
Abstract
Whitefly, Bemisia tabaci (Gennadius) is a cryptic species complex that infests various plant species and act as a vector for many plant viruses all over the world. To understand the B. tabaci cryptic species diversity in Bihar more comprehensively, interhost and interlocation surveys were conducted during the year 2020-2021. The genetic variability among 29 populations (16 interhost and 13 interlocation) was explored using nuclear markers viz. Random Amplified Polymorphic DNA (RAPD), Simple Sequence Repeats (SSR) along with mitochondrial marker, mtCOI. The dissimilarity coefficients of 29 populations clustered in a dendrogram with RAPD and SSR primers showed that interlocation populations were less diverged than the interhost populations. Bayesian phylogenetic analysis of 657 bp mtCOI sequences identified the presence of four cryptic species viz. Asia I, Asia II 1, Asia II 7 and China 3 belonging to two genetic groups (Asia and China) with high variations in interhost unlike in interlocation. Among the four cryptic species, Asia I was the most prevalent in Bihar, establishing 86.20% of all the sequenced samples and Asia II 7 and China 3 were reported for the first time in Bihar region. We believe that the information generated in this study is important from the perspective of identifying cryptic species diversity and to develop long term pest management strategies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Silverleaf whitefly, Bemisia tabaci (Gennadius), is a polyphagous pest that causes severe damage to more than 600 plant species, directly by feeding and excreting honeydew that causes sooty mould, and indirectly by transmitting more than 200 plant viruses (De Lima et al. 2021). In 1889 it was first discovered in cotton fields in Greece (Cock 1993), and on cotton in Pusa (Bihar, India) during 1905 (Misra and Lamba 1929). Due to its remarkable ability to shift, develop, adapt, and monopolise in new environments, it is regarded as one of the top 100 invasive alien species in the world (Ramos et al. 2018). Studies have also revealed the existence of B. tabaci cryptic species, which are morphologically ambiguous but does have distinctive biological, physiological, and genetic variations that have caused its prominence to shift its nomenclature from biotypes (Costa et al. 1991), to races (De Barro et al. 2005), to genetic groups (Boykin et al. 2007) and species (Tay et al. 2013).
To date, globally 46 cryptic species of B. tabaci with 4% genetic divergence have been identified under 11 genetic groups (Mugerwa et al. 2018; Lestari et al. 2021; Rehman et al. 2021). While 10 cryptic species (Asia I, Asia II 1, Asia II 5, Asia II 7, Asia II 8, Asia II 11, Asia IV, China 3, China 7 and Middle East Asia Minor 1 (MEAM 1)) have been reported from India. Understanding the genetic variation and distribution of B. tabaci cryptic species has become incredibly important in light of the current climate change, increase in the domestic transportation of agricultural products, and the intensive pest control techniques.
Therefore, in the current study we firstly conducted an interhost survey and learned that Brinjal host had the highest B. tabaci infestation, allowing us to conduct interlocation survey in the Brinjal crop of Bihar. Molecular markers viz. RAPD, SSR, mtCOI were used to study the genetic variability and also for characterizing the morphologically indistinguishable B. tabaci cryptic species (Mugerwa et al. 2018).
Materials and methods
Whitefly collection
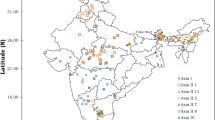
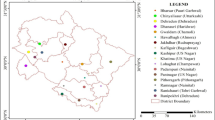
During the survey, adult whiteflies were randomly collected using a handheld aspirator from underside of the leaves from different hosts in the Pusa region of Bihar and also from brinjal host of different locations in Bihar (Kothia, Pusa, Dhrubgama, Mandai Dih, Mirapur, Madhurapur, Jhakra, Alipur Bihta, Faridpur, Charuipar and Dariapur); one location each from Telangana (Tadikonda) and Andhra Pradesh (Bapatla) states as check to explore the diversity (Table 1; Fig. 1). Adults taken from the same host plant at a sampling location were kept in the same tube, whilst those taken from different host plants were kept in separate tubes with 99.9% ethanol. Samples were identified for confirmation of B. tabaci using morphological keys (Calvert et al. 2001; Baig et al. 2015).

Survey during the year 2020-2021
Genomic DNA extraction
Genomic DNA was extracted based on the method given by Frohlich et al. (1999) with several modifications. Individual adult whitefly from each population (interhost and interlocation) were selected randomly and inserted into 1.5 μL eppendorf tube and a total of 100 μL of 2% cetyltrimethyl ammonium bromide (CTAB), 10 μL of 20% sodium dodecyl sulphate (SDS), 10 μL of 0.1% 2-mercaptaethanol and 10 μL of proteinase K were added and homogenized using the plastic rods. After being homogenized 400 μL of 2% CTAB was added and incubated at 65 °C for 45 min. After the incubation phenol: choloroform: isoamyl alcohol (25:24:1) was added and shaken gently for 10 sec and centrifuged for 10 min at 14000 rpm. A total of 400 μL of supernatant was taken, transferred into a new tube and added equal volume of chilled Isopropanol and put in a −20 °C for 1 hr. After that, it was centrifuged at 12000 rpm for 8 min. The supernatant was then discarded, added 400 μL of 70% chilled ethanol and centrifuged at 12000 rpm for 5 min. The supernatant was then discarded and dried at room temperature, added 50 μL of TE buffer and stored at −4 °C until used as template for PCR amplification.
DNA amplification
Thermocycler was executed with a 20 μl reaction mixture comprising of 2 μl template DNA (100 ng), 1.5 μl of forward and reverse primers, 8 μl of Taq mixture and 8.5 μl of nuclease free water. The following conditions were used to run the thermo cycler: initial denaturation at 95°C for 2.30 sec, 35 cycles of denaturation at 94°C for 45 sec, annealing at (Supplementary 1) for 30 sec and extension at 72°C for 5 min followed by final extension of 72°C for 10 min. The PCR results were then electrophoresed using a 2% agarose gel suspension in TAE buffer with 5 µL ethidium bromide at 100 V for 30 min and PCR products were visualized under UV light by Syngene gel documentation system.
Amplicon scoring and data analysis of RAPD and SSR primers
Gel images acquired with the Syngene gel documentation system were employed to score the data matrix (one and zero for the presence and absence of bands, respectively) with AlphaView SA software. The scored marker data matrix was further used to generate a dendrogram (Sneath and Sokal 1973) based on genetic dissimilarity in DARwin 6 software (Perrier et al. 2003). As a result, total amplified bands, number of polymorphic bands, Percentage of Polymorphic Bands (PPB), Polymorphism Information Content (PIC) (Anderson et al. 1993); Resolving Power (RP) (Prevost and Wilkinson 1999); Effective Multiplex Ratio (EMR) (Kumar et al. 2009); Marker Index (MI) (Powell et al. 1996) for each RAPD and SSR marker were computed to determine the informativeness of primers. The genetic variations among interhost and interlocation B. tabaci populations analyzed using RAPD and SSR primers (1st tier) were promoted to the 2nd tier (Universal primer: mtCOI analysis).
Sequencing and phylogenetic analysis
The PCR products (mtCOI) were purified using a QIA quick PCR purification kit (Qiagen Inc. Valencia, CA and USA) and then directly sequenced by ABI 3130XL genetic analyzer at Eurofins Genomics Bengaluru, Karnataka. The obtained sequences (657 bp) were aligned in Molecular Evolutionary Genetic Analysis Version X (MEGA X) using ClustalW to look for duplicates, gaps, indels and pseudogenes (Tamura et al. 2011). By performing maximum likelihood fits of 24 different nucleotide substitutions, the best model for phylogenetic tree construction was estimated with help of Bayesian Information Content (BIC) value (Felsenstein 1981). Further, estimates of evolutionary divergence between sequences and maximum composite likelihood of nucleotide substitution pattern were computed in MEGA X. To ensure proper reading frame, the sequences were translated into their corresponding amino acids using ExPASy translate (Gasteiger et al. 2003), then aligned with ClustalW to observe for conserved, semi-conserved, and fully conserved regions (Sievers et al. 2011). Neutrality tests, such as Fisher's (Fisher 1935) and Tajima's (Tajima 1989) were employed to determine whether the COI fitted to the neutrality requirements. The mtCOI generated sequences were found to be 100% identical to B. tabaci and were submitted in National Center for Biotechnology Information (NCBI) GenBank database (Altschul et al. 1997) and accession numbers were retrieved for all the populations (Table 1).
Results and discussion
RAPD analysis (1st tier)
RAPD polymorphism
The genetic variation of B. tabaci populations was explored with 11 RAPD primers, which were amplified with polymorphism ranging from 90 to 100% and produced 110 bands altogether with mean number of total bands and polymorphic bands per primer was 10.00 and 9.09 respectively. Higher PIC (0.81) in F2 and higher EMR (14.00), MI (10.42) and RP (8.07) in F12 primers revealed there greater informativeness and low EMR (7.00), RP (2.82) in OPA-15; low PIC (0.49), MI (4.45) in OPA-5 revealed there lesser informativeness in examining variation of B. tabaci populations (Table 2). Among all the primers, OPA-11 was identified as a potential genetic marker owing to its single monomorphic band with 90% polymorphism, because if there was no monomorphic band, then population would be deliberated as a distinct species (Maurya et al. 2020). Queiroz et al. (2017) observed more than 70% polymorphism in OPA-05 (70.0), OPA-10 (77.9), OPA-11 (73.8), OPA-13 (77.3), OPA-15 (70.8) and these observations are in line with our findings. Similarly, Hameed et al. (2012) and Hopkinson et al. (2020) employed RAPD primers to identify variations in B. tabaci populations.
UPGMA clustering and dendrogram
The data were clustered using the methodology of the Unweighted pair-group method with arithmetic averages (UPGMA) in a dendrogram, the dissimilarity coefficients of 29 populations. There were ten distinct clusters in the interlocation population, with Bapatla (cluster IV) having the highest dissimilarity coefficient of 46% and Madhurapur and Mirapur (cluster I) having the closest relationships with a dissimilarity coefficient of 30%. Interhost populations of Okra and Dolichos bean (cluster VI) are most closely related with a dissimilarity coefficient of 36% and Common jasmine (cluster X) populations shared higher dissimilarity coefficient of 57%. Cluster I of interlocation population viz. Madhurapur, Mandai Dih, Mirapur, Dhrubgama, Jhakra and Kothia (Northern Bihar) (Fig. 2) and cluster II of Charuipar, Faridpur and Dariapur (Southern Bihar) demonstrates that populations were differentiated based on their geographical locations. Cluster V of interhost populations belong to the Solanaceae family shared a lesser dissimilarity coefficient (37%). Results clearly demonstrated that interlocation populations were less diverged than the interhost populations, due to the fact that they were primarily collected from the same host viz. brinjal. Similar pattern studies were conducted with RAPD primers in B. tabaci (De Barro and Driver 1997); Myzus persicae (Zitoudi et al. 2001); Helicoverpa armigera (Lopes et al. 2017); Leucinodes orbonalis (Murali et al. 2021).

Dendrogram deduced from matrix of pair wise distances in RAPD analysis of B. tabaci interhost and interlocation using UPGMA
SSR analysis (1st tier)
SSR polymorphism
The genetic diversity of B. tabaci populations was investigated using nine SSR primers, which amplified with 100% polymorphic bands and generated a total of 60 bands, with 6.66 mean number of total bands and polymorphic bands. Higher PIC (0.878), EMR (9.0), MI (7.902) and RP (5.79) in Btls1-2 revealed there greater informativeness and low PIC (0.664) in Bta1; EMR (4.00) and MI (3.196) in Btls 1-6 and RP (1.238) in Bta 11 revealed less informativeness in examining the variability of B. tabaci populations (Table 3). Similar to our studies, De Barro et al. (2003), Simón et al. (2007), Gauthier et al. (2008) and Ben Abdelkrim et al. (2017) used these primers for examining genetic variability among B. tabaci populations. Contrarily, Valle et al. (2012) observed lowest polymorphism percentage in Bta11 primer, which showed 100% in our study.
UPGMA clustering and dendrogram
The UPGMA technique was used to cluster the data into a dendrogram using the dissimilarity coefficients of 29 populations. There were ten distinct clusters in the interlocation population, with Alipur Bihta, Dhrubgama, and Mirapur (cluster V) having the highest dissimilarity coefficient of 42% (cluster VIII) and being the most closely connected with a dissimilarity value of 30%. However, interhost populations of Cucumber and Potato (cluster I) are most closely related, sharing a dissimilarity coefficient of 38%, while, Common jasmine was the most divergent with higher dissimilarity coefficient of 56% (Fig. 3). According to Fakrudin et al. (2004), this augmented genetic variability might aid species in evolving and adapting to new environment more quickly. The lower dissimilarity coefficient observed between B. tabaci populations from interlocation can be enlightened by the certainty that they were all collected from the same host (Brinjal), whereas the higher dissimilarity coefficient observed among B. tabaci populations from interhost could be due to their collection from different hosts. Similar pattern of differentiation studies were conducted by Valle et al. (2012) and Reddy et al. (2022) using SSR primers in Bemisia tabaci and Helicoverpa armigera populations, respectively.

Dendrogram deduced from matrix of pair wise distances in SSR analysis of B. tabaci interhost and interlocation using UPGMA
mtCOI analysis (2nd tier)
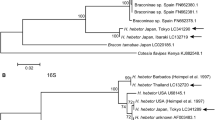
To construct a B. tabaci cryptic species phylogenetic tree, we first collected 29 mtCOI sequences from interhost and interlocation of Bihar along with Andhra Pradesh (Bapatla) and Telangana (Tadikonda) produced an amplicon of 657 bp mtCOI region (Supplementary 1). The phylogenetic tree was built based on Hasegawa-Kishino-Yano with Gamma distribution model (HKY+G) for 29 sequences along with 44 reference sequences and Bemisia atriplex, Bemisia afer and Trialeurodes vaparorium as out groups (Fig. 4) (Supplementary 2).

Phylogenetic tree inferred from 657 bp sequences of 29 mtCOI genes, 44 cryptic species of B. tabaci and three out groups
It was noted that four cryptic species viz. Asia I, Asia II 1, Asia II 7 and China 3 were found to cluster with 29 B. tabaci populations (Fig. 5). The sequences of interhost (Okra, Dolichos bean, Pointed gourd, Tomato, Potato, Mexican marigold, Cucumber, French bean, Indian jujube, Congress grass, White fig, Cluster fig and Common jasmine); interlocation (Pusa, Bapatla, Dariapur, Charuipar, Faridpur, Mirapur, Jhakra, Mandai Dih, Dhrubgama, Alipur Bihta, Madhurapur, Kothia) clustered with Asia I was found to be the major cryptic species accounting for 25 of the 29 (86.20%) B. tabaci populations. And therefore, Asia I had a greater potential to inflate and adapt in Bihar among four reported cryptic species. Similarly, Roopa et al. (2015) sequenced 71 B. tabaci samples and found Asia I cryptic species to be the most predominant accounting for 44 out of the 71 samples (61.97%).

Phylogenetic tree inferred from 657 bp sequences of 29 mtCOI genes, four cryptic species of B. tabaci and three out groups
Asia II is a genetically diverse group consisting of 13 sub cryptic species, Asia II (1–13) (Kanakala and Ghanim 2019) among them, only Asia II 1 and Asia II 7 were detected from the collected B. tabaci samples. Interestingly, Black nightshade and Chinese hibiscus collected from single location (Pusa region) clustered with Asia II 1 and Asia II 7, respectively shows that cryptic species add a impact on host plant selection. Roopa et al. (2015) discovered Asia II 7 cryptic species on Chinese hibiscus, which supports with our findings. Chowda-Reddy et al. (2012) stated that Asia II 7 was primarily found in Southern and Western India, which is in contrast to our results as Asia II 7 cryptic species was found in Bihar (Eastern India). Moreover, Barberton daisy grouped with China 3 (Fig. 4) has been discovered for the first time in Bihar, which borders West Bengal and is where Ellango et al. (2015) first discovered the China 3 cryptic species.
In contrast to the prior data given by Misra and Lamba (1929), Chowda-Reddy et al. (2012), Roopa et al. (2015) and Rangaswamy et al. (2019), it was evident that among the four cryptic species (Asia I, Asia II 1, Asia II 7, and China 3); Asia II 7 and China 3 were reported for the first time in the Bihar region. Given the wide-spread occurrence of numerous cryptic species, the abundance of suitable hosts, climate change, the overall domestic transportation of agricultural products, and intensive pest control strategies, B. tabaci has a high likelihood of acquiring an adaptive advantage in various parts of the nation.
Multiple sequence alignment
Multiple alignment of 29 B. tabaci nucleotide sequences revealed 305 completely conserved residues (Supplementary 5) and 105 Single Nucleotide Polymorphisms (SNPs). Furthermore, in multiple alignment of amino acid sequences 75 fully conserved residues, 46 conserved residues and 23 semi conserved residues were identified (Supplementary 6). This greater level of nucleotide similarity indicates that they evolved from a common origin. Similarly, Wosula et al. (2017); Kunz et al. (2019) found 7453 SNPs and 125 conserved amino acid residues, respectively with amplification of mtCOI region of B. tabaci.
Pair wise genetic distance
The pair wise genetic distance of B. tabaci populations ranged from 0.00 to 0.47 (Supplementary 4) with an over mean distance of 0.08. Similarly, Dinsdale et al. (2010) reported zero to 34% genetic distance among 198 B. tabaci populations.
Patterns of nucleotide substitution in mtCOI
As nucleotide composition is a crucial aspect of nucleic acids, the study revealed that thiamine (T) (43.37%) and guanine (G) (19.00%) had the highest and lowest numbers of nucleotide bases, respectively. Similarly, Roopa et al. (2015) observed highest nucleotide base in thiamine (43.10%) and lowest in guanine (13.22%) with B. tabaci mtCOI sequences. Furthermore, the base composition of mtCOI gene fragment was biased towards Adenine (A) and Thymine (T) with an overall 67.54% which was a universal feature of nucleotide diversity (Lynch 2008).
Neutrality tests
The negative Tajima's D = -0.726310 (Table 4) and Fisher’s exact test with P value <0.05 (Common jasmine, Congress grass, Chinese hibiscus, Black nightshade, Cucumber, Kothia, Madhurapur, Dhrubgama, Alipur Bihta, White fig, Chinese hibiscus and Tadikonda) (Supplementary 7) indicated excess of low frequency polymorphisms. Thus, both tests supported the neutral theory of evolution and these findings were supported by Tocko-Marabena et al. (2017) who concluded that B. tabaci was found to be significant with Tajima's D (-2.45317).
Conclusion
In the present study, we were able to confirm the existence of four B. tabaci cryptic species (Asia I, Asia II 1, Asia II 7, and China 3) in Bihar. In particular, Asia II 7 and China 3 were discovered for the first time in the Bihar region, while Asia I cryptic species dominated all interhost and interlocation populations. Overall, this study contributes to the characterization of various B. tabaci cryptic species for assessing ongoing changes in genetic diversity, evolutionary history, and potential spread that enable effective pest management while avoiding overuse of insecticides and lowering environmental pressure.
Data availability
All data relevant to the study are included in the article.
References
Anderson A, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME (1993) Optimizing parental selection for genetic linkage maps. Genome 36:181–186. https://doi.org/10.1139/g93-024
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. https://doi.org/10.1093/nar/25.17.3389
Baig MM, Dubey AK, Ramamurthy VV (2015) Biology and morphology of life stages of three species of whiteflies (Hemiptera: Aleyrodidae) from India. Pan-Pac Entomol 91:168–183. https://doi.org/10.3956/2015-91.2.168
Ben Abdelkrim A, Hattab T, Fakhfakh H, Belkadhi MS, Gorsane F (2017) A landscape genetic analysis of important agricultural pest species in Tunisia: The whitefly Bemisia tabaci. PLoS One 12:e0185724. https://doi.org/10.1371/journal.pone.0185724
Boykin LM, Shatters RG Jr, Rosell RC, McKenzie CL, Bagnall RA, De Barro P, Frohlich DR (2007) Global relationships of Bemisia tabaci (Hemiptera: Aleyrodidae) revealed using Bayesian analysis of mtCOI DNA sequences. Mol Phylogenet Evol 44:1306–1319. https://doi.org/10.1016/j.ympev.2007.04.020
Calvert LA, Cuervo M, Arroyave JA, Constantino LM, Bellotti A, Frohlich D (2001) Morphological and mitochondrial DNA marker analyses of whiteflies (Homoptera: Aleyrodidae) colonizing cassava and beans in Colombia. Ann Entomol Soc Am 94:512–519. https://doi.org/10.1603/0013-8746(2001)094[0512:MAMDMA]2.0.CO;2
Chowda-Reddy RV, Kirankumar M, Seal SE, Muniyappa V, Valand GB, Govindappa MR, Colvin J (2012) Bemisia tabaci phylogenetic groups in India and the relative transmission efficacy of Tomato leaf curl Bangalore virus by an indigenous and an exotic population. J Integr Agric 11:235–248. https://doi.org/10.1016/S2095-3119(12)60008-2
Cock MJW (1993) Bemisia tabaci: An update 1986-1992 on the cotton whitefly with an annotated bibliography. International Institute of Biological Control. Ascot, UK
Costa HS, Brown JK, Byrne DN (1991) Life history traits of the whitefly, Bemisia tabaci (Homoptera: Aleyrodidae) on six virus-infected or healthy plant species. Environ Entomol 20:1102–1107. https://doi.org/10.1093/ee/20.4.1102
De Barro PJ, Driver F (1997) Use of RAPD PCR to distinguish the B biotype from other biotypes of Bemisia tabaci (Gennadius)(Hemiptera: Aleyrodidae). Aust J Entomol 36:149–152. https://doi.org/10.1111/j.1440-6055.1997.tb01447.x
De Barro PJ, Scott KD, Graham GC, Lange CL, Schutze MK (2003) Isolation and characterization of microsatellite loci in Bemisia tabaci. Mol Ecol Notes 3:40–43. https://doi.org/10.1046/j.1471-8286.2003.00344.x
De Barro PJ, Trueman JWH, Frohlich DR (2005) Bemisia argentifolii is a race of Bemisia tabaci (Hemiptera: Aleyrodidae): the molecular genetic differentiation of Bemisia tabaci populations around the world. Bull Entomol Res 95:193–203. https://doi.org/10.1079/BER2004351
De Lima TCA, da Silva PF, Oliveira MD, Aires ES, Seabra S Jr, Lima GPP, de Oliveira RC (2021) Change in the physiological and biochemical aspects of tomato caused by infestation by Cryptic species of Bemisia tabaci MED and MEAM1. Insects 12:1105. https://doi.org/10.3390/insects12121105
Dinsdale A, Cook L, Riginos C, Buckley YM, De Barro P (2010) Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase I to identify species level genetic boundaries. Ann Entomol Soc Am 103:196–208. https://doi.org/10.1603/AN09061
Ellango R, Singh ST, Rana VS, Gayatri PN, Raina H, Chaubey R, Naveen NC, Mahmood R, Ramamurthy VV, Asokan R, Rajagopal R (2015) Distribution of Bemisia tabaci genetic groups in India. Environ Entomol 44:1258–1264. https://doi.org/10.1093/ee/nvv062
Fakrudin B, Prakash SH, Krishnareddy KB, Prasad PB, Patil BV, Kuruvinashetti MS (2004) Genetic variation of cotton bollworm, Helicoverpa armigera (Hübner) of South Indian cotton ecosystem using RAPD markers. Curr Sci 87:1654–1657
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376
Fisher RA (1935) The fiducial argument in statistical inference. Ann Eugen 6:391–398. https://doi.org/10.1111/j.1469-1809.1935.tb02120.x
Frohlich DR, Torres-Jerez I, Bedford ID, Markham PG, Brown JK (1999) A phylogeographical analysis of the Bemisia tabaci species complex based on mitochondrial DNA markers. Mol Ecol 8:1683–1691. https://doi.org/10.1046/j.1365-294x.1999.00754.x
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31:3784–3788. https://doi.org/10.1093/nar/gkg563
Gauthier N, Dalleau-Clouet C, Bouvret ME (2008) Twelve new polymorphic microsatellite loci and PCR multiplexing in the whitefly, Bemisia tabaci. Mol Ecol Resour 8:1004–1007. https://doi.org/10.1111/j.1755-0998.2008.02163.x
Hameed S, Hameed S, Sadia M, Malik SA (2012) Genetic diversity analysis of Bemisia tabaci populations in Pakistan using RAPD markers. Electron J Biotechnol 15:6. https://doi.org/10.2225/vol15-issue6-fulltext-5
Hopkinson J, Pumpa S, van Brunschot S, Fang C, Frese M, Tay WT, Walsh T (2020) Insecticide resistance status of Bemisia tabaci MEAM1 (Hemiptera: Aleyrodidae) in Australian cotton production valleys. Austral Entomol 59:202–214. https://doi.org/10.1111/aen.12436
Kanakala S, Ghanim M (2019) Global genetic diversity and geographical distribution of Bemisia tabaci and its bacterial endosymbionts. PLoS One 14:e0213946. https://doi.org/10.1371/journal.pone.0213946
Kumar M, Mishra GP, Singh R, Kumar J, Naik PK, Singh SB (2009) Correspondence of ISSR and RAPD markers for comparative analysis of genetic diversity among different apricot genotypes from cold arid deserts of trans-Himalayas. Physiol Mol Biol Plants 15:225–236. https://doi.org/10.1007/s12298-009-0026-6
Kunz D, Tay WT, Elfekih S, Gordon KHJ, De Barro PJ (2019) Take out the rubbish–removing NUMTs and pseudogenes from the Bemisia tabaci cryptic species mtCOI database. BioRxiv 724765. https://doi.org/10.1101/724765
Lestari SM, Hidayat P, Hidayat SH, Shim JK, Lee KY (2021) Bemisia tabaci in Java, Indonesia: genetic diversity and the relationship with secondary endosymbiotic bacteria. Symbiosis 83:317–333. https://doi.org/10.1007/s13199-021-00752-w
Lopes HM, Bastos CS, Boiteux LS, Foresti J, Suinaga FA (2017) A RAPD-PCR-based genetic diversity analysis of Helicoverpa armigera and H. zea populations in Brazil. Genet Mol Res 16:gmr16038757. https://doi.org/10.4238/gmr16038757
Lynch M (2008) Estimation of nucleotide diversity, disequilibrium coefficients, and mutation rates from high-coverage genome-sequencing projects. Mol Biol Evol 25:2409–2419. https://doi.org/10.1093/molbev/msn185
Maurya RP, Brijwal L, Patwal H, Suyal P (2020) Genetic variability in predatory stink bug Andrallus spinidens (F.) from North-west Himalaya. Indian J Entomol 3:523–527
Misra CS, Lamba KS (1929) The cotton whitefly (Bemisia gossypiperda n.sp.). Bulletin of the Agricultural Research Institute of Pusa, Bihar 196:1–7
Mugerwa H, Seal S, Wang HL, Patel MV, Kabaalu R, Omongo CA, Colvin J (2018) African ancestry of New World, Bemisia tabaci-whitefly species. Sci Rep 8:1–11. https://doi.org/10.1038/s41598-018-20956-3
Murali P, Hilda K, Ramakrishnan M, Ganesh A, Bhuvaragavan S, Janarthanan S (2021) Molecular genotypic diversity of populations of brinjal shoot and fruit borer, Leucinodes orbonalis and development of SCAR marker for pesticide resistance. Mol Biol Rep 48:7787–7800
Perrier X, Flori A, Bonnot F (2003) Data Analysis Methods. Hamon P, Seguin PM, Perrier X, Glaszmann JC, [Eds] Genetic Diversity of Cultivated Tropical Plants. Science Publishers, Enfield, pp 43–76
Powell W, Morgante M, Andre C, Hanafey M, Vogel J, Tingey S, Rafalski A (1996) The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol Breed 2:225–238
Prevost A, Wilkinson MJ (1999) A new system of comparing PCR primers applied to ISSR fingerprinting of Potato cultivars. Theor Appl Genet 98:107–112. https://doi.org/10.1007/s001220051046
Queiroz PR, Lima LH, Sujii ER, Monnerat RG (2017) Description of the molecular profiles of Bemisia tabaci (Hemiptera: Aleyrodidae) in different crops and locations in Brazil. J Entomol Nematol 9:36–45. https://doi.org/10.5897/JEN2017.0170
Ramos RS, Kumar L, Shabani F, Picanço MC (2018) Mapping global risk levels of Bemisia tabaci in areas of suitability for open field tomato cultivation under current and future climates. PLoS One 13:e0198925. https://doi.org/10.1371/journal.pone.0198925
Rangaswamy KT, Nagaraju N, Prameela HA, Achari R, Jagadish KS, Govin K, Karkale B, Shankarappa KS, Ramegowda GK, Rathod TS, Babu DR (2019) First report of the invasive MEAM1 whitefly [Bemisia tabaci (Gennadius)], vector of tomato leaf curl viruses in a major tomato-growing region of Maharashtra, India. In: VI International Symposium on Tomato Diseases: Managing Tomato Diseases in the Face of Globalization and Climate Change, pp 113–120. https://doi.org/10.17660/ActaHortic.2021.1316.16
Reddy MSS, Agnihotri M, Jaiswal JP, Subbanna AR, Karthik S (2022) Intra species diversity of Helicoverpa armigera (Hübner)(Lepidoptera: Noctuidae) in relation to geography and host plants affiliation in Uttarakhand Himalayan population, India. Phytoparasitica 50:359–373. https://doi.org/10.1007/s12600-021-00972-2
Rehman M, Chakraborty P, Tanti B, Mandal B, Ghosh A (2021) Occurrence of a new cryptic species of Bemisia tabaci (Hemiptera: Aleyrodidae): an updated record of cryptic diversity in India. Phytoparasitica 49:869–882. https://doi.org/10.1007/s12600-021-00909-9
Roopa HK, Asokan R, Rebijith KB, Hande RH, Mahmood R, Kumar NK (2015) Prevalence of a new genetic group, MEAM-K, of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae) in Florida Entomol Karnataka, India, as evident from mtCOI sequences. Fla Entomol 98:1062–1071
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. https://doi.org/10.1038/msb.2011.75
Simón B, Cenis JL, La De, Rúa P (2007) Distribution patterns of the Q and B biotypes of Bemisia tabaci in the Mediterranean basin based on microsatellite variation. Entomol Exp Appl 124:327–336. https://doi.org/10.1111/j.1570-7458.2007.00586.x
Sneath PH, Sokal RR (1973) Numerical taxonomy. The principles and practice of numerical classification, p 573
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. – Genetics 123:585–595. https://doi.org/10.1093/genetics/123.3.585
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. https://doi.org/10.1093/molbev/msr121
Tay WT, Evans GA, Boykin LM, De Barro PJ (2013) Demonstrating the value of voucher specimens to help resolve modern whitefly taxonomy. In: Abstract 1st International Whitefly Symposium, p 71
Tocko-Marabena BK, Silla S, Simiand C, Zinga I, Legg J, Reynaud B, Delatte H (2017) Genetic diversity of Bemisia tabaci species colonizing cassava in Central African Republic characterized by analysis of cytochrome c oxidase subunit I. PloS One 12:e0182749. https://doi.org/10.1371/journal.pone.0182749
Valle GE, Lourenção AL, Zucchi MI, Pinheiro JB (2012) Low polymorphism revealed in new microsatellite markers for Bemisia tabaci (Hemiptera: Aleyrodidae). Genet Mol Res 11:3899–3903. https://doi.org/10.4238/2012.November.12.7
Wosula EN, Chen W, Fei Z, Legg JP (2017) Unravelling the genetic diversity among Cassava Bemisia tabaci whiteflies using NextRAD sequencing. Gen Biol Evol 9:2958–2973. https://doi.org/10.1093/gbe/evx219
Zitoudi K, Margaritopoulos JT, Mamuris Z, Tsitsipis JA (2001) Genetic variation inMyzus persicae populations associated with host-plant and life cycle category. Entomol Exp Appl 99:303–311. https://doi.org/10.1046/j.1570-7458.2001.00829.x
Acknowledgement
The authors are grateful to Department of Entomology, Department of Genetics and Plant Breeding, and Department of Biotechnology, Dr. Rajendra Prasad Central Agricultural University for their unwavering support throughout the research. This research is part of Gummudala Yashaswini M.Sc dissertation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yashaswini, G., Karthik, S., Reddy, B.D. et al. Genotyping of Bemisia tabaci (Hemiptera: Aleyrodidae) affirmed a new record of Asia II 7, China 3 and dominance of Asia I cryptic species in Bihar, India. Int J Trop Insect Sci 43, 1123–1133 (2023). https://doi.org/10.1007/s42690-023-01004-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42690-023-01004-8