
Abstract
CeO2 nanospheres with the core–shell nanostructure have been successfully synthesized by a template-free hydrothermal method. The structures, morphologies and optical properties of core–shell CeO2 nanospheres were analyzed by X-ray diffraction (XRD), TG, Fourier transform infrared spectroscopy, XRD, EDS, SAED, scanning electron microscopy and transmission electron microscopy, UV–Vis diffuse reflectance spectra, Raman analyses. The degradation efficiencies of core–shell CeO2 nanospheres for methyl orange were as high as 93.49, 95.67 and 98.28% within 160 min, and the rates of photo degradation of methyl orange by core–shell CeO2 nanospheres under UV-light were 0.01693, 0.01782 and 0.02375 min−1. Methyl orange was degraded in photocatalytic oxidation processes, which mainly gave the credit to a large number of reactive species including h+, surface superoxide species ·O2−, and ·OH radicals. The core–shell structure, small crystallite size and the conversion between Ce3+ and Ce4+ of CeO2 nanospheres were of importance for its catalytic activity. These results demonstrated the possibility of improving the efficient catalysts of the earth abundant CeO2 catalysts.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The development of nanotechnology has created a renewed interest in nano materials, which has the ability to improve the material properties by controlling the nanostructure. Nano-particulate transition metal oxides are of technological importance in various areas of chemistry and materials science, such as catalysis, energy storage and electronics [1,2,3]. It is well known that CeO2 is a major compound in the useful rare-earth family with predominant physical and chemical properties. Recently, CeO2 nanoparticles and their composites have attracted extensive attention in various fields, such as polishing materials [4], catalysts [5], oxygen gas sensors [6], and irradiation [7].
Semiconductor photocatalysts such as TiO2, ZnO and CeO2 are promising materials for photodecomposition of organic pollutants for green technology because of their self-cleaning functionality driven by sun light [8, 9]. Moreover, compared to TiO2 (3.27 eV) and ZnO (3.37 eV), CeO2 has a relatively small band gap (3.19 eV), a higher transparency to visible light, and better UV absorption properties in the UV wavelength range. Particularly, cerium dioxide (CeO2) has drawn considerable photocatalysis for degradation of dyes with high photocatalytic efficiency, low cost, high stability and environmental friendliness [10, 11].
Cerium oxides are another group of effective catalysts in oxidation of organic matters, such as phenol, cyclohexane and cyclohexanone [12]. They are especially effective for dye removal since they have large pore size that can easily accommodate dye molecules [13]. The surface oxygen of CeO2 can be reduced to form oxygen vacancies under reduction atmosphere. Oxygen vacancies are crucial in catalytic reactions, especially in the catalytic reduction reaction [14]. In the series of rare earth oxides, cerium oxide (CeO2) has the following activity: Firstly, it is specific adsorption material for O2, because the reduction reaction is happened easily between Ce4+ and Ce3+ which produces a large amount of oxygen vacancies in the lattice of CeO2, so CeO2 is an ideal material with strong capacity for oxygen storage and release [15, 16]. Many studies have also confirmed that the reducibility and forming ability of the oxygen vacancies of CeO2 can be improved by introducing other metal elements [17]. Moreover, photocatalytic activity not only depends on the morphologies but the active sites, surface area, composition and surface modification of the material.
In this study, mono-disperse CeO2 nanospheres with core–shell structure were prepared by a facile template-free hydrothermal method. Scanning electron microscopy (SEM) was used to observe the surface morphologies of the CeO2 nanospheres. Furthermore, photocatalysis properties of the products under ultraviolet (UV) irradiation are conducted, which presents some new insights for the photocatalysis of core–shell nanospheres. The photocatalytic activity of CeO2 nanospheres were evaluated through the photodegradation of methyl orange under UV radiation. The more obvious core–shell structure of cerium oxide nanospheres exhibits better photocatalytic activity. Our research will be useful for the preparation of catalytic materials, especially mono-disperse multifunctional core–shell microspheres, which have profound significance in the synthesis of materials, the study of optical and photocatalytic properties.
2 Experimental Procedure
All the chemicals utilized during the synthesis process were of analytical grade without any further purification. The detailed synthetic process of the synthesis of Ce(OH)CO3 precursors was as follows. 0.347 g cerium nitrate hexahydrate (Ce(NO3)3·6H2O) and 1.6 g polyethylene glycol (= 20,000) were dissolved in 30 mL distilled water under vigorous magnetic stirring for 0.5 h at room temperature. 3.6 g urea ((NH2)2CO) was dissolved in 10 mL distilled water under vigorous magnetic stirring for 0.5 h. And then, 10 mL urea aqueous solution was rapidly added to 30 mL cerium nitrate and polyethylene glycol mixed aqueous solution under continuous stirring. After stirring for 1 h, the mixed solution was transferred into a 50 ml Teflon-lined autoclave and heated at 100 °C for 3 h. After the solvothermal treatment, the powdered product was separated by centrifugation, washed with distilled water and absolute ethylalcohol three times, and then dried at 70 °C for 12 h. The other samples were synthesized by keeping a similar procedure except changing a single parameter, namely, the concentration of urea.
Powder X-ray diffraction (XRD, SmartLab 9KW, Japan) analysis using Cu Kα radiation (λ = 0.154 nm) was employed to identify the crystalline phases of the prepared powder at room temperature, with an accelerating voltage of 40 kV and tube current of 100 mA. The thermal behavior of the as-synthesized products was measured by simultaneous thermogravimetric analysis apparatus (TG-DTA, STA 449F3, Germany) heating from room temperature to 1000 °C (10 °C/min) in air atmosphere. The Fourier transform infrared (FTIR, Vertex 80 Fourier transform infrared spectrometer, Bruker) spectra of both precursors and as-synthesized CeO2 powders were recorded on infrared spectrometer. The morphologies of synthesized products were characterized by transmission electron microscope (TEM, JEM-2100, Japan) and scanning electron microscope (SEM, S-4800, China) equipped with an attached energy dispersive spectroscopy (EDS) system. The elemental chemical state was analyzed by X-ray photo-electron spectroscopy (XPS, Escalab 250xi, USA), and the mono Al Ka radiation was used as the excitation source. The Raman spectra were measured by a Raman spectrometer system (Raman, inVia-Reflex, England) with the excitation laserwave length of 532 nm at room temperature. The ultraviolet–visible (UV–Vis) diffuse reflectance spectra was recorded on a spectrophotometer (U-4100, China) equipped with the analyzed range was 200–800 nm.
The photocatalytic activities of the as-prepared ceria were evaluated by photodegradation of methyl orange under simulated UV lamp illumination at ambient temperature. All experiments were conducted in a 7.0 cm culture dish. The experimental details were as follows: 0.05 g sample was dispersed into a 15 mL methyl orange aqueous solution with a concentration of 10 mg/L. In the pre-adsorption catalytic ultrasonic process, methyl orange solution with catalyst was irradiated for different time. After reaction, the mixture was centrifuged at 3000 rpm for 5 min, and the supernatant was used for methyl orange detection.
3 Results and Discussion
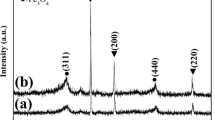
Figure 1 illustrates the XRD patterns and reference pattern (JCPDS No. # 41-0013) of the nanospheres synthesized at 100 °C for 3 h doped urea of 1 mmol and 60 mmol. All the detectable peaks can be readily indexed to a pure orthorhombic structure of CeCO3OH (JCPDS No. # 41-0013), indicating that the experimental condition difference does not affect the phase composition of the samples.

XRD patterns and reference pattern (JCPDS No. # 41-0013) of the as-synthesized precursors at 100 °C for for 3 h doped urea of 1 mmol and 60 mmol
Typical SEM images with different magnification of the as-synthesized precursors are presented in Fig. 2. The morphology of the precursors can be readily controlled by changing experimental parameters such as the concentration of urea. Figure 2a, c, e are the panoramic SEM image and Fig. 2b, d, f the higher magnification SEM image of the precursors. Figure 2a, b show the precursor synthesized at 100 °C for 3 h doped urea of 20 mmol has a spherical structure with the random distribution sizes of 50–145 nm. Figure 2c, d show the precursor has a spherical structure with some holes on the surface and the uniform sizes of 200 nm. Figure 2e, f show the precursor synthesized at 100 °C for 3 h doped urea of 60 mmol still shows the spherical structure with sizes of 150 nm, but The nanospheres were suspected to be core–shell structure form the broken nanospheres.

SEM images of the precursor synthesized at 100 °C for 3 h doped urea of a, b 20 mmol, c, d 40 mmol and e, f 60 mmol
To further confirm the core–shell structure and elementary composition of the precursor, the TEM have been operated. Figure 3 shows the panoramic and the higher magnification TEM images of the precursor synthesized at 100 °C for 3 h doped urea of 60 mmol. It can be clearly seen that the CeCO3OH nanospheres is the core–shell structure. The thickness of shell is around 10–15 nm, and the size of the core is around 60–120 nm.

a The panoramic and b the higher magnification TEM images of the precursor synthesized at 100 °C for 3 h doped urea of 60 mmol
Before calcination, the thermal behavior of the as-synthesized nanopowder was analyzed by means of thermogravimetric/differential thermal analysis (TG/DTG) from 40 to 1000 °C with the heating rate of 20 °C/min in an air atmosphere, as shown in Fig. 4. TG curve indicates that there are two weight loss steps at the temperature ranges 60–275 and 275–500 °C. The first weight loss of 2.5% is mainly attributed to the evaporation of H2O, whereas the second one of 21.2% may be ascribed to the decomposition of the precursor, which is consistent with the theoretical value of 20.7%, according to the decomposition reaction (1):

TG-DTG curves of the as-synthesized Ce(OH)CO3 synthesized at 100 °C for 3 h doped urea of 60 mmol
The weight-loss process ceases at 500 °C, and the stable residue can be reasonably ascribed to CeO2. The data of DTG curve, which corresponds to the the date of TG curve, shows that a significant weight loss process occurs in the range of 294 °C and the mass of the sample remains constant after 500 °C. According to the above analysis, the calcination temperature for the precursor was chosen to be at 500 °C.
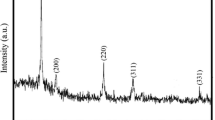
After the calcination of the obtained precursor at 500 °C for 4 h in air accompanied by a color change from white to slight yellow according to the TG data, the precursors synthesized at 100 °C for 3 h doped urea of different quantity were converted into CeO2, as shown in Fig. 5. From the typical XRD pattern of as-prepared calcined samples, some broad peaks at diffraction angles of 28.5°, 33.1°, 47.5°, 56.3°, 59.1° and 69.4°, which are related to CeO2 (111) (200), (220), (311), (222) and (400) reflections respectively, can be indexed to the pure fluorite cubic structures (JCPDS Card # 34-0394). The crystallite size of CeO2 was calculated by using the Scherrer’s formula.

XRD patterns and reference pattern (JCPDS Card # 34-0394) of CeO2 obtained by calcining the CeCO3OH samples at 500 °C for 4 h
where K is 0.89, λ is 0.15406 nm, β is the full-width at half maximum of the diffraction peak and θ is the diffraction angle. The crystal is made up of many small grains, and its crystallite size of 13.1, 15.2 and 13.7 nm were calculated by Scherrer’s formula. All the sharp diffraction peaks indicate that as-obtained CeO2 samples possess a good crystallinity.
The formation of as-generated CeO2 was further verified by FIIR spectroscopy. Figure 6 shows that the Fourier transform infrared (FTIR) spectra of the CeOHCO3 and the samples calcined at 500 °C. As shown in Fig. 6a, the band centered at 3454 cm−1 of the precursors obtained at 100 °C for 5 h was normally ascribed to the O–H bending vibrations bound to the Ce atoms and O–H stretching vibration of the OH− groups, respectively [18]. The absorbance at 1508 and 1417 cm−1 were assigned to the υ3 mode of the CO32− groups, and the intense peaks at 1074, 857, 808 and 723 cm−1 are attributed to the C–O, νC–O, δCO32− and νasCO2, respectively [19]. The intense peak at 482 cm−1 is ascribed to the vibration of CeO2 [20]. After calcination, the peaks of the residual water or carbonate species become weaker or disappear except for the bands of Ce–O stretching vibration as shown in Fig. 6b. The absorbance at 3381 cm−1 can be assigned to surface hydroxyl groups and absorbed water molecules on the CeO2 surface, The peaks at 555 cm−1 is assigned to the Ce–O stretching vibration [21]. The FTIR band at 1531 cm−1 in the sample corresponds to O–H stretching and vibrations, and these absorbed OH groups and water molecules play significant roles in photocatalytic activity because they act as oxidizers to decompose pollutants [22]

FTIR spectra of (a) the core–shell CeOHCO3 nanosphere synthesized at 100 °C for 5 h and (b) the calcined products
Figure 7 clearly shows the TEM images of the CeO2 nanospheres synthesized at 100 °C for 3 h doped urea of different quantities, including the panoramic TEM image and the higher magnification TEM image. Figure 7a, b show the CeO2 nanospheres synthesized at 100 °C for 3 h doped urea of 20 mmol have a loosened spherical structure with the random distribution sizes of 40–145 nm. Figure 7c, d show the CeO2 nanospheres have a hazy core–shell structure with the sizes of 100–200 nm. Figure 7e, f show the CeO2 nanospheres synthesized at 100 °C for 3 h doped urea of 60 mmol still show an obvious core–shell structure with sizes of 150 nm, which further confirms the surmise of SEM. The core of the core–shell nanospheres is 40–80 nm, and the thickness of the shell is about 8 nm. From this, the thickness of the shell becames smaller, and the space between the core and shell becames bigger. During the calcination process, the contraction force exceeded the adhesion force, resulting in the inner core shrinkage and leaving a outer shell. Spherical morphology has advantageous features with packing efficiencies and high volumetric specific energy, which has promising in commercial applications.

TEM image of the samples synthesized at 100 °C for 3 h doped urea of a, b 20 mmol, c, d 40 mmol and e, f 60 mmol
HRTEM images of the powders are shown in Fig. 8a–c, respectively. Measured interplanar spacings of 0.264, 0.271, and 0.308 nm are in good consistent with lattice distance between (200), (200) and (111) planes of fluorite cubic phase. The crystalline nature of the CeO2 nanospheres can be verified by the selected area electron diffraction (SAED) pattern (Fig. 8d, e) which is taken from an area of 30 nm in diameter containing many nanospheres and is basically a ring pattern, suggesting that the CeO2 nanospheres are polycrystalline. And the diffraction rings can be indexed using the cubic fluorite crystal structure, which is in agreement with the XRD results [23]. Figure 8f is a typical energy dispersive X-ray spectrum (EDX) of the core–shell CeO2 nanosphere synthesized at 100 °C for 3 h doped urea of 60 mmol. The EDX spectrum shows peaks for O, C, Cu and Ce, which confirms the impregnation of O and Ce on the nanospheres, apart from the C and Cu peak, which derive from the surface of sample sprayed with carbon and cuprum plates used in EDX instrument, respectively. These results show that the samples as-prepared is CeO2, indicating no other element was introduced into the ceria during the synthesized process of the sample.

HRTEM images and SAED pattern of the samples synthesized at 100 °C for 3 h doped urea of a 20 mmol, b, d 40 mmol, c, e 60 mmol and f EDS spectrum of the core–shell nanosphere
Surfactant dissolved in water, surfactants can reduce the surface tension of the water, and increase of soluble organic compounds [24]. Complexing agent with multifunctional group has a strong binding ability, which also has the effect of dispersive and suspension without contamination textile products and equipment. This experiment was carried out using HO(CH2CH2O)nH as surfactant and urea as complexing agent, and mainly studies the effect of the complexing agent on the sample. To verify the role of urea, the concentration of urea in the synthesis mixture was increased by double while keeping other parameters unchanged. Urea-hydrothermal synthesis has been used to synthesize spherical nanostructures where released ammonia by urea hydrolysis plays an essential role in determining the morphology of product, acting as a structure directing agent. Here, a possible chemical reaction for the CeCO3OH formation was proposed as follows:
Precipitation of Ce3+ ions with CO23− and OH− species formed Ce(OH)CO3 nanocrystals, and the CeO2 was obtained by calcining Ce(OH)CO3 nanocrystals, as shown in Eq. (1). Figure 9 shows the SEM images of the precursor synthesized at 100 °C for 3 h doped urea of different quantity. Based on the characterization data and the description of our experiments, we suggest that the whole hydrothermal synthetic process of the core–shell CeO2 nanospheres can be divided into three steps. At the very beginning, when the reaction began in the hydrothermal system, the CeO2 nanocrystals with irregular shapes are formed in the solution through a nucleation process (Fig. 9a). The formed CeO2 nanoparticles then agglomerate and self-assemble to form nanospheres, but the size is about 50–145 nm and has a random distribution (Fig. 9b). Since the size of the ammonia bubble is randomly distributed, non-uniform CeO2 spheres are produced. With a decreased amount of urea, the self-generated pressure in the synthesis system drops, increasing the size of ammonia bubble. From TEM and Fig. 9c, It can be observed that the outer part of the nanospheres is packed much more loosely than the inside, indicating the appearance of core–shell structure. The core–shell structure appears through the hollow interior spaces between the cores and the shells, and the hollow interior space expanded, due to the partial dissolution of cores (Fig. 9d). The formation process of the core–shell structure has been vividly described, through schematic illustration for the possible growth processes of the different CeO2 nanospheres, as shown in Fig. 10. Therefore, a core–shell structure was formed with a thick shell and thin core in the Ce(OH)CO3 precursor. Subsequently, calcination of this precursor causes a spontaneous transformation of the morphology and produces core–shell CeO2 nanospheres. This phenomenon confirms that urea determines the morphology and size of the CeO2 materials through a structure confining effect.

SEM images of the precursor synthesized at 100 °C for 3 h doped urea of a 1 mmol, b 20 mmol, c 40 mmol and d 60 mmol

Schematic illustration for the possible growth processes of the different CeO2 nanospheres
X-ray photo-electron spectroscopy (XPS) is powerful to examine the chemical states and composition of pure ceria. Figure 11a, b show the survey scan indicating the presence of C and O along with Ce. Figure 11e, f show XPS Ce 3d core levels spectra for CeO2 samples synthesized at 100 °C for 3 h added urea of 40 and 60 mmol. XPS spectra of Ce 3d can be deconvoluted into two series of peaks (u and v) corresponding to spin-orbital doublet peaks of 3d3/2 and 3d5/2. According to the literature, two peaks labeled v′ (883.88 eV) and u′ (903.38 eV) are related to the 3d104f1 electronic state of Ce3+ ions, and the other six peaks marked as v (882.83 eV), v″ (889.30 eV), v‴ (898.78 eV), u (901.3 eV), u″ (907.86 eV) and u‴ (917.13 eV) are ascribed to the 3d104f0 electronic state of Ce4+ ions [25]. The presence of Ce3+ implies the defect of samples and reflects the concentration of oxygen vacancies. From the area of these peaks, the contents of Ce3+ species with respect to total Ce concentration are calculated according to the following equations:
where Ai is the integrated area corresponding to peak i. The integrated area of Ce 3d has been showed in Table 1. The content of Ce3+ has decreased from 18.2 to 17.5% with the increase of the reaction time.

XPS survey scan, C1s, Ce3d and O1s core levels spectra taken on CeO2 nanospheres synthesized at 100 °C for 3 h added urea of a, c, e, g 40 mmol and b, d, f, h 60 mmol
Further support is provided by O 1s XPS spectrum shown in Fig. 11g, h. O 1s exhibits three kinds of surface oxygen species, which depicts three peaks corresponding to binding energies of 531.95, 531.31 and 529.54 eV, respectively. The peak at 529.54 eV is assigned to the lattice oxygen ions in CeO2 (Ce4+–O), and that at 531.31 eV is ascribed to the surface adsorbed oxygen species (such as O2−, O−, and –OH) [26]. Generally, the surface adsorbed oxygen species is reported to be active for catalytic reactions. The peak at 531.95 eV is assigned to the lattice oxygen ions in Ce2O3 (Ce3+–O) [27]. The conversion of Ce4+ to Ce3+ will create more oxygen vacancies, hence benefiting the improvement of catalytic activity (Table 2).
In order to investigate if the trivalent Ce3+ has the form of Ce2O3 is due to oxygen vacancies, we calculated the oxygen content in the CeO2 assuming that the total oxygen content is the sum of the required oxygen to fully oxidize Ce4+ and Ce3+ and to form CeO2 and Ce2O3, respectively. The stoichiometric ratio (O/Ce) of CeO2 and Ce2O3 is 2 and 1.5. The ratio of the required oxygen is determined from the concentrations [Ce4+] and [Ce3+] according to the following equations:
where the AO and ACe are corresponding to the peak area of O1s and Ce3d, respectively. The SCe, SO are corresponding to sensitivity factor of cerium and oxygen atom, respectively (SCe = 7.399, SO = 0.711). Table 1 has shows the concentrations of Ce3+ and Ce4+ ions and the stoichiometry variations of CeO2 nanopheres synthesized at 100 °C for 3 h added urea of 40 and 60 mmol. The stoichiometric ratio of oxygen (absorbed oxygen isn’t included) Eq. (6) is 0.034–0.344 bigger than stoichiometric ratio of Eq. (7). The results of the two methods are not in agreement for CeO2 samples synthesized within different urea, meaning that in these nanopheres part of Ce3+ content is consumed in forming Ce2O3, and part of Ce3+ is forming oxygen vacancies, suggesting that Ce2O3 and oxygen vacancies coexist in these nanopheres. The oxygen vacancies and Ce3+ ions exist in CeO2 deposits as proven by XPS, while Ce2O3 is not observed from XRD. Therefore, the Ce2O3 phase is amorphous. The amorphous character of Ce2O3 is an indication that this phase is located at the grain surface and boundaries. These XPS results demonstrate the presence of oxygen vacancies and the surface adsorbed oxygen species in the CeO2 nanospheres, which can act as donors for the charge carriers and trap sites of electrons [28].
In order to better understand the CeO2 structure, Raman scattering of the samples prepared under different conditions was carried out and the corresponding results were exhibited in Fig. 12. The Raman spectroscopy shows that all the samples exhibit a strong band at 462 cm−1 attributed to the F2g vibration mode of cubic fluorite-type structure, which is viewed as symmetric breathing mode of the oxygen atoms surrounding each cation. [29].The Raman line broadening of CeO2 nanostructures can be described by the dependence of the half-width (Γ) on the inverse of grain size (d g ), which follows a linear behavior as Eq. (13).

Raman spectra of CeO2 nanospheres synthesized at 100 °C for 3 h added urea of different quantity
The strong peaks become sharper and narrower with the increasing of the urea concentration, which is consistent with the XRD measurements. The second-order features at 1177 cm−1 are very prominent for all samples, which can be attributed to the second-order Raman mode of surface superoxide species (\(\cdot{\text{O}}_{2}^{ - }\)), and has little additional contributions from F2g symmetry. In addition, the peak at 595 cm−1 can be attributed to intrinsic oxygen vacancies caused by the existence of Ce3+ [30], and the peak near 262 cm−1 can be contributed to oxygen vacancies and a transverse acoustic mode in the ceria lattice in the system [31]. Hydrothermal method is the most feasible way to synthesize nanomaterials with more bulk phase oxygen vacancies and surface active oxygen, which is conducive to light catalytic properties.
The effect of CeO2 on the light sorption properties is investigated, which may play a critical role in determining the photo catalytic performance, especially for the UV-light photo degradation of contaminants [32]. The absorbance of the as-prepared samples is measured by UV–Vis absorption spectra, and the results are recorded in Fig. 13. UV–visible absorption of CeO2 nanospheres shows an excellent absorption in UV region (465 nm) certifying an appropriate photocatalyst in UV light. The band gap energy of the as-prepared samples can be calculated through the following equation [33, 34]:
where A, a, v, Eg and h are a constant, absorption coefficient, light frequency, band gap energy and Planck constant, respectively. For the value of n, it was determined by the type of optical transition of semiconductors (n = 1 for direct transition and n = 4 for indirect transition). For CeO2, the value of n is 1 [35]. As shown in the inset, the band gap energy of CeO2 is estimated to be 2.77, 3.05 and 2.87 eV according to a plot of (ahv)2 versus energy (hv). The change in band gap has been attributed to oxygen vacancies, which is responsible for the unique property for photo catalytic applications [36]. Because band gap narrowing and the photocatalytic activity are closely related, CeO2 nanospheres are expected to show more activity in the visible region of the electromagnetic spectrum.

UV–Vis diffuse reflectance spectrum of as-synthesized CeO2 nanospheres and the inset are the corresponding plots of (ahv)2
The photocatalytic activity of the as-synthesized core–shell CeO2 nanospheres evaluate by measuring the photodegradation of methyl orange under UV-light illumination, as shown in Fig. 14. By plotting Ct/C0 changing over irradiation time (Fig. 14a), the changing of the normalized organic compounds concentration can be gained. From Fig. 14a, without a catalyst, the photodegradation of methyl orange is very low even after 160 min. The methyl orange could hardly be self-photosensitized and self-decomposed without photocatalysts and the loss of methyl orange by the adsorption process could be ignored in this experiment [37]. In the presence of the catalyst, the color of the suspension faded away gradually with the increase of the irradiation time in the experiment, and the photodegradation is obviously enhanced. In this study, within the last 20 min, the degradation rate constant diminishes gradually due to the reducing of the normalized organic compounds concentration and the catalysts made up of spherical particles have the tendency to agglomerate into larger particles. The degradation efficiencies of CeO2 nanospheres for methyl orange were as high as 93.49, 95.67 and 98.28% within 160 min. In order to quantitatively understand the reaction rates of the methyl orange degradations by CeO2 nanospheres, they can be expressed by the pseudo-first order model as follows:
where Ct and C0 are absorbance of the dyes at time t and at time 0. K is the first order rate constant in min−1 and t is the time in min [38, 39]. Figure 14b shows the plots of ln(C0/Ct) versus the irradiation time for the photo degradation of methyl orange. The rates of photo degradation of methyl orange by CeO2 nanospheres under UV-light are 0.01693, 0.01782 and 0.02375 min−1. The degradation efficiency and degradation rate of core–shell CeO2 nanospheres are very significant due to the unique property.

a Photocatalytic degradation of methyl orange and b Kinetic relationship of ln(C0/Ct) versus irradiation time using as-synthesized CeO2 nanospheres synthesized at 100 °C for 3 h doped urea of different quantity
On the basis of the above-mentioned experimental results and analysis, the schematic mechanism of the photocatalytic activity of the CeO2 composite photocatalyst is proposed and illustrated in Fig. 15. In general, organic dyes such as methyl orange are degraded through photocatalytic oxidation processes, which involve a large number of reactive species including h+, ·O2−, and ·OH radicals [40]. Under the irradiation of UV-light, CeO2 can be both excited and generate the photo-induced electron–hole pairs [Eq. (16)], and electrons (e−) are excited from valence band (VB) to conduction band (CB). The created holes (h+) in the VB combine with H2O and OH− to form ·OH radicals and superoxide radical anion (·O2−) [Eqs. (17) and (18)]. The photoexcited electrons (e−) in the CB react with adsorbed O2 on the CeO2 nanospheres to form ·O2− radical anions [Eq. (19)]. Ce4+ ion acts as a stronger Lewis acid than O2 [41]. The Ce4+ ion is superior to O2 in its capability of trapping electrons (Ce4+ + e− → Ce3+). The electrons trapped in Ce4+ sites can transfer to the adsorbed O2 via an oxidation process (Ce3+ + O2 → Ce4+ + ·O2−) and produce a superoxide radical anion (·O2−) [42, 43]. In this process, the photoexcited electron in the CB of CeO2 (4f0 5d0 6 s0) immediately reacts with Ce4+ and forms Ce3+ (4f1 5d0 6s0), which is stabilized by creating an oxygen vacancy state [44], further resulting in the enhanced photocatalytic activity. The created holes (·O2−) combined with h+ to form HO2· radicals, which will break down into O2 and H2O2 [Eqs. (20) and (21)]. The H2O2 combined with ·O2− to form·OH, OH−, and O2· [Eq. (22)]. Subsequently, methyl orange were decomposed to CO2, H2O and other inorganic molecules, as shown in Eq. (23). The process is detailedly described as follows:

Schematic diagram of reaction mechanism in catalytic process
In a word, the core–shell structure, small crystallite size and the conversion between Ce3+/Ce4+ of CeO2 nanospheres are of importance for its catalytic activity.
4 Conclusions
In summary, core–shell CeO2 nanospheres have been successfully synthesized by a template-free hydrothermal method. The concentration of the precipitator was systematically studied, which was key parameter for controlling the final morphology. The existence and concentration of Ce4+ and Ce3+ were confirmed by Raman and XPS. The UV absorption spectra of core–shell nanostructure clearly indicated the variation in the energy. From the test of the photocatalytic ability of the nanospheres, the conclusion were that the degradation efficiencies for methyl orange are 93.49, 95.67 and 98.28%, and the rates of photodegradation were 0.01693, 0.01782 and 0.02375 min−1, respectively. Photodegradation of methyl orange mainly gave the credit to a large number of reactive species including h+, surface superoxide species ·O2−, and ·OH radicals. Consequently, some factors played an important role in its catalytic activity, such as the core–shell structure, small crystallite size, and the conversion between Ce3+/Ce4+. Finally, it is hoped that the current work will stimulate further interest in this intriguing topic
References
Zhang, B., Li, D., Wang, X.: Catalytic performance of La-Ce-O mixed oxide for combustion of methane. Catal. Today 158, 348 (2010)
Ren, Y., Liu, Z., Pourpoint, F., Armstrong, A.R., Grey, C.P., Bruce, P.G.: Nanoparticulate TiO2(B): an anode for lithium-ion batteries. Angew. Chem. 51, 2164 (2012)
Hope, M.A., Halat, D.M., Magusin, P.C.M.M., Paul, S., Pengc, L., Grey, C.P.: Surface-selective direct 17O DNP NMR of CeO2 nanoparticles. Chem. Commun. 53, 2142 (2017)
Auffan, M., Rose, J., Orsiere, T., Meo, M.D., Thill, A., Zeyons, O., Proux, O., Masion, A., Chaurand, P., Spalla, O., Botta, A., Wiesner, M.R., Bottero, J.Y.: Original CeO2 nanoparticles induce DNA damage towards human dermal fibroblasts in vitro. Nanotoxicology 3, 161 (2009)
Zabilskiy, M., Djinovic, P., Tchernychova, E., Tkachenko, O.P., Kustov, L.M., Pintar, A.: Nanoshaped CuO/CeO2 materials: effect of the exposed ceria surfaces on catalytic activity in N2O decomposition reaction. ACS Catal. 5, 5357 (2015)
Liu, Y., Lei, Y.: Pt-CeO2 nanofibers based high-frequency impedancemetric gas sensor for selective CO and C3H8 detection in high-temperature harsh environment. Sens. Actuators B 188, 1141 (2013)
Lu, Z., Mao, C., Meng, M., Liu, S., Tian, Y., Yu, L., Li, C.M.: Fabrication of CeO2 nanoparticle-modified silk for UV protection and antibacterial applications. J. Colloid Interface Sci. 435, 8 (2014)
Kameya, Y., Yamaki, H., Ono, R., Motosuke, M.: Fabrication of micropillar TiO2 photocatalyst arrays using nanoparticle-microprinting method. Mater. Lett. 175, 262 (2016)
Ou, M., Zhong, Q., Zhao, Y., Xue, Y., Song, F.: Graphene-decorated 3D BiVO4 photocatalysts with controlled size and shape for efficient visible-light-induced photocatalytic performance. Mater. Lett. 184, 227 (2016)
Lin, W., Zhang, S., Wang, D., Zhang, C., Sun, D.: Ultrasound-assisted synthesis of high-efficiency Ag3PO4/CeO2 heterojunction photocatalyst. Ceram. Int. 41, 8956 (2015)
Khan, S.B., Faisal, M., Rahman, M.M., Jamal, A.: Exploration of CeO2 nanoparticles as a chemi-sensor and photo-catalyst for environmental applications. Sci. Total Environ. 409, 2987 (2011)
Lin, S.S., Chen, C.L., Chang, D.J., Chen, C.C.: Catalytic wet air oxidation of phenol by various CeO2 catalysts. Water Res. 36, 3009 (2002)
Sonawane, S.H., Chaudhari, P.L., Ghodke, S.A., Parande, M.G., Bhandari, V.M., Mishra, S., Kulkarni, R.D.: Ultrasound assisted synthesis of polyacrylic acid-nanoclay nanocomposite and its application in sonosorption studies of malachite green dye. Ultrason. Sonochem. 16, 351 (2009)
Anicic, B., Trop, P., Goricanec, D.: Comparison between two methods of methanol production from carbon dioxide. Energy 77, 279 (2014)
Plakas, K.V., Karabelas, A.J., Sklari, S.D., Zaspalis, V.T.: Toward the Development of a novel electro-fenton system for eliminating toxic organic substances from water, part 1, in situ generation of hydrogen peroxide. Ind. Eng. Chem. Res. 52, 13948 (2013)
Skorodumova, N.V., Simak, S.I., Lundqvist, B.I., Abrikosov, I.A., Johansson, B.: Quantum origin of the oxygen storage capability of ceria. Phys. Rev. Lett. 89, 166601 (2002)
Bhargava, S.K., Tardio, J., Prasad, J., Foger, K., Akolekar, D.B., Grocott, S.C.: Wet oxidation and catalytic wet oxidation. Ind. Eng. Chem. Res. 45, 1221 (2006)
Dai, Q.G., Bai, S.X., Li, H., Liu, W., Wang, X.Y., Lu, G.Z.: Template-free and non-hydrothermal synthesis of CeO2 nanosheets via a facile aqueous-phase precipitation route with catalytic oxidation properties. Cryst. Eng. Commun. 16, 9817 (2014)
Yue, L., Zhang, X.M.: Structural characterization and photocatalytic behaviors of doped CeO2 nanoparticles. J. Alloys Compd. 475, 702 (2009)
Liu, Y.C., Chen, X.Y., Yao, R., Liao, S.Y., Chen, W.P., Nie, D.H., Wang, Y.M., Zheng, F.: Mechanical properties of 1 mol% CeO2/10 mol% Sc2O3 co-doped and stabilized ZrO2 synthesized through hydro/solve-thermal method. Int. J. Appl. Ceram. Technol. 14, 474 (2017)
Tian, G.H., Chen, Y.J., Zhou, W., Pan, K., Dong, Y.Z., Tian, C.G., Fu, H.G.: Facile solvothermal synthesis of hierarchical flower-like Bi2MoO6 hollow spheres as high performance visible-light driven photocatalysts. J. Mater. Chem. 21, 887 (2011)
Wang, T., Zhao, S.S., Lin, B., Xue, C., Yang, G.D., Ding, S.J., Ma, C.S., Yang, G., Yang, G.R.: A facile one-step synthesis of three-dimensionally ordered macroporous N-doped TiO2 with ethanediamine as the nitrogen source. J. Mater. Chem. A 2, 15611 (2014)
Zhang, D.S., Niu, F.H., Yan, T.T., Shi, L.Y., Du, X.J., Fang, J.H.: Ceria nanospindles Template-free solvothermal synthesis and shape-dependent catalytic activity. Appl. Surf. Sci. 257, 10161 (2011)
Wright, J.R.: Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 5, 58 (2005)
Wang, Y., Shen, X., Chen, F.: Improving the catalytic activity of CeO2/H2O2 system by sulfation pretreatment of CeO2. J. Mol. Catal. A-Chem. 381, 38 (2014)
Zhao, H., Dong, Y., Jiang, P., Wang, G., Zhang, J., Li, K., Feng, C.: An α-MnO2 nanotube used as a novel catalyst in ozonation: performance and the mechanism. New J. Chem. 38, 1743 (2014)
Guo, R.T., Chen, Q.L., Ding, H.L., Wang, Q.S., Pan, W.G., Yang, N.Z., Lu, C.Z.: Preparation and characterization of CeOx@MnOx core–shell structure catalyst for catalytic oxidation of NO. Catal. Commun. 69, 165 (2015)
Rana, A.M., Akbar, T., Ismail, M., Ahmad, E., Hussain, F., Talib, I., Imran, M., Mehmood, K., Iqbal, K., Endurance, M.Y.N.: Endurance and cycle-to-cycle uniformity improvement in tri-Layered CeO2/Ti/CeO2 resistive switching devices by changing top electrode material. Sci. Rep. 7, 39539 (2017)
Lu, X.H., Huang, X., Xie, S.L., Zheng, D.Z., Liu, Z.Q., Liang, C.L., Tong, Y.X.: Facile electrochemical synthesis of single crystalline CeO2 octahedrons and their optical properties. Langmuir 26, 7569 (2010)
Singh, K., Singh, S.K., Kaur, D.: Tunable multiferroic properties of Mn substituted BiFeO3 thin films. Ceram. Int. 42, 13432 (2016)
Maiti, S., Dhawa, T., Mallik, A.K., Mahanty, S.: CeO2@C derived from benzene carboxylate bridged metal–organic frameworks: ligand induced morphology evolution and influence on the electrochemical properties as a lithium-ion battery anode. Sustain. Energy Fuels 1, 288 (2017)
Zhang, Y.W., Si, R., Liao, C.S., Yan, C.H., Xiao, C.X., Kou, Y.: Facile alcohothermal synthesis, size-dependent ultraviolet absorption, and enhanced CO conversion activity of ceria nanocrystals. J. Phys. Chem. B 107, 10159 (2003)
Wang, L., Ding, J., Chai, Y.Y., Liu, Q.Q., Ren, J., Liu, X., Dai, W.L.: CeO2 nanorod/g-C3N4/N-rGO composite: enhanced visible-light-driven photocatalytic performance and the role of N-rGO as electronic transfer media. Dalton Trans. 44, 11223 (2015)
Patil, P.B., Mali, S.S., Kondalkar, V.V., Pawar, N.B., Khot, K.V., Hong, C.K., Patil, P.S., Bhosale, P.N.: Single step hydrothermal synthesis of hierarchical TiO2 microflowers with radially assembled nanorods for enhanced photovoltaic performance. RSC Adv. 4, 47278 (2014)
Vignesh, K., Suganthi, A., Min, B.K., Rajarajan, M., Kang, M.: Designing of YVO4 supported β-AgI nano-photocatalyst with improved stability. RSC Adv. 5, 576 (2014)
Atitar, M.F., Ismail, A.A., Sayari, S.A.A., Bahnemann, D., Afanasev, D., Emeline, A.V.: Mesoporous TiO2 nanocrystals as efficient photocatalysts: Impact of calcination temperature and phase transformation on photocatalytic performance. Chem. Eng. J. 264, 417 (2015)
Fan, Z.H., Meng, F.M., Zhang, M., Wu, Z.Y., Sun, Z.Q., Li, A.X.: Solvothermal synthesis of hierarchical TiO2 nanostructures with tunable morphology and enhanced photocatalytic activity. Appl. Surf. Sci. 360, 298 (2016)
Yayapao, O., Thongtem, T., Phuruangrat, A., Thongtem, S.: Ultrasonic-assisted synthesis of Nd-doped ZnO for photocatalysis. Mater. Lett. 90, 83 (2013)
Huang, C., Wun, X.Q., Ren, W., Shi, P.: Preparation of CeO2 micro/nanostructure and their photo catalytic properties. Ceram. Int. 41, 47 (2015)
Balestrieri, M., Colis, S., Gallart, M., Schmerber, G., Ziegler, M., Gilliot, P., Dinia, A.: Photoluminescence properties of rare earth (Nd, Yb, Sm, Pr)-doped CeO2 pellets prepared by solid-state reaction. J. Mater. Chem. C 3, 7014 (2015)
Islam, M.J., Reddy, D.A., Choi, J., Kim, T.K.: Surface oxygen vacancy assisted electron transfer and shuttling for enhanced photocatalytic activity of a Z-scheme CeO2–AgI nanocomposite. RSC Adv. 6, 19341 (2016)
Li, W., Li, D., Lin, Y., Wang, P., Chen, W., Fu, X., Shao, Y.: Structural, electronic, and optical properties of Ag-doped ZnO nanowires: first principles study. J. Phys. Chem. C 115, 3552 (2011)
Lin, Y., Li, D., Hu, J., Xiao, G., Wang, J., Li, W., Fu, X.: Highly efficient photocatalytic degradation of organic pollutants by PANI-Modified TiO2 composite. J. Phys. Chem. C 116, 5764 (2012)
Coronado, J.M., Maira, A.J., Arias, A.M., Conesa, J.C., Soria, J.: EPR study of the radicals formed upon UV irradiation of ceria-based photocatalysts. J. Photochem. Photobiol. A 150, 213 (2002)
Acknowledgements
This work was supported by the Anhui Provincial Natural Science Foundation of China (1508085SME219).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, H., Meng, F., Gong, J. et al. Template-Free Hydrothermal Synthesis, Mechanism, and Photocatalytic Properties of Core–Shell CeO2 Nanospheres. Electron. Mater. Lett. 14, 474–487 (2018). https://doi.org/10.1007/s13391-018-0049-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13391-018-0049-7