
Abstract
Belonging to genus Bandavirus in Phenuiviridae family, Guertu bandavirus (GTV) is a potential pathogen closely related to severe fever with thrombocytopenia syndrome virus (SFTSV) and heartland virus (HRTV) associated with human diseases. Although the medical significance of GTV is not clear, there was serological evidence suggesting past infection with this virus has occurred, indicating its potential threat to human health. So, it is important to prepare for detection of GTV infection so as to control virus transmission and promote disease diagnosis and treatment. This study is aimed at obtaining monoclonal antibodies (mAbs) against GTV nucleoprotein (NP) and evaluating their activities in recognizing viral antigens from genetic-related bandaviruses, SFTSV and HRTV. Eight mAbs were obtained and four of them (22G1, 25C2, 25E2, and 26F8) recognize linear epitopes of GTV NP. The four mAbs showed cross-reactivity to SFTSV but were unable to react with HRTV. Two fine epitopes were identified by the four mAbs, ENP1 (194YNSFRDPLHAAV205) and ENP2 (226GPDGLP231), which are highly conserved in the NPs of GTV and SFTSV but are distinct in HRTV NP. The features of epitopes, including their hydrophilicity, antibody accessibility, flexibility, antigenicity, and spatial locations, were predicted and analyzed, and their potential functional impacts on virus infection and replication and their use for virus detection were discussed. Our results promote the understanding of the molecular basis of GTV and SFTSV NP in inducing antibody responses. The NP-specific mAbs generated in this study are promising fundamental materials for developing viral antigen detection methods for GTV and SFTSV.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Emerging pathogenic tick-borne viruses (TBVs) that can infect animals and humans have attracted considerable attention because of the increasing incidence of tick-borne viral diseases and their significant impact on the human healthcare system [1,2,3]. With the rapid development of metagenomics, an increasing number of novel arboviruses have been identified and isolated from various hosts [4], indicating increasing potential threats from emerging arbovirus spillover to different hosts. In recent years, two novel tick-borne viruses (TBVs), severe fever with thrombocytopenia syndrome virus (SFTSV) and heartland virus (HRTV), have been shown to be associated with severe human diseases, resulting in fatal cases in Eastern Asian countries and in the United States, respectively [5,6,7,8,9,10]. In China, SFTSV was first isolated from patients with severe fever and thrombocytopenia syndrome disease (SFTS). The clinical signs of SFTS include fever, thrombocytopenia, and leukocytopenia, among others [8]. From 2010 to 2020, over 10000 SFTS cases were reported in 23 provinces in China, resulting in mortality rates ranging from 5 to 30% [1]. Additionally, SFTSV was identified and isolated from patients in Japan and South Korea [11], and there was serological evidence of SFTSV prevalence in Vietnam and Pakistan [12, 13], suggesting the wide distribution of this virus. In 2009, patients in the United States with clinical symptoms similar to those observed for SFTS were found to have infection with HRTV, a novel TBV with a close genetic relationship to SFTSV [9]. To date, over 50 HRTV cases, including three deaths, have been reported in the United States [14].
In 2018, Shen et al. isolated a novel virus that displayed a strong evolutionary relationship with SFTSV from Dermacentor nuttalli ticks in Xinjiang, China [15]. The phylogenetic tree constructed for this virus showed that it belongs to the group SFTS/heartland of genus Bandavirus in the family Phenuiviridae and is named Guertu bandavirus (GTV) [15, 16]. Two human cases with neutralization of this virus have been identified in serological surveys, indicating that GTV is a potential pathogen to humans [15]. In Xinjiang, GTV is widely distributed along with its tick vectors; thus, it may pose a threat to residents. In addition, a case report suggested that SFTSV is prevalent in Xinjiang [17]. To prepare for emerging diseases associated with SFTSV, GTV, or other related viruses, it is important to develop fundamental materials for establishing effective techniques for virus detection and identification as well as strategies for rapid response to emerging outbreaks.
Similar to SFTSV and HRTV, GTV contains three segmented negative-sense single-stranded RNAs (ssRNAs) [15]. The S segment encodes a nonstructural protein (NSs, 285 amino acids (AAs)) and nucleoprotein (NP, 245 AAs), whereas the M and L segments encode glycoprotein (GP) and RNA-dependent RNA polymerase, respectively. The ability of SFTSV NP to bind viral RNA and form a ribonucleoprotein complex is an important role in RNA transcription and replication [18]. Detected NP expression in cells commonly represents virus invasion [19]. SFTSV NP is the primary antigen detected in infected cells and patient or animal tissues [20]. To date, a panel of SFTS patient-derived human monoclonal antibodies (mAbs) has been obtained, and epitopes have been identified, indicating the important role of NP in inducing strong humoral responses to SFTSV infection and the ideal molecular basis for establishing methods for the detection and diagnosis of SFTSV infection using mAbs against NP [21]. A sandwich antigen-capture enzyme-linked immunosorbent assay (ELISA) for detecting SFTSV has been developed and evaluated using mAbs [22], which provides an additional method for the virological diagnosis of SFTS besides viremia detection. Murine mAbs against HRTV NP were developed and showed potential for inclusion in future HRTV diagnostic assays [23]. However, there is still a lack of GTV NP mAb preparations, which may hinder the development of diagnostic strategies for this virus. Moreover, the mAbs against NP may have serological cross-reactions among the three bandaviruses, which share close phylogenetic and serological correlations. Therefore, identifying fine antigenic epitopes of viral proteins could provide a research basis to improve the specificity of detection methods using mAbs and to promote the design of detection strategies to distinguish bandaviruses.
In this study, we obtained mAbs against GTV NP using hybridoma cell screening techniques and identified the epitopes recognized by mAbs. Further, we assessed the cross-recognition ability of the anti-GTV NP mAbs against SFTSV and HRTV and analyzed the fine epitopes mapped in NPs of GTV, SFTSV, and HRTV, respectively, through multiple sequence alignment and three-dimensional (3D) structure models. Our results provide foundation materials and suggest their promising use in developing specific and broad-spectrum methods for detecting bandaviruses.
Materials and methods
Cells and viruses
Vero (ATCC number: CCL-81), HeLa (ATCC number: CCL-2), and mouse myeloma cells (ATCC number: PTA-9396) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in Dulbecco’s Modified Eagle’s medium–high glucose (DMEM, NZK Biotech, Wuhan, China) containing 10% fetal bovine serum (FBS, Gibco, Australia). The National Virus Resource Center (NVRC, Wuhan, China) preserved GTV strain DXM (GenBank accession number: KT328591), SFTSV strain WCH (GenBank accession number: AFJ15061), and HRTV strain patient 1 (GenBank accession number: NC024496). Infection assays for GTV, SFTSV, and HRTV were conducted in a biosafety level (BSL)-2 laboratory.
Plasmids and antibodies
The eukaryotic expression plasmid pCAGGS and the prokaryotic expression plasmid pXXGST-3 expressing short peptide fused with GST-188 protein were preserved at the NVRC. An anti-His antibody (Abcam, Shanghai, China) was used as the primary antibody to verify the expression of GTV/SFTSV/HRTV NP. Goat anti-mouse IgG H&L (Alexa Fluor ®488) (Abcam) was used as the secondary antibody to verify the expression of GTV/SFTSV/HRTV NP and to detect mAb binding. Rabbit polyclonal antibodies against SFTSV nucleoprotein (anti-SFTSV NP PAbs) were used as positive controls [24], and goat anti-rabbit IgG H&L conjugated with horseradish peroxidase (HRP) (Proteintech, Rosemont, USA) or goat anti-rabbit IgG H&L (Alexa Fluor ®488) (Abcam) were used as secondary antibodies. Serum collected from healthy C57/BL6 mice was used as the negative control.
Production of mAbs
Vero cells were infected with GTV at a multiplicity of infection (MOI) of 1. Three days post-infection, culture supernatants containing viral particles were harvested and concentrated using ultrafiltration centrifugation as previously described [25], which was followed by an endpoint dilution assay to determine virus titers. Six C57/BL6 mice aged 6–8 weeks were immunized with 100 μL of supernatant containing GTV (8 × 107 TCID50 per mouse). On day 10, serum samples from mice were collected and used to detect antibody responses to GTV using indirect immunofluorescence assay (IFA). On day 14, two mice that had anti-GTV NP antibodies were selected, and their spleens were harvested. Hybridoma cells were produced as previously described by the fusion of SP2/0 myeloma cells with mouse spleen cells using polyethylene glycol (Glpbio, Montclair, USA) [25]. The culture supernatants of hybridoma cells were screened for antibodies against GTV NP by IFA and Western blot. Positive cells were diluted to approximately 1 cell/well by limiting dilution, and positive single stable clones were subsequently transferred to culture flasks. All experiments were conducted in a biosafety level (BSL)-2 laboratory. Reverse transcriptase polymerase chain reaction (RT-PCR) was used to detect GTV in the supernatants of the single stable clones collected when the cell density reached 80% to rule out the presence of this virus, as previously described [15].
Eukaryotic expression of GTV/SFTSV/HRTV NP
The NP genes of GTV, HRTV, and SFTSV were amplified from viral cDNA using PCR and cloned into the vector pCAGGS in fusion with a 6 × His-tag to generate NP-expressing plasmids (pCAGGS-GTV-NP, pCAGGS-SFTSV-NP, and pCAGGS-HRTV-NP), which were verified by Sanger sequencing. HeLa cells were transfected with plasmids using Lipofectamine™ 3000 transfection reagent (Thermo Fisher Scientific, USA) for IFAs.
Indirect immunofluorescence assay
Using virus-infected or plasmid-transfected cells, IFAs were performed to detect antibody responses from mAbs. Briefly, Vero cells were seeded in 96-well plates and infected with GTV (MOI = 1.0), and HeLa cells were transfected with the GTV-, SFTSV-, or HRTV-NP-expressing plasmids (100 ng/well). Cells were harvested three days after infection and two days after transfection and were fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized with 0.2% Triton X-100, and blocked with phosphate-buffered saline (PBS) containing 5% bovine serum albumin. The supernatants of hybridoma cells were inactivated at 56 °C for 30 min and then incubated with the fixed cells for 60 min at 37 °C. The polyclonal antibody anti-SFTSV NP was used as a positive control, and the serum of healthy mice was used as a negative control. Binding of antibodies was detected using Alexa Fluor ®488 and observed under an immunofluorescence microscope.
Determination of epitopes in GTV NP recognized by mAbs
To identify fine antigenic epitopes in GTV NP recognized by mAbs, the GTV NP ORF (238 amino acids) residues were truncated into thirty-two 16mer-peptides numbered P1–32 with overlaps of eight AAs between adjacent peptides. Fragments of DNA encoding each short peptide were cloned into the pXXGST-3 vector fused with GST188-tagged protein, which was transformed into Escherichia coli (E. coli) BL21 cells [26]. Peptide expression was induced at 42 °C, cells were harvested, and peptide expression was verified by SDS-PAGE. Western blotting was used to identify the reaction between mAbs and truncated peptides. Then, 8mer-peptides or 9–12mer-peptides with overlaps of 7 or 8–11 AA between adjacent peptides were generated according to the sequence of the 16mer-peptides which were blotted by mAbs. The AA sequences of the expressed 16/12/8mer peptides and their positions on GTV NP are listed in Tables S1 and S2.
SDS-PAGE and Western blot analysis
The collected E. coli cells expressing truncated peptides were immersed in a water bath at 95 °C for 10 min. Proteins were resolved by 12% SDS-PAGE. The gel was stained with Coomassie brilliant blue R-250 to analyze whether the proteins were expressed correctly, and the confirmed r-clones were further verified by Sanger sequencing. Culture supernatants of GTV, SFTSV, and HRTV were treated with β-propiolactone (SERVA Electrophoresis GmbH, Heidelberg, Germany) inactivation, removal of cell debris, and ultracentrifugation. The purified virions, together with the proteins expressed correctly, were resolved by 12% SDS-PAGE gel electrophoresis and then electrotransferred onto a 0.2 μM polyvinylidene fluoride (PVDF) membrane for Western blotting. For the specific antigen–antibody reaction, the PVDF membrane was blocked with 5% skimmed milk powder in PBS, incubated with anti-GTV NP mAbs or anti-SFTSV NP PAbs (1:2000 dilution) as the primary antibody overnight at 4 °C, and then incubated with goat anti-mouse/rabbit IgG H&L conjugated with HRP at 1:2000 dilution as the secondary antibody after washing with Tris-buffered saline-Tween 20 (TBS-T). Finally, the PVDF membrane was washed with TBS-T, and blotted bands were developed using the enhanced chemiluminescence color reagent. A hypersensitive chemiluminescence imager was used to visualize immunoblots.
Cellular ELISA
The supernatants from hybridoma cells containing anti-GTV NP mAbs were serially diluted from 1:1 to 1:213. Each dilution was incubated with fixed cells infected with GTV or SFTSV for 60 min at 37 °C, followed by incubation with HRP-conjugated goat anti-mouse IgG (1:4,000 dilution) at 37 °C for 1 h. Then, 3,3′,5,5′-tetramethylbenzidine and chromogen solution was added, and the optical density (OD) value was measured by dual wavelength detection at 450/630 nm. Each test was performed in dual mode, and OD values higher than twice the negative control were considered positive [25]. The dose–response curves of mAb-antigen reactions were fitted using GraphPad Prism (version 8) by the log(agonist) vs. normalized response-variable slope mode.
Bioinformatic analyses
To analyze the similarity of each mapped fine antigenic epitope among homologous proteins, NP sequences of GTV (GenBank accession number: KT328591), SFTSV (GenBank accession number: AFJ15061), and HRTV (GenBank accession number: NC024496) were downloaded from GenBank. The location of the experimentally identified fine epitopes in the 3D structure of the NP protein was analyzed using PyMOL™ software (https://pymol.org/2/) based on the crystal structure of SFTSV NP (PDB:4J4U). The secondary structure was analyzed using the phyre2 website (http://www.sbg.bio.ic.ac.uk/phyre2/) and the DNAStar-Protean software. Sequences of GTV NP, SFTSV NP, and HRTV NP were aligned using MEGA software (https://megasoftware.net/) and the ESPript website (https://espript.ibcp.fr/ESPript/ESPript/).
Results
Eight murine mAbs specific to GTV NP were identified
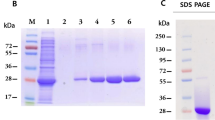
Eight mAbs against GTV NP were generated from different clones of hybridoma cells, designated as 17F9, 21B7, 22G1, 23B9, 24D9, 25C2, 25E2, and 26F8. Due to the inability of RT-PCR to detect GTV RNA, GTV presence was excluded from these clones (Fig. S1). According to the IFA results, these mAbs could react with GTV-infected cells (Fig. 1A) and GTV NP-expressing plasmid transfected cells (Fig. 1B). The results of Western blotting showed that GTV NP was blotted by four (22G1, 25C2, 25E2, and 26F8) of the eight mAbs (Fig. 1C), suggesting that these four mAbs recognize linear epitopes of GTV NP, while the other four recognize spatial structures.

Eight mAbs against GTV NP were identified by IFAs and Western blot assays. IFA assays identified eight mAbs reactive with GTV infected cells (A) and with pCAGGS-GTV-NP transfected cells (B). (C) Western blot assays identified four of the eight mAbs (22G1, 25C2, 25E2, and 26F8) reactive with GTV NP using the purified GTV virions as antigen. PC, the positive control which was blotted by GTV NP antibody; NC, the negative control
Two different epitopes recognized by mAbs were identified in GTV NP
Expression plasmids containing overlapping 16mer-peptides fused with the GST188 tag were generated as described in the Materials and Methods section, and their expression was validated by SDS-PAGE (Fig. S2A and B). Subsequently, Western blotting results indicated that the four mAbs, which react with linearized GTV NP, specifically recognize two 16mer-petides (Fig. 2A). 22G1 and 26F8 reacted with P31 (223–238 AA), and 25C2 and 25E2 reacted with P27 (191–206 AA). Nine sets of overlapping 8mer-petides (P33–P50) were then constructed (Fig. S2 B). Two mAbs (22G1 and 26F8) reacted with three peptides, P43 (224–231 AA), P44 (225–232 AA), and P45 (226–233 AA), which are derived from the 16mer peptide P31 (Fig. 2B). This suggests that 22G1 and 26F8 recognize a consensus epitope among the three peptides. However, reactions of the mAbs 25C2 and 25E2 were not found with any of the 8mer-peptides. Five 12mer-peptides with an overlap of 11 AAs derived from P27 were constructed. As expected, mAbs 25C2 and 25E2 could react with the same peptide P54 (Fig. 2C), since they were derived from the same cell clone in the last dilution.

Western blot assays for mapping the epitopes recognized by the four mAbs which react with linearized GTV NP antigen. (A) Western blot analysis for mapping reactive 16mer-peptides in P26–P32 (negative results of P1–P25 were not shown). (B) Western blot analysis for mapping reactive 8mer-peptides in P42–P50. (C) Western blot analysis for mapping reactive 12mer-peptides in P51–P55
Therefore, the sequence of the 12mer peptide P54 (YNSFRDPLHAAV, 194–205 AA) recognized by mAbs 25C2 and 25E2 was designated as epitope 1 (ENP1), and the consensus sequence of the 8mer peptides P43, P44, and P45 recognized by mAbs 22G1 and 26F8 was designated as epitope 2 (ENP2) (Table 1).
MAbs had a cross reaction with SFTSV and not HRTV
We further evaluated whether the GTV-derived mAbs can cross-react with the NPs of SFTSV and HRTV because GTV is closely related to SFTSV and HRTV. As shown in the results of IFAs and Western blotting, the four mAbs cross-reacted with SFTSV NP but not with HRTV NP (Fig. 3A and B).

Evaluation of the cross-recognition of mAbs against GTV NP with the GTV-related viruses, SFTSV, and HRTV. (A) Western blot assays showed that the four selected mAbs (22G1, 25C2, 25E2, and 26F8) reactive with GTV NP had cross-reaction with SFTSV NP, but not with HRTV NP using cells transfected with pCAGGS-GTV NP, pCAGGS- SFTSV NP, or pCAGGS–HRTV NP plasmids. (B) Western blot assays showed the four selected mAbs reactive with GTP NP were cross-reactive with linear epitopes of SFTSV NP but not with HRTV NP by using purified virions of GTV, SFTSV, and HRTV as antigens, respectively. M, the protein molecular marker; PC, positive control; NC, negative control
The sequences of ENP1 and ENP2 in the NPs of GTV, SFTSV, and HRTV were compared by aligning these NP proteins. The sequence similarity between GTV NP and SFTSV NP (88.6%) was higher than that between HRTV NP and GTV NP (61.4%). Based on the secondary structure of the SFTSV NP crystal, ENP1 and ENP2 are located in the helix α11 and a short turn between helices α12 and α13, respectively. (Fig. 4A). ENP1 is highly conserved between GTV and SFTSV, whereas in HRTV NP, two amino acids (NS, 195–196 AA) in this epitope are replaced by AK. The antigenicity of this epitope was predicted to be greater in GTV and SFTSV than in HRTV (Fig. 4B), suggesting that the N195S196 in GTV and SFTSV mutated to A195K196 in HRTV, resulting in the change of polarity from neutral to alkaline in these positions. This may affect the antigenicity and antibody accessibility to this epitope.

Graphic representation of the protein sequence alignment of GTV-, SFTSV-, and HRTV-NPs (A) and the bio-properties of these proteins (B). (A) Sequence alignment of GTV NP, SFTSV NP, and HRTV NP shows the locations of the ENP1 and ENP2. The secondary structures were illustrated according to SFTSV NP crystal structure. (B) Prediction of the bio-properties of ENP1 and ENP2 in GTV/SFTSV/HRTV NP, including the hydrophilicity, flexibility, antigenicity, and surface probability
Except for L230 in GTV NP, which was mutated to V230 in SFTSV NP, ENP2 is conserved among the five amino acids in SFTSV and GTV NPs. In HRTV NP, the sequence of this epitope is completely distinct from that of GTV NP and SFTSV NP (Fig. 4A), which may form a differential structure that is not accessible to GTV-derived mAbs. ENP2 exhibited better hydrophilicity and higher flexibility than ENP1 (Fig. 4B). L230 in GTV mutated to V230 while in SFTSV it did not change the polarity of this position (Table 2), which did not affect GTV deriving mAb recognition of ENP2 in SFTSV. In addition, ENP2 of GTV and SFTSV had stronger antigenicity than that of HRTV NP (Fig. 4B), as the epitope of HRTV NP had different amino acids and altered AA features at three positions (T228K229E230) (Table 2).
MAb-based cellular ELISA showed efficient reaction with SFTSV and GTV NPs
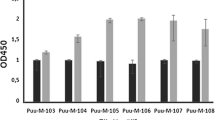
We developed a cellular ELISA method to investigate the capacity of these mAbs to recognize GTV and SFTSV antigens. The titers of mAbs were expressed as the reciprocal of the dilutions that reacted with cellular antigens by end-point dilution assays. 22G1 and 26F8 were slightly more efficient (both titers were 212) in reacting with GTV antigen than SFTSV (both titers were 211). 25C2 and 25E2 showed comparable efficiencies in reacting with GTV and SFTSV antigens (titers of 212 and 211, respectively) (Table 3). The dose–response curves of mAbs-antigens reactions fitted by GraphPad Prism were shown in Fig. 5. By analyzing the efficiencies of the four mAbs in serial dilutions to react with GTV and SFTSV antigens, we found that all four mAbs showed higher OD values at low dilutions to detect GTV than SFTSV, which suggested different efficiencies of GTV NP deriving mAbs reacting with their own antigen and SFTSV antigen. Nevertheless, this difference was not obvious when the dilution fold of the mAb increased (Fig. 5).

The dose-dependent response curves of mAbs reacting with GTV and SFTSV tested by ELISA. The mAbs were serially diluted from 1:1 to 1:213. The cut-off value was set at 0.024. The dose–response curves of mAb-antigen reactions were fitted using GraphPad Prism (version 8) by the log(agonist) vs. normalized response-variable slope mode
Discussion
Due to their high purity and strong specificity, mAbs have been widely used to establish detection methods for protein antigens from pathogens or disease-associated biomarkers [27, 28]. Due to their ability to block virus binding to hosts and prevent virus infection, neutralizing mAbs, which target viral GP, are promising candidates for new antiviral drugs and/or vaccines for improving disease prevention and treatment, more than their use in detecting viral antigens [29, 30]. As for bunyaviruses containing three RNA segments, the NP encoded by the S segment generally has the most abundant expression and has been shown to be the immunodominant protein of phleboviruses, such as Rift Valley fever virus, Toscana virus, and SFTSV [19, 31, 32]. By generating and screening the Fab antibody phage library from humans recovered from SFTSV infection, over 400 mAbs were found to react with NP, and 16 with GP. This showed an important role of SFTSV NP in inducing humoral responses and suggested the advantage of developing methods for detecting viruses using NP as a target. Developing mAbs against this viral antigen would promote understanding of the molecular basis of virus antigenicity and provide fundamental materials for developing detection methods to facilitate infection diagnosis and epidemiological investigations. In a previous study [22], the mAb-based IgG sandwich ELISA and IgM capture ELISA methods for the diagnosis of SFTSV were established. This method could be used to detect IgG or IgM antibodies separately, which helps to distinguish a previous infection from a recent one in humans. Another antigen capture ELISA using mAbs was developed to detect SFTSV in serum samples from suspected SFTS patients and was further applied to detect SFTSV antigens in autopsied materials [21]. Murine derived mAbs were generated; however, these mAbs did not show any neutralizing activity against HRTV. Instead, they were reactive to linear epitopes in HRTV NP, and one of them was found to be cross-reactive with SFTSV. These HRTV mAbs could be applied in ELISAs for the detection of HRTV-specific IgM and IgG in human infections [23].
The notorious human disease-associated viruses SFTSV and HRTV share a very close evolutionary relationships with GTV, which is a potential tick-borne viral pathogen [15]. It is essential to establish rapid and sensitive methods to detect GTV as well as its related viruses in order to prepare for the first-aid response to identify emerging infections and distinguish them from other agents. Before this study, there have been no reports on the generation and characterization of GTV mAbs. In this study, we generated eight murine mAbs specific to GTV NP. Four (17F9, 21B7, 23B9, and 24D9) of the eight mAbs were found to react only to spatial structures, whereas the other four mAbs (22G1, 25C2, 25E2, and 26F8) could recognize linear epitopes. Two distinct linear epitopes were identified, ENP1 (194YNSFRDPLHAAV205) and ENP2 (226GPDGLP231). As the four mAbs recognizing linear epitopes could cross-react with SFTSV NP other than HRTV NP, we analyzed the sequence similarities of the three viruses. The results showed that ENP1 is conserved in GTV and SFTSV while two amino acids changed in HRTV, and GTV and SFTSV ENP2 had one amino acid difference from each other, while the sequence of this epitope is completely distinct in HRTV. To better understand the differential amino acids affecting the reaction of mAbs to these viruses, the 3D structures of GTV NP and HRTV NP were predicted based on the crystal structure of SFTSV NP (Fig. 6). Three domains make up SFTSV NP: an N-terminal arm (M1–D34), an N-lobe (P35–L111), and a C-lobe (P112–L245) [33]. The GTV, SFTSV, and HRTV share similar protein structures. The root-mean-square deviation values between GTV NP and SFTSV NP, GTV NP and HRTV NP, and SFTSV NP and HRTV NP were 0.006, 0.035, and 0.294, respectively, and ENP1 and ENP2 were located in the C-lobe of NPs (Fig. 6A). Although the AA change in the two epitopes did not significantly affect protein structures, their significant difference in AA features had an impact on the accessibility of GTV-derived mAbs to HRTV NP. This indicates that the molecular basis of HRTV NP induces a humoral response different from that of SFTSV and GTV.

The 3D structures showing the locations of ENP1 and ENP2 in the GTV, SFTSV, and HRTV NPs. (A) The localizations of each epitope in the monomer of GTV/SFTSV/HRTV NPs. GTV NP and HRTV NP were predicted based on the crystal structure of SFTSV NP. The N-terminal arm, N lobe, and C lobe are indicated in white, light blue, and green, respectively; ENP1 and ENP2 were indicated in purple and red, respectively. (B) The structures of monomer and hexamer of GTV, SFTSV, and HRTV NPs showing ENP1 locating in the RNA binding cavity and buried in the inner side of hexamers and ENP2 exposed on the surface of the outer side of hexamers. The RNA binding cavity was indicated by gray; the conserved residues (P200 and A203) in both ENP1 and the RNA binding cavity were indicated in green; the epitopesENP1 and ENP2 were indicated in purple and red, respectively
In the viral replication cycle of bunyaviruses, NP plays a critical role. It binds to viral RNA and polymerizes into large oligomers to protect the entire length of the viral RNA genome [34]. The N-terminal arm and N-lobe of SFTSV NP are mainly responsible for the oligomerization of NPs, whereas the C-lobe, which contains ENP1 and ENP2, is responsible for binding viral RNA. We found that the conserved P200 and A203 located in ENP1 of all the three viruses are strictly conserved residues located in the RNA-binding cavity critical for RNA binding (Fig. 6B) [35]. Therefore, we speculated that mAbs against ENP1 might have the potential to block the RNA binding of GTV and SFTSV, which could be further clarified by in vitro and in vivo assays. Furthermore, the SFTSV NP oligomer exhibited a ring-shaped hexameric architecture [33]. Based on the NP hexameric structure analysis, unlike ENP1 located on the inner side of the NP, ENP2 is present on the external side of the bottom (Fig. 6B), which may provide great antibody accessibility to this epitope. Thus, mAbs against this epitope could become ideal materials for developing new methods to detect GTV and SFTSV antigens. Moreover, we analyzed their efficiencies to react with GTV and SFTSV NP antigens using cellular ELISA. It was found that reactions to GTV and SFTSV could be distinguished using mAbs diluted in low folds and were commonly identified using mAbs of high-fold dilutions. This suggests that these mAbs are promising candidates for developing methods to detect GTV and SFTSV infections.
In conclusion, this study reported the generation of eight mAbs against GTV NP, of which four mAbs recognized linear epitopes of GTV NP and showed cross-reactivity with SFTSV NP other than HRTV. Moreover, two new epitopes were identified, distinct from the SFTV epitopes identified in previous studies [21]. Overall, these results improve our understanding of the antigenic characteristics of NPs in bandaviruses. It also provides fundamental materials (mAbs) for developing new antigen detection methods for bandaviruses, such as SFTSV and GTV, which may promote the diagnosis and early invention of associated diseases.
Data availability
All data generated or analyzed during this study were included in this published article. Further details are available on request from the authors if required.
References
Li J, Li S, Yang L, Cao P, Lu J (2021) Severe fever with thrombocytopenia syndrome virus: a highly lethal bunyavirus. Crit Rev Microbiol 47:112–125. https://doi.org/10.1080/1040841X.2020.1847037
Lu DH, Jiang H, Lian JQ (2021) Hantavirus infection during pregnancy. Virol Sin 36:345–353. https://doi.org/10.1007/s12250-020-00300-8
Petrova V, Kristiansen P, Norheim G, Yimer SA (2020) Rift valley fever: diagnostic challenges and investment needs for vaccine development. BMJ Glob Health 5. https://doi.org/10.1136/bmjgh-2020-002694
Shi M, Lin XD, Tian JH et al (2016) Redefining the invertebrate RNA virosphere. Nature 540:539–543. https://doi.org/10.1038/nature20167
Kim KH, Yi J, Kim G et al (2013) Severe fever with thrombocytopenia syndrome, South Korea, 2012. Emerg Infect Dis 19:1892–1894. https://doi.org/10.3201/eid1911.130792
Kurihara S, Satoh A, Yu F et al (2016) The world first two cases of severe fever with thrombocytopenia syndrome: an epidemiological study in Nagasaki, Japan. J Infect Chemother 22:461–465. https://doi.org/10.1016/j.jiac.2016.04.001
Takahashi T, Maeda K, Suzuki T et al (2014) The first identification and retrospective study of severe fever with thrombocytopenia syndrome in Japan. J Infect Dis 209:816–827. https://doi.org/10.1093/infdis/jit603
Yu XJ, Liang MF, Zhang SY et al (2011) Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med 364:1523–1532. https://doi.org/10.1056/NEJMoa1010095
McMullan LK, Folk SM, Kelly AJ et al (2012) A new phlebovirus associated with severe febrile illness in Missouri. N Engl J Med 367:834–841. https://doi.org/10.1056/NEJMoa1203378
Fill MA, Compton ML, McDonald EC et al (2017) Novel clinical and pathologic findings in a heartland virus-associated death. Clin Infect Dis 64:510–512. https://doi.org/10.1093/cid/ciw766
Yoshikawa T, Shimojima M, Fukushi S et al (2015) Phylogenetic and geographic relationships of severe fever with thrombocytopenia syndrome virus in China, South Korea, and Japan. J Infect Dis 212:889–898. https://doi.org/10.1093/infdis/jiv144
Zohaib A, Zhang J, Saqib M et al (2020) Serologic evidence of severe fever with thrombocytopenia syndrome virus and related viruses in Pakistan. Emerg Infect Dis 26:1513–1516. https://doi.org/10.3201/eid2607.190611
Tran XC, Kim SH, Lee JE et al (2022) Serological evidence of severe fever with thrombocytopenia syndrome virus and IgM positivity were identified in healthy residents in Vietnam. Viruses 14. https://doi.org/10.3390/v14102280
Dupuis AP 2nd, Prusinski MA, O’Connor C et al (2021) Heartland virus transmission, Suffolk County, New York, USA. Emerg Infect Dis 27:3128–3132. https://doi.org/10.3201/eid2712.211426
Shen S, Duan X, Wang B et al (2018) A novel tick-borne phlebovirus, closely related to severe fever with thrombocytopenia syndrome virus and heartland virus, is a potential pathogen. Emerg Microbes Infect 7:95. https://doi.org/10.1038/s41426-018-0093-2
Abudurexiti A, Adkins S, Alioto D et al (2019) Taxonomy of the order bunyavirales: update 2019. Arch Virol 164:1949–1965. https://doi.org/10.1007/s00705-019-04253-6
Zhu L, Yin F, Moming A et al (2019) First case of laboratory-confirmed severe fever with thrombocytopenia syndrome disease revealed the risk of SFTSV infection in Xinjiang, China. Emerg Microbes Infect 8:1122–1125. https://doi.org/10.1080/22221751.2019.1645573
Liu D, Li Y, Zhao J et al (2014) Fine epitope mapping of the central immunodominant region of nucleoprotein from Crimean-Congo hemorrhagic fever virus (CCHFV). PLOS ONE 9:e108419. https://doi.org/10.1371/journal.pone.0108419
Yu F, Du Y, Huang X, Ma H, Xu B, Adungo F, Hayasaka D, Buerano CC, Morita K (2015) Application of recombinant severe fever with thrombocytopenia syndrome virus nucleocapsid protein for the detection of SFTSV-specific human IgG and IgM antibodies by indirect ELISA. Virol J 12:117. https://doi.org/10.1186/s12985-015-0350-0
Wu X, Qi X, Liang M, Li C, Cardona CJ, Li D, Xing Z (2014) Roles of viroplasm-like structures formed by nonstructural protein NSs in infection with severe fever with thrombocytopenia syndrome virus. FASEB J 28:2504–2516. https://doi.org/10.1096/fj.13-243857
Yu L, Zhang L, Sun L et al (2012) Critical epitopes in the nucleocapsid protein of SFTS virus recognized by a panel of SFTS patients derived human monoclonal antibodies. PLOS ONE 7:e38291. https://doi.org/10.1371/journal.pone.0038291
Fukuma A, Fukushi S, Yoshikawa T et al (2016) Severe fever with thrombocytopenia syndrome virus antigen detection using monoclonal antibodies to the nucleocapsid protein. PLOS Negl Trop Dis 10:e0004595. https://doi.org/10.1371/journal.pntd.0004595
Calvert AE, Brault AC (2015) Development and characterization of monoclonal antibodies directed against the nucleoprotein of heartland virus. Am J Trop Med Hyg 93:1338–1340. https://doi.org/10.4269/ajtmh.15-0473
Zhang Y, Shen S, Shi J et al (2017) Isolation, characterization, and phylogenic analysis of three new severe fever with thrombocytopenia syndrome bunyavirus strains derived from Hubei Province, China. Virol Sin 32:89–96. https://doi.org/10.1007/s12250-017-3953-3
Zhang M, Du Y, Yang L, Zhan L, Yang B, Huang X, Xu B, Morita K, Yu F (2022) Development of monoclonal antibody based IgG and IgM ELISA for diagnosis of severe fever with thrombocytopenia syndrome virus infection. Braz J Infect Dis 26:102386. https://doi.org/10.1016/j.bjid.2022.102386
Xu WX, Wang J, Tang HP et al (2017) A simpler and more cost-effective peptide biosynthetic method using the truncated GST as carrier for epitope mapping. PLOS ONE 12:e0186097. https://doi.org/10.1371/journal.pone.0186097
Umeki K, Yasuda A, Umekita K et al (2020) Detection of anti-SFTSV nuclear protein antibody in the acute phase sera of patients using double-antigen ELISA and immunochromatography. J Virol Methods 285:113942. https://doi.org/10.1016/j.jviromet.2020.113942
Wang G, Chang H, Jia B et al (2019) Nucleocapsid protein-specific IgM antibody responses in the disease progression of severe fever with thrombocytopenia syndrome. Ticks Tick-Borne Dis 10:639–646. https://doi.org/10.1016/j.ttbdis.2019.02.003
Wu X, Li Y, Huang B et al (2020) A single-domain antibody inhibits SFTSV and mitigates virus-induced pathogenesis in vivo. JCI Insight 5. https://doi.org/10.1172/jci.insight.136855
Guo X, Zhang L, Zhang W et al (2013) Human antibody neutralizes severe Fever with thrombocytopenia syndrome virus, an emerging hemorrhagic fever virus. Clin Vaccine Immunol 20:1426–1432. https://doi.org/10.1128/CVI.00222-13
Martín-Folgar R, Lorenzo G, Boshra H, Iglesias J, Mateos F, Borrego B, Brun A (2010) Development and characterization of monoclonal antibodies against Rift Valley fever virus nucleocapsid protein generated by DNA immunization. mAbs 2:275–284. https://doi.org/10.4161/mabs.2.3.11676
Magurano F, Nicoletti L (1999) Humoral response in Toscana virus acute neurologic disease investigated by viral-protein-specific immunoassays. Clin Diagn Lab Immunol 6:55–60. https://doi.org/10.1128/CDLI.6.1.55-60.1999
Zhou H, Sun Y, Wang Y et al (2013) The nucleoprotein of severe fever with thrombocytopenia syndrome virus processes a stable hexameric ring to facilitate RNA encapsidation. Protein Cell 4:445–455. https://doi.org/10.1007/s13238-013-3901-4
Ruigrok RW, Crépin T, Kolakofsky D (2011) Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr Opin Microbiol 14:504–510. https://doi.org/10.1016/j.mib.2011.07.011
Jiao L, Ouyang S, Liang M et al (2013) Structure of severe fever with thrombocytopenia syndrome virus nucleocapsid protein in complex with suramin reveals therapeutic potential. J Virol 87:6829–6839. https://doi.org/10.1128/JVI.00672-13
Acknowledgements
We thank the Center for Experimental Animals and the Center for Instrumental Analysis and Metrology in Wuhan Institute of Virology, Chinese Academy of Sciences for the technical support. We also thank Miss Min Zhou, Mr. Tao Zhang, Dr. Abilimiti Moming, and Ms. Yanfang Zhang from the National Virus Resource Center for assisting cell culture and mAb preparation.
Funding
This work was supported by the National Key Research and Development project of China (No. 2018YFE0200400), the National Natural Science Foundation of China (No. U20A20135), and the EVAg-Global project (871029).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethical approval
Animal experiments were approved by the ethics committee of Wuhan Institute of Virology, Chinese Academy of Sciences under the approval number WIVA33202004.
Competing interests
The authors of this manuscript have the following competing interests: Jin Qian, Shu Shen, Fei Deng, Liyan Fu, Xiaoli Wu, Zhiying Wang, and Guirong Cheng are the inventors in a pending patent application filed on the reported antibodies. The other authors have no relevant financial or non-financial interests to disclose.
Additional information
Responsible Editor: Flavio Guimaraes Fonseca
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Qian, J., Fu, L., Wu, X. et al. Developing and characterizing monoclonal antibodies of Guertu bandavirus nucleoprotein for developing methods of Guertu bandavirus and severe fever with thrombocytopenia syndrome virus detection. Braz J Microbiol 54, 1433–1445 (2023). https://doi.org/10.1007/s42770-023-00982-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-023-00982-8