
Abstract
American foulbrood (AFB) caused by Paenibacillus larvae is the most destructive honeybee bacterial disease and its dissemination via commercial bee pollen is an important mechanism for the spread of this bacterium. Because Mexico imports bee pollen from several countries, we developed a tRNACys-PCR strategy and complemented that strategy with MALDI-TOF MS and amplicon-16S rRNA gene analysis to evaluate the presence of P. larvae in pollen samples. P. larvae was not detected when the tRNACys-PCR approach was applied to spore-forming bacterial colonies obtained from three different locations and this result was validated by bacterial identification via MALDI-TOF MS. The genera identified in the latter analysis were Bacillus (fourteen species) and Paenibacillus (six) species. However, amplicon-16S rRNA gene analysis for taxonomic composition revealed a low presence of Paenibacillaceae with 0.3 to 16.2% of relative abundance in the commercial pollen samples analyzed. Within this family, P. larvae accounted for 0.01% of the bacterial species present in one sample. Our results indicate that the tRNACys-PCR, combined with other molecular tools, will be a useful approach for identifying P. larvae in pollen samples and will assist in controlling the spread of the pathogen.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The economic importance of bees for the pollination of many high-value crops, fruits, and wild flowers has led to the commercial production of over a million bee colonies per year in Europe, North America, South America, and Asia [1,2,3]. However, these insects are attacked by numerous pathogens and parasites including viruses, bacteria, fungi, and metazoans [4]. Therefore, maintenance of healthy colonies is crucial not only for apiculture but also for agriculture and food safety, and to reduce the risk that the bees will act as vectors for the dissemination of various pathogens during pollination or through the use of pollen and honey as a foodstuff by humans and insects [3].
Bee pollen is a favorable microhabitat for many spore-forming bacteria, such as Paenibacillus larvae, which is one of the most important bacterial pathogens of honeybees [5, 6]. P. larvae is the causal agent of American foulbrood (AFB). The larval stage of honeybees becomes infected through the ingestion of P. larvae spores, which germinate in the midgut lumen. Vegetative bacteria proliferate within the midgut, breaching the epithelium, and invading the hemocele, ultimately leading to the death of the infected organisms [4, 7,8,9].
P. larvae detection plays an important role in the efficient control of dissemination of the pathogen during commercial production of hive products and many countries have implemented mandatory disease screening for imported materials [4, 10]. However, control measures for this bee pathogen are not implemented in all countries. In Mexico, for example, regulatory requirements are in general not well defined or do not include detailed instructions for their application. Although American foulbrood has been reported in Mexico since 1994 and a few officially acknowledged cases have been detected or isolated by the Mexican Agriculture Ministry [11, 12], there are no available data quantifying disease screening for P. larvae or the detection of the pathogen in imported hive products.
Although various sterilization methods have been used to eliminate microorganisms present in pollen, some bacteria survive these treatments because of their ability to form spores [13, 14]. In consequence, several approaches have used gamma radiation as a control method for eradication of spore-forming P. larvae [13]. Nevertheless, there is ample reason for concern about the possibility that preparations of commercial bee pollen may be contaminated with pathogenic bacteria such as P. larvae, despite the implementation of sterilization methods.
Several approaches have been used to identify P. larvae, including molecular typing methods based on 16S rRNA gene sequences [6, 15,16,17,18,19] and methods based on the repetitive-element polymerase chain reaction (PCR) (rep-PCR) using enterobacterial repetitive intergenic consensus (ERIC) primers [6]. Interestingly, ERIC genotypes can also be distinguished via matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) [20]. The utilization of multilocus sequence typing schemes (MLST) for P. larvae has allowed the establishment of global patterns in the population structure [21]. Another molecular approach, high-throughput sequencing of the 16S rRNA gene, allows recognition of the diversity of bacteria present in a sample, facilitating the identification of pathogens. This methodology has allowed the identification of P. larvae in worker honeybees, and thus has a high potential for use in the diagnosis of AFB [22].
While these molecular strategies for detecting P. larvae have their applications, their limitations and the fact that Mexico imports bee pollen from a number of different countries led us to consider alternative molecular approaches for the identification of P. larvae. In this study, we take advantage of the observation that some tRNAs, such as tRNACys, are encoded by a single copy gene in some Firmicutes [23, 24]. The position of these genes and their association with rRNA and tRNA operons or constitutive genes in the P. larvae genome recommends their use in molecular approaches for P. larvae detection. Thus, we report here the evaluation of the presence of P. larvae in commercial bee pollen, based on tRNACys-PCR amplification, complemented by MALDI-TOF MS and amplicon-16S rRNA gene analysis. Application of this approach should help to reduce the risk of the spread of P. larvae through commercial bee pollen.
Materials and methods
Bacterial strains
Bacterial strains obtained from the LMUAQ-collection (Laboratory of Microbiology, University Autonomous of Queretaro, Mexico) and primers used in this study are described in Table 1. Paenibacillus spp. strains were grown in MYPGP medium, consisting of 1.5% yeast extract (Difco Laboratories), 1.0% Mueller-Hinton broth (Difco Laboratories), 0.2% glucose, 0.3% K2HPO4, 0.1% sodium pyruvate, and 2.0% agar [25]. All other bacterial strains were grown in tryptic soy agar medium (Difco Laboratories; Detroit, MI. USA). P. polymyxa, P. odorifer, and P. peoriae were isolated in a screening of pollen samples and identified by analyzing 16S ribosomal deoxyribonucleic acid (rDNA) sequences and by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF MS) mass spectrometry using a MicroFlex LT mass spectrometer (Bruker Daltonics, Bremen, Germany).
tRNA sequences analysis
The tRNA sequences used in our analysis were aligned and extracted from complete Paenibacillus genomes using the tRNAscan-SE program [26]. From predicted genes, tRNA counts per iso-acceptor were obtained for each genomic sequence and cluster analyses were done using the software R, version 3.0.2.
Amplification conditions
PCR primers were designed to amplify the tRNACys region in the Paenibacillus larvae genome. Pan1 and Pan3 were designed to target the hydrolase-like gene, while Pan2 and Pan4 were designed to target the tRNACys gene. The hydrolase and tRNACys genes are located relatively close to each other in the P. larvae genome (See Fig. 3). The various primer combinations were predicted to yield products of the following sizes: Pan1-Pan2, 1268 bp; Pan3-Pan4, 907 bp; Pan1-Pan4, 1218 bp; and Pan2-Pan3, 957 bp. For PCR, a single colony (or in some cases, a pool of several colonies) of each bacterial strain tested (see Table 1) was suspended in 50 μL of distilled water and heated to 95 °C for 10 min and 1 μL of the bacterial suspension was used in a 30-μL PCR mix using Phusion high-fidelity DNA polymerase (Thermo Scientific; Waltham, MA, USA). PCR conditions were as follows: after a hot start (2 min, 95 °C), 35 cycles at 95 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min, and final elongation step at 72 °C for 5 min. PCR products (5 μL) were analyzed by electrophoresis in 1.0% agarose (Sigma-Aldrich; St. Louis, MO, USA) and visualized on a UV transilluminator.
Paenibacillus larvae detection
Commercial bee pollen samples destined for production facilities for bumblebee rearing in Mexico were obtained. We analyzed five samples from Europe, one sample from Mexico, and one sample from Chile. Each replicate of 1 g of bee pollen sample was homogenized in 10 mL of peptone and treated at 80 °C for 10 min to select spore-forming bacteria. Serial dilutions were then inoculated onto MYPGP agar medium and incubated at 37 °C during 48 h. Bacterial colonies obtained by this procedure (> 1000) were tested by tRNACys-PCR. Additionally, bacterial colonies were analyzed by MALDI-TOF MS using a MicroFlex LT mass spectrometer (Bruker Daltonics, Bremen, Germany) for species identification. The MALDI-TOF mass spectrometry method uses colonies directly after their treatment with 2 μL of MALDI matrix (a saturated solution of α-cyano-4-hydroxycinnamic acid in 50% acetonitrile and 2.5% trifluoroacetic acid). Spectra were analyzed by using the Bruker Biotyper 2.0 software and library (version 2.0, 3740 entries; Bruker Daltonics). The identification score criteria used were those recommended by the manufacturer: a score ≥ 2.0 indicated species-level identification, a score between 1.7 and 1.9 indicated identification at the genus level, and a score < 1.7 was interpreted as no identification.
For amplicon-16S rRNA gene analysis, DNA was extracted from the pollen samples themselves or from microbial enrichment cultures obtained from those samples. Enriched cultures were used to select spore-forming bacteria from bee pollen. To prepare enrichment cultures, 1-L flasks containing 200 mL of MYPGP medium were inoculated with 0.2 g of the pollen samples from Europe, Mexico, and Chile previously treated at 80 °C for 10 min as described above. Culture flasks were incubated at 37 °C for 48 h. DNA was extracted using a ZymoBIOMICS DNA Mini Kit (Zymo Research; Irvine, CA. USA).
Amplicons for 16S rRNA genes were sequenced using the Illumina MiSeq platform at Macrogen Inc. (Seoul, Republic of Korea). Amplicon sequencing data were downloaded in Fastq format and each library was analyzed with the FastQC program (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to validate library quality. All libraries were trimmed in Geneious v.9 with the Trim Ends tool and sequences with an error probability limit of 0.05 were removed in regions with average quality lower than Q30. Reads with sizes larger than 50 bases were merged in paired reads. For the generation of contigs, consensus sequences were used to search the 16S Microbial Database from NCBI (downloaded 05-2017) with Megablast using the following parameters: E-value 0.000001; word size 64; gap cost linear. The Megablast output was then used as the input for the classify sequences tool in Geneious v.9 to perform the taxonomic assignments and the descriptive analysis. The sequences merged in paired reads were used for analysis of bacterial diversity in environmental samples with the Naive Bayesian classifier and the Ribosomal Database Project (RDP) Classifier in the 16S biodiversity tool in Geneious v.9 [27]. DNA sequences of bacteria in bee pollen samples were deposited in GenBank under the accession numbers; SRP132301 (Chile sample), SRP132302 (Mexico sample), and SRP132303 (Europe samples).
DNA sequences from the amplicon-16S rRNA gene analyses were compared with sequences of 16S rRNA from sixteen species of Paenibacillus (31 strains), three species of Fontibacillus and one of Cohnella using the NCBI databases (http://www.ncbi.nlm.nih.gov/genbank/). The 16S rRNA sequence of Aneurinibacillus aneurinilyticus was designated as the outgroup taxon. Sequence data were analyzed using Bioedit v7.0.9.0 [28] and aligned using the Clustal W algorithm in Clustal X v2.1 [29]. Alignments were edited in Seaview v4.6 [30]. Identification of species was performed by phylogenetic inference methods of maximum likelihood using the online platform PhyML (http://www.atgc-montpellier.fr/phyml/) [31]. A best fit nucleotide substitution model was calculated using Smart Model Selection (SMS) [32]. Phylogenetic trees were constructed and edited using Geneious v.9 [27].
Results
tRNA sequences analysis
In the present study, we aimed to establish a reliable method to detect the presence of P. larvae in commercial bee pollen and thus, to avoid the dissemination of this pathogen in pollen samples. The initial approach employed a PCR strategy based on tRNA genes present in single copy in the genome of P. larvae. The organization of tRNA genes is expected to be relatively stable through evolutionary time, so that tRNA gene counts were predicted using tRNAscan-SE and compared with sequence data from 58 Paenibacillus species (Fig. 1).

Distribution and copy number of tRNA genes in genomes of Paenibacillus spp. The tRNA genes were mapped and extracted using the tRNAscan-SE program [26]. From predicted genes, tRNA counts per iso-acceptor were obtained and cluster analyses were done using the software R package, version 3.0.2. The color key refers to numbers of copies of the tRNA genes
It is apparent from Fig. 1 that some tRNA iso-acceptors displayed relatively low copy numbers consistently across all genomes analyzed, such as tRNACys, tRNAIle, and tRNATrp. These tRNAs are represented by a light green color in Fig. 1. Other tRNAs displayed relatively high copy numbers consistently across all genomes analyzed, e.g., tRNAArg, tRNAGly, tRNALeu, tRNAMet, tRNASer, and tRNAVal group. Particularly relevant to the present study, there were tRNAs decoding triplets for a given amino acid in P. larvae with a single copy gene, for example, tRNACys, tRNAIle, and tRNATrp. We decided to use the trnC gene (tRNACys) to develop a diagnostic method for P. larvae detection.
In P. larvae, the gene for tRNACys, is located within an operon consisting of 18 tRNA genes, 23S rDNA, and 16S rDNA genes (genome region plv: ERIC2_c30780–plv: ERIC2_c30970). For the development of our PCR strategy, we used the hydrolase-like gene located upstream from tRNACys (genome region plv: ERIC2_c30770). After the alignment of the selected sequences (from the hydrolase-like gene to tRNACys), DNA sequences were found to be specific for P. larvae and used for the design of specific primers (Fig. 2).

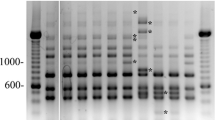
Organization of the hydrolase/tRNACys region from P. larvae (upper panel) and tRNACys-PCR products (lower panel). Primers were designed to amplify the hydrolase-tRNACys region in P. larvae. Lane 1, primer combination Pan1-Pan2 (1268 bp product), lane 2, Pan3-Pan4 (907 bp product), lane 3, Pan1-Pan4 (1218 bp product), and lane 4, Pan2-Pan3 (957 bp product). Lane M contains a Thermo Scientific GeneRuler 100 bp DNA ladder
tRNACys-PCR analyses revealed the presence of specific amplicons for P. larvae (Fig. 2, lanes 1, 2, 3, and 4) according to the predicted sizes. The amplicon of primers Pan1 and Pan2 in P. larvae was sequenced and it was found that the amplicon is unique to the P. larvae species (Fig. 3). To test further for the presence of the tRNACys-PCR region in other bacteria for cross-reactions, we tested both Gram-negative and Gram-positive bacteria (Table 1) using the PCR conditions used for P. larvae. No other bacterial strain generated any similar amplicon under the specified PCR conditions.

Phylogenetic dendrogram of amplicon Pan1-Pan2. The amplicon was sequenced and compared to the NCBI database using Blast to identify highly similar sequences with E-values < 10−40. Sequences were aligned and a distance tree was generated using the Blast pairwise alignments. The phylogenetic analysis placed the Pan1-Pan2 amplicon in the clade with sequences corresponding to Paenibacillus larvae subsp. larvae
Paenibacillus larvae detection
tRNACys-PCR analysis
To test for the presence of P. larvae in commercial bee pollen from Europe, Mexico, and Chile, we recovered more than one thousand colonies of spore-forming bacteria from MYPGP agar medium and analyzed them by tRNACys-PCR with the Pan1 and Pan2 primers. No specific PCR product was obtained from any of these bacterial colonies (data not shown). Thus, by this measure, P. larvae was absent from the commercial pollen samples we analyzed.
MALDI-TOF MS analysis
To validate these negative results, the bacterial colonies obtained from the commercial bee pollen were identified by MALDI-TOF MS (Table 2). This analysis identified members of the genus Bacillus as the most common species found in association with bee pollen from Europe, followed by Paenibacillus species. In the samples from Mexico, the genus Bacillus presented eight species and Paenibacillus just one species. The samples from Chile presented eight species for the genus Bacillus. Confirming the results of the PCR analyses, P. larvae was not observed among the bacterial species associated with the bee pollen samples. This result could mean either that P. larvae was absent from these samples or was present in amounts too low to be detected by these methods. Therefore, we decided to obtain massive amplicon-16S rRNA gene sequencing data using the pollen samples themselves and liquid cultures enriched to select spore-forming bacteria.
Bacterial taxonomic diversity in commercial bee pollen
The amplicon-16S rRNA gene analysis generated in Geneious with RPD yielded the following results. For each direct sequencing sample, we obtained 36,704 to 39,031 high-quality reads representing 6 to 7 operational taxonomic units (OTUs) at a 97% level of similarity. Members of the Firmicutes and Proteobacteria were found in all pollen samples. In all enriched samples, the Firmicutes were the most abundant phylum, with a count of 152,320 high-quality reads. Figure 4 shows the relative abundance of bacterial families belonging to the order Bacillales. The high-quality reads were dominated by Bacillaceae (3.7 to 46.3%), Planococcaceae (7.5 to 48.6%), and Thermoactinomycetaceae (9.9 to 88.1%). The most representative families observed in the enriched cultures from the order Bacillales were Bacillaceae (93 to 94%) and Paenibacillaceae (5 to 7%) (Fig. 4).

Relative abundance of different Families within the order Bacillales obtained from commercial bee pollen from three different regions, indicated along the bottom of the figure (five samples from Europe, indicated by 1 to 5, and one sample from Mexico and Chile). Data from enrichment culture samples are indicated by an asterisk (*). 16S rRNA genes were amplified and sequenced as described in “Materials and Methods”
Figure 5 shows the classification of genera from the family Paenibacillaceae. In general, 7 OTUs were present: Oxalophagus, Brevibacillus, Ammoniibacillus, Paenibacillus, Fontibacillus, Saccharibacillus, and Cohnella. The sequences generated from all the pollen samples for the genus Paenibacillus were extracted and compared with the NCBI database with Megablast, identifying the following species: P. motobuensis, P. aceti, P. vini, P. telluris, P. faecis, P. barengoltzii, P. xylanilyticus, P. typhae, P. pini, P. wynnii, P. odorifer, P. taiwanensis, P. apiarius, P. larvae, P. alvei, P. kribbensis, P. forsythiae, P. polymyxa, P. jamilae, and P. durus.

Relative abundance of different genera from the family Paenibacillaceae observed in commercial bee pollen from three different regions, indicated along the bottom of the figure (four samples from Europe, indicated with number, and one sample from Mexico and Chile). Data from enrichment culture samples are indicated by an asterisk (*). 16S rRNA genes were amplified and sequenced as described in “Materials and Methods”
Phylogenetic analyses were performed using 16S rRNA from members of the Paenibacillaceae family. The phylogenetic tree was robust with the following species: P. lautus, P. amylolyticus, P. chibensis, P. glucanolyticus, P. validus, P. peoriae, P. abekawaensis, Fontibacillus aquaticus, F. panacisegetis, F. phaseoli, Cohnella thermotolerans, P. xylanilyticus, Bacillus aporrhoeus, P. lautus, P. chibensis, and P. abekawaensis. A. aneurinilyticus was used as the outgroup. The analysis showed that the three contigs from the Europe5 sample were identified as Paenibacillus larvae. The best scoring PhyML tree is shown in Fig. 6 and it is rooted to A. aneurinilyticus.

Phylogenetic inference analysis of the three contigs related to P. larvae. Maximum likelihood inference was performed with the GTR nucleotide substitution model, estimated from an analysis of the 16S rRNA sequence alignment. Bootstrap support values from 100 replicates are shown at the nodes only for branches supported by more than 50%. The three contigs generated from the Europe5 sample in this study indicate a phylogenetic relationship with P. larvae. The tree was rooted to A. aneurinilyticus. The best nucleotide substitution model obtained was GTR. Maximum likelihood analysis (Akaike, − l nL = 3148.09696, Gamma shape parameter 0.304, freqA = 0.23493, freqC = 0.21230, freqG = 0.34438, freqT = 0.20839)
The phylogenetic analysis confirmed the result obtained from the Megablast database classification, where only one pollen sample (Europe5) contained a bacterial species identified as P. larvae, with a relative abundance of 0.01%.
Discussion
In order to identify P. larvae in commercial pollen samples and as a tool for controlling the spread of the pathogen, we developed the tRNACys-PCR approach, based on the amplification of the tRNACys gene region. The results presented indicate that consensus tRNACys gene, with a single copy gene in the P. larvae genome, is an excellent molecular marker associated with an upstream hydrolase gene and could be used as a molecular diagnostic. This property makes the method suitable to the identification of P. larvae and recommends tRNACys-PCR as a rapid and straightforward approach that complements identification methods that already exist. Moreover, tRNACys-PCR is technically less demanding and time-consuming than DNA sequencing when 16S rRNA-PCR is used. tRNACys-PCR could be the method of choice when large numbers of commercial bee pollen samples are to be screened or as a first step when identifying species based on genomic sequences.
Applying the tRNACys-PCR method to several samples of commercial bee pollen showed P. larvae to be absent from all colonies of spore-forming bacteria analyzed from those samples. This result was verified by MALDI-TOF MS analysis which also identified a number of bacterial species which were present as microbial contaminants of the pollen samples; some of those species were related to Paenibacillus. However, our amplicon-16S rRNA gene analysis revealed that Firmicutes and Proteobacteria were the most abundant phyla in pollen samples. Our amplicon-metagenomic analysis did reveal a low abundance (0.01%) of P. larvae in one pollen sample from Europe. Additionally, in enrichment culture samples, the Paenibacillaceae family was detected in lower abundance (5 to 7%). This observation establishes a lower limit for the tRNACys-PCR method for the detection of P. larvae. Recent evidence shows that detection limits of spores of P. larvae using a multiplex PCR revealed threshold values of 0.4 spore/mL in samples of honey, 0.8 spore/g in samples of bee pollen, and in samples of royal jelly 1.6 spores/g, these relevant results supporting the idea that PCR is useful to detect spore of P. larvae in bee pollen [33]. Due to regulatory restrictions, we were not able to test bee pollen samples from honeybee colonies with AFB symptoms in this study. We feel, nevertheless, that our method will be extremely useful in evaluating the potential for the transmission of AFB via P. larvae contamination of pollen samples.
Based on our results, in addition to microbiological and other molecular methods, amplicon 16S-rRNA gene analysis should be useful for detection of P. larvae on commercial bee pollen. A metagenomic approach has been used recently to analyze the occurrence of P. larvae in affected apiaries, in colonies with clinical signs of AFB infection, in asymptomatic colonies and control colonies. These studies revealed P. larvae relative abundances of 50%, 5%, and 3% respectively [22]. Thus, in the apiaries, this pathogenic bacterium could be transmitted by honeybees or as a component of the microbiota present on pollen. These results are relevant to the interpretation of our metagenomics data which showed a relatively low abundance of P. larvae in commercial bee pollen. In both contexts, there is a risk of spread of P. larvae during the acquisition of commercial bee pollen and therefore more emphasis should be placed on its detection.
Pollen microbiome studies have increased because of their recognized ecological, commercial, and medical importance. Due to its structure and nutritive composition, pollen provides a unique microhabitat for microorganisms and a reservoir for these microbes in insects that are fed with pollen. Therefore, the diet of an insect and its gut microbiome are tightly linked. Some studies of bacterial community profiles based on 16S rRNA gene sequences show that gut microbiota of Apis mellifera and some Bombus species consistently harbor a specific microbial species. Recent evidence shows that many endogenous bacteria have a mutualistic relationship with their insect host and play a key role in digestion, nutrient production, and pathogen protection [34,35,36]. It is apparent that additional study is necessary to characterize the microbiomes in commercial bee pollen, with the specific goal of identifying pathogenic species to avoid their spread.
Change history
23 April 2019
In the article mentioned above an author’s name was misspelled.
References
De Luca PA, Vallejo-Marín M (2013) What’s the “buzz” about? The ecology and evolutionary significance of buzz-pollination. Curr Opin Plant Biol 16(4):429–435
Roldán Serrano A, Guerra-Sanz JM (2006) Quality fruit improvement in sweet pepper culture by bumblebee pollination. Sci Hortic 110(2):160–166
Velthuis HHW, Doorn AV (2006) A century of advances in bumblebee domestication and the economic and environmental aspects of its commercialization for pollination. Apidologie 37(4):421–451
Graystock P, Yates K, Evison S, Darvill B, Goulson D, Hughes W (2013) The Trojan hives: pollinator pathogens, imported and distributed in bumblebee colonies. J Appl Ecol 50(5):1207–1215
Hansen H, Brødsgaard CJ (1999) American foulbrood: a review of its biology, diagnosis and control. Bee World 80(1):5–23
Genersch E, Forsgren E, Pentikäinen J et al (2006) Reclassification of Paenibacillus larvae subsp. pulvifaciens and Paenibacillus larvae subsp. larvae as Paenibacillus larvae without subspecies differentiation. Int J Syst Evol Microbiol 56(3):501–511
Yue D, Nordhoff M, Wieler LH, Genersch E (2008) Fluorescence in situ hybridization (FISH) analysis of the interactions between honeybee larvae and Paenibacillus larvae, the causative agent of American foulbrood of honeybees (Apis mellifera). Environ Microbiol 10(6):1612–1620
Hutchison EA, Miller DA, Angert ER (2014) Sporulation in bacteria: beyond the standard model. Microbiol Spectr 2(5)
Ebeling J, Knispel H, Hertlein G, Fünfhaus A, Genersch E (2016) Biology of Paenibacillus larvae, a deadly pathogen of honey bee larvae. Appl Microbiol Biotechnol 100(17):7387–7395
Bisson LF, Walker G, Ramakrishnan V et al (2016) The two faces of Lactobacillus kunkeei: wine spoilage agent and bee probiotic. Catalyst
Djordjevic S, Ho-Shon M, Hornitzky M (1994) DNA restriction endonuclease profiles and typing of geographically diverse isolates of Bacillus larvae. J Apic Res 33(2):95–103
Peréz de la Rosa D, Pérez de la Rosa JJ, Cossio-Bayugar R et al (2015) Complete genome sequence of Paenibacillus larvae MEX14, isolated from honey bee larvae from the Xochimilco quarter in Mexico City. Genome Announc 3(4):e00968–e00915
De Guzman ZM, Cervancia CR, Dimasuay KG et al (2011) Radiation inactivation of Paenibacillus larvae and sterilization of American foul brood (AFB) infected hives using co-60 gamma rays. Appl Radiat Isot 69(10):1374–1379
Graystock P, Jones JC, Pamminger T, Parkinson JF, Norman V, Blane EJ, Rothstein L, Wäckers F, Goulson D, Hughes WOH (2016) Hygienic food to reduce pathogen risk to bumblebees. J Invertebr Pathol 136:68–73
Alippi AM, Aguilar OM (1998) Unique DNA fingerprint patterns of Paenibacillus larvae subsp. larvae strains. J Apic Res 37(4):273–280
Govan VA, Allsopp MH, Davison S (1999) A PCR detection method for rapid identification of Paenibacillus larvae. J Appl Environ Microbiol 65(5):2243–2245
Dobbelaere W, Graaf DC, Peeters JE (2001) Development of a fast and reliable diagnostic method for American foulbrood disease (Paenibacillus larvae subsp. larvae) using a 16S rRNA gene based PCR. Apidologie 32(4):363–370
Genersch E, Otten C (2003) The use of repetitive element PCR fingerprinting (rep-PCR) for genetic subtyping of German field isolates of Paenibacillus larvae subsp. larvae. Apidologie 34(3):195–206
Antúnez K, D'Alessandro B, Piccini C, Corbella E, Zunino P (2004) Paenibacillus larvae larvae spores in honey samples from Uruguay: a nationwide survey. J Invertebr Pathol 86(1):56–58
Schäfer MO, Genersch E, Fünfhaus A, Poppinga L, Formella N, Bettin B, Karger A (2014) Rapid identification of differentially virulent genotypes of Paenibacillus larvae, the causative organism of American foulbrood of honey bees, by whole cell MALDI-TOF mass spectrometry. Vet Microbiol 170(3):291–297
Morrissey BJ, Helgason T, Poppinga L, Fünfhaus A, Genersch E, Budge GE (2015) Biogeography of Paenibacillus larvae, the causative agent of American foulbrood, using a new multilocus sequence typing scheme. Environ Microbiol 17(4):1414–1424
Erban T, Ledvinka O, Kamler M, Nesvorna M, Hortova B, Tyl J, Titera D, Markovic M, Hubert J (2017) Honeybee (Apis mellifera)-associated bacterial community affected by American foulbrood: detection of Paenibacillus larvae via microbiome analysis. Sci Rep 7:5084
Campos-Guillén J, Arvizu-Gómez JL, Jones GH, Olmedo-Alvarez G (2010) Characterization of tRNACys processing in a conditional Bacillus subtilis CCase mutant reveals the participation of RNase R in its quality control. Microbiology 156(7):2102–2111
Kanaya S, Yamada Y, Kudo Y, Ikemura T (1999) Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene 238(1):143–155
Dingman DW, Stahly DP (1983) Medium promoting sporulation of Bacillus larvae and metabolism of medium components. Appl Environ Microbiol 46(4):860–869
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25(5):955–964
Wang Q, Garrity G, Tiedje J, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948
Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27(2):221–224
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59(3):307–321
Lefort V, Longueville JE, Gascuel O (2017) SMS: smart model selection in PhyML. Mol Biol Evol 34(9):2422–2424
Guimarães-Cestaro L, Serrão JE, Message D, Martins MF, Teixeira EW (2016) Simultaneous detection of Nosema spp., Ascosphaera apis and Paenibacillus larvae in honey bee products. J Hymenopt Res 49:43–50
Koch H, Schmid-Hempel P (2012) Gut microbiota instead of host genotype drive the specificity in the interaction of a natural host-parasite system. Ecol Lett 15(10):1095–1103
Engel P, Moran NA (2013) The gut microbiota of insects – diversity in structure and function. FEMS Microbiol Rev 37(5):699–735
Cariveau DP, Elijah PJ, Koch H, Winfree R, Moran NA (2014) Variation in gut microbial communities and its association with pathogen infection in wild bumble bees (Bombus). ISME J 8(12):2369–2379
Funding
This study was partially financed by the Universidad Autónoma de Querétaro (PFCE 2018 and FOPER 2017).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible Editor: LUCY SELDIN
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Andrade, V.D.M., Flores, J.L.H., López, M.A.R. et al. Evaluation of the presence of Paenibacillus larvae in commercial bee pollen using PCR amplification of the gene for tRNACys. Braz J Microbiol 50, 471–480 (2019). https://doi.org/10.1007/s42770-019-00039-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-019-00039-9