
Abstract
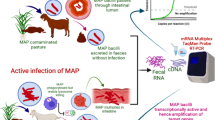
Mycobacterium avium subsp. paratuberculosis (MAP) is the etiological agent of Johne’s disease in ruminants and a probable pathogen of Crohn’s disease in humans. Accurate, cost-effective, and time-relevant diagnostics are the basis for efficient control programs. This study was conducted as an attempt to re-evaluate MAP detection improvement by coupling solid media enrichment to a more specific IS900 conventional PCR and a very specific F57/IC real-time PCR. In a spiking experiment, we investigated the improvement of molecular-based MAP detection in feces after a culture-based enrichment step into Herrold’s egg yolk media with mycobactin J (HEYM-MJ) for different time intervals, when compared to traditional culture. Detection limit of culture was 0.33 × 104 bacteria × g−1 (33 CFU g−1), while that of IS900 PCR when coupled with an enrichment step for 2, 4, and 6 weeks was 0.33 × 105 (0.33 × 103 CFU g−1), 0.33 × 104 (33 CFU g−1), and 33 (>3.3 CFU g−1) bacteria × g−1, respectively. Whereas the detection limits of F57/IC real-time PCR after the enrichment step for the same time intervals were 0.33 × 105 (0.33 × 103 CFU g−1), 0.33 × 103 (3.3 CFU g−1), and 33 (>3.3 CFU g−1) bacteria × g−1, respectively. Altogether, enrichment of bovine fecal samples into solid media increased the sensitivity of specific molecular detection of MAP using IS900 conventional PCR and duplex F57/IC real-time PCR and offers an expedited and accurate alternative for MAP detection in bovine feces. Validation of these results is further recommended using field bovine fecal samples.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Johne’s disease (JD) is an important disease in ruminants today. The disease is caused by Mycobacterium avium subsp. paratuberculosis (MAP), a slow-growing, acid-fast, and mycobactin-dependent microorganism which is widely distributed throughout the world (Dreier et al. 2006). The disease was reported in different tropical countries and is considered of great economic importance (Fernandez-Silva et al. 2014). The economic losses of the affected dairy farms in the USA, for example, were estimated to be about US$ 100 per cow mainly due to drop in milk production and premature culling (Losinger 2005). Many countries have adopted different programs for control and eradication of paratuberculosis. Highly accurate diagnostics are usually required for such programs to avoid false-positive reactions that would cost a lot in a culling strategy and false-negative ones that allow continuing of herd exposure by low shedders. Beside accuracy, cost-effectiveness and time relevance are other important aspects of control programs.
Culture is the gold standard of pathogen detection in living animals, which is more sensitive than indirect serologic tests and might allow earlier detection of infected cattle (Slana et al. 2008). Liquid culture systems provide faster and more sensitive MAP detection compared to traditional solid cultures (Whittington 2009); however, the high costs of automation and the radioactive hazards associated with those systems pushed researchers to develop inexpensive modified Middlebrook-based liquid cultures of comparable performance (Pozzato et al. 2011; Whittington et al. 2013).
In contrast to type I/III (sheep) MAP strains, most type II (cattle) strains grow well on Herrold’s egg yolk media with mycobactin J (HEYM-MJ) slope agars (de Juan et al. 2006). Those agars are relatively more cost-effective than Middlebrook-based media that require additional commercial Oleic Albumin Dextrose Catalase (OADC) enrichment and a specific mixture of antibiotics (PANTA PLUS). Some studies suggested that Middlebrook-based liquid cultures are more sensitive than HEYM-MJ in detecting MAP strains in bovine fecal samples (Pozzato et al. 2011). However, the sensitivity of HEYM-MJ in such studies seems to be underestimated, since molecular confirmation was only performed upon visible growth. Invisible MAP growth on solid agars was already proven by molecular methods in previous studies (Secott et al. 1999; Whittington 2009). However, in the light of recent advances in molecular methods, limited data are available about that approach.
The aim of the present work is to re-evaluate MAP detection improvement by coupling HEYM-MJ enrichment to specific conventional IS900 PCR (Bull et al. 2003) and F57/IC real-time PCR (Schönenbrücher et al. 2008). In a spiking experiment using negative bovine fecal samples, detection limits of each couple at different time points post-inoculation, compared to the traditional culture, were determined.
Materials and methods
Preparation of MAP isolate
A field bovine isolate of MAP (no. 16451) from the strain collection of the central veterinary diagnostic laboratory (Landesbetrieb Hessisches Landeslabor, Giessen, Germany), confirmed by the growth characteristics on HEYM-MJ, ZN staining, and molecular confirmation via both IS900 and F57 gene detection by PCRs, was prepared for spiking experiments as described previously (Kralik et al. 2012). Briefly, it was inoculated into Middlebrook 7H9 broth supplied with Middlebrook OADC and 2 μg/ml mycobactin J (MJ) (Sigma-Aldrich, Germany) and then cultured for up to 5 weeks at 37 °C. The bacteria were harvested by centrifugation at 7000×g for 2 min, resuspended, and vortexed with zirconia silica beads (0.1 mm diameter, Carl Roth GmbH+Co. KG, Karlsruhe, Germany) for 10 s to minimize clumping. Bacterial concentration was then measured via optical density at 550 nm according to Hughes et al. (2001). An optical density of 0.25 at 550 nm was equivalent to approximately 108 organisms/ml, which was confirmed by counting using Neubauer hemocytometer. Eight tenfold serial dilutions were prepared in Middlebrook 7H9 medium, and each dilution was divided into three aliquots. This MAP strain was also used as a positive control in PCR.
Determination of MAP CFU in serial dilutions by culture
In order to determine detection limits in the proposed spiking experiment, in terms of colony forming units (CFU), 100 μl of the prepared serial dilutions were inoculated into HEYM-MJ agars and incubated at 37 °C for 3 months.
Spiking experiments
Detection limits of the proposed enrichment approach as well as traditional culture were assessed by testing spiked negative bovine fecal samples with the prepared serial dilutions of MAP in three independent experiments. Those samples were obtained from a paratuberculosis-free herd and were additionally proven to be MAP free after being negative by culture, IS900 PCR, and F57 PCR. Two milliliters of each aliquot was mixed with 4 g of dry feces (to obtain nearly 6 g), mixed well, and divided into two equal sets in order to carry out traditional culture and enrichment approach separately. Non-spiked controls were included in both cases.
Traditional culture
Isolation of MAP from one set of spiked feces was performed as a modification of a technique used before by our group (Eisenberg et al. 2013). Three grams of the fecal samples was suspended in 30 ml freshly prepared 0.75 % hexadecyl pyridinium chloride (HPC) (Sigma, Germany) in a 50-ml plastic tube that was shaken at 200 rpm for 30 min then kept for 1 min at room temperature. The supernatant was transferred into new serial plastic tube without disturbing the fecal sediment. The sample was kept for 18 h at room temperature in the dark and centrifuged at 2.500×g for 15 min. The supernatant was descanted, and the pellet was resuspended in 1 ml HPC; then, 200 μl were inoculated into one tube in each experiment of HEYM-MJ, incubated at 37 °C, and checked for growth and/or contamination every week for up to 16 weeks.
Coupling culture as enrichment step with PCRs
The other set of spiked feces was processed exactly like the traditional culture protocol as an enrichment step with final inoculation of three tubes in each experiment in order to be able to assess the enrichment effect at different time intervals post-inoculation (one tube for each time point). The culture surfaces were harvested from each of the three tubes by gentle scraping and investigated by IS900 and F57/IC real-time PCRs at different time intervals (2, 4, and 6 weeks) of incubation at 37 °C.
Polymerase chain reactions
DNA extraction procedures
DNA was extracted from culture surface harvests collected from three independent experiments at three different time points (2, 4, and 6 weeks) after incubation (totally nine times) as described previously (Whittington et al. 1999). Harvesting of inoculums from HEYM-MJ tubes was carried out by gentle scraping of the whole surface of a tube followed by resuspension in 200 μl of distilled water. Absolute ethanol (500 μl) was added, and the tube was left for 2 min in room temperature, vortexed, and then centrifuged at 8×g for 10 min. The supernatant was transferred to a clean tube and centrifuged at 18,000×g for 5 min. The pellet was washed twice in 200 μl PBS, resuspended in 50 μl of distilled water, and boiled at 100 °C for 20 min.
Conventional IS900 PCR
The optimized 20 μl PCR mixture contained 0.6 μl of the primers TJ1 (GCT GAT CGC CTT GCT CAT) and TJ2 (CGG GAG TTT GGT AGC CAG TA) (10 pmol/μl) (Bull et al. 2003), 10.0 μl HotStarTaq Master Mix (Qiagen, Germany), and a 2 μl aliquot of the DNA sample. The reaction mixture was optimized according to instructions of HotStarTaq Master Mix manual. PCR tubes were then placed in a thermocycler (TC-3000, Applied Biosystem, USA) with a pre-heated lid. They were subjected to initial denaturation at 95 °C for 15 min and then to 35 cycles of denaturation at 94 °C for 1 min, annealing at 58 °C for 1 min, and extension at 72 °C for 1 min, and final extension was at 72 °C for 10 min. The PCR product was analyzed in a 2 % agarose gel at 100 V for 1 h employing a gel documentation system (UV star, Biometra, Germany) after being stained with ethidium bromide. Representative 357 bp banding pattern indicative for MAP was identified when compared to 100 bp DNA ladder.
Qualitative F57/IC duplex real-time PCR
Duplex F57/IC real-time PCR was developed as a modification of the method published by Schönenbrücher et al. (2008). The modified 20 μl PCR mixture for the duplex real-time PCR assay contained 1 μl of the primers F57-F (5′ TAC GAG CAC GCA GGC ATTC 3′)/F57-R (5′ CGG TCC AGT TCG CTG TCAT 3′) (10 pmol/ μl), 0.63 μl F57 TaqMan probe (5′ CY5- CCT GAC CAC CCT TC-BHQ1 3′) (10 pmol/μl), 125 copies of a heterologous internal amplification control (IAC) DNA based on enhanced green fluorescent protein (EGFP) gene (Hoffmann et al. 2006), 0.5 μl EGFP-Mix4 oligonucleotide mix, 10.0 μl 2X QuantiTect Multiplex Master Mix No Rox (QIAGEN, Germany), and 5 μl aliquot of the DNA sample. EGFP-Mix4 oligonucleotide mix contained 2.5 pmol/μl of each primer EGFP-11-F (5′ CAG CCA CAA CGT CTA TAT CAT G 3′) and EGFP-10-R (5′ CTT GTA CAG CTC GTC CAT GC 3′) and 1.25 pmol/μl EGFP-1HEX probe (5′ HEX-AGC ACC CAG TCC GCC CTG AGC A-BHQ1 3′).
The PCR tubes were then placed in the thermocycler (Mx3005P, Stratagen, Germany) with a pre-heated lid and subjected to initial denaturation at 95 °C for 15 min and then to 50 cycles of denaturation at 94 °C for 1 min and annealing and extension at 60 °C for 90 s. Analysis of the fluorescence was carried out using Mx3000P software (Stratagen, Germany). Amplification curves were carefully analyzed after checking raw fluorescence values, baseline settings, and the threshold as recommended by the manufacturer. Positive signals were recorded as Ct (threshold cycle) values.
Results
Determination of MAP CFU in serial dilutions by culture
According to results of serial dilutions plating, the CFU in stock solution is estimated to be 106 (Table 1).
Spiking experiments
The limit of detection (LOD) of each method was determined as the lowest dilution that was successfully detected by that method (visible growth in case of culture and a positive PCR signal in case of enrichment method) and was expressed both as colony forming units per gram and bacterial cells per gram feces (Table 1). According to the rate of successful detection of LOD in one, two, or three independent experiments, a new parameter, lowest detection probability (LDP), was calculated and expressed as 100, 66.7, or 33.3 %.
Culture detection limits
The detection limit of culture was 0.33 × 104 bacteria × g−1 (33 CFU g−1) with 100 % LDP (Table 2).
Detection limits of coupling culture with PCRs
The detection limits of IS900 PCR when coupled with an enrichment step on HEYM-MJ tubes increased from 0.33 × 105 bacteria × g−1 (0.33 × 103 CFU g−1) (100 % LDP) at 2 weeks to 0.33 × 104 bacteria × g−1 (33 CFU g−1) (66.7 % LDP) at 4 weeks and then 33 bacteria × g−1 (>3.3 CFU g−1) (66.7 % LDP) at 6 weeks post-inoculation.
The detection limit of F57/IC real-time PCR was the same as the IS900 PCR at 0.33 × 105 bacteria × g−1 (0.33 × 103 CFU g−1) (100 % LDP) at 2 weeks. However, the sensitivity of the real-time PCR increased relative to the IS900 PCR at 4 weeks at 0.33 × 103 bacteria × g−1 (3.3 CFU × g−1) (66.7 % LDP). At 6 weeks, the real-time PCR sensitivity was similar to the IS900 PCR at 33 bacteria × g−1 (>3.3 CFU × g−1) but with an LDP of 100 % (Table 2).
Discussion
In Johne’s disease control programs, there are many aspects of the used diagnostics to be considered. A perfect diagnostic assay would combine specificity, sensitivity, rapidity, and cost-effectiveness. In our study, we investigated the improved limit of detection, compared to traditional culture, of two specific PCRs (IS900 and F57) of agar-enriched MAP cells for different time points in spiked bovine feces. The detection limits of IS900 and F57 PCRs at 4 weeks (0.33 × 104 MAP g−1 (33 CFU g−1)), (0.33 × 103 MAP g−1 (3.3 CFU g−1)) and 6 weeks (33 MAP g−1 (>3.3 CFU g−1)) post-inoculation were usually more than traditional culture (0.33 × 104 MAP g−1 (0.33 × 102 CFU g−1)) (Table 2).
The low detection sensitivity of traditional culture detected in our study was in agreement with other studies (Clark et al. 2007; Norton et al. 2010) and is hypothesized to be caused by inhibitory effects from HPC solutions or by fecal inhibitors on MAP growth (Secott et al. 1999). The almost similar detection limits of the multicopy element IS900 and the single-copy element F57 PCRs may be due to the use of one MAP isolate in the spiking experiment. Sohal et al. (2009) have reported the possibility that some MAP strains are either low copy number or IS900 negative. Moreover, the real-time PCRs are known to be more sensitive than conventional ones.
Molecular detection of invisible MAP growth on solid agars has been already carried out (Secott et al. 1999). They combined a 6-week solid culture enrichment step (without visible growth) to IS900-based PCR in an attempt to increase sensitivity and decrease time-to-detection of MAP. However, Cousins et al. (1999), later, discovered the presence of IS900-like sequences in other mycobacteria, which makes the improved MAP detection in that study, in terms of specificity, somewhat unreliable. The sensitivity of the proposed method seems to be also overestimated, especially by observing the inability to detect MAP in 11 specimens that were only positive by culture 12 weeks post-inoculation.
Due to the high cost and safety problems associated with radioactivity of liquid culture systems together with the discontinuing of the supply of Bactec 12B medium in 2012, some research groups have developed new non-automated more cost-effective Middlebrook-based alternatives of comparable performance (Pozzato et al. 2011; Whittington et al. 2013). However, HEYM-MJ solid agars used in our work are considered relatively less expensive than those Middlebrook-based liquid media that require additional costly commercial ingredients such as OADC enrichment and a special antibiotic mixture (PANTA PLUS). Pozzato et al. (2011) have indicated that HEYM-MJ agars require a long incubation period until a visible growth is detectable and are much less sensitive than liquid cultures. However, the sensitivity of HEYM-MJ agars in their work seems to be underestimated since the definition of a positive result depended only on visible colonies confirmed molecularly, while invisible growth could have occurred without being detected.
As expected, the sensitivity of molecular detection of MAP DNA after 6 weeks of incubation into HEYM-MJ tubes in our study ((33 MAP g−1) (>3.3 CFU g−1)) was higher than traditional interpretation of visible growth in traditional cultures ((0.33 × 104 MAP g−1) (33 CFU g−1)) and had a comparable sensitivity to that reported by Pozzato et al. (2011) for the liquid cultures (1 CFU g−1) and that of Whittington et al. (2013) for cattle strains (24 MAP g−1). The contamination rate and the favored growth of irrelevant microorganism may inhibit the molecular detection of MAP DNA in liquid cultures, and double incubation-centrifugation processing is usually required before inoculation that add also to the overall cost of culture protocol (Whittington 2009). On the other side, using solid agars in our work makes it obligatory to detect the growth at only one time point after incubation, while two different successive readings could be obtained from liquid cultures as recommended by Pozzato et al. (2011).
In regard to direct molecular diagnosis, although saving time, the complexity of fecal matrix that contains a lot of PCR inhibitors affecting amplification efficiency had led to the discrepancy in data about detection sensitivity with some authors reporting sensitivity higher than culture (Soumya et al. 2009; Kralik et al. 2011) and others reporting the opposite (Taddei et al. 2004; Schönenbrücher et al. 2008). Anyway, the proposed approach in this study provides a more sensitive detection of MAP as it increases the bacterial number in solid agars before molecular detection.
Altogether, the previous results, collectively, indicate that detection of MAP DNA using specific duplex F57/IC real-time PCR or IS900 conventional PCR after an enrichment step on HEYM-MJ for 4 or 6 weeks offers rapid alternatives for accurate diagnosis of Johne’s disease with a usually increased sensitivity compared to traditional culture. Further validation of the results of our study using a large panel of field bovine fecal samples is recommended. It would be also interesting to compare our approach with liquid cultures under the same conditions in future studies.
References
Bull, T. J., McMinn, E. J., Sidi-Boumedine, K., Skull, A., Durkin, D., Neild, P., Rhodes, G., Pickup, R., Hermon-Taylor, J., 2003. Detection and verification of Mycobacterium avium subsp. paratuberculosis in fresh ileocolonic mucosal biopsy specimens from individuals with and without Crohn’s disease. Journal of Clinical Microbiology, 41(7), 2915–2923.
Clark, D. L., Koziczkowski, J. J., Radcliff, R. P., Clarson, R. A., Ellingson, J. L. E., 2007. Detection of Mycobacterium avium subsp. paratuberculosis: comparing fecal culture versus serum enzyme-linked immunosorbent assay and direct fecal polymerase chain reaction. Journal of Dairy Science, 91, 2620–2627.
Cousins, D. V., Whittington, R., Marsh, I., Masters, A., Evans, R. J., Kluver, P., 1999. Mycobacteria distinct from Mycobacterium avium subsp. paratuberculosis isolated from the feces of ruminants possess IS900-like sequences detectable IS900 polymerase chain reaction: implications for diagnosis. Molecular and Cellular Probes, 13, 431–442.
De Juan, L., Alvarez, J., Romero, B., Bezos, J., Castellanos, E., Aranaz, A., Mateos, A., Dominguez, L., 2006. Comparison of four different culture media for isolation and growth of type II and type I/III Mycobacterium avium subsp. paratuberculosis strains isolated from cattle and goats. Applied and Environmental Microbiology, 72 (9), 5927–5932.
Dreier, S., Khol, J. L., Stein, B., Fuchs, K., Guetler, S., Baumgartner, W., 2006. Serological, bacteriological and molecular biological survey of paratuberculosis (Johne’s disease) in Austrian cattle. Journal of Veterinary Medicine. B, Infectious Diseases and Veterinary Public Health, 53, 477–481.
Eisenberg, T., Wolter, W., Lenz, M., Schlez, K., Zschöck, M., 2013. Boot swabs to collect environmental samples from common locations in dairy herds for Mycobacterium avium ssp. paratuberculosis (MAP) detection. Journal of Dairy Research, 80(4), 485–489.
Fernandez-Silva, J. A., Correa-Valencia, N. M., Ramirez, N. F., 2014. Systematic review of the prevalence of paratuberculosis in cattle, sheep, and goats in Latin America and the Caribbean. Tropical Animal Health and Production, 46, 1321–1340.
Hoffmann, B., Depner, K., Schirrmeier, H., Beer, M., 2006. A universal heterologous internal control system for duplex realtime RT-PCR assays used in a detection system for pestiviruses. Journal of Virological Methods, 136(1–2), 200–209.
Hughes, V. M., Stevenson, K., Sharp, J. M., 2001. Improved preparation of high molecular weight DNA for pulsed-field gel electrophoresis from mycobacteria. Journal of Microbiological Methods, 44, 209–215.
Kralik, P., Slana, I., Kralova, A., Babak, V., Whitlock, R. H., Pavlik, I., 2011. Development of a predictive model for detection of Mycobacterium avium subsp. paratuberculosis in faeces by quantitative real time PCR. Veterinary Microbiology, 149 (1–2), 133–138.
Kralik, P., Beran, V., Pavlik, I., 2012. Enumeration of Mycobacterium avium subsp. paratuberculosis by quantitative realtime PCR, culture on solid media and optical densitometry. BMC Research Notes, 5, 114.
Losinger, W. C., 2005. Economic impact of reduced milk production associated with Johne’s disease on dairy operations in the USA. Journal of Dairy Research, 72(4), 425–432.
Norton, S., Johnson, W. O., Jones, G., Heuer, C., 2010. Evaluation of diagnostic tests for Johne’s disease (Mycobacterium avium subsp. paratuberculosis) in New Zealand dairy cows. Journal of Veterinary Diagnostic Investigation, 22, 341–351.
Pozzato, N., Gwozdz, J., Gastaldelli, M., Capello, K., Dal Ben, C., Stefani, E., 2011. Evaluation of a rapid and inexpensive liquid culture system for the detection of Mycobacterium avium subsp. Paratuberculosis in bovine feces. Journal of Microbiological Methods, 84(3), 413–417.
Schönenbrücher, H., Abdulmawjood, A., Failing, K., Bülte, M., 2008. New triplex realtime PCR assay for detection of Mycobacterium avium subsp. paratuberculosis in bovine feces. Applied and Environmental Microbiology, 74, 2751–2758.
Secott, T. E., Ohme, A. M., Barton, K. S., Wu, C. C., Rommel, F. A., 1999. Mycobacterium paratuberculosis detection in bovine feces is improved by coupling agar culture enrichment to an IS900-specific polymerase chain reaction assay. Journal of Veterinary Diagnostic Investigation, 11, 441–447.
Slana, I., Kralik, P., Kralova, A., Pavlik, I., 2008. On-farm spread of Mycobacterium avium subsp. paratuberculosis in raw milk studied by IS900 and F57 competitive real time quantitative PCR and culture examination. International Journal of Food Microbiology, 128, 250–257.
Sohal, J. S., Singh, S. V., Subodh, S., Sheoran, N., Narayanasamy, K., Singh, P. K., Singh, A. V., Maitra, A., 2009. Mycobacterium avium subsp. paratuberculosis diagnosis and geno-typing: genomic insights. Microbiological research, 164 (3), 330–337.
Soumya, M. P., Pillai, R. M., Antony, P. X., Mukhopadhyay, H. K., Rao, V. N., 2009. Comparison of faecal culture and IS900 PCR assay for the detection of Mycobacterium avium subsp. paratuberculosis in bovine faecal samples. Veterinary Research Communications, 33 (7), 781–791.
Taddei, S., Robbi, C., Cesena, C., Rossi, I., Schiano, E., Arrigoni, N., Vicenzoni, G., Cavirani, S., 2004. Detection of Mycobacterium avium subsp. paratuberculosis in bovine fecal samples: comparison of three polymerase chain reaction-based diagnostic tests with a conventional culture method. Journal of Veterinary Diagnostic Investigation, 16, 503–508.
Whittington, R. J., 2009. Factors affecting isolation and identification of Mycobacterium avium subsp. paratuberculosis from fecal and tissue samples in a liquid culture system. Journal of Clinical Microbiology, 47 (3), 614–622.
Whittington, R. J., Marsh, I., McAllister, S., Turner, M. J., Mashall, D. J., Fraser, C. A., 1999. Evaluation of modified BACTEC 12B radiometric medium and solid media for culture of Mycobacterium avium subsp. paratuberculosis from sheep. Journal of Clinical Microbiology, 37(4), 1077–1083.
Whittington, R. J., Whittington, A., Waldron, A., Begg, D. J., de Silva, K., Purdie, A. C., Plain, K. M., 2013. Development and validation of a liquid medium (M7H9C) for routine culture of Mycobacterium avium subsp. paratuberculosis to replace modified Bactec 12 medium. Journal of Clinical Microbiology, 51 (12), 3993–4000.
Acknowledgments
This work was financed by a grant from Hessian Ministry for the Environment, Climate Change, Agriculture and Consumer Protection (HMUKLV). It was also carried out in cooperation with the German Academic Exchange Service (DAAD).
Ethical standards
The manuscript does not contain clinical studies or patient data.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fawzy, A., Eisenberg, T., El-Sayed, A. et al. Improvement of sensitivity for Mycobacterium avium subsp. paratuberculosis (MAP) detection in bovine fecal samples by specific duplex F57/IC real-time and conventional IS900 PCRs after solid culture enrichment. Trop Anim Health Prod 47, 721–726 (2015). https://doi.org/10.1007/s11250-015-0784-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11250-015-0784-9