrasonic power for adsorption and desorption processes, adsorption time, and elution solvent type and concentration was performed by the ultrasonic-assisted magnetic solid-phase extraction. Under optimal conditions, linear ranges were found to be 20–1000 μg L–1 for Ag(I) and Cd(II) ions and 200–10,000 μg L–1 for Pb(II) ions with a regression coefficient of R2 > 0.99. The limits of detection for Ag, Pb, and Cd ions were 1.7 μg L–1, 14.1 μg L–1, and 0.9 μg L–1, respectively, and the limits of quantification for Ag, Pb, and Cd ions were 5.9 μg L–1, 47.3 μg L–1, and 3.2 μg L–1, respectively. The recovery rate of these metal ions for water samples (drinking water, tap water, swamp water, and wastewater from plating plants) ranged between 80 and 115%. Therefore, the proposed method can potentially be used for the simultaneous preconcentration and determination of heavy metal ions from real water samples, and its reusability can be easily carried out by the deionized water treatment.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Ultrasonic-assisted preconcentration extraction is a popular sample preparation method due to its numerous advantages, including small sample volume, short extraction time, and high extraction efficiency [1,2,3]. Moreover, this preconcentration method is widely used for the enrichment of trace metals by increasing the dispersive mass transfer of adsorbents and analytical molecules under ultrasonic irradiation [4, 5]. Hence, the ultrasonic-assisted technique has a promising extraction performance. To increase the analytical concentration through the removal of sample matrixes or interferences, different preconcentration methods, such as dispersive liquid–liquid microextraction (DLLME) [6], cloud point extraction (CPE) [7], hollow fiber solid/liquid microextraction [8], ion-exchange chromatography [9], and solid-phase microextraction (SPME) [10,11,12], have been applied. SPME has the advantages of easy operation, short contact time, small eluent volume, low laboratory cost, and high preconcentration factor, and thus, it is an effective method for the preconcentration of analytes. However, SPME still faces some separation limitations for nanosorbents; therefore, magnetic materials are introduced to solve this problem. Magnetic (Fe3O4) is the most common iron oxide used in biomedicine due to its low toxicity, relative ease of functionalization, and high magnetization at room temperature [13,14,15]. It can improve the separation of nanosorbents and expand the use of the conventional SPE limit. Sorbents are not required to be packed into cartridges, and phase separation can be easily realized under an external magnetic field. In general, nanosorbents have a high surface area, a good adsorption capacity, and a short adsorption rate; thus, rapid equilibrium time and facile analyte extraction could be achieved by incorporating a small amount of sorbent.
Among several nanosorbents, graphene oxide (GO) attracts considerable attention due to its high preconcentration capability for different analytes, such as metal ions [16, 17]. GO is a two-dimensional structure of a single carbon layer [18]. GO manifests numerous interesting properties, such as low weight, high porosity, high surface area, non-toxic, high chemical and thermal stability, excellent electrical properties, high adsorption capacity, and low cost [19,20,21], and thus, it has an excellent ability for the preconcentration of heavy metals. Moreover, it contains abundant reactive oxygen-containing functional groups, such as hydroxyl, epoxy, and carboxylic [17]. In an effort to increase the chemical versatility, chemical functionalization is an easy way to modify GO. Avashthi et al. explored the use of 2-amino-1,3,4-thidiazole functionalized GO for photoluminescence and nanocytotoxicity activities [22]. It appeared as an advanced material which can be used for development of various biomedical applications. The nitrogen functionalized GO has proven useful for thermally stable bioscaffold [23]. Moreover, the surface modification of GO by nitrogen species showed potential optically active photoluminescence properties [24], in vitro cytotoxicity, and antioxidant evaluations [25]. Furthermore, the functionalization of GO by thiol groups increases its adsorption capacity [26]. The thiol group is an excellent ligand due to its strong affinity to several metal ions, such as Cd(II), Ni(II), Cu(II), Cr(II), Hg(II), and Pb(II) [27,28,29], and it can be attributed to Lewis acid–base interactions based on Pearson’s Hard Soft Acid–Base theory [28]. Due to its soft base characteristics, the thiol group can interact with soft acid metals or borderline metal ions [29]. Therefore, the combination of thiol-functionalized GO with Fe3O4 as an adsorbent for ultrasonic-assisted magnetic solid-phase extraction (UA-MSPE) can improve the preconcentration efficiency of Ag(I), Pb(II), and Cd(II) metal ions in water samples and reduce the contact time in the preconcentration process. In addition, the ultrasonic technique can also be used for adsorbent synthesis. Furthermore, a small absorbent particle size leads to a short synthesis time and fast mass transfer [30,31,32]. Therefore, the UA-MSPE technique can improve the performance of simultaneous preconcentration and determination of Ag(I), Pb(II), and Cd(II) metal ions in water samples.
In the present work, the preparation, characterization, and application of Fe3O4–GO–SH as a new magnetic nanosorbent for UA-MSPE were studied. Fe3O4–GO–SH was synthesized from 3-mercaptopropyl trimethoxysilane by a facile co-precipitation method under ultrasonic irradiation. The resultant magnetic nanosorbent was characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD), attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FT-IR), and vibrating sample magnetometry (VSM). The UA-MSPE based on Fe3O4–GO–SH, was used as an efficient sample preconcentration method for the determination of selected metal ions [Ag(I), Pb(II), and Cd(II)] in ten different types of water samples, followed by atomic absorption spectrometry (AAS). Alternatively, this approach can be considered green, because we present a simple, efficient as well as green way to synthesize Fe3O4–GO–SH using ultrasonic technology. Then UA-MSPE was carried out under the synergistic effect of ultrasound action and vibration, which significantly improved extraction and preconcentration efficiency, enhanced sample throughput, and accelerated the process of extraction.
2 Experimental section
2.1 Chemicals and reagents
All chemicals used in the present work were of analytical grade. Ferric chloride hexahydrate, ferrous sulfate heptahydrate, nitric acid, and sodium acetate were purchased from QRёC (Auckland, New Zealand). 3-mercaptopropyl trimethoxysilane and graphite powder were procured from Sigma-Aldrich (Saint Louis, USA). Lead nitrate and cadmium acetate dihydrate were supplied by Fluka Chemie AG (Buchs, Switzerland). Silver nitrate and ammonium hydroxide were purchased from Carlo Erba (Milan, Italy) and AnalaR (Poole, England), respectively. A water purification system (Molsheim, France) was used to produce deionized water with a resistivity of 18.2 MΩ cm.
2.2 Characterization
ATR-FT-IR spectra were measured by a TENSOR 27 system (Bruker Optics, Germany) in the range of 500–4000 cm–1. EDX spectra and surface images were recorded by a HITACHI S–3000 N SEM (Hitachi Co. Ltd., Japan). XRD patterns were characterized by a PANalytical Empyrean instrument (Netherlands) under monochromatic Cu–Kα radiation (λ = 0.15406 nm) with 2θ = 15°–70° and a scanning speed of 4 min–1. A Lake Shore VSM 7403 (USA) was used to examine magnetic properties. TEM images were recorded by a Tecnai G2 20 FEI (USA) operating at 200 kV. An ultrasonic bath (Model GT Sonic, China) with a tank size of 36 cm × 65 cm × 40 cm, a frequency of 40 kHz, the maximum ultrasonic power of 720 W, and a heater operating at 20–80 °C with a heating power of 800 W was applied for UA-MSPE. The solution pH was measured by a UB-10 Ultra Basic pH meter. The detection of Ag(I), Cd(II), and Pb(II) ions in water samples were performed by a Perkin Elmer Instrument AA Analyst 100 (USA).
2.3 Synthesis of Fe3O4–GO and Fe3O4–GO–SH
Magnetic graphene oxide (Fe3O4–GO) was synthesized by the co-precipitation method under ultrasonic irradiation. FeCl3·6H2O (0.2 g) and FeSO4·7H2O (0.1 g) at a ratio of 2:1 (w/w) [33] were dissolved in deionized water (9 mL) and sonicated in an ultrasonic bath at 0.2 kW for 5 min. Ammonia solution (1 mL, 30%, w/v) and GO were then dropped into the mixture. A black precipitate of Fe3O4 magnetic nanoparticles (MNPs) appeared. The obtained Fe3O4 MNPs were washed with deionized water until the solution pH became neutral and dried in an oven at 50 °C for 12 h.
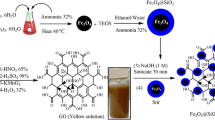
Thiol-functionalized magnetic graphene oxide (Fe3O4–GO–SH) was prepared by the reaction of 3-mercaptopropyl trimethoxysilane (MPTMS) with Fe3O4–GO. The Fe3O4–GO powder was dispersed in ethanol (1 mL) and sonicated in an ultrasonic bath at 0.2 kW for 3 h. MPTMS (300 µL) was then added to the mixture, heated to 45 °C, and sonicated at 0.2 kW for 20 min. Finally, Fe3O4–GO–SH was dried in an oven at 50 °C for 12 h. The resultant sorbent was also characterized by SEM, TEM, EDX, XRD, FT-IR, and VSM. The synthesis route of Fe3O4–GO–SH is illustrated in Scheme 1.

The synthesis of Fe3O4–GO–SH as a novel adsorbent by ultrasonic-assisted method
2.4 Preparation of standard solutions and samples
For each metal analyte, 25 mL stock solutions at concentrations of 1 g L−1 were prepared in deionized water. Working standards were prepared daily from the stock solutions by dilution with deionized water to a final concentration of calibration curves. Optimization of the UA-MSPE procedure was carried out with stock solutions diluted with 0.01 M acetate buffer (pH 6.5).
Real water samples used in the present study included drinking water, tap water, and swamp from a local area of Khon Kaen, Thailand. All real water samples were pretreated by filtration and adjusting the desired pH with acetate buffer (pH 6.5) before performing the UA-MSPE process.
2.5 UA-MSPE
The determination of Ag(I), Pb(II), and Cd(II) metal ions by thiol-functionalized magnetic graphene oxide/iron oxide was carried out under optimum conditions of UA-MSPE. Scheme 2 shows the experimental procedure. First, the sorbent 0.03 g in 20 mL aqueous sample solution containing different concentrations of analyst with an acetate buffer solution of pH = 6.5 were added into a tube. The mixture was then sonicated in an ultrasonic bath at 0.4 kW for 25 min. Subsequently, the supernatant was removed by a magnet. The elution of adsorbed metal ions was performed by 10 mL of 1 M H2SO4 under sonication at 0.2 kW for 10 min. The supernatant was separated by the same magnet prior to the determination of metal ions by AAS.

Picture illustration of UA-MSPE procedures
The determination of the studied metal ions in the solution was performed by AAS and expressed in terms of the equilibrium adsorption capacity (qe; µg g–1) [34, 35]:
where qe is the equilibrium adsorption capacity (µg g–1), C0 is the initial concentration (μg mL–1), and Ce is the residual concentration after the equilibrium of metal ions (μg mL–1), and m is the magnetic sorbent mass (g), and V is the volume of metal ions in the acetate buffer solution (mL).
The relative percentage recovery was calculated by Eq. (2) [36]:
where Cfound, Creal, and Cadded are, respectively the metal ion concentration found after the addition of a known amount of standard metal ions in real samples, the real concentration of metal ions in the samples, and the standard metal ion concentration in real samples.
3 Results and discussion
3.1 Characterization of Fe3O4–GO–SH
The TEM and SEM images of Fe3O4, Fe3O4–GO, and Fe3O4–GO–SH are displayed in Figs. 1 and 2, respectively. The average particle sizes of Fe3O4, Fe3O4–GO, and Fe3O4–GO–SH were around 10 nm. The face-centered cubic structure of Fe3O4 was confirmed by TEM images and selected area electron diffraction patterns (SAED) (Fig. 1) [37]. The presence of GO ripples and sphere-shaped Fe3O4 particles was noticed in Fe3O4–GO. The morphology and thickness of Fe3O4–GO changed after thiol-functionalization; thus, the as-synthesized Fe3O4–GO-SH acted as a magnetic sorbent.

TEM images of Fe3O4, Fe3O4–GO, Fe3O4–GO–SH, and its SAED from XRD of Fe3O4

SEM images of Fe3O4, Fe3O4-GO, and Fe3O4–GO–SH
The composition of Fe3O4–GO–SH MNPs was analyzed by EDX spectrometry (Fig. 3). The contents of O and Fe in Fe3O4 MNPs were found as 41.1 wt% and 58.79 wt%, respectively. The C, O, and Fe contents in Fe3O4–GO were calculated as 52.90 wt%, 23.22 wt%, and 23.87 wt%, respectively [38]. A new peak of C from GO appeared at 0.277 keV. In addition, the EDX spectrum of Fe3O4–GO–SH had the peaks of C (23.34%), O (32.40%), Si (3.84%%), S (9.80%), and Fe (30.60%). Both the peaks of S and Si in MPTMS were also present in this spectrum. Therefore, the successful thiol-modification on Fe3O4–GO was confirmed.

EDX spectra and their wt% element composite of Fe3O4, Fe3O4–GO, and Fe3O4–GO–SH
Figure 4 displays the XRD patterns of Fe3O4, Fe3O4–GO, and Fe3O4–GO–SH in the 2θ range of 15°–70°. The diffraction peaks at 2θ = 30.4°, 35.6°, 43.4°, 57.3°, and 62.9° originated from the (220), (311), (400), (422), (511), and (440) planes of Fe3O4, respectively [39]; thus confirming the presence of pure Fe3O4 particles with a face-centered cubic structure [40]. Furthermore, the diffraction peak intensities of Fe3O4–GO and Fe3O4–GO–SH decreased due to the interactions between GO and SH.

XRD patterns of Fe3O4, Fe3O4–GO, and Fe3O4–GO–SH
The FT-IR spectra of GO, Fe3O4–GO, Fe3O4–GO–SH, and MPTMS are demonstrated in Fig. 5. The absorption band around 570 cm–1 appeared from Fe–O, suggesting the presence of iron oxide (Fe3O4) deposition [41]. In the FT-IR spectrum of Fe3O4–GO, the O–H-stretching band at 3340 cm–1 and the C–O-stretching band at 1043 cm–1 indicate the formation of Fe3O4–GO. In the IR spectrum of Fe3O4–GO–SH, the peaks at 2974 cm–1, 2900 cm–1, 1080 cm–1, and 2567 cm–1 appeared from C–H stretching (CH3 and CH2), Si–O stretching, and S–H stretching of the thiol group, respectively [42], therefore, confirming the successful synthesis of Fe3O4–GO–SH.

FT-IR spectra of GO, Fe3O4–GO, Fe3O4–GO–SH, and MPTMS in range of 4000–500 cm−1
The magnetic properties of the nanosorbent were evaluated by VSM. The magnetic hysteresis loops of Fe3O4, Fe3O4–GO, Fe3O4–GO–SH, and GO are exhibited in Fig. 6. GO exhibited no magnetic properties. The saturation magnetization values for Fe3O4, Fe3O4–GO, and Fe3O4–GO–SH were 91.58 emu g–1, 31.18 emu g–1, and 17.51 emu g–1, respectively. The saturation magnetization of Fe3O4–GO and Fe3O4–GO–SH decreased due to the formation of Fe3O4–GO and Fe3O4–GO–SH. The separation of metal ions in the aqueous dispersion of Fe3O4–GO–SH under the external magnetic field was achieved within 10 s. Therefore, Fe3O4–GO–SH could be used for the rapid magnetic separation of metal ions.

Magnetization curves of Fe3O4, Fe3O4–GO, Fe3O4–GO–SH, and GO
3.2 pH at the point of zero charge (pHpzc)
The point of zero charge (pHpzc) is an important factor to characterize surface charges of materials. First, a 0.01 M NaCl solution was added to sealed vials (10 mL). The pH of each vial was adjusted to between 1 and 12 with 0.1 M HCl or NaOH solutions. A 30 mg portion of the Fe3O4–GO–SH was added to each vial and shaken for 48 h at room temperature. pH was determined using a pH meter and pHpzc is the point where the curve pHfinal vs. pHinitial crosses the line pHinitial = pHfinal. Generally, solution pH has some effects on the adsorption capacity of an adsorbent. Therefore, functional groups of Fe3O4–GO–SH could dissociate as either positively or negatively charged species depending on solution pH. At pH < pHpzc, the adsorbent surface was protonated by hydrogen ions (H+) in the solution; therefore, the surface became positively charged. In contrast, the surface was negatively charged by hydroxide ions (OH–) at pH > pHpzc [43]. The pHpzc of Fe3O4–GO–SH with the relationship between initial and final pH is demonstrated in Fig. 7. The presence of the plateau (acted as a buffer zone) indicates that the material exhibited amphoteric properties. pHinitial–pHfinal refers to the pH range in which the buffering capacity of the adsorbent occurred; therefore, for all pH values in this, the pHfinal value was almost the same and corresponded to pHpzc. The pHpzc value of Fe3O4–GO–SH was found as 5.60, suggesting that the adsorbent surface was predominantly positive at pH < 5.60 and negative at pH > 5.60. Therefore, pH = 5.60 was the pHpzc of Fe3O4–GO–SH.

pHpzc determination of Fe3O4–GO–SH nanoadsorbent
3.3 Optimal conditions for the UA-MSPE method
For the simultaneous determination of Ag(I), Pb(II), and Cd(II) in the solution using Fe3O4–GO–SH as the nanosorbent by UA-MSPE method, different experimental parameters, such as solution pH, sorbent amount, sample volume, sonication time, ultrasonic power, elution solvent type, eluent concentration, and enrichment factor, were optimized. In addition, the adsorbent reusability was also investigated in detail. The nature of the extraction solution, ultrasonic power, and sonication time were the most important parameters. Therefore, high extraction efficiency was achieved under optimized extraction conditions. Each experiment was performed at least three times.
3.3.1 Effect of solution pH
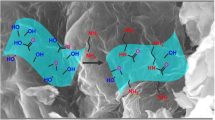
The test was carried out for the pH range of 3–8 using 10 mL of the solution containing 21.58 mg L–1 of Ag(I), 207.2 mg L–1 of Pb(II), and 11.2 mg L–1 of Cd(II) and 0.02 g of the adsorbent for 5 min at 0.2 kW, and the obtained results are exhibited in Fig. 8. The simultaneous adsorption of metal ions increased with increasing pH. At pH = 3, the adsorption of Ag(I), Pb(II), and Cd(II) was 973 µg g–1, 590 µg g–1, and 1208 µg g–1, respectively. Due to competition between proton–adsorbed area interaction and the metal ion–adsorbed area interaction, the qe of metal ions decreased in the acidic solution. The qe of metal ions continuously increased from pH = 3 to pH = 5.6 (pHpzc = 5.6) and then remained constant till pH = 8. The sorbent surface was negatively charged at pH > 5.6 because carboxyl and hydroxyl groups in Fe3O4–GO–SH acted as anions. Negatively charged hydroxyl, carboxyl, and thiol groups of Fe3O4–GO–SH electrostatically interacted with metal ions based on the hard–soft acid–base (HSAB) (Scheme 3); therefore, pH = 6.5 was selected as the optimum condition.

Effect of the solution pH of these metal ions on their adsorption capacity

The adsorption mechanism of metal ions using Fe3O4–GO–SH as adsorbent
3.3.2 Effect of adsorbent dosage
The test was carried out with 0.03–0.06 g of Fe3O4–GO–SH and 30 mL of the deionized water sample containing of Ag(I), Pb(II), and Cd(II) for 5 min at pH = 6.5 and 0.2 kW, and the corresponding results are displayed in Fig. 9a. It is noticeable that 0.03 g of the adsorbent was sufficient for metal ions adsorption, and the further addition of the adsorbent decreased the qe values of metal ions. Higher amounts of MNPs caused the aggregation of metal ions; thus, the surface area available to metal ions decreased [44]. Therefore, the adsorbent amount was selected as 0.03 g for the adsorption process.

UA-MSPE of a amount of adsorbent, b volume of sample, c adsorption time, and d ultrasonic power and e amount of salt
3.3.3 Effect of sample volume
The test was performed with the initial sample volume of 5–30 mL for 5 min at 0.2 kW, and the obtained results are presented in Fig. 9b. The equilibrium adsorption capacity increased between 5 and 20 mL and then remained constant. Therefore, 20 mL was selected as a suitable sample volume, because this was the minimum dose of the initial sample volume to achieve the highest qe for the simultaneous adsorption of metal ions.
3.3.4 Effect of adsorption time
The effect of adsorption time on the qe of the studied metal ions was investigated using Fe3O4–GO–SH as an adsorbent. The test was carried out in an ultrasonic bath for the adsorption time of 5–30 min at 0.2 kW, and the corresponding results are displayed in Fig. 9c. The curve first continuously increased and then remained constant. Due to the equilibrium state of the sample and the adsorbent in the solution, the simultaneous adsorption of metal ions reached within 25 min; hence, the optimum adsorption time was selected as 25 min.
3.3.5 Effect of ultrasonic power
Ultrasonic power is also a key parameter to evaluate the qe of the studied metal ions. The experiment was conducted in the range of 0.1–0.5 kW. Figure 9d reveals that the qe of metal ions continuously increased up to 0.4 kW, and an indistinguishable recovery rate was noticed. As the adsorbent was well dispersed by ultrasonication, a high surface area of the adsorbent and a high metal ion adsorption rate were achieved; therefore, the optimum ultrasonic power was selected as 0.4 kW.
3.3.6 Effect of salt addition
The effect of ionic strength on the efficiency of metal ions adsorption was investigated for 0–5% (w/v) NaCl. Figure 9e expresses that salt addition had a negative effect on metal ions adsorption. Due to the competition between metal ions and Na+ ions under electrostatic interactions reduced the surface area of the adsorbent for metal ions adsorption; thus, no salt addition was needed for further study.
3.3.7 Effect of elution solvent
To achieve a high enrichment factor and percent recovery, the desorption process was carried out using 10 mL of 1 M HCl, HNO3, H2SO4, and CH3COOH, each with 0.03 g of the sorbent for 25 min at 0.4 kW. Metal ions were then extracted to the liquid phase by each elution, followed by AAS. At pH lower than 4, metal ions were replaced by H+ from the acid; thus, an acidic medium should be preferred for metal ion desorption. Figure 10a indicates that H2SO4 yielded the highest recovery of the studied metal ions.

UA-MSPE of a elution solvent, b eluent concentration, c desorption time, d ultrasonic power, e sample volume toward EF, f sample volume toward recovery, and g reusability
3.3.8 Effect of eluent concentration
The effect of H2SO4 concentration on the desorption of metal ions was studied. The test was carried out using 10 mL of 0.2–1.2 M H2SO4 and 0.03 g of the adsorbent for 25 min at 0.4 kW, and the corresponding results are presented in Fig. 10b. It is evident that 1 M H2SO4 could elute all metal ions simultaneously with a high percent recovery; therefore, 1 M H2SO4 was sufficient for the elution of the studied metal ions.
3.3.9 Effect of desorption time
The effect of sonication time on metal ions desorption was studied for 5–10 min using 10 mL of 1 M H2SO4 as the eluent under an ultrasonic power of 0.4 kW. The highest recovery of metal ions was found after 10 min (Fig. 10c). As the adsorbent had a high surface area, the desorption equilibrium state was achieved within a short period. Therefore, the optimal sonication time for the elution of metal ions was 10 min.
3.3.10 Effect of ultrasonic power
The effect of ultrasonic power for the desorption of metal ions was also investigated. The ultrasonic power for the desorption process varied in the range of 0–0.5 kW for 10 min (Fig. 10d). It is evident that the application of ultrasonic waves for metal ion desorption could produce higher percent recovery. Moreover, the desorption process was successfully carried out for 10 min at 0.2 kW in an ultrasonic bath, and an indistinguishable recovery rate was achieved. Therefore, the ultrasonic power of 0.2 kW for 10 min was the optimized condition for metal ions desorption.
3.3.11 Effects of sample volume and enrichment factor (EF)
Sample volume is an important parameter to obtain a high enrichment factor. The sample volume varied from 10 to 200 mL, and the enrichment factor was determined by Eq. 3 [45]:
where EF is the enrichment factor, Cext is the analyte concentration in the extraction phase, and Cini is the initial analyte concentration. Under optimal conditions, the EF value increased with the increasing sample volume from 10 to 200 mL. The EF values ranged between 1.0 and 20.5 for Ag(I), 0.99–20.1 for Pb(II), and 0.92–19.2 for Cd(II) (Fig. 10e). The recovery rates of Ag(I), Pb(II), and Cd(II) were above 90% (Fig. 10f); thus, the UA-MSPE method could be operated with the above-mentioned sample volume. Higher sample volumes were not considered due to the difficulty in sample preparation. Therefore, 200 mL of a real sample was used for sample extraction to achieve the highest EF.
3.4 Reusability of the magnetic nanoadsorbent
The reusability of the adsorbent was repeated eight times, and the corresponding results are presented in Fig. 10g. It is evident that Fe3O4–GO–SH MNps were stable after six times of the reusability test without any significant loss in the sorption capacity, indicating a good efficacy of the as-synthesized Fe3O4–GO–SH as an adsorbent for the simultaneous preconcentration of Ag(I), Pb(II), and Cd(II) metal ions. The highest recovery rate was found as > 80% and then decreased to 72% and 67% respectively; thus, the studied metal ions were also reusable for at least six times.
3.5 Analytical performance
The analytical performance parameters used in the present study are summarized in Table 1. The analysis was performed based on the calibration curves for five different concentration levels. Linear ranges were found to be 20–100 μg L–1 for Ag(I) and Cd(II) and 200–10,000 μg L–1 for Pb(II) with the regression coefficients of 0.9983, 0.9970, and 0.9967 for Ag(I), Pb(II), and Cd(II), respectively. The limits of detection (LOD; 3 × SD/m) and limits of quantification (LOQ; 10 × SD/m) (SD is the standard deviation of metal ions at low concentrations and m is the slope of a calibration curve) for Ag, Pb, and Cd were found as 1.7 μg L–1 and 5.9 μg L–1, 14.1 μg L–1 and 47.3 μg L–1, and 0.9 μg L–1 and 3.2 μg L–1 respectively. The repeatability (data collected from three independent standard preparations, intra-day %RSD) and reproducibility (data collected from 3 consecutive days, inter-day %RSD) for Ag, Pb, and Cd were found as 1.28% and 2.76%, 5.04% and 3.87%, and 0.77% and 6.34% respectively (%RSD = percentage of relative standard deviation); therefore, these results indicate the high sensitivity and precision of the proposed method. Moreover, the EF values for Ag(I), Pb(II), and Cd(II) were found as 20.5, 20.1, and 19.2, respectively. The previous results of other relative techniques based on the utilization of SPE combined with the FAAS for Ag(I), Pb(II), and Cd(II) analysis are summarized in Table 2. The proposed method provides high performance in terms of recovery, LOD and LOQ. In addition, this method is robust as it requires less adsorbent, and is also cost-effective and simple. It also offers high sensitivity and rapid preconcentration with satisfied recovery.
3.6 Analysis of real water samples
The demonstrated process was applied for the simultaneous preconcentration and determination of Ag(I), Pb(II), and Cd(II) ions in real water samples (three drinking water samples, three wastewater samples from plating plants, and three swamp water and tap water samples from Khon Kaen, Thailand). It is evident from Table 3 that all metal ions in drinking water and tap water samples were not detectable. Some swamp sample water samples contained a trace amount of Cd (1.9 ± 3.3 mg L–1). Industrial wastewater samples contained Pb (25.3 ± 9.7 mg L–1–298.4 ± 9.2 mg L–1) and Cd (1.9 ± 2.6 mg L–1). The accuracy of the proposed method was performed by studying the recovery rates of metal ions in the real water samples. Each sample was spiked with 2 mg L–1 of Ag(I), Pb(II), and Cd(II) standard solution under optimum conditions by the UA-MSPE method for preconcentration. The obtained recovery values are the mean value of three independent determinations. The recovery rates for Ag(I), Pb(II), and Cd(II) ranged between 96.6 ± 6.6% and 104.6 ± 6.7%, 80.4 ± 7.2% and 105.4 ± 6.7%, and 83.8 ± 9.1%and 111.1 ± 6.0% respectively. Ag(I), Pb(II), and Cd(II) were effectively adsorbed and preconcentrated by the UA-MSPE process, and their recovery rates ranged between 80 and 115%. In addition, the preconcentration efficacy was expressed in terms of EF for the UA-MSPE method. The EF values were found to be approximately 20 for Ag(I), Pb(II), and Cd(II) ions and 10 for Cr ions.
4 Conclusion
In the present study, an ultrasonic-assisted magnetic solid-phase extraction (MSPE) method was applied for the simultaneous preconcentration and determination of trace amounts of metal ions [Ag(I), Pb(II), and Cd(II)] in water samples. Fe3O4–GO–SH NPs as a novel adsorbent were synthesized from 3-mercaptopropyl trimethoxysilane by the co-precipitation method under ultrasonic irradiation. Fe3O4–GO–SH NPs have the advantages of high surface area and facile reuse of adsorbents using deionized water. Ultrasonication is a beneficial tool to increase the dispersion of an adsorbent into a solution. Therefore, the combination of sonication with MSPE improved the performance of simultaneous preconcentration and determination of metal ions. According to Pearson’s Hard–Soft Acid–Base theory, the thiol group has soft base characteristics; thus, it can easily interact with soft acid metals or borderline metal ions. Hence, Fe3O4–GO–SH easily adsorbed Ag(I), Pb(II), and Cd(II) ions from water sample, because Ag(I) and Cd(II) are soft acids, and Pb(II) is a borderline metal ion. In addition, high recovery rates, good precision, and high enrichment factors were obtained. Therefore, the proposed method can effectively be used for the simultaneous preconcentration and determination of metal residues in water samples.
References
Z. Lianfu, L. Zelong, Optimization and comparison of ultrasound/microwave assisted extraction (UMAE) and ultrasonic assisted extraction (UAE) of lycopene from tomatoes. Ultrason. Sonochem. 15, 731–737 (2008)
F. Chemat, N. Rombaut, A.-G. Sicaire, A. Meullemiestre, A.-S. Fabiano-Tixier, M. Abert Vian, Ultrasound assisted extraction of food and natural products. Mechanisms, techniques, combinations, protocols and applications. A review. Ultrason. Sonochem. 34, 540–560 (2017)
G. Pan, G. Yu, C. Zhu, J. Qiao, Optimization of ultrasound-assisted extraction (UAE) of flavonoids compounds (FC) from hawthorn seed (HS). Ultrason. Sonochem. 19, 486–490 (2012)
P. Sricharoen, N. Lamaiphan, P. Patthawaro, N. Limchoowong, S. Techawongstien, S. Chanthai, Phytochemicals in Capsicum oleoresin from different varieties of hot chilli peppers with their antidiabetic and antioxidant activities due to some phenolic compounds. Ultrason. Sonochem. 38, 629–639 (2017)
S. Bahrani, M. Ghaedi, M.J.K. Mansoorkhani, A. Asfaram, A.A. Bazrafshan, M.K. Purkait, Ultrasonic assisted dispersive solid-phase microextraction of Eriochrome cyanine R from water sample on ultrasonically synthesized lead (II) dioxide nanoparticles loaded on activated carbon: experimental design methodology. Ultrason. Sonochem. 34, 317–324 (2017)
E.A. Dil, M. Ghaedi, A. Asfaram, Optimization and modeling of preconcentration and determination of dyes based on ultrasound assisted-dispersive liquid–liquid microextraction coupled with derivative spectrophotometry. Ultrason. Sonochem. 34, 27–36 (2017)
M. Khan, T.G. Kazi, H.I. Afridi, M. Bilal, A. Akhtar, N. Ullah, S. Khan, S. Talpur, Application of ultrasonically modified cloud point extraction method for simultaneous enrichment of cadmium and lead in sera of different types of gallstone patients. Ultrason. Sonochem. 39, 313–320 (2017)
S. Bahar, Z. Es’haghi, A. Nezhadali, A. Banaei, S. Bohlooli, An innovative method for analysis of Pb (II) in rice, milk and water samples based on TiO2 reinforced caprylic acid hollow fiber solid/liquid phase microextraction. Food Chem. 221, 1907–1910 (2017)
N.A. Pinchukova, A.Y. Voloshko, M.A. Merko, Y.A. Bondarenko, V.A. Chebanov, Intensification of ion exchange desorption of thiamine diphosphate by low-powered ultrasound. Ultrason. Sonochem. 41, 261–266 (2018)
N.A. Al-Dhabi, K. Ponmurugan, P. Maran Jeganathan, Development and validation of ultrasound-assisted solid-liquid extraction of phenolic compounds from waste spent coffee grounds. Ultrason. Sonochem. 34, 206–213 (2017)
A. Asfaram, M. Ghaedi, A. Goudarzi, Optimization of ultrasound-assisted dispersive solid-phase microextraction based on nanoparticles followed by spectrophotometry for the simultaneous determination of dyes using experimental design. Ultrason. Sonochem. 32, 407–417 (2016)
M. Hemmati, M. Rajabi, A. Asghari, Ultrasound-promoted dispersive micro solid-phase extraction of trace anti-hypertensive drugs from biological matrices using a sonochemically synthesized conductive polymer nanocomposite. Ultrason. Sonochem. 39, 12–24 (2017)
J.U. Hur, J.S. Choi, S.-C. Choi, G.S. An, Highly dispersible Fe3O4 nanoparticles via anionic surface modification. J. Korean Ceram. Soc. 57, 80–84 (2020)
Y. Seo, H.J. Choi, Core–shell-structured Fe3O4 nanocomposite particles for high-performance/stable magnetorheological fluids: preparation and characteristics. J. Korean Ceram. Soc. (2020). https://doi.org/10.1007/s43207-020-00070-9
L.F. Sukhodub, L.B. Sukhodub, A.D. Pogrebnjak, A. Turlybekuly, A. Kistaubayeva, I. Savitskaya, D. Shokatayeva, Effect of magnetic particles adding into nanostructured hydroxyapatite–alginate composites for orthopedics. J. Korean Ceram. Soc. 57, 557–569 (2020)
B. Zawisza, R. Sitko, E. Malicka, E. Talik, Graphene oxide as a solid sorbent for the preconcentration of cobalt, nickel, copper, zinc and lead prior to determination by energy-dispersive X-ray fluorescence spectrometry. Anal. Methods. 5, 6425–6430 (2013)
A. Keramat, R.Z. Dorabei, Ultrasound-assisted dispersive magnetic solid phase extraction for preconcentration and determination of trace amount of Hg (II) ions from food samples and aqueous solution by magnetic graphene oxide (Fe3O4@GO/2-PTSC): Central composite design optimization. Ultrason. Sonochem. 38, 421–429 (2017)
S. Gong, J. Lee, H.S. Kim, Development of electrode architecture using Sb–rGO composite and CMC binder for high-performance sodium-ion battery anodes. J. Korean Ceram. Soc. 57, 91–97 (2020)
P. Sricharoen, S. Chanthai, N. Lamaiphan, C. Sakaew, N. Limchoowong, P. Nuengmatcha, W.-C. Oh, Sono-synthesized Fe3O4–GO–NH2 nanocomposite for highly efficient ultrasound-assisted magnetic dispersive solid-phase microextraction of hazardous dye Congo red from water samples. J. Korean Ceram. Soc. (2020). https://doi.org/10.1007/s43207-020-00089-y
A. Saha, S. Basiruddin, C.S. Ray, S.S. Roy, R.N. Jana, Functionalized graphene and graphene oxide solution via polyacrylate coating. Nanoscale 2, 2777–2782 (2010)
S. Akbarzade, M. Chamsaz, H.G. Rounaghi, Highly selective preconcentration of ultra-traceamounts of lead ions in real water and food samples by dispersive solid phase extraction using modified magnetic graphene oxide as a novel sorbent. Anal. Methods. 10, 2081–2087 (2018)
G. Avashthi, S.S. Maktedar, M. Singh, in Sonochemically Covalent Functionalized Graphene Oxide Towards Photoluminescence and Nanocytotoxicity Activities. ed. by A. Khan, M. Jawaid, B. Neppolian, A. Asiri (Springer, Singapore, 2019), p. 105
G. Avashthi, S.S. Maktedar, M. Singh, Nitrogen-functionalized graphene oxide as thermally stable bioscaffold. AIP Conf. Proc. 2220, 020175 (2020)
G. Avashthi, S.S. Maktedar, M. Singh, Sonochemically N-functionalized graphene oxide towards optically active photoluminescent bioscaffold. Ultrason. Sonochem. 58, 104651 (2019)
G. Avashthi, S.S. Maktedar, M. Singh, Surface-induced in situ sonothermodynamically controlled functionalized graphene oxide for in vitro cytotoxicity and antioxidant evaluations. ACS Omega 4, 16385–16401 (2019)
L. Chen, D. Zhao, S. Chen, X. Wang, C. Chen, One-step fabrication of amino functionalized magnetic graphene oxide composite for uranium(VI) removal. J. Colloid. Interf. Sci. 472, 99–107 (2016)
J. Liu, H. Du, S. Yuan, W. He, Z. Liu, Synthesis of thiol-functionalized magnetic graphene as adsorbent for Cd(II) removal from aqueous systems. J. Environ. Chem. Eng. 3, 617–621 (2015)
M. Tahergorabi, A. Esrafili, M. Kermani, M.S. Siboni, Application of thiol-functionalized mesoporous silica-coated magnetite nanoparticles for the adsorption of heavy metals. Desal. Water Treat. 57, 19834–19845 (2016)
G. Li, Z. Zhao, J. Liu, G. Jiang, Effective heavy metal removal from aqueous systems by thiol functionalized magnetic mesoporous silica. J. Hazard. Mater. 192, 277–283 (2011)
J. Li, Y. Song, Z. Ma, N. Li, S. Niu, Y. Li, Preparation of polyvinyl alcohol graphene oxide phosphonate film and research of thermal stability and mechanical properties. Ultrason. Sonochem. 43, 1–8 (2018)
G. Mohammadnezhad, S. Abad, R. Soltani, M. Dinari, Study on thermal, mechanical and adsorption properties of amine-functionalized MCM-41/PMMA and MCM-41/PS nanocomposites prepared by ultrasonic irradiation. Ultrason. Sonochem. 39, 765–773 (2017)
A.D.A. Rojo, L.M.C. Lopez, R.R. Villagrana, M.H. Jimenez, I.A.G. Galicia, Ultrasound and meat quality: a review. Ultrason. Sonochem. 55, 369–382 (2019)
N. Limchoowong, P. Sricharoen, Y. Areerob, P. Nuengmatcha, T. Sripakdee, S. Techawongstien, S. Chanthai, Preconcentration and trace determination of copper (II) in Thai food recipes using Fe3O4@Chi–GQDs nanocomposites as a new magnetic adsorbent. Food Chem. 230, 388–397 (2017)
N. Limchoowong, P. Sricharoen, S. Chanthai, A novel bead synthesis of the Chiron-sodium dodecyl sulfate hydrogel and its kinetics-thermodynamics study of superb adsorption of alizarin red S from aqueous solution. J. Polym. Res. 26, 265 (2019)
P. Sricharoen, N. Limchoowong, P. Nuengmatcha, S. Chanthai, Ultrasonic-assisted recycling of Nile tilapia fish scale biowaste into low-cost nano-hydroxyapatite: ultrasonic-assisted adsorption for Hg2+ removal from aqueous solution followed by “turn-off” fluorescent sensor based on Hg2+-graphene quantum dots. Ultrason. Sonochem. 63, 104966 (2020)
C. Sakaew, P. Sricharoen, N. Limchoowong, P. Nuengmatcha, C. Kukusamude, S. Kongsri, S. Chanthai, Green and facile synthesis of water-soluble carbon dots from ethanolic shallot extract for chromium ion sensing in milk, fruit juices, and wastewater samples. RSC Adv. 10, 20638–20645 (2020)
C. Yang, J. Wu, Y. Hou, Fe3O4 nanostructures: synthesis, growth mechanism, properties and applications. Chem. Commun. 47, 5130–5141 (2011)
H.T. Naeem, K.S. Mohammed, Microstructural evaluation and mechanical properties of an Al–Zn–Mg–Cu-alloy after addition of nickel under RRA conditions. Mater. Sci. Appl. 4, 704–711 (2013)
W. Cheng, K. Tang, Y. Qi, J. Sheng, Z. Liu, One-step synthesis of superparamagnetic monodisperse porous Fe3O4 hollow and core-shell spheres. J. Mater. Chem. 20, 1799–1805 (2010)
C. Ding, W. Cheng, Y. Sun, X. Wang, Novel fungus-Fe3O4 bio-nanocomposites as high performance adsorbents for the removal of radionuclides. J. Hazard. Mater. 295, 127–137 (2015)
P. Sricharoen, N. Limchoowong, Y. Areerob, P. Nuengmatcha, S. Techawongstien, S. Chanthai, Fe3O4/hydroxyapatite/graphene quantum dots as a novel nano-sorbent for preconcentration of copper residue in Thai food ingredients: optimization of ultrasound-assisted magnetic solid phase extraction. Ultrason. Sonochem. 37, 83–93 (2017)
P. Nuengmatcha, R. Mahachai, S. Chanthai, Adsorption of functionalized thiol-graphene oxide for removal of mercury from aqueous solution. Asian J. Chem. 27, 4167–4170 (2015)
P. Nuengmatcha, P. Porrawatkul, S. Chanthai, P. Sricharoen, N. Limchoowong, Enhanced photocatalytic degradation of methylene blue using Fe2O3/graphene/CuO nanocomposites under visible light. J. Environ. Chem. Eng. 7, 103438 (2019)
E. Akar, A. Altinisik, Y. Seki, Using of activated carbon produced from spent tea leaves for the removal of malachite green from aqueous solution. Ecol. Eng. 52, 19–27 (2013)
M.S. Arain, T.G. Kazi, H.I. Afridi, J. Ali, A. Akhtar, Ultrasonic energy enhanced the efficiency of advance extraction methodology for enrichment of trace level of copper in serum samples of patients having neurological disorders. Ultrason. Sonochem. 37, 23–28 (2017)
E. Yavuz, S. Tokalioglu, S. Sahan, FAAS determination of Ag(I) in water, anode slime, rock and cream samples by solid phase extraction method based on sepabeads SP207/5-(p-dimethylaminobenzylidene) rhodanine combination. J. Braz. Chem. Soc. 24, 736–742 (2013)
M.A. Karimi, S.Z. Mohammadi, A. Mohadesi, A. Hatefi-Mehrjardi, M. Mazloum-Ardakani, L. Sotudehnia Korani, A. Askarpour Kabir, Determination of silver(I) by flame atomic absorption spectrometry after separation/preconcentration using modified magnetite nanoparticles. Sci. Iran. 18, 790–796 (2011)
M.R. Fathi, N. Pourreza, S. Purweis, Determination of silver by flame atomic absorption spectrometry after preconcentration on naphthalene modified with dithizone. J. Chin. Chem. Soc. 56, 725–728 (2009)
C.-G. Yuan, P. Liang, Y.-Y. Zhang, Determination of trace silver in environmental samples by room temperature ionic liquid-based preconcentration and flame atomic absorption spectrometry. Microchim. Acta 175, 333–339 (2011)
A.A. Ensafi, A. Zendegi-Shiraz, Combination of solid phase extraction and flame atomic absorption spectrometry for trace analysis of cadmium. J. Braz. Chem. Soc. 19, 11–17 (2008)
M. Payehghadr, S. Esmaeilpour, M. KazemRofouei, L. Adlnasab, Determination of trace amount of cadmium by atomic absorption spectrometry in table salt after solid phase preconcentration using octadecyl silica membrane disk modified by a new derivative of pyridine. J. Chem. 2013, 417085 (2013)
A.A. Ensafi, A.R. Ghaderi, On-line solid phase selective separation and preconcentration of Cd(II) by solid-phase extraction using carbon active modified with methyl thymol blue. J. Hazard. Mater. 148, 319–325 (2007)
A. Tadjarodi, A. Abbaszadeh, M. Taghizadeh, N. Shekari, A.A. Asgharinezhad, Solid phase extraction of Cd(II) and Pb(II) ions based on a novel functionalized Fe3O4@ SiO2 core-shell nanoparticles with the aid of multivariate optimization methodology. Mater. Sci. Eng. C 49, 416–421 (2015)
W. Aeungmaitrepirom, W. Ngeontae, T. Tuntulani, Silica gel chemically modified with ethyl-2-benzothiazolylacetate for selective extraction of PB(II) and Cu(II) from real water samples. Anal. Sci. 25, 1477–1482 (2009)
S. Baytak, E. Kendüzler, A.R. Türker, N. Gök, Penicillium digitatum immobilized on pumice stone as a new solid phase extractor for preconcentration and/or separation of trace metals in environmental samples. J. Hazard. Mater. 153, 975–983 (2008)
Acknowledgements
The authors would like to thank the Materials Chemistry Research Center, Department of Chemistry, Faculty of Science, Khon Kaen University and the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Department of Chemistry, Faculty of Science, Srinakharinwirot University and Nuclear Research and Development Division, Thailand Institute of Nuclear Technology (Public Organization) for financial support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have declared no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lamaiphan, N., Sakaew, C., Sricharoen, P. et al. Highly efficient ultrasonic-assisted preconcentration of trace amounts of Ag(I), Pb(II), and Cd(II) ions using 3-mercaptopropyl trimethoxysilane-functionalized graphene oxide–magnetic nanoparticles. J. Korean Ceram. Soc. 58, 314–329 (2021). https://doi.org/10.1007/s43207-020-00094-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43207-020-00094-1