
Abstract
The Mycobacterium avium subspecies paratuberculosis (MAP) causes paratuberculosis (Johne’s disease), a systemic and chronic inflammation of intestine that affects bovine, small ruminants like goat and sheep. The disease has a greater economic importance in cattle and in small ruminants. But its effective control is impeded due to lack of rapid and accurate diagnostics. The present study is aimed at developing a LAMP-coupled lateral flow device (LFD) for rapid detection of paratuberculosis in livestock animal species such as cattle and in small ruminants at resource-limited areas. LAMP primers with biotin and FITC end tags were designed for IS900 gene specific for MAP. To determine sensitivity of LAMP assay, 10-fold serial dilutions were made from 10 ng/μl MAP stock DNA and were compared with PCR. The detection limits of LAMP-coupled LFD were defined and reactions were repeated for reproducibility. The specificity was evaluated using other infectious bacteria such as M. bovis, M. tuberculosis, Brucella abortus, Leptospira interrogan, Yersinia enterocolitica, Salmonella typhimurium, Listeria monocytogens, and Staphylococcus aureus. A total of 95 samples turned positive for LAMP-coupled LFD out of 389 fecal samples. All the cultural-positive and PCR-positive samples showed positive in LAMP-coupled LFD. Nine samples with negative cultures turned positive in LAMP assay. The overall sensitivity and specificity of the LAMP-coupled LFD assays were 100% and 97.02% respectively in comparison with the culture as the gold standard method. The sensitivity detection limit of developed assay was 10 fg/μl and specificity was 100%. This assay successfully detected MAP not only by using bacterial DNA but also in clinical fecal samples. The clear band formation at control and test positions was observed on LAMP-coupled LFD. The developed assay is a simple, rapid, easy to perform, and is very useful in early diagnosis of Mycobacterium avium subsp. paratuberculosis at point of care resource-limited areas.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mycobacterium avium subspecies paratuberculosis is an intracellular pathogen that causes Johne’s disease in ruminants and Crohn’s disease in humans [1,2,3]. There is a huge economic loss in dairy cattle industry worldwide due to paratuberculosis [4,5,6]. The route of transmission occurs through fecal, oral route, or in contact with infected animals [1]. The seroprevalence of Mycobacterium avium subsp. paratuberculosis of 29% is reported in northern India [7]. The prevalence rate in buffalo is 28.6%, in cattle 29.8%, and in sheep, goat, and other ruminants in between 18.2 and 23%. This is alarming and a major sensitive issue [7,8,9]. So, there is a need to develop high throughput, sensitive diagnostic methods for the detection of infected animals and animal products. The clinically infected animals can shed 106 to 108 CFU/g of fecal material that can easily spread the infection to newborn babies, for which the infectious dose is 103 CFU/animal [10]. At present, the diagnosis of paratuberculosis is based on direct and indirect methods of diagnostic techniques. The direct method of diagnosis is based on the isolation of MAP from feces culture [11]. Gold standard method is used generally for diagnosis of paratuberculosis [12]. Isolation and identification of MAP-specific organisms is carried out by culturing the feces followed by gold standard method. But, due to lack of qualified veterinary microbiologists and long incubation period (8 to 16 weeks) along with chance of high contamination rate makes it difficult to diagnose paratuberculosis [5, 13, 14].
Indirect diagnostic methods are mainly based on serological techniques such as complement fixation test (CFT), Johnin gamma interferon skin test, and enzyme-linked immunosorbent assay (ELISA). All these existing serological techniques have shown less sensitivity and specificity for the detection of paratuberculosis [15,16,17,18]. In whole-herd, the diagnostic sensitivity of serological assays is less than 50% as compared with fecal culture. Recently, IS900 PCR-based detection of MAP from white blood cells (WBCs) has been described [3], but its use in animals is still limited. The discovery of IS900 insertion gene sequence in the MAP genome has offered an alternative tool for rapid detection and improvement in the detection of MAP bacterial DNA in molecular level using clinical samples. After infection, the MAP circulates via blood stream to various parts; therefore, the detection of MAP by IS900 PCR in blood samples may help in diagnosis of paratuberculosis in young animals and chances of false positive detection due to passive infection will be very less. PCR assays using primers specific for F57, ISMav2, ISMAP02, and ISMAP04 elements have been used for specific detection of MAP DNA [3, 19,20,21]. Several investigations revealed that detection of IS900 gene sequence by nested PCR increases the diagnostic sensitivity and specificity [22]. The development of PCR for the detection of paratuberculosis displayed similar results compared with fecal culture [23]. However, higher copy number of IS900 in comparison with other IS elements makes IS900-based detection very sensitive. Therefore, many studies proposed IS900 as the most specific sequence for various PCR-based molecular tests with higher sensitivity. Although these PCR techniques are gold standard for the detection of paratuberculosis, they require highly trained personals, expensive instrumentation with highly equipped lab facilities, and require several other operations for the amplification process of bacterial DNA. Therefore, there is a need to establish a proper diagnostic method for the detection of paratuberculosis at field level. The recent advancement of loop-mediated isothermal amplification of nucleic acids makes molecular diagnostics more robust and easier.
Loop-mediated isothermal amplification (LAMP) amplifies the target IS900 DNA rapidly at isothermal conditions with higher sensitivity and specificity [24]. The LAMP method relies on auto-cycling of DNA synthesis performed by the Bst DNA polymerase enzyme with high strand displacement activity. The specially designed primers for paratuberculosis LAMP assay were designed using Premier Biosoft 1.15 LAMP designer software. Three sets of two inner and two outer and loop primers were used. The loop primers were tagged with biotin and FITC at its 5′ end. The reaction is highly specific for the target IS900 gene sequence. The specificity of the LAMP assay is attributable to recognition of the target sequence by six independent sequences in the initial stage and by four independent sequences during the later stages of the LAMP assay. The entire process of amplification was performed at 63 °C isothermal temperature conditions. With the addition of loop primers into the reaction mixture, the sensitivity of the LAMP assay was typically increased. The present study aims at developing a LAMP-coupled LFD for the detection of MAP infection very effectively and rapidly at resource-limited point of care areas. Our results proved that the LAMP-coupled LFD assay is useful for the detection of MAP from fecal samples with increased sensitivity and specificity and suitable for the field level detection of Mycobacterium avium subsp. paratuberculosis at resource-limited point of care areas.
Materials and methods
Study site and sample collection
The assay development and the clinical-related work were performed at Genomix Molecular Diagnostics Pvt. Ltd., Hyderabad, India. A total of 389 fecal samples from bovine, sheep, and goat origin were used in this study. Out of 389 samples, 143 fecal samples were procured from AMITY Center for Mycobacterial Division, AMITY University, Jaipur, India. In that, 79 samples were positive and 64 fecal samples were negative for MAP by fecal culture and by PCR assays. Out of 79 positive samples, 31 were from heavy shedders (> 50 colony forming units (CFU)/slant), 19 from moderate shedders (10–50 CFU/slant), and 29 from low shedders (< 10 CFU/slant). The remaining 64 fecal samples were collected from a dairy herd greater than 2 years of age and considered free from MAP on the basis of fecal culture, and PCR-negative results. Two hundred and forty-six fecal samples were also analyzed on-site by LAMP-coupled lateral flow assay. Fecal culture and PCR assays were performed off-site from the same samples. All these field samples were collected from 10 dairy farms in southern parts of India. The collected fecal samples were used for cultural conformation and the remaining volumes were stored at − 80 °C for further use.
Processing of fecal samples
Fecal samples were used for cultural confirmation as well as DNA extraction followed by PCR and LAMP assay. The fecal sample processing for culture confirmation was performed using a sedimentation protocol described by Whipple et al. [14] briefly; 1 g of fresh fecal sample was suspended in 20 ml of sterile distilled water. Fecal suspensions were agitated by horizontal shaking for 30 min at room temperature and then left to settle for 15 min. After the process of settlement, 3.5 ml of upper supernatant was transferred into a new 50-ml falcon tube containing hexadecyl pyridinium chloride (0.75% final concentration) to eliminate fungal and nonmycobacterial contaminants. The tubes were incubated at room temperature for overnight decontamination and used for further analysis.
Cultural confirmation
Sediment (0.1 ml) from the processed fecal samples was inoculated on agar slants containing Harold egg yolk medium with 0.0002% (weight/volume) mycobactin J. The inoculated slants were incubated at 37 °C for 8 to 16 weeks [25, 26]. The inoculated slants in which the bacteria could merely grow on HEYM containing mycobactin J were considered as positive for MAP. The samples were characterized based on the average CFU/slant, 1–10, 11–50, and 51 or more as low, medium, and heavy shedders respectively. Tubes were examined every 2 weeks. The colonies were confirmed by mycobactin dependence, colony morphology, and IS900 PCR assay.
Extraction of DNA
Supernatant (0.1 ml) from the processed fecal samples was plotted onto a biosample collection membrane filters (Genomix Molecular Diagnostics Pvt.Ltd., Hyderabad, India). The membranes were allowed to air dry at room temperature for 30 min. The dried sample collection membranes were cut in to small pieces and transferred on to a 1.5-ml Eppendorf tube containing 200 μl of sterile PBS. The mixture was crushed into fine pieces with the help of sterile pipette tip and incubated at 75 °C for 3 min. The tubes were allowed to centrifuge at 12000×g for 5 min. Supernatant was allowed to collect into a fresh tube. Supernatant (2.0 μl) was used as a template for PCR and LAMP assay and the remaining volume was stored at − 20 °C for further use.
PCR assay
To compare LAMP with PCR, the sensitivity and specificity of PCR were tested with the same template DNA. Reactions were performed with established primer sequences of MAP IS900 gene [27]. The reactions were performed in a 25-μl final volume reaction mixture containing 1.25 U of Taq DNA polymerase (Promega, USA), 25 mM dNTPs (Promega, USA), 0.5 μM forward and reverse primers, and 2 μl of template obtained from filter paper extraction method. PCR was performed in a thermocycler (Biorad model T100) machine. The reaction consists of an initial denaturation step at 95 °C for 5 min, final denaturation 94 °C for 1 min, annealing at 60 °C for 1.35 s and extension at 72 °C for 2 min, followed by 35 amplification cycles, and a final extension step at 72 °C for 10 min. The size of the PCR-amplified product (229 bp) was determined by comparison with a molecular weight marker using 1.5% agarose gel stained with ethidium bromide (0.5 μg/ml) followed by UV light visualization.
LAMP primer design
Six primers were used for MAP LAMP assay. These primers were designed against IS900 insertion sequence gene of MAP. The gene sequence of IS900 (Gene Accession no. AF416985.1) was obtained from NCBI GenBank. The primers were designed using Premier Biosoft LAMP primer designing software 1.13 version (PREMIER Biosoft International., USA). The designed primers were synthesized at Eurofins Inc., USA. The primer sequences are listed in Table 1. Out of six primers, two inner primers (FIP, BIP), two outer primers (F3 and B3), and the remaining two primers were loop primers. The 5′ end of these loop primers LPF and LPB was tagged with biotin and fluorescein isothiocyanate (FITC) [28] respectively as shown in Table 1.
LAMP assay
The LAMP was carried out in a 25-μl reaction mixture containing 6 μl of 6 primers (FIP and BIP 1.6 μM, LPF and LPB 0.8 μM, and F3 and B3 0.2 μM), 12.5 μl of reaction buffer, 1 μl of Bst DNA polymerase enzyme (New England Biolabs, MA, USA), 2 μl of template DNA and distilled water to a total volume of 25 μl. The reaction buffer consists of 40 mM Tris-HCl, 20 mM NH4SO4, 20 mM KCl, 16 mM MgSO4, 0.2% Triton X-100, 1.6 M glycine betaine, and 2.8 mM dNTPs in each [29]. Out of the six primers used in the reaction mix set up, two loop primers, LPF and LPB, were labeled with biotin and FITC at 5′ ends that play a major role in detecting the LAMP-amplified samples on lateral flow test devices (LFD). The whole amplification process was carried out at 63 °C for about an hour in ESE Tube Scanner (QIAGEN Lake Constance, Germany). After the completion of reaction process, amplicons were tested using LAMP-coupled LFD kits.
Development of LAMP-coupled LFD
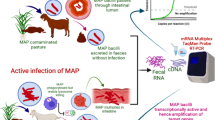
The LAMP-coupled lateral flow devices (LFD) were developed to detect the amplified products after the completion of paratuberculosis LAMP assay. The LAMP-coupled LFD contains a nitrocellulose membrane (MDI membrane technologies, India) coated with a control and test lines. The LFD test strip consists of sample pad, conjugated pad, nitrocellulose membrane, and absorbent pad (MDI membrane technologies, India). Sample pad is for loading the amplified sample and the conjugate pad holds the conjugate matrix, contains gold nano-particles conjugated with streptavidin and mouse IgG antibody (Arista Biologicals Inc., USA). Nitrocellulose membrane was coated with captured components such as anti-FITC antibody at test line and anti-mouse IgG at control line that are specific for detecting components [28]. The complete setup of test strip was kept in a plastic device having well for sample addition and detector window labeled with control “C” and test “T” positions. The primers were designed with specific biotin and FITC tags that bind to 5′ end of amplified products. The forward loop primer (LPF) having a biotin tag at 5′ end binds to streptavidin in conjugate matrix and the reverse loop primer (LPB) with a FITC tag at 5′ end binds to anti-FITC component at test region as shown in Fig. 1. The unamplified sample does not react, so, there is no reaction at test position.

Diagrammatic representation of LAMP-coupled lateral flow test device (LFD)
Analysis of the endpoint assessment
The LAMP reaction causes turbidity in the reaction tube corresponding to the amount of amplified DNA. Hence, the turbidity of the reaction was observed with naked eye. The turbidity was based on the involvement of magnesium pyrophosphate as a by-product of the reaction. For further validation, all reactions were analyzed by 1.5% agarose gel electrophoresis and positive results defined by the appearance of typical ladder bands of various sizes and in negative wells, no amplicon observed under UV light with ethidium bromide staining. The amplified products were further confirmed by LAMP-coupled lateral flow device.
Confirmation using LAMP-coupled LFD
The amplified products of LAMP assay were further confirmed by LAMP-coupled lateral flow test device which shows the reactivity at control “C” and test “T” lines on a plastic cassette platform. The cassette contains a nitrocellulose membrane strip which is coated with capture components at test “T” region. The amplified samples show colored lines at both control and test lines which denotes positive sample, whereas the unamplified samples display signal only at the control region. This indicates that negative sample gives the visual confirmation of amplification to our naked eye.
Sensitivity of LAMP-coupled LFD
To determine the sensitivity of LAMP-coupled LFD in terms of detection limits, 10-fold serial dilutions [10-1 to 10-7] of 10 ng of M. avium subsp. paratuberculosis (MAP 316F) genomic DNA were used as templates for biotin- and FITC-labeled LAMP reactions performed under optimized conditions. Nuclease free water was used as negative control. The amplified DNA with biotin and FITC end tags was detected using 1.5% agarose gel electrophoresis and ethidium bromide staining followed by visualization of amplified product under UV transilluminator and further confirmed by LAMP-coupled LFD.
Specificity of LAMP-coupled LFD
The specificity of LAMP primers was examined using 10 ng of genomic DNA extracted from other infectious non-MAP bacterial strains. Specificity of LAMP assay was evaluated by M. avium subsp. paratuberculosis strain MAP 316F genomic DNA and other infectious bacterial species including Mycobacterium bovis AN5, Mycobacterium tuberculosis DT, Brucella abortus, Leptospira interrogan, Yersinia enterocolitica, Salmonella typhimurium, Listeria monocytogenes, and Staphylococcus aureus. The specificity of these results was analyzed by 1.5% agarose gel electrophoresis followed by the use of LAMP-coupled LFD.
Statistical analysis
Statistical analysis was performed using SPSS software version 16.0 (SPSS, USA). For independent samples, t test was used to evaluate the results. The diagnostic performances of fecal culture, PCR, and LAMP-coupled LFD were evaluated by calculating the sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and efficiency.
Results
Culture method
In the present study, the fecal culture was considered as the gold standard method in the diagnosis of MAP infection. The inoculated samples with bacterial growth on HEYM with mycobactin J supplemented media were considered as positive for MAP and the samples with no bacterial growth after 6 months were considered as negative. After 8 weeks of inoculation, small colonies were observed on the cultural slants supplemented with mycobactin. From 389 collected fecal samples, 86 were found positive and 303 negative. All colonies grown on HEYM with mycobactin J resulted positive in PCR and LAMP-coupled LFD.
Colony morphology
Colonies observed on HEYM supplemented with mycobactin J cultured slants were initially small in size and grown to 3 mm in diameter over time. They appeared as white to pale yellow transparent smooth surfaces.
PCR assay
PCR assay was performed using the primers designed against the IS900 insertion sequence gene of MAP. The DNA extracted from the fecal samples using bio-sample collection membranes were used as template. Out of 389 fecal samples, 93 were found positive and 296 displayed negative. From 86 fecal samples with positive culture, PCR analysis was found positive. Moreover, seven fecal samples with negative culture were found positive by PCR assay.
LAMP assay coupled with LFD
A total of 95 fecal samples turned positive for LAMP-coupled LFD assay out of 389 fecal samples. All the samples with positive culture and PCR positive results turned positive in LAMP-coupled LFD assay. The amplified LAMP products turned turbid after adding Syber Green I, and clear color differentiation was noticed between positive and negative samples as shown in Fig. 2. LAMP-coupled LFD results demonstrated a clear-colored line reaction at control and test lines with amplified products termed as positive reaction (Fig. 3). The evaluation of LAMP-amplified products with the naked eye under natural light revealed a white turbidity.

LAMP products for visual conformation using Syber Green I. 1—Negative control, 2—positive for MAP with control DNA, 3 and 4—positive for MAP with fecal samples

LAMP-coupled LFD developed with LAMP products. 1—Negative control, 2—positive for MAP with control DNA, 3 and 4—positive for MAP with fecal samples
Sensitivity of LAMP and PCR
Using equivalent quantities of control DNA extracted from MAP culture samples as template at various dilutions, detection limits of both LAMP-coupled LFD and PCR are shown in Table 2.
The primers for both LAMP and PCR were designed against IS900 insertional sequence for MAP. Tenfold serial dilutions [10−1 to 10−7] of 10 ng of M. avium sub sp. paratuberculosis (MAP 316F) genomic DNA were used as templates. The LAMP primers were able to detect 10 fg of DNA per reaction volume as shown in Fig. 4. This detection limit of sensitivity was greater than that for conventional PCR assay, which can detect only 100 pg of DNA per reaction.

The sensitivity in terms of detection limit of the developed LAMP assay. M-1 kb DNA ladder, 1—negative control, 2–10 ng MAP DNA, 3–1000 pg MAP DNA, 4–100 pg MAP DNA, 5–10 pg MAP DNA, 6–1 pg MAP DNA, 7–0.1 pg MAP DNA, 8–0.01 pg MAP DNA, 9–0.001 pg or 1 fg MAP DNA, 10–0.1 fg MAP DNA, 11–0.01 fg MAP DNA, 12–0.001 fg MAP DNA
Specificity of LAMP
The LAMP-coupled LFD specificity test was performed using 10 ng each of M. avium subsp. paratuberculosis strain MAP 316F genomic DNA and other infectious bacterial species like M. bovis, M. tuberculosis, B. abortus, L. interrogan, Y. enterocolitica, S. typhimurium, L. monocytogens, and S. aureus. The data revealed no cross reactions from LAMP gel electrophoresis and LAMP-coupled LFD as shown in Fig. 5. The specificity of these results was analyzed by 1.5% agarose gel electrophoresis followed by the use of LAMP-coupled LFD.

Specificity test for LAMP assay and LFD using DNA templates of MAP and other bacterial species. 5(a) Agarose gel electrophoresis and by 5(b) LAMP-coupled LFD. Lane M-DNA ladder, 1–9 represent the assay conducted with the DNAs of MAP 316F, M. bovis, M. tuberculosis, B. abortus, L. interrogan, Y. enterocolitica, S. typhimurium, L. monocytogens, and S. aureus respectively in Fig. 5(A) and in Fig. 5(B)
Statistical analysis
Overall, 389 fecal samples were used for the evaluation of culture, PCR, and LAMP-coupled LFD assays for the detection of M. avium subsp. paratuberculosis. Out of 389 samples, 86 fecal samples were positive and 303 negative for MAP. But, 93 samples were found positive and 296 negative in PCR assay. From the 86 fecal samples with positive culture, PCR results revealed positive. Further, 7 fecal samples with negative culture were also found PCR positive.
A total of 95 fecal samples turned positive for LAMP-coupled LFD assay out of 389. All the cultural-positive and PCR-positive samples also displayed positive in LAMP assay analysis. Nine samples with negative culture turned positive in LAMP assay. The overall sensitivity and specificity of the LAMP-coupled LFD assay for the diagnosis of MAP were 100% and 97.02% respectively in comparison with the culture result as the gold standard. The sensitivity and specificity of PCR were 100% and 97.68% respectively. The PPV, NPV, and efficiencies of all these diagnostic assays for MAP are shown in Table 3.
Discussion
The culture of the Mycobacterium avium subspecies paratuberculosis for the diagnosis of Johne’s disease is not suitable for the routine diagnosis because of its slower growth conditions in media. The control of paratuberculosis is becoming difficult and interpretation of test results challenging due to the lack of proper diagnostic tests and lack of adequate knowledge about a variety of MAP species [30, 31]. The existing common methods for the diagnosis of Mycobacterium avium subspecies paratuberculosis include fecal culture, microscopic tests, and several molecular assays. The fecal culture for the diagnosis of MAP is gold standard technique with specificity of 100% but sensitivity of 50% [32]. The lower sensitivity is due to slow growth rate and higher culture contamination rate. Due to these drawbacks, the culture method is only preferred in reference laboratories. Therefore, this technique is not recommended for routine diagnostic purposes. All the culture-positive results should be further confirmed by PCR assay. Several PCR methods were used for the molecular detection of MAP. Though PCR has promising sensitivity and specificity, it requires highly equipped and sophisticated laboratory with well-trained technical persons. Because of these limitations with molecular assays, the PCR methods are used only in fully equipped laboratories with trained staff and cannot be performed at resource-limited point of care areas [33, 34]. Because of all these limitations and lack of field applicable diagnostic methods, there is a need to adopt highly specific, sensitive, rapid, simpler, and efficient diagnostic method that can be performed in both well-equipped laboratories and onsite at point of care areas for the detection of Johne’s disease.
In the present study, MAP was detected using LAMP-coupled LFD technique, in which the target primers were designed specifically for IS900 insertion sequence gene. Several molecular studies proposed IS900 as the most specific and highly sensitive target sequence for the detection of MAP [35]. The 5′ end of these loop primers LPF and LPB was tagged with biotin and fluorescein isothiocyanate (FITC) respectively [28]. The LAMP-coupled LFD takes an hour which is much shorter compared with other PCR assays. The limits of detection of this method are calculated as 10−7. On the other hand, the sensitivity of LAMP-coupled LFD in terms of limits of detection was 0.01 pg, i.e., 10 fg and the analysis specificity of LAMP-coupled LFD was 100% with other bacterial controls. Although there are many insertion sequences similar to IS900, with the use of LAMP method based on the region of IS900 specific to MAP, it is possible to specifically detect MAP. The specifically designed primer sets for MAP LAMP were able to detect 0.01 pg of DNA per reaction mixture. On the other hand, the limits of detection of LAMP-coupled LFD were greater than that for conventional PCR assay, which can detect 100 pg of DNA/tube as pointed out by Van der Giessen et al. (1992) [35].
Fecal samples were used for evaluation of all the three methods, i.e., culture, PCR, and LAMP-coupled LFD for the detection of Mycobacterium avium subsp. paratuberculosis. Out of 389, 86 fecal samples were positive and 303 negative for MAP using culture method. On the other hand, 93 samples were found positive and 296 resulted negative in PCR assay and 95 fecal samples turned positive for LAMP-coupled LFD. These observations showed a clinical sensitivity of 100% with LAMP-coupled LFD assay compared with the gold standard culture method. Low positive results in fecal cultures were due to the intermittent shedding of bacteria, and low detection sensitivity in conventional culture method [36]. Recent studies on MAP suggest high sensitivity for various IS900 molecular-based tests. The ineffectiveness of PCR could be due to the presence of few unknown inhibiting factors in feces, but the same may not be affecting the LAMP assay.
Recently, the use of LAMP in the detection of MAP was developed and similar LAMP results were reported [37]. In the present study, the LAMP-coupled LFD demonstrated a detection limit of 10 fg/μl of MAP DNA from the LAMP-amplified fecal samples and the developed assay successfully detected MAP not only in the bacterial cultures but also in the clinical fecal samples and the specificity was 100%. Similar results were reported by Safi et al. (2015) [37] and the LAMP assay was performed under isothermal conditions and no special apparatus is needed. They demonstrated that LAMP exhibits a 10-fold higher detection limit than conventional PCR by using genomic DNA samples from MAP and our results also corroborate the same. The endpoint assessment of LAMP end product can be monitored via addition of the colorimetric, fluorescent agents, turbid meter, and gel electrophoresis. Most of the LAMP end products were assessed using turbidity of the LAMP reaction due to the accumulation of pyrophosphate ion by-products in the final reaction solution. These pyrophosphate ions react with Mg2+ ions to form an insoluble end product magnesium pyrophosphate that can be detected by colorimetric assay. For example, by adding the hydroxyl naphthol blue (HNB) dye to a reaction, the color for a positive reaction changes from ultra violet to sky blue, whereas calcein and Syber Green results in a color change from orange to parrot green [29, 37]. Similar studies in the recent years [38] managed to detect T. gondii oocysts down to 25 oocysts/50 g in ready-to-eat baby lettuce [39]. Wang et al. [39] reported a multiplex LAMP-coupled LFD for the simultaneous detection of Staphylococcus aureus and Enterococcus faecalis using FITC and digoxigenin-modified primers in the LAMP assay. Similar studies from Nurul Najian et al. [40] revealed that a multiplex LAMP lateral flow test can be effectively used for the detection of pathogenic Leptospira spp. The LAMP assay used by Sange et al. [41] for the detection of MAP revealed a sensitivity detection limit of 10 fg/μl with an accuracy of 100% and no cross-reactivity with any of the other related Mycobacterium species. The assay showed 100% specificity which strengthens the results obtained in the present study. The LAMP-coupled LFD employed in the present study could detect the LAMP-amplified products following the completion of a MAP LAMP assay. This is the major difference of LAMP assay when compared with the previous studies. The 5′ ends of the LAMP end product were tagged with specific biotin and FITC. The LAMP end product with a biotin tag at one end binds to a streptavidin present in the colloidal gold conjugate matrix and the other end with FITC tag builds the complex with IgG fraction of anti-FITC specific antibody coated at test region as shown in Fig. 1. The results could be obtained by loading the LAMP end product into a LFD cassette without the need for extra incubation time and no other external instrumentation. The immune complexes diffuse further through the nitrocellulose membrane of the LFD and get entrapped resulting in the formation of a purple color bands at the test line “T” and control line “C” positions. The results can be read within 10 min. A positive reaction is indicated if a purple-colored line appears at both “T” and “C” positions on the LFD cassette. The unamplified LAMP sample does not react at the test position. This result is in accordance with other studies employed in LAMP lateral flow dipstick and in analysis of LAMP amplicons [28, 38,39,40]. Use of LAMP-coupled lateral flow device in the detection of LAMP amplicons is not only to eliminate the post amplification manipulations and use of ethidium bromide but also to strengthen the specificity of LAMP assay by hybridization of the specific probe to the LAMP amplicons.
Our results proved that the LAMP-coupled LFD for the detection of MAP is a simple, rapid and easy to perform point of care assay for the detection of MAP in livestock animal species such as bovine and in small ruminants like goat and sheep at resource-limited areas for point of care diagnosis.
Conclusion
In conclusion, the LAMP-coupled LFD developed in the present study provides a robust diagnostic method for the detection of MAP which is more advanced than the currently available molecular diagnostic assays. The LAMP-coupled LFD requires a minimal facility and the results are easily interpreted even by a non-technical personnel. The overall sensitivity and specificity of the LAMP-coupled LFD assay for the diagnosis of MAP were 100% and 97.02% respectively in comparison with the culture result as the gold standard. LAMP-coupled lateral flow assay not only improves sensitivity and rapidity but also reduces the time to diagnose M. avium subsp. paratuberculosis from livestock feces and can potentially be used to aid in characterization of the transmission of Johne’s disease in dairy farms. Our preliminary analyses suggest that this approach is of considerable use as an effective point of care diagnostic assay for the easy and rapid detection of MAP infections at resource-limited areas.
References
Collins DM, Radford AJ, De Lisle GW, Billman Jacobe H (1994) Diagnosis and epidemiology of bovine tuberculosis using molecular biological approaches. Vet Microbiol 40:83–94
Momotani E, Ozaki H, Hori M, Yamamoto S, Kuribayashi T, Eda S, Ikegami M (2012) Mycobacterium avium subsp. paratuberculosis lipophilic antigen causes Crohn’s disease-type necrotizing colitis in mice. Springerplus. 1:47
Naser SA, Ghobrial G, Romero C, Valentine JF (2004) Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn’s disease. Lancet. 364:1039–1044
Cetinkaya B, Egan K, Harbour DA, Morgan KL (1996) An abattoir based study of the prevalence of subclinical Johne’s disease in adult cattle in south west England. Epidemiol Infect 116:373–379
Chiodini RJ, Van Kruiningen HJ, Merkal RS (1984) Ruminant paratuberculosis (Johne’s disease): the current status and future prospects. Cornell Vet 74:218–262
De Lisle GW, Yates GF, Montgomery H (2003) The emergence of Mycobacterium paratuberculosis in farmed deer in New Zealand. A review of 619 cases. N Z Vet J 51:58–62
Singh SV, Singh AV, Singh R, Sharma S, Shukla N, Misra S, Singh PK, Sohal JS, Kumar H, Patil PK, Misra P, Sandhu KS (2008) Sero-prevalence of bovine Johne’s disease in buffaloes and cattle population of North India using indigenous ELISA kit based on native Mycobacterium avium subspecies paratuberculosis 'Bison type' genotype of goat origin. Comp Immunol Microbiol Infect Dis 31:419–433
Singh AV, Singh SV, Singh PK, Sohal JS, Mahour K (2010) Sero-surveillance of Mycobacterium avium subspecies paratuberculosis infection in domestic livestock in North India using indigenous absorbed ELISA test. J Adv Lab Res Biol 1:1–5
Singh SV, Singh PK, Singh AV, Sohal JS, Kumar N, Chaubey KK, Gupta S, Rawat KD, Kumar A, Bhatia AK, Srivastav AK, Dhama K (2014) Bio-load and bio-type profiles of Mycobacterium avium subspecies paratuberculosis infection in the domestic livestock population endemic for Johne’s disease: a survey of 28 years (1985-2013) in India. Transbound Emerg Dis 61(Suppl 1):43–55
Whittington RJ, Sergeant ESG (2001) Progress towards understanding the spread, detection and control of Mycobacterium avium subsp. paratuberculosis in animal populations. Aust Vet J 79:267–278
Ristow P, Marassi CD, Rodrigues ABF, Oelemann WMR, Rocha F, Santos ASO, Carvalho ECQ, Carvalho CB, Ferreira R, Fonseca LS, Lilenbaum W (2004) Investigation of Bovine Paratuberculosis on a Brazilian dairy herd through serological, bacteriological, Molecular & Anatomopathological tools. Anais XVII Congr. Latinoamericano e X Congr. Argentino de Microbiologia, Buenos Aires
Stabel JR (1997) An improved method for cultivation of Mycobacterium paratuberculosis from bovine faecal samples and comparison to three other methods. J Vet Diagn Investig 9:375–380
Merkal RS (1984) Paratuberculosis. In: Kubica GP, Wayne LG (eds) The Mycobacteria: a source book. Marcel. Dekker Inc, New York, NY, pp 1237–1249
Whipple DL, Calihan DR, Jarnagin JL (1991) Cultivation of Mycobacterium Paratuberculosis from bovine fecal specimens and a suggested standardized procedure. J Vet Diagn Investig 3:368–373
Hilbink F, West DM, de Lisle GW, Kittelberger R, Hosie BD, Hutton J, Cooke MM, Penrose M (1994) Comparison of a complement fixation test, a gel diffusion test and two absorbed and unabsorbed ELISAs for the diagnosis of paratuberculosis in sheep. Vet Microbiol 41:107–116
McDonald WL, Ridge SE, Hope AF, Condron RJ (1999) Evaluation of diagnostic tests for Johne’s disease in young cattle. Aust Vet J 77:113–119
Sockett DC, Conrad TA, Thomas CB, Collins MT (1992) Evaluation of four serological tests for bovine paratuberculosis. J Clin Microbiol 30:1134–1139
Stabel JR, Whitlock RH (2001) An evaluation of a modified interferon-gamma assay for the detection of paratuberculosis in dairy herds. Vet Immunol Immunopathol 79:69–81
Strommenger B, Stevenson K, Gerlach GF (2001) Isolation and diagnostic potential of ISMav2, a novel insertion sequence-like element from Mycobacterium avium subspecies paratuberculosis. FEMS Microbiol Lett 196:31–37
Rodriguez-Lazaro D, Lloyd J, Herrewegh A, Ikonomopoulos J, D’Agostino M, Pla M, Cook N (2004) A molecular beacon-based real-time NASBA assay for detection of Mycobacterium avium subsp. paratuberculosis in water and milk. FEMS Microbiol Lett 237:119–126
Singh PK, Singh SV, Kumar H, Sohal JS, Singh AV (2010) Diagnostic application of IS900 PCR using blood as a source sample for the detection of Mycobacterium avium subspecies Paratuberculosis in early and subclinical cases of caprine paratuberculosis. Vet Med Int 2010:748621
Collins DM, Stephens DM, De Lisle GW (1993) Comparison of polymerase chain reaction tests and fecal culture for detecting Mycobacterium Paratuberculosis in bovine feces. Vet Microbiol 36:289–299
Fang Y, Wu WH, Pepper JL, Larsen JL, Marras SA, Nelson EA, Epperson WB, Christopher Hennings J (2002) Comparison of real-time, quantitative PCR with molecular beacons to nested PCR and culture methods for detection of Mycobacterium avium subsp. paratuberculosis in bovine fecal samples. J Clin Microbiol 40:287–291
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T (2000) Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28:E63–E663
OIE Terrestrial manual (2014) Paratuberculosis (Johne’s disease). Chapter 2.1.15
Taddei S, Robbi C, Cesena C, Rossi I, Schiano E, Arrigoni N, Vicenzoni G, Cavirani S (2004) Detection of Mycobacterium avium subsp. paratuberculosis in bovine fecal samples: comparison of three polymerase chain reaction-based diagnostic tests with a conventional culture method. J Vet Diagn Investig 16:503–508
Vary PH, Andersen PR, Green E, Hermon Taylor J, Mcfadden JJ (1990) Use of highly specific DNA probes and the polymerase chain reaction to detect Mycobacterium Paratuberculosis in Johne's disease. J Clin Microbiol 28:933–937
Mallepaddi PC, Lai MY, Podha S, Ooi CH, Liew JW, Polavarapu R, Lau YL (2018) Development of loop mediated isothermal amplification based lateral flow device method for the detection of malaria. Am J Trop Med Hyg 99:704–708
Surabattula R, Vejandla MP, Mallepaddi PC, Faulstich K, Polavarapu R (2013) Simple, rapid, inexpensive platform for the diagnosis of malaria by loop mediated isothermal amplification (LAMP). Exp Parasitol 134:333–340
Sohal JS, Singh SV, Subodh S, Singh AV, Singh PK, Sheoran N, Sandhu K, Narayanasamy K, Maitra A (2007) Mycobacterium avium subspecies paratuberculosis diagnosis and strain typing present status and future developments. Indian J Exp Biol 45:843–852
Whittington RJ (2009) Factors affecting isolation and identification of Mycobacterium avium subsp. paratuberculosis from fecal and tissue samples in a liquid culture system. J Clin Microbiol 47:614–622
Whitlock RH, Wells SJ, Sweeney RW, Van Tiem J (2000) ELISA and fecal culture for Paratuberculosis (Johne’s disease): sensitivity and specificity of each method. Vet Microbiol 77:387–398
Castellanos E, de JL, Dominguez L, Aranaz A (2012) Progress in molecular typing of Mycobacterium avium subspecies paratuberculosis. Res Vet Sci 92:169–179
Pinedo PJ, Rae DO, Williams JE, Donovan GA, Melendez P, Buergelt CD (2008) Association among results of serum ELISA, fecal culture and nested PCR on milk, blood and faeces for the detection of paratuberculosis in dairy cows. Transbound Emerg Dis 55:125–133
Van Der Giessen JWB, Haring RM, Vauclare E, Eger A, Haagsma J, Van Der Zeijst BAM (1992) Evaluation of the abilities of three diagnostic tests based on the polymerase chain reaction to detect Mycobacterium paratuberculosis in cattle: application in a control program. J Clin Microbiol 30:1216–1219
Mobius P, Hotzel H, Rassbach A, Kohler H (2008) Comparison of 13 single-rounds and nested PCR assays targeting IS900, ISMav2, f57 and locus 255 for detection of Mycobacterium avium subsp. paratuberculosis. Vet Microbiol 25:324–333
Safi S, Heidarnejhad O, Mosavari N, Sakha M, Afshar D, Moazami L, Meshkat M, Keshavarz R, Taheri M, Pajoohi RA, Tadayon K, Reshadi S, Dashtipour S (2015) Comparative evaluation of LAMP and nested-PCR for the diagnosis of bovine paratuberculosis. Intern J Mycobacteriol 4:98–102
Lalle M, Possenti A, Dubey JP, Pozio E (2018) Loop-mediated isothermal amplification-lateral-flow dipstick (LAMP-LFD) to detect Toxoplasma gondii oocyst in ready-to-eat salad. Food Microbiol 70:137–142
Wang Y, Li H, Wang Y, Zhang L, Xu J, Ye C (2017) Loop-mediated isothermal amplification label-based gold nano particles lateral flow biosensor for detection of Enterococcus faecalis and Staphylococcus aureus. Front Microbiol 8:192
Nurul Najian AB, Engku Nur Syafirah EA, Ismail N, Mohamed M, Yean CY (2016) Development of multiplex loop mediated isothermal amplification (m-LAMP) label-based gold nano particles lateral flow dipstick biosensor for detection of pathogenic Leptospira. Anal Chim Acta 903:142–148
Sange MD, Becker A, Hassan AA, Bulte M, Ganter M, Siebert U, Abdulmawjood A (2019) Development and validation of a loop-mediated isothermal amplification assay - a rapid and sensitive detection tool for Mycobacterium avium subsp. paratuberculosis in small ruminants. J Appl Microbiol 127(1):47–58
Funding
This work was supported by the Department of Biotechnology, Government of India, New Delhi, India. The authors gratefully acknowledge the financial support under the CRS project on “Shielding the livestock from paratuberculosis using point of care diagnostics (PoCD)” with grant sanction order no: BIRAC/BT/CRS0226/CRS-11/17.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Ethical approval
All the experimental procedures were performed with the approval of the Institute Animal Ethics Committee, Genomix, Hyderabad, India. All applicable institutional guidelines for the care and use of animals were followed.
Additional information
Responsible Editor: Roxane Piazza
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
First Author: Ms. Rudrama Devi Punati.
Rights and permissions
About this article

Cite this article
Punati, R.D., Mallepaddi, P.C., Poonati, R. et al. Development and evaluation of LAMP-coupled lateral flow device for the detection of MAP in livestock at point of care resource-limited areas. Braz J Microbiol 50, 1105–1114 (2019). https://doi.org/10.1007/s42770-019-00116-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-019-00116-z