
Abstract
A composite photocatalyst of zinc oxide (ZnO) nanoparticles decorated with different content of reduced graphene oxide (rGO) was prepared via a simple and facile one-step method in this paper. X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), Raman spectra, and UV–Vis diffuse reflection spectroscopy (UV–Vis DRS) were used to characterize the crystal structure, morphology and optical properties of the rGO–ZnO composite photocatalyst. The photocatalytic properties of the composites were investigated using methyl orange (MO), a typical orange compound, as a test pollutant. The results showed that rGO–ZnO composites displayed significantly enhanced photocatalytic activity in MO degradation than pure ZnO, and the pseudo-first-order kinetic constant on the optimal rGO–ZnO composite was 14 times as great as that on pure ZnO. The enhanced photocatalytic ability of the rGO-ZnO composites was mainly benefited from the high specific surface area and high conductivity of rGO, which facilitated efficient charge separation in the rGO-ZnO nanocomposite.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
At present, water pollution is one of the most serious challenges to human survival [1]. Photocatalytic technology has been paid worldwide attention for water pollution treatment, because solar energy is an inexhaustible, low-cost, environmentally friendly energy resource. To date, a large number of semiconductor materials like metal sulfides, metal oxides and complex oxides (such as SnS2, Cu2O, TiO2, CeO2, Zn2SnO4) have been identified as active photocatalysts for the photodegradation of organic contaminants in water pollution [2,3,4,5,6]. Among them, zinc oxide (ZnO) has been widely used as a photocatalyst for the degradation of organic pollutants due to its low cost, large exciton binding energy (60 meV), and environmentally friendly feature [7]. However, only the ultraviolet light energy in solar light could be absorbed for generating electron–hole pairs by ZnO due to its comparatively higher bandgap energy (ca. 3.37 eV). Besides, the rapid recombination of photogenerated electron–hole pairs also limits its practical applications. Therefore, numerous methods have been developed to improve its photocatalytic ability, including metal doping [8,9,10,11], and coupling with other semiconductor materials [12,13,14,15,16]. For example, Pt nanoparticles were introduced on the surface of ZnO nanowires, obtaining excellent photocatalytic performance [17].
Graphene, a one-atom-thick sheet with a two-dimensional honeycomb arrangement of carbon atoms, possessing excellent electron-transport ability and large surface area makes it a potential support material to load functional materials. So far, a lot of research interest has been devoted to designing graphene-based composite materials, aiming to enhance photocatalytic activity [18,19,20]. Xu et al. synthesized graphene-modified ZnO flower composite via a simple one-pot hydrothermal method and the results showed that the composite exhibited a prominent enhancement photocatalytic performance for methylene blue (MB) than pure ZnO [21]. Chen and coworkers presented a two-step method to fabricate a ZnO/rGO composite, and the introduction of graphene improved the photocatalytic activity of ZnO [22]. In the above-published works, although ZnO-graphene composites have been obtained, the synthesis methods have some shortcomings, such as long reaction time, complicated preparation procedures, high cost, requiring high pressure and high temperature. Thus, a simple and efficient method to fabricate a high-performance photocatalyst of graphene–ZnO is challenging but highly desirable.
In this paper, we synthesized rGO–ZnO composites through a fast, easy and efficient alcohol-assisted method. And the effect of different contents of rGO in the rGO/ZnO composites for photocatalytic activity has been studied in detail. At last, the possible mechanism for the enhanced photocatalytic activity over the rGO–ZnO composite photocatalyst was also proposed.
2 Experiment
2.1 Materials
All reactants were analytical purity and used as received without further purification. Natural graphite powder (325 mesh) was commercially obtained from Qingdao Tianyuan Company. Hydrogen peroxide (30%, H2O2), methyl orange (MO), hydrazine hydrate (80%, N2H4), potassium hydroxide (KOH) and absolute ethyl alcohol (99.5%, CH3CH2OH) were purchased from Tianjin Hengxing Chemical Preparation Co., Ltd (China). Sodium nitrate (NaNO3) was purchased from Taishan Chemical Factory Co., Ltd (China). Concentrated sulfuric acid (98%, H2SO4), zinc nitrate hexahydrate (Zn(NO3)2·6H2O), potassium permanganate (KMnO4) were purchased from Shanghai Chemical Reagent Co., Ltd (China). Double-distilled water was used during the experimental process. The experiments were carried out at room temperature and humidity.
2.2 Preparation of graphene oxide sheets (GO)
GO was synthesized following the modified Hummers’ method [23]. In detail, 2 g of natural graphite, and 2 g of NaNO3 were put into 96 mL of concentrated H2SO4 under continuous stirring. Then, the mixture was put in an ice bath, and 12 g of KMnO4 was slowly added to this mixture and stirred for 90 min. Then, the suspension was stirred continuously in a water bath at 35 °C for 2 h and diluted with 100 mL of deionized water. The temperature of the water bath was raised to 95 °C and was kept for 15 min. Finally, 10 mL of H2O2 (30%) was added to the prepared suspension to reduce residual permanganate to soluble manganese ions. The color of the solution changed from dark to bright yellow. The mixture was then repeatedly washed with 5% HCl solution and water until a constant pH of 6 was obtained. Finally, the black GO was harvested using centrifugation and dried at 60 °C for 24 h in a vacuum.
2.3 Preparation of ZnO
3.677 g of Zn(NO3)2·6H2O was dispersed in 100 mL of ethanol under mechanical stirring to obtain a transparent liquid. The pH of the suspension was adjusted to 10 with the aid of 1 mol L−1 KOH-alcohol solution. After being kept at 80 °C for 4 h, the final products were collected and washed with distilled water and ethanol three times and then dried under vacuum conditions at 80 °C.
2.4 Preparation of rGO–ZnO composite
GO was used as the precursor to synthesize rGO–ZnO composites due to its reactive sites which are favorable to the attachment of metal oxides. The oxygenated functional groups (such as hydroxyl, carbonyl and epoxide) of GO can act as nucleation centers to anchor active materials [24]. First, a suitable amount of GO prepared above was dispersed in 100 mL of ethanol under ultrasonication at a frequency of 40 kHz with the power of 250 W for 5 h to produce a well-dispersed suspension. Then, 3.677 g of Zn(NO3)2·6H2O was added to the GO suspension under mechanical stirring. The pH of the suspension was also adjusted to 10. Then, N2H4·H2O (80%, 0.5 mL) was added to the suspension under vigorous stirring. The suspension was heat-treated at 80 °C for 4 h to complete the reaction. The product was filtered and washed several times with water and ethanol, and dried in a vacuum at 80 °C for further characterization. For comparison, rGO was also obtained using the same methods under the same conditions just without the addition of Zn(NO3)2·6H2O.
To evaluate the effect of various rGO contents on the photocatalysis properties of the rGO/ZnO composites, we fabricated several types of devised products with an increasing weight ratio of rGO. The weight ratio of GO: ZnO = 0.025, 0.05, 0.075, 0.1, 0.125, were used as precursors and were labeled as 0.25% rGO–ZnO, 0.5% rGO–ZnO, 0.75% rGO–ZnO, 1.0% rGO-ZnO, 1.25% rGO–ZnO, respectively.
2.5 Material characterization
The XRD patterns of the samples were obtained on the Bruker D8 diffractometer using Cu Kα radiation (λ = 0.15418 nm) at room temperature, and the 2θ range from 5° to 80° with a step size of 0.004°. Field emission scanning electron microscopy (SEM; Hitachi S-4800) was used to investigate the morphology of the samples with an acceleration voltage of 10 kV and emission current of 20 μA. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HR-TEM) were performed on a JEM-3010 transmission electron microscopy at an accelerating voltage of 200 kV. Raman spectra were measured by Labram-010 micro-Raman spectrometer by employing a laser of 632 nm as the incident light. UV–Vis diffuse reflectance spectroscopy (UV–Vis DRS) was measured on a UV–Vis spectrometer (U-4100). Herein, BaSO4 was used as the reflectance standard.
2.6 Measurements of photocatalytic activity
The photocatalytic activities of the as-prepared samples were evaluated by photodegradation of the methyl orange (MO) under UV–Vis irradiation with a 350 W xenon lamp. MO was chosen as a representative reactant to evaluate the photocatalytic performance, which showed a major absorption band at 464 nm. In a typical procedure, 0.2 g of the as-prepared rGO–ZnO composite sample or pure ZnO was dispersed into 400 mL of MO aqueous solution with the concentration of 10 mg L−1 in a 500 mL quartz beaker. Before the photocatalytic reaction, the mixture was first stirred in darkness for 30 min to ensure the establishment of adsorption–desorption equilibrium. Afterward, at given irradiation time intervals (10 min), 10 mL of the suspension was taken out from the quartz vessel and immediately centrifuged at 5000 rpm for 5 min to separate any suspended solids. Then, the supernatant was directly analyzed by recording the absorption spectrum with the help of a UV–Vis spectrophotometer (UV-6000). The photocatalytic capability of the rGO–ZnO composite was evaluated based on the decomposition efficiency of the maximum absorption band of MO (λmax = 464 nm).
3 Results and discussion
3.1 Crystal structures
Figure 1a gives the XRD patterns of as-obtained GO and rGO. GO shows a strong diffraction peak centered at 2θ = 11.03° belonging to (002) planes of GO with a layer–layer distance of 0.8017 nm, which is much larger than that of natural graphite powder (0.334 nm) [25]. The expansion of the d-spacing for GO sheets can be ascribed to oxide-induced oxygen-containing groups such as hydroxyl (–OH), carboxyl (–COOH), epoxy groups, inserted H2O molecules, and other structural defects [26]. The addition of hydrazine hydrate into the GO solution and the subsequent thermal treatment were taken to guarantee the reduction of GO to rGO [27]. It can be seen that the diffraction peak of GO at 11.03° disappears and a small broad peak at 26.53° appears in the XRD pattern of rGO (Fig. 1a), due to the removal of the oxygen functional groups between the layers of carbon nanosheets [28].

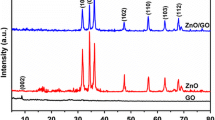
XRD patterns of a GO and rGO; b ZnO and rGO–ZnO nanocomposites
For the XRD spectra of the as-prepared ZnO and rGO-ZnO composites shown in Fig. 1b, the sharp diffraction peaks indicate the good crystallization of ZnO when the weight percent of rGO is 0%. The peaks at 31.8°, 34.4°, 36.3°, 47.5°, 56.6°, 62.8°, 66.4° and 67.9° were indexed to (100), (002), (101), (102), (110), (103), (112) and (201) crystalline planes of hexagonal wurtzite-structured ZnO (JCPDS card 36-1451). No diffraction peaks of other phases were observed, indicating the formation of pure ZnO. It is observed that the XRD patterns of all the rGO–ZnO composite samples with different rGO contents are very similar to that of pure ZnO, implying that rGO has no influence on the crystal structure of ZnO. However, there are almost no typical diffraction peaks of GO or rGO in the XRD patterns, which can be ascribed to two probable reasons. For one reason, the quantity of rGO is much less than that of ZnO. And the other possible reason is that the regular layered structure of rGO has been destroyed due to the growth of ZnO into the sheets of rGO [29]. The average crystallite size of the ZnO particles is about 40 nm estimated through the Debye–Scherrer formula [30]:
where D is the apparent particle size (nm), K is a constant (0.9), λ is the wavelength of the X-ray radiation (0.15148 nm), β is the full-width half-maximum, and θ is the half diffraction angle.
3.2 Morphology characterizations
The morphology and microstructure of the representative samples were examined by using SEM and TEM analysis. Figure 2a shows the SEM image of pure GO. Some light-gray thin films indicate the layered structure of GO sheets. And the dark areas may be the agglomerated GO sheets. The atoms located on the surface of the nanoscale-thick GO sheets occupy a considerable proportion, making it a thermodynamically unstable system. In this case, the GO sheets tend to agglomerate to reduce the energy of the system. Figure 2b reveals that the average diameter of ZnO particles is about 40–50 nm, which is consistent with the calculated results of XRD. The SEM images of different rGO–ZnO composites are shown in Fig. 2c, d and Fig. S1. The nanoparticle structure of ZnO could be found in 0.25%, 1.0% and 1.25% rGO-ZnO composites in Fig. S1 and the particle size gradually increased with the increase of rGO content. It is worth noting that ZnO particles are well dispersed on the surface of rGO sheets in the 1.0% rGO–ZnO composite (Fig. 2c, d), which may be favorable for the enhancement of photocatalytic performance.

SEM images of the representative samples: a GO; b pure ZnO; c, d 1.0% rGO–ZnO composite
The TEM image shown in Fig. 3a further proves that the ZnO nanoparticles are well dispersed on rGO, which agrees well with the SEM results. HR-TEM image (Fig. 3b) displays two groups of distinct lattice fringes with lattice spacing of 0.513 nm and 0.278 nm corresponding to the interplanar spacings of {0001} and {0110} planes of hexagonal ZnO, respectively.

a The TEM image of 1.0% rGO–ZnO composite; b The HR-TEM image of a ZnO fragment marked with the circle in a
3.3 Raman spectra analysis
Raman spectra were used to further characterize the composition of rGO–ZnO composites. Figure 4 reveals the Raman results of pure GO and 1.0% rGO–ZnO composite. In the Raman spectrum of pure GO, there are two main features at 1344 cm−1 and 1589 cm−1 corresponding to the D and G band of GO, respectively. The D band is the A1g symmetry vibration mode of κ-point phonons and suggests the presence of sp3 defects. While the G band is the E2g symmetry vibration mode of all sp2 bonded carbons [25, 26, 31]. The peaks at 2669 cm−1 and 2908 cm−1 are associated with the 2D and D + G bands, respectively [32, 33]. The sample of 1.0% rGO–ZnO composite presents a similar spectrum to GO. However, in the case of 1.0% rGO–ZnO composite, the D and G bands are located at 1352 and 1596 cm−1 with additional peaks (431 and 580 cm−1) that are due to ZnO nanoparticles. The intensity ratio of the D band G band (ID/IG) reflects the degree of defects in GO. Compared with pure GO, an increased ID/IG value of the rGO–ZnO composite is found (0.95 for pure GO and 1.07 for 1.0% rGO–ZnO, respectively), indicating that the reduction of GO and the growth of ZnO leads to the fragmentation of sp2 graphitic network and the formation of new graphitic domains, which is consistent with XRD results [31, 34]. For the rGO–ZnO composite, the G band shifts from 1589 to 1596 cm−1 compared with that of GO. And the D band also blueshifts to 1352 cm−1, which results from the charge transfer caused by chemical interaction between rGO nanosheets and ZnO nanoparticles [35]. These results testify that there is an intimate interaction formed between the components in the rGO–ZnO composites.

The Raman spectra of GO and the 1.0% rGO–ZnO composite
3.4 UV–Vis DRS analysis
The UV–Vis diffuse reflectance spectra of pure ZnO and rGO–ZnO composites are shown in Fig. 5a. ZnO shows a characteristic spectrum with an absorption edge of 400 nm. The rGO–ZnO composites show more intensive absorption in the visible region owing to the absorption contribution of rGO and the increase of surface electric charge of the oxides in virtue of the introduction of rGO [36, 37], which is beneficial to improve the utilization of sunlight. When the content of rGO increased from 0.25 to 0.75%, the absorbance did not enhance significantly. The bandgap of the prepared samples can be estimated by the formula [38, 39]:

a The UV–Vis DRS of pure ZnO and rGO-ZnO composites; b The calculated bandgaps of ZnO and rGO–ZnO composites
where α is the absorption coefficient, h is Planck’s constant and ν is the frequency of UV–Visible light, Eg is the gap between the bottom of the conduction band and the top of the valence band, n is determined by the type of optical transition of a semiconductor (n = 1 for direct transition and n = 4 for indirect transition). For ZnO, the n value is 1. By plotting (αhν)2 versus hν, the bandgap energies are estimated from the energy axis at α = 0. The bandgaps of these samples are 3.17 eV, 2.86 eV, 2.67 eV and 2.75 eV for pure ZnO, 0.25% rGO–ZnO, 0.75% rGO–ZnO and 1.0% rGO–ZnO, respectively (Fig. 5b). The bandgaps of the composites decreased compared with pure ZnO, which can be attributed to the hybridization of C 2p orbitals in graphene and O 2p orbitals in ZnO forming a new valence band [40].
3.5 Photocatalytic activity
To evaluate the photocatalytic activity of rGO–ZnO composites, all obtained samples were investigated via MO photodegradation experiments under UV–Vis light irradiation and the results are shown in Fig. 6. Figure 6a displays the change of the absorption spectra of MO aqueous solution over 1.0% rGO–ZnO composite under UV–Visible light irradiation. With increasing irradiation time, the absorption peak intensity at 464 nm diminishes gradually. Meanwhile, the intense orange color of the initial solution disappears with increasing longer exposure times.

a The time-dependent absorption spectra of MO solution for 1.0% rGO–ZnO composite; b Photocatalytic degradation of MO over ZnO and rGO–ZnO composite samples under UV–Vis light irradiation; c First-order kinetics plot for the photodegradation of MO by pure ZnO and various rGO–ZnO composites; d Degradation rate constant k (min−1) for the photodegradation of MO by pure ZnO and various rGO–ZnO composites
Figure 6b shows the photocatalytic activities of the rGO–ZnO composites with different rGO contents as well as pure ZnO under UV–Vis light irradiation. As a reference, the photodegradation of MO without catalysts was also carried out under the same condition, and the result confirmed that MO was only slightly degraded in the absence of catalysts, indicating that its self-photodegradation process can be neglected. The pure ZnO showed a relatively poor photocatalytic activity, with only 9.2% MO degraded under irradiation for 60 min. Compared with the pristine ZnO, the photocatalytic properties of all composites were improved to some extent due to the introduction of rGO. The degradation efficiencies were 40.01%, 53.43%, 72.44%, 76.78% and 63.95% for 0.25% rGO–ZnO, 0.5% rGO–ZnO, 0.75% rGO–ZnO, 1.0% rGO–ZnO and 1.25% rGO–ZnO, respectively.
Photocatalytic reactions on the surface of rGO–ZnO composites can be described in terms of the Langmuir-Hinshelwood (L-H) model, and the formula can be expressed by the following equation [41]:
where k is the rate constant (min−1), C0 and C are the initial concentration and concentration at time t of MO aqueous solution, respectively. The kinetic linear curves of photocatalytic degradation of MO with pure ZnO and rGO–ZnO composites are presented in Fig. 6c, respectively. It can be found that the photocatalytic degradation reaction indeed fits well with the first-order equation. The kinetic constant k was calculated and collected in Fig. 6d, and the constants are 0.00133 min−1, 0.00706 min−1, 0.01009 min−1, 0.0198 min−1, 0.02017 min−1 and 0.01159 min−1 for ZnO, 0.25% rGO–ZnO, 0.5% rGO–ZnO, 0.75% rGO–ZnO, 1.0% rGO–ZnO and 1.25% rGO–ZnO, respectively. The samples showed gradually enhanced activities when the rGO concentration increased from 0 to 1.0%. However, the photocatalytic performance of the rGO–ZnO composite declined when the content of rGO increased to 1.25%. The kinetic constant of the optimal sample (1.0% rGO–ZnO) is 0.02017 min−1, which is 14 times as great as that of pure ZnO particles.
According to the photocatalytic results above, the 1.0% rGO–ZnO possesses the highest photocatalytic performance. However, the photocatalytic performance of the rGO–ZnO composite sample decreases when the content of rGO exceeds 1.0%. This phenomenon may be explained as follows: (a) although the special structure of rGO can transmit photoinduced electron–hole pairs and thus improve the photocatalytic efficiency of the composites, too much rGO will serve as a kind of recombination center and diminish the photocatalytic performance of rGO–ZnO [42]. (b) The excess addition of rGO may reduce the irradiation intensity through the reaction suspension and thus diminish the photocatalytic efficiency of rGO–ZnO composites. Therefore, 1.0% rGO is the optimal ration for rGO–ZnO catalysts in this study.
A comparison with the other photocatalysts prepared by different methods is given in Table 1. The results depict that the composite materials in this study could be synthesized in a milder reaction through a simpler procedure, and supplied efficient photocatalytic properties compared to other photocatalysts.
The results of photocatalytic experiment show that the powder samples of rGO–ZnO composites express remarkable adsorption properties in MO solution. The adsorption constants of MO over catalysts were evaluated by Eq. (4):
where qt is the amount adsorbed per gram of adsorbent (mg/g), mc is the mass of adsorbent used (g), ma is the adsorption mass of MO (mg). And the calculated adsorption constants are collected in Fig. 7. From this figure, it can be clearly seen that the adsorption capability of rGO–ZnO composites obviously enhances with increasing content of rGO because of the large intrinsic specific surface of rGO. The enhanced adsorption capacity could provide abundant active sites during the photocatalytic process, which is conductive to improve the photocatalytic performance.

The adsorption performance of rGO–ZnO composites with different GO contents as well as pure ZnO. The inset is the color change of the samples
3.6 Photocatalytic mechanism
A possible mechanism of photocatalytic degradation of MO over rGO-ZnO composite is proposed in Fig. 8. When the light energy is equal to or larger than the bandgap energy of the photocatalyst, ZnO can capture the photons and generate photoinduced electron–hole pairs. Photoinduced electron (e−) can be injected into the conduction band of ZnO, leaving holes (h+) in the valence band of ZnO, and then those photoinduced electrons transfer from the conduction band of ZnO onto the surface of rGO [50,51,52]. Those photoinduced electrons could rapidly react with O2 adsorbed on the surface of rGO to generate superoxide (·O2−) and then the activated ·O2− further produces hydroxyl radical (·OH) [51]. Meanwhile, the holes (h+) can be captured by water molecules to produce active ·OH [53]. The photoinduced holes and the products of these oxidation reactions (such as ·O2− and ·OH) have the strong ability to oxidize organic pollutants. At last, MO can be eventually decomposed into CO2, H2O and other small molecules [54, 55]. These involved reactions are expressed as follows:

The proposed mechanism for photocatalytic degradation of MO molecules by the rGO–ZnO composites under UV–Visible light irradiation
Based on the above analysis, we may conclude that the photocatalytic activity of rGO-ZnO could be greatly improved compared with pure ZnO because the high conductivity of rGO could accelerate the electron transfer so as to promote the separation of photogenerated charge carriers and effectively prolong the lifetime of photogenerated charge carriers [44].
4 Conclusions
Highly effective rGO–ZnO composite photocatalysts with different content of rGO have been successfully prepared via a fast and facile one-step method. Due to the introduction of rGO, rGO–ZnO composites exhibit much higher photocatalytic efficiency than pure ZnO, and the optimal loading content of rGO in the rGO–ZnO composites is 1.0 wt%. The enhanced photocatalytic performance of rGO–ZnO composites can be attributed to the effective separation of photoinduced electron–hole pairs with rGO as the conducting plane. Therefore, the rGO–ZnO composite photocatalysts should be potential candidate materials for pollutant elimination.
References
Zhou M, Yang T, Hu W, He X, Xie J, Wang P, Jia K, Liu X (2019) Scalable fabrication of metallopolymeric superstructures for highly efficient removal of methylene blue. Nanomaterials 9:1001. https://doi.org/10.3390/nano9071001
Damkale SR, Arbuj SS, Umarji GG, Panmand RP, Khore SK, Sonawane RS, Rane SB, Kale BB (2019) Two-dimensional hexagonal SnS2 nanostructures for photocatalytic hydrogen generation and dye degradation. Sustain Energy Fuels 3:3406–3414. https://doi.org/10.1039/c9se00235a
Mohammed AM, Mohtar SS, Aziz F, Mhamad SA, Aziz M (2021) Review of various strategies to boost the photocatalytic activity of the cuprous oxide-based photocatalyst. J Environ Chem Eng 9:105138. https://doi.org/10.1016/j.jece.2021.105138
Zhang B, Cao S, Du M, Ye X, Wang Y, Ye J (2019) Titanium dioxide (TiO2) mesocrystals: synthesis, growth mechanisms and photocatalytic properties. Catalysts 9:91. https://doi.org/10.3390/catal9010091
Li H, Meng F, Gong J, Fan Z, Qin R (2018) Template-free hydrothermal synthesis, mechanism, and photocatalytic properties of core–shell CeO2 nanospheres. Electron Mater Lett 14:474–487. https://doi.org/10.1007/s13391-018-0049-7
Qi Y, Meador WE, Xiong J, Abbaszadeh M, Thirumala RVKG, Delcamp JH, Kundu S, Hill GA Jr, Dai Q (2021) Structural, optical, photocatalytic, and optoelectronic properties of Zn2SnO4 nanocrystals prepared by hydrothermal method. Nanotechnology 32:145702. https://doi.org/10.1088/1361-6528/abd509
Han W, Kim J, Park HH (2019) Control of electrical conductivity of highly stacked zinc oxide nanocrystals by ultraviolet treatment. Sci Rep 9:6244. https://doi.org/10.1038/s41598-019-42102-3
Aydin EB (2021) Preparation, characterization and immobilization of Ag-doped ZnO-nanorods into Ca and Cu alginate beads and their application in the photodegradation of methylene blue. ChemistrySelect 6:11653–11663. https://doi.org/10.1002/slct.202102863
Esgin H, Caglar Y, Caglar M (2022) Photovoltaic performance and physical characterization of Cu doped ZnO nanopowders as photoanode for DSSC. J Alloys Compd 890:161848. https://doi.org/10.1016/j.jallcom.2021.161848
Šutka A, Käämbre T, Pärna R, Juhnevica I, Maiorov M, Joost U, Kisand V (2016) Co doped ZnO nanowires as visible light photocatalysts. Solid State Sci 56:54–62. https://doi.org/10.1016/j.solidstatesciences.2016.04.008
Yang X, Qiu L, Luo X (2018) ZIF-8 derived Ag-doped ZnO photocatalyst with enhanced photocatalytic activity. RSC Adv 8:4890–4894. https://doi.org/10.1039/C7RA13351K
Jung H, Pham T-T, Shin EW (2019) Effect of g-C3N4 precursors on the morphological structures of g-C3N4/ZnO composite photocatalysts. J Alloys Compd 788:1084–1092. https://doi.org/10.1016/j.jallcom.2019.03.006
Han F, Song Z, Nawaz MH, Dai M, Han D, Han L, Fan Y, Xu J, Han D, Niu L (2019) MoS2/ZnO-heterostructures-based label-free, visible-light-excited photoelectrochemical sensor for sensitive and selective determination of synthetic antioxidant propyl gallate. Anal Chem 91:10657–10662. https://doi.org/10.1021/acs.analchem.9b01889
Liu Y, Li G, Mi R, Deng C, Gao P (2014) An environment-benign method for the synthesis of p-NiO/n-ZnO heterostructure with excellent performance for gas sensing and photocatalysis. Sens Actuators B Chem 191:537–544. https://doi.org/10.1016/j.snb.2013.10.068
Zhou T, Wang J, Chen S, Bai J, Li J, Zhang Y, Li L, Xia L, Rahim M, Xu Q, Zhou B (2020) Bird-nest structured ZnO/TiO2 as a direct Z-scheme photoanode with enhanced light harvesting and carriers kinetics for highly efficient and stable photoelectrochemical water splitting. Appl Catal B-Environ 267:118599. https://doi.org/10.1016/j.apcatb.2020.118599
Li J, Yuan H, Li J, Zhang W, Liu Y, Liu N, Cao H, Jiao Z (2021) The significant role of the chemically bonded interfaces in BiVO4/ZnO heterostructures for photoelectrochemical water splitting. Appl Catal B-Environ 285:119833. https://doi.org/10.1016/j.apcatb.2020.119833
Zhang Y, Xu J, Xu P, Zhu Y, Chen X, Yu W (2010) Decoration of ZnO nanowires with Pt nanoparticles and their improved gas sensing and photocatalytic performance. Nanotechnology 21:285501. https://doi.org/10.1088/0957-4484/21/28/285501
Tian H, Liu M, Zheng W (2018) Constructing 2D graphitic carbon nitride nanosheets/layered MoS2/graphene ternary nanojunction with enhanced photocatalytic activity. Appl Catal B-Environ 225:468–476. https://doi.org/10.1016/j.apcatb.2017.12.019
Zhang Y, Park M, Kim H-Y, Park S-J (2016) In-situ synthesis of graphene oxide/BiOCl heterostructured nanofibers for visible-light photocatalytic investigation. J Alloys Compd 686:106–114. https://doi.org/10.1016/j.jallcom.2016.06.004
Chandu B, Kurmarayuni CM, Kurapati S, Bollikolla HB (2020) Green and economical synthesis of graphene-silver nanocomposite exhibiting excellent photocatalytic efficiency. Carbon Lett 30:225–233. https://doi.org/10.1007/s42823-019-00091-3
Xu S, Fu L, Pham TSH, Yu A, Han F, Chen L (2015) Preparation of ZnO flower/reduced graphene oxide composite with enhanced photocatalytic performance under sunlight. Ceram Int 41:4007–4013. https://doi.org/10.1016/j.ceramint.2014.11.086
Chen Y-L, Zhang C-E, Deng C, Fei P, Zhong M, Su B-T (2013) Preparation of ZnO/GO composite material with highly photocatalytic performance via an improved two-step method. Chin Chem Lett 24:518–520. https://doi.org/10.1016/j.cclet.2013.03.034
Hummers WS, Offeman RE (1958) Preparation of graphitic oxide. J Am Chem Soc 80:1339–1339. https://doi.org/10.1021/ja01539a017
Hsu KC, Chen DH (2014) Green synthesis and synergistic catalytic effect of Ag/reduced graphene oxide nanocomposite. Nanoscale Res Lett 9:484. https://doi.org/10.1186/1556-276x-9-484
Wang H, Wang L, Qu C, Su Y, Yu S, Zheng W, Liu Y (2011) Photovoltaic properties of graphene oxide sheets beaded with ZnO nanoparticles. J Solid State Chem 184:881–887. https://doi.org/10.1016/j.jssc.2011.02.025
Pan D, Wang S, Zhao B, Wu M, Zhang H, Wang Y, Jiao Z (2009) Li storage properties of disordered graphene nanosheets. Chem Mater 21:3136–3142. https://doi.org/10.1021/cm900395k
Park S, Ruoff RS (2009) Chemical methods for the production of graphenes. Nat Nanotechnol 4:217–224. https://doi.org/10.1038/nnano.2009.58
Khan M, Al-Marri AH, Khan M, Shaik MR, Mohri N, Adil SF, Kuniyil M, Alkhathlan HZ, Al-Warthan A, Tremel W, Tahir MN, Siddiqui MRH (2015) Green approach for the effective reduction of graphene oxide using Salvadora persica L. root (miswak) extract. Nanoscale Res Lett 10:281. https://doi.org/10.1186/s11671-015-0987-z
Zhang Y, Chen Z, Liu S, Xu Y-J (2013) Size effect induced activity enhancement and anti-photocorrosion of reduced graphene oxide/ZnO composites for degradation of organic dyes and reduction of Cr(VI) in water. Appl Catal B-Environ 140–141:598–607. https://doi.org/10.1016/j.apcatb.2013.04.059
Qazi SJS, Rennie AR, Cockcroft JK, Vickers M (2009) Use of wide-angle X-ray diffraction to measure shape and size of dispersed colloidal particles. J Colloid Interface Sci 338:105–110. https://doi.org/10.1016/j.jcis.2009.06.006
Gu F, Nie R, Han D, Wang Z (2015) In2O3–graphene nanocomposite based gas sensor for selective detection of NO2 at room temperature. Sens Actuators B Chem 219:94–99. https://doi.org/10.1016/j.snb.2015.04.119
Wang F, Zhang K (2011) Reduced graphene oxide-TiO2 nanocomposite with high photocatalystic activity for the degradation of rhodamine B. J Mol Catal A Chem 345:101–107. https://doi.org/10.1016/j.molcata.2011.05.026
Fu D, Han G, Chang Y, Dong J (2012) The synthesis and properties of ZnO-graphene nano hybrid for photodegradation of organic pollutant in water. Mater Chem Phys 132:673–681. https://doi.org/10.1016/j.matchemphys.2011.11.085
Park H, Lee KH, Kim YB, Ambade SB, Noh SH, Eom W, Hwang JY, Lee WJ, Huang J, Han TH (2018) Dynamic assembly of liquid crystalline graphene oxide gel fibers for ion transport. Sci Adv 4:eaau2104. https://doi.org/10.1126/sciadv.aau2104
Zhang L, Zhang X, Zhang G, Zhang Z, Liu S, Li P, Liao Q, Zhao Y, Zhang Y (2015) Investigation on the optimization, design and microwave absorption properties of reduced graphene oxide/tetrapod-like ZnO composites. RSC Adv 5:10197–10203. https://doi.org/10.1039/c4ra12591f
Liu X, Pan L, Zhao Q, Lv T, Zhu G, Chen T, Lu T, Sun Z, Sun C (2012) UV-assisted photocatalytic synthesis of ZnO–reduced graphene oxide composites with enhanced photocatalytic activity in reduction of Cr(VI). Chem Eng J 183:238–243. https://doi.org/10.1016/j.cej.2011.12.068
Xu T, Zhang L, Cheng H, Zhu Y (2011) Significantly enhanced photocatalytic performance of ZnO via graphene hybridization and the mechanism study. Appl Catal B-Environ 101:382–387. https://doi.org/10.1016/j.apcatb.2010.10.007
Chen Y, Huang W, He D, Situ Y, Huang H (2014) Construction of heterostructured g-C3N4/Ag/TiO2 microspheres with enhanced photocatalysis performance under visible-light irradiation. ACS Appl Mater Interfaces 6:14405–14414. https://doi.org/10.1021/am503674e
Huang H, Liu K, Zhang Y, Chen K, Zhang Y, Tian N (2014) Tunable 3D hierarchical graphene–BiOI nanoarchitectures: their in situ preparation, and highly improved photocatalytic performance and photoelectrochemical properties under visible light irradiation. RSC Adv 4:49386–49394. https://doi.org/10.1039/C4RA07533A
Bilgin Simsek E, Kilic B, Asgin M, Akan A (2018) Graphene oxide based heterojunction TiO2–ZnO catalysts with outstanding photocatalytic performance for bisphenol-A, ibuprofen and flurbiprofen. J Ind Eng Chem 59:115–126. https://doi.org/10.1016/j.jiec.2017.10.014
Wang H-J, Sun Y-Y, Wang C-F, Cao Y (2012) Controlled synthesis, cytotoxicity and photocatalytic comparison of ZnO films photocatalysts supported on aluminum matrix. Chem Eng J 198–199:154–162. https://doi.org/10.1016/j.cej.2012.05.088
Dai K, Lu L, Liang C, Dai J, Zhu G, Liu Z, Liu Q, Zhang Y (2014) Graphene oxide modified ZnO nanorods hybrid with high reusable photocatalytic activity under UV-LED irradiation. Mater Chem Phys 143:1410–1416. https://doi.org/10.1016/j.matchemphys.2013.11.055
He J, Niu C, Yang C, Wang J, Su X (2014) Reduced graphene oxide anchored with zinc oxide nanoparticles with enhanced photocatalytic activity and gas sensing properties. RSC Adv 4:60253–60259. https://doi.org/10.1039/c4ra12707b
Ranjith KS, Manivel P, Rajendrakumar RT, Uyar T (2017) Multifunctional ZnO nanorod-reduced graphene oxide hybrids nanocomposites for effective water remediation: effective sunlight driven degradation of organic dyes and rapid heavy metal adsorption. Chem Eng J 325:588–600. https://doi.org/10.1016/j.cej.2017.05.105
Ong CB, Mohammad AW, Ng LY, Mahmoudi E, Azizkhani S, Hayati Hairom NH (2017) Solar photocatalytic and surface enhancement of ZnO/rGO nanocomposite: degradation of perfluorooctanoic acid and dye. Process Saf Environ Prot 112:298–307. https://doi.org/10.1016/j.psep.2017.04.031
Ramos PG, Flores E, Luyo C, Sánchez LA, Rodriguez J (2019) Fabrication of ZnO-RGO nanorods by electrospinning assisted hydrothermal method with enhanced photocatalytic activity. Mater Today Commun 19:407–412. https://doi.org/10.1016/j.mtcomm.2019.03.010
Prabhu S, Megala S, Harish S, Navaneethan M, Maadeswaran P, Sohila S, Ramesh R (2019) Enhanced photocatalytic activities of ZnO dumbbell/reduced graphene oxide nanocomposites for degradation of organic pollutants via efficient charge separation pathway. Appl Surf Sci 487:1279–1288. https://doi.org/10.1016/j.apsusc.2019.05.086
Nguyen CH, Tran ML, Tran TTV, Juang R-S (2020) Enhanced removal of various dyes from aqueous solutions by UV and simulated solar photocatalysis over TiO2/ZnO/rGO composites. Sep Purif Technol 232:115962. https://doi.org/10.1016/j.seppur.2019.115962
Elumalai N, Prabhu S, Selvaraj M, Shanavas S, Navaneethan M, Harish S, Ramu P, Ramesh R (2021) Investigation on synergistic effect of rGO and carbon quantum dots-embedded ZnO hollow spheres for improved photocatalytic aqueous pollutant removal process. J Mater Sci Mater Electron 32:28633–28647. https://doi.org/10.1007/s10854-021-07239-w
Gao P, Li A, Sun DD, Ng WJ (2014) Effects of various TiO2 nanostructures and graphene oxide on photocatalytic activity of TiO2. J Hazard Mater 279:96–104. https://doi.org/10.1016/j.jhazmat.2014.06.061
Verma N, Ananthakrishnan R (2019) Riboflavin-immobilized CeO2–RGO nanohybrid as a potential photoredox catalyst for enhanced removal of organic pollutants under visible light. J Phys Chem C 124:404–415. https://doi.org/10.1021/acs.jpcc.9b08326
Qiu B, Li Q, Shen B, Xing M, Zhang J (2016) Stöber-like method to synthesize ultradispersed Fe3O4 nanoparticles on graphene with excellent Photo-Fenton reaction and high-performance lithium storage. Appl Catal B-Environ 183:216–223. https://doi.org/10.1016/j.apcatb.2015.10.053
Wu M, Gu L, Wang Q, Wang C, Zhang H (2018) Interfacial assembly of robust TiO2 nanosheets onto silica-modified reduced graphene oxide for highly efficient degradation of organic dyes. ChemNanoMat 4:387–393. https://doi.org/10.1002/cnma.201700369
Li X, Zheng S, Zhang C, Hu C, Chen F, Sun Y, Duo S, Zhang R, Hu Q, Li W, Kang Y (2017) Synergistic promotion of photocatalytic performance by core@shell structured TiO2/Au@rGO ternary photocatalyst. Mol Catal 438:55–65. https://doi.org/10.1016/j.mcat.2017.05.016
Kubo W, Tatsuma T (2006) Mechanisms of photocatalytic remote oxidation. J Am Chem Soc 128:16034–16035. https://doi.org/10.1021/ja066041y
Acknowledgements
No funding was received to assist with the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ma, Y., Liu, Y. Preparation of graphene–ZnO composite with enhanced photocatalytic performance. Carbon Lett. 32, 1265–1275 (2022). https://doi.org/10.1007/s42823-022-00349-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42823-022-00349-3